

Abstract
Background
ATR, which signals DNA damage to S/G2 cell cycle checkpoints and for repair, is an attractive target in cancer therapy. ATR inhibitors are being developed and a pharmacodynamic assay is needed to support clinical studies.
Methods
Phosphorylation of ATR targets, Chk1 and H2AX, was evaluated in MCF7 and K562 cells, human volunteer PBMCs and whole blood by Western blot, immunofluorescence microscopy and flow cytometry after DNA damage. The effect of cell cycle phase, ATR knockdown and inhibition on these phosphorylation events was determined.
Results
Hydroxyurea, UV and 4NQO induced Chk1 and H2AX phosphorylation in MCF7 and K562 cells. UV/4NQO activation of ATR was detectable in non‐cycling cells. Chk1 phosphorylation was reduced by ATR knockdown and reflects ATR activity for 3 h, H2AX phosphorylation after UV/4NQO is ATR‐dependent for 1 h but increasingly ATM and DNA‐PK‐dependent at later time points. In isolated PBMCs both phospho‐targets were detectable after UV/4NQO but in PBMCs from whole blood treated with 4NQO only H2AX was detectable.
Conclusion
PhosphoChk1 and H2AX are useful biomarkers for ATR inhibition using a variety of immuno‐detection methods, but timing may be critical. Importantly, ATR activity is detectable in non‐cycling PBMCs allowing them to be used as a surrogate tissue for biomarker measurement. In PBMCs from whole blood treated with 4NQO phosphoH2AX was the most useful biomarker of ATR activity and a clinically viable pharmacodynamic assay for ATR inhibitors has been developed.
Keywords: ATR, pChk1, γH2AX, PBMC, UV, 4NQO, Biomarker
Highlights
Chk1 and H2AX phosphorylation shortly after UV or 4NQO exposure is ATR dependent.
Phospho Chk1 and H2AX are detectable in PBMCs after 4NQO exposure.
A clinically viable pharmacodynamic biomarker of ATR inhibition has been developed.
1. Introduction
ATR (ataxia telangiectasia and Rad3‐related) is a DNA damage signalling kinase activated by single stranded (ss)–double stranded (ds) DNA junctions generated by DNA replication stress, nucleotide excision repair (NER) intermediates or from resection of a DNA double strand break (DSB) (Smith et al., 2010; Chen et al., 2012). Activated ATR phosphorylates Chk1 and other proteins, resulting in S or G2 cell cycle arrest and suppression of mitotic entry. Most cancer cells are defective in the G1 checkpoint and therefore rely on the S and G2 checkpoints (Massague, 2004), making inhibitors of the S/G2 checkpoint signaling pathway attractive agents for cancer‐specific sensitization to DNA‐damaging chemotherapy and radiotherapy.
Proof of principle data that ATR inactivation leads to abrogation of DNA damage‐induced G2/M arrest, and sensitization to multiple classes of DNA‐damaging agents was generated using genetic approaches, such as knockdown or overexpressing a kinase‐dead mutant ATR (ATR‐kd) (Nghiem et al., 2001; Cliby et al., 2002). A number of highly potent and selective inhibitors of ATR have recently been identified: NU6027, VE‐821, ETP46464, VE‐822, AZ‐20 and AZD‐6738 (Peasland et al., 2011; Reaper et al., 2011; Toledo et al., 2011; Fokas et al., 2012; Foote et al., 2013; Jones et al., 2013). These inhibitors sensitise human cancer cell lines to a variety of DNA‐damaging chemotherapy and radiotherapy (Pires et al., 2012; Prevo et al., 2012; Huntoon et al., 2013; Sultana et al., 2013) and some have shown single agent cytotoxic activity in certain cancer cell lines (Foote et al., 2013). Two of these agents, VE‐822 and AZD‐6738, have recently entered clinical trial (EudraCT number 2012‐003126‐25 and Clinical Identifier NCT01955668 respectively), both having demonstrated pre‐clinical in vivo activity either in combination with damaging agents or as a single agent (Fokas et al., 2012; Jacq et al., 2012; Jones et al., 2013).
There is much debate as to whether maximum tolerated dose (MTD) or optimum biological dose (OBD) should be used to guide clinical trials of molecularly targeted drugs. For ATR inhibitors OBD may be a better guide. Therefore, a pharmacodynamic (PD) biomarker is required to provide molecular confirmation of target inhibition and to direct dose escalation. Chk1 is a primary ATR target and can be phosphorylated at Ser317 and Ser345 (Zhao and Piwnica‐Worms, 2001). Chk1 Ser317 has been shown to be phosphorylated by kinases other than ATR, however the Ser345 site appeared to be solely phosphorylated by ATR (Peasland et al., 2011). ATR also phosphorylates other proteins including H2AX at Ser139 (γH2AX), which is a commonly used biomarker for DSB and stalled replication forks (Redon et al., 2010). There is ambiguity regarding the role of ATR in H2AX phosphorylation. Exposure to hydroxyurea (HU) or UV leads to ATR‐dependent γH2AX formation (Ward and Chen, 2001; Ward et al., 2003). Consistent with this, VE‐821 (10 μM) inhibited γH2AX shortly after treatment with HU and inhibition was ATR‐specific as the ATM and DNA‐PK inhibitors KU55933 and NU7026 had no impact on phosphorylation (Reaper et al., 2011). However, knockdown of Chk1 or ATR resulted in an increase in H2AX phosphorylation in response to replication stress (Gagou et al., 2010), which was thought to be due to an increase in ATM and DNA‐PK activation (Chanoux et al., 2009).
Tumour is the obvious tissue in which to measure drug effect, but collecting tumour biopsies is often risky for the patient, therefore normal tissues need to be identified that can act as a surrogate. Peripheral blood mononuclear cells (PBMCs) are easy to obtain and have been used as sources for PD biomarkers in clinical studies. In this report, we tested the potential of pChk1Ser345 and γH2AX as PD biomarkers for ATR inhibition in replicating cultured human cancer cells and importantly, for the first time, in non‐replicating PBMCs using a variety of immunological techniques (western blotting, flow cytometry and immunofluorescence microscopy). With depletion of ATR by siRNA/shRNA or using the selective ATR inhibitors VE‐821 and VE‐822, we found that pChk1Ser345 was ATR‐dependent in MCF7 and K562 cells, and could be detected in PBMCs after exposure to UV or the UV mimetic drug, 4NQO. UV‐ and 4NQO‐induced H2AX phosphorylation was ATR‐dependent in K562 cells and human peripheral blood mononuclear cells (PBMCs) and in MCF7 cells at early time points after DNA damage. Moreover, 4NQO treatment of whole blood induced H2AX phosphorylation in PBMCs which was inhibited by VE‐822.
2. Materials & methods
2.1. Chemicals and reagents
All chemicals and reagents were obtained from Sigma (Poole, UK) unless otherwise stated. VE‐821 and VE‐822 (Vertex Pharmaceuticals (Europe) Ltd, Oxford, UK), 4‐nitroquinoline‐1‐oxide (4NQO), the ATM inhibitor, KU55933 and the DNA‐PK inhibitor, NU7441 (kind gift from Celine Cano, School of Chemistry, Newcastle University, UK), were dissolved in DMSO and stored at −20 °C. Both gemcitabine and HU were dissolved in water and stored at −20 °C.
2.2. Cell lines and surrogate normal human tissue
K562 (human chronic myelogenous leukaemia) and MCF7 (human breast cancer) cell lines were obtained from the American Type Culture Collection (ATCC, Manassus, VA, USA), and were cultured in RPMI 1640 medium containing l‐glutamine supplemented with 10% foetal bovine serum. GM847kd cells containing a doxycycline‐inducible, dominant negative ATR kinase‐dead mutant (Cliby et al., 1998) were kindly provided by William Cliby (Mayo Clinic, Rochester, Minnesota, USA), which was grown in Dulbecco's Modified Eagle's Medium supplemented with 400 μg/ml G418 sulphate (Geneticin, Life Technologies, Paisley, UK). Cell lines, authenticated by STR profiling (LGC Standards, Teddington, UK) and confirmed mycoplasma‐free (MycoAlert, Lonza, Basel, Switzerland) were incubated at 37 °C in 5% CO2. Blood from healthy volunteers was collected by venipuncture into tubes containing anticoagulant (EDTA or heparin) according to institutional ethical procedures. The PBMCs separated and collected using a Ficoll‐Paque Plus (GE healthcare, Pittsburgh, USA) gradient either before or after treatment with DNA‐damaging agents. The collection of fresh PBMCs was approved by Newcastle University. For UV treatment, cells in uncovered tissue culture dishes were irradiated with 254‐nm UV light in a UV cross‐linker (Stratalinker‐2400, Stratagene, La Jolla, CA, USA).
2.3. Cell elutriation and EdU labeling
K562 cells were separated by a Beckman JE‐6B elutriating rotor system at room temperature into different cell cycle phases. Briefly, the pump speed was increased from 25 ml/min to 65 ml/min by 5 ml/min each time, and each fraction was collect into an individual tube. Cell cycle phase was determined by flow cytometry analysis of DNA staining with propidium iodide. After fixing with 70% ethanol and staining with 400 μg/ml propidium iodide, cell samples were run on a FACSCalibur flow cytometer (Becton Dickinson Franklin Lakes, USA). 10,000 events were analysed using the CELLQUEST (Becton Dickinson Franklin Lakes, USA) programme. For EdU labeling, cells were incubated with 25 mM EdU for 30 min and stained using a Click‐iT EdU Alexa Fluor 546 Imaging kit (Life Technologies, Paisley, UK).
2.4. Knock‐down of ATR expression
For siRNA treatment, Lipofectamine™ RNAiMAX (Life technologies, Paisley, UK) was added to ATR siRNA diluted in Opti‐MEM I medium without serum to give a final concentration of 10 nM. After incubating for 10–20 min at room temperature, MCF7 cells in complete growth medium without antibiotics were added to the mix and incubated for 48 h at 37 °C in a CO2 incubator before being harvested. The scrambled siRNA (5′‐UUCUCCGAACGUGUCACGUdtdt) and ATR siRNA (5′‐CAUCUUAUCCCAUGCGUGUdtdt) sequences were synthesized by Sigma. For shRNA knockdown, ATR shRNA virus was made by transfection of HEK293T cells with the packaging vector pCMVΔ8.91, the envelope vector pMD2.G and the pTRIPZ inducible lentiviral vector containing ATR shRNA (Thermo Fisher Scientific, Northumberland, UK). Viral particles were purified and enriched using Lenti‐X Concentrator (Clontech, Mountain View, USA), which was then transduced into MCF7 cells. Stable MCF7 cell clones with shATR were generated by selection at 1 μg/ml puromycin (Life technologies, Paisley, UK). The expression of ATR shRNA was induced by supplementing media with 1 μg/ml of doxycycline. Cells grown in the absence of doxycycline were used as a control.
2.5. Western blotting
Exponentially‐growing, treated cells were lyzed using PhosphoSafe Extraction Reagent (EMD Millipore, Merck KGaA, Darmstadt, Germany). Protein concentrations were determined using a Pierce BCA Protein Assay kit (Thermo Fisher Scientific, Northumberland, UK). After addition of sample buffer (Bio‐Rad, Hemel Hempstead, UK), protein samples (20 μg for cell lines, 30 μg for PBMCs) were resolved on 4–15% polyacrylamide tris/glycine gels (Bio‐Rad, Hemel Hempstead, UK) and blotted onto Hybond C‐extra membrane (GE Healthcare UK Ltd., Buckinghamshire, UK). Proteins were detected using primary antibodies: goat anti‐ATR (1:300), mouse anti‐Chk1 (1:300), rabbit anti‐DNA‐PKcs (1:1000) (Santa Cruz Biotech, Heidelberg, Germany); rabbit anti‐pChk1Ser345 (1:300; Cell Signalling Tech., Danvers, MA, USA); Mouse anti‐γH2AX (1:1000 EMD Millipore, Billerica, MA, USA), Mouse anti‐Actin (1:10,000 Sigma). Secondary antibodies were donkey anti‐goat (1/2000, Insight Biotechnology, Wembley, UK), goat anti‐rabbit HRP (1/1000, Dako UK Ltd, Ely, UK) and goat anti‐mouse HRP (1/2000, Dako UK Ltd).
2.6. Flow cytometry
To measure γH2AX in isolated PBMCs by flow cytometry 5 × 105 PBMCs per sample were incubated with VE‐822 for 10 min at 37 °C, then stimulated with 1.5 μM 4NQO (Sigma) for 1 h at 37 °C. Cells were fixed with 4% paraformaldehyde (Polysciences Warrington PA USA) in PBS for 10 min then permeabilised for 10 min using 0.5% Triton‐X‐100 (Sigma Poole, UK). Cells were stained with γH2AX antibody (1/200, EMD Millipore, Billerica MA USA) for 1 h at room temperature, and with goat anti‐rabbit Alexafluor 488 secondary antibody (1/500, Life Technologies, Paisley, UK) for 1 h room temperature in the dark. Cells were then analysed using a FacsCanto II (Becton Dickinson Franklin Lakes USA) collecting 10,000 events. To determine H2AX phosphorylation in PBMCs in whole blood it was incubated with VE‐822 for 10 min at 37 °C, then stimulated with 62 μM 4NQO (Sigma Poole, UK) for 1 h at 37 °C. The samples were subsequently treated with BD Lyse/Fix solution (Becton Dickinson Franklin Lakes USA) and the PBMCs were permeabilised and processed as described above.
2.7. Immunofluorescence
MCF7 cells were seeded onto coverslips and left to attach for 48 h before treatment. K562 cells and PBMCs were attached to microscope slides by centrifugation for 3 min at 450 rpm in a Cytospin 2 centrifuge (Shandon, Frankfurt, Germany). Cells were then fixed with 4% paraformaldehyde in PBS and permeabilized with 0.5% Triton‐X‐100 in PBS. Permeabilized cells were left in 5% BSA and 10% goat serum in PBS for 1 h. The cells were then incubated with rabbit anti‐pChk1Ser345 (Cell Signaling Tech, Danvers, USA) at 1:300; Mouse anti‐γH2AX at 1:1000 (EMD Millipore) in 1% BSA in PBS overnight at 4 °C. Coverslips were washed with PBS and incubated in a 1:1000 dilution of AlexaFluor 488 goat anti‐rabbit IgG, or AlexaFluor 546 goat anti‐mouse IgG secondary antibodies (Life technologies, Paisley, UK) for 1 h in darkness. After washing with PBS for 3 times, the coverslips were mounted onto slides with VectaShield with DAPI (Vector Laboratories, Peterborough, UK), sealed with nail varnish and viewed by confocal microscopy (Carl Zeiss, Cambridge, UK). Images were quantified using Image J software (National Institute of Health, USA, http://rsb.info.nih.gov/ij/) with the PZFociEZ macro (www.pzfociez.com).
2.8. Statistical analysis
Data analysis was performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Western blotting results were analysed using paired Students t tests. Statistically significant changes for immunofluorescence results were determined by using one‐way ANOVA followed by Tukeys Multiple Comparison Test.
3. Results
3.1. Phosphorylation of Chk1 and H2AX following DNA damage and reduction by ATR depletion in MCF7 cells
To identify a potential biomarker for ATR activity based on the known targets of ATR, we investigated Chk1 and H2AX phosphorylation in MCF7 cells treated with HU and UV, the doses used being selected on the basis of previous studies with this cell line (Ferguson et al., 2003; Peasland et al., 2011). We also investigated cisplatin induction in these cells but no induction was observed after 1 h and even after 15 h exposure there was only a modest induction in comparison to HU (data not shown). Chk1Ser345 phosphorylation was induced within 1 h after UV and HU and ATR depletion by siRNA/shRNA reduced both UV‐ and HU‐induced Chk1Ser345 phosphorylation by >85%, confirming that this phosphorylation is ATR‐dependent (Figure 1A, B, Supplementary Table 1). In contrast, H2AX phosphorylation was only induced by UV, and ATR depletion by siRNA and shRNA only reduced the UV‐induced increase in γH2AX by 13% (p = 0.41) and 20% (p = 0.45), respectively, in these studies (Figure 1A, B, Supplementary Table 1). Because ex vivo UV irradiation may not be feasible in many clinical facilities, we investigated whether the UV mimetic 4NQO would also stimulate ATR‐dependent phosphorylation events. Similar to UV, exposure to 4NQO for 2 h induced Chk1 and H2AX phosphorylation but ATR knockdown only reduced Chk1 phosphorylation (Figure S1A). The specificity of pChk1Ser345 as a measure of ATR activity 1 h after UV exposure was also confirmed in GM847‐ATRkd cell lines (Figure S1B).
Figure 1.
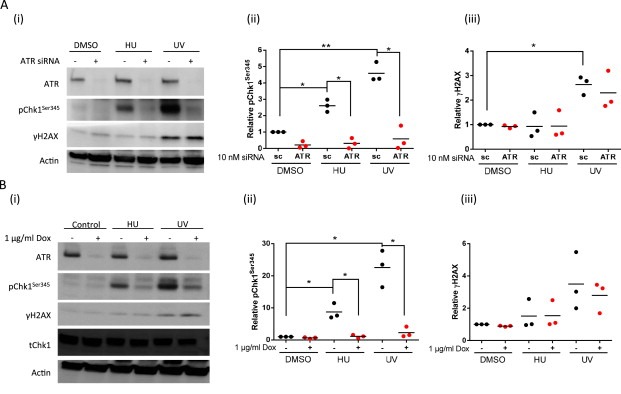
Chk1 Ser345 and H2AX phosphorylation in response to DNA damage and reduction by ATR knockdown in MCF7 cells. Analysis of pChk1Ser345 and γH2AX levels by western blot following 1 h exposure to HU (10 mM) or 1 h after exposure to UV (10 J/m2) in control MCF7 cells and following ATR depletion (A) after 2 days siRNA (10 nM) treatment and (B) in MCF7 cells transduced with doxycycline‐inducible ATR shRNA after 3 days treatment with or without doxycycline (1 μg/ml). Data are: representative blot (i) and scatter plot of data from 3 independent experiments, each spot representing data from one experiment and represents quantitation of pChk1Ser345 (ii) and γH2AX (iii) normalized to the actin loading control and then expressed relative to untreated control. Significance is given by: ns, not significant, *, p < 0.05; **, p < 0.01.
3.2. Effect of ATR inhibitor VE‐821 on HU and UV‐induced pChk1Ser345 and γH2AX in MCF7 and K562 cells
The effect of the small molecule ATR inhibitor, VE‐821, on ATR activity was tested in MCF7 cells. Western blotting results showed that VE‐821 (10 μM) inhibited HU‐ and UV‐induced pChk1Ser345 by >60% in MCF7 cells (Figure 2A, Supplementary Table 2). This was concentration‐dependent with the IC50 being around 2.3 μM VE‐821 (Figure S2A). Inhibition was rapidly reversed on removal of the drug (Figure S2A). VE‐821 (1 μM) also inhibited 4NQO‐induced Chk1 phosphorylation (Figure S2B) indicating that 4NQO may be suitable to activate ATR in biomarker assays. There was no increase in γH2AX after HU treatment, and the UV‐induced increase in γH2AX was not significantly inhibited by VE‐821 (Figure 2A).
Figure 2.
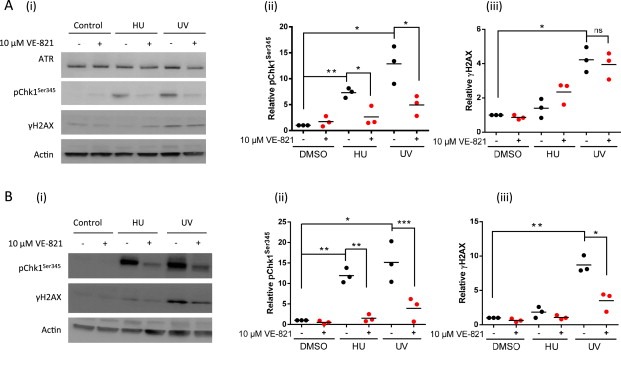
The effect of the ATR inhibitor VE‐821 on DNA‐damage induced Chk1 Ser345 and H2AX phosphorylation in MCF7 and K562 cells. Western blot analysis of the effect of VE‐821 on HU and UV‐induced pChk1Ser345 and γH2AX. MCF7 cells (A) and K562 cells (B) were exposed to HU (10 mM) for 1 h in the presence or absence of VE‐821 (10 μM) or cultured in control or VE‐821‐containing medium for 2 h with exposure to UV (10 J/m2) after the first hour. Data are: representative blot (i) and scatter plot of data from 3 independent experiments, each spot representing data from one experiment and represents quantitation of pChk1Ser345 (ii) and γH2AX (iii) normalized to the actin loading control and then expressed relative to untreated control. Significance is given by: ns, not significant, *, p < 0.05; **, p < 0.01, ***, p < 0.001.
Next we evaluated ATR inhibition in a cell line of haematopoietic origin (K562) that might better reflect human PBMCs (a likely surrogate normal tissue for PD biomarker development) Both HU and UV caused a ≥10‐fold induction of pChk1Ser345. Although substantial γH2AX induction was observed by western blot following UV treatment, induction of γH2AX was modest with HU (Figure 2B). VE‐821 inhibited UV‐induced pChk1Ser345 and, in contrast to its effect in MCF7 cells, VE‐821 also significantly inhibited UV‐induced γH2AX in K562 cells (Figure 2B).
3.3. The effect of VE‐821 on UV and 4NQO induced ATR activity in surrogate tissues
PBMCs represent an easily available source of human material, accessible with minimally invasive procedures, to use as a surrogate tissue for pharmacodynamic evaluation. Since UV‐induced and 4NQO‐induced Chk1 phosphorylation was ATR‐dependent in both MCF7 and K562 cells and H2AX phosphorylation was ATR‐dependent in cells of haematological origin, we investigated whether these phosphorylation events could be detected in human PBMCs, as a potential clinical PD biomarker for ATR activity. ATR and total Chk1 signals were weak but detectable in PBMCs by Western blot and pChk1Ser345 was detectable after UV treatment (Figure S3). Cell lysates from hair follicles and buccal scrapes proved difficult to obtain and gave highly variable results (data not shown).
3.4. Immunofluorescence detection of biomarkers in cancer cell lines
Since the levels of ATR and pChk1Ser345 were near the limit of detection in surrogate normal tissues by western blot, we assessed whether immunofluorescence microscopy would be a more sensitive method to detect ATR‐dependent phosphorylation events. We first investigated the effect of VE‐821 on UV‐induced and 4NQO‐induced ATR activity in individual K562 and MCF7 cells (Figure 3). In both cell lines UV and 4NQO caused a robust induction of γH2AX signals and a significant induction of pChk1Ser345 in almost all the cells (Figure 3A and B). VE‐821 significantly inhibited UV and 4NQO induced pChk1Ser345 and γH2AX in both cell lines. This contrasts with a lack of effect on γH2AX as observed by Western Blot. To investigate potential causes of these discrepancies, and to determine the specificity of pChk1Ser345 and γH2AX for ATR over the related kinases ATM and DNA‐PK, we looked at the effects of ATR, ATM and DNA‐PK inhibitors (VE‐821, KU55933 and NU7441 respectively) on UV‐induced Chk1Ser345 and H2AX phosphorylation with respect to time (Figure 4). A substantial increase in Chk1Ser345 phosphorylation was detectable at 30 min and remained at this high level for the next 2.5 h, whereas γH2AX increased steadily over the 3 h time course (Figure 4A). Inhibition of Chk1Ser345 phosphorylation by VE‐821 was greatest (70%) at 30 min and gradually declined to 50% after 3 h. In contrast inhibition of ATM and DNA‐PK by KU55933 and NU7441, blocked the signal by only 25% at 30 min but this increased over time thereafter. Similarly, VE‐821 caused substantial inhibition of H2AX phosphorylation at early time points (80% at 30 min) but this rapidly declined over the next 2 h (<20% at 3 h). KU55933 and NU7441 made an increasing contribution to the reduction in H2AX phosphorylation from 50% at 30 min to 65% at 3 h (Figure 4A). This increase in H2AX phosphorylation may reflect the accumulation of collapsed replication forks and replication‐associated DSB. Similar results were obtained with 4NQO (Supplementary Figure S4). The contribution of ATR versus ATM and DNA‐PK to the phosphorylation of Chk1 and H2AX was further investigated in MCF7 cells over 24 h using Western blot analysis (Figure 4B). This showed that VE‐821 preferentially inhibited Chk1Ser345 phosphorylation by >85% for up to 3 h, declining to 40% at 7 h, with no inhibition by 24 h. Inhibition of ATM and DNA‐PK had little impact on Chk1Ser345 phosphorylation at 1 h but made an increasing contribution with time such that inhibition was around 80% at 24 h. VE‐821 had a modest impact (30–40% inhibition) on γH2AX at early time points (1–3 h) but this had declined to <10% inhibition at 7 and 24 h whereas KU55933 + NU7441 caused >50% inhibition of γH2AX at all time points.
Figure 4.
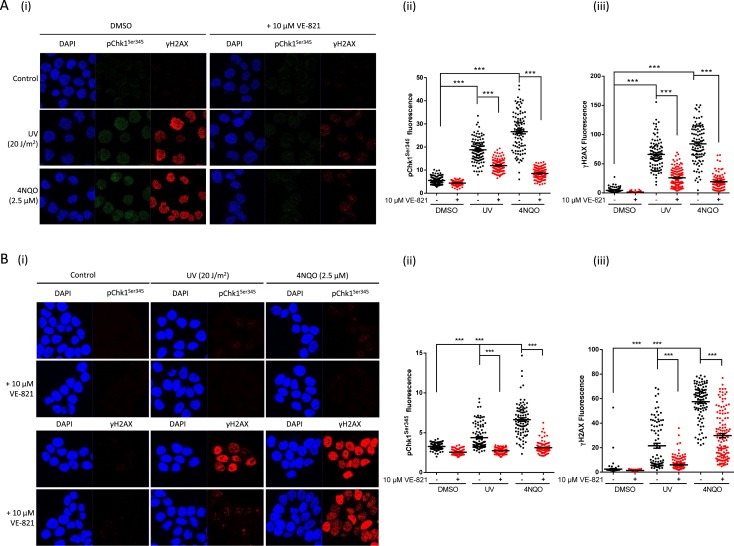
Chk1 Ser345 and H2AX phosphorylation in response to DNA damage and reduction by ATR inhibition in K562 and MCF7 cells. pChk1Ser345 and γH2AX by immunofluorescence microscopy in K562 cells (A) or MCF7 cells (B) with or without VE‐821 (10 μM). K562 cells were harvested 1 h after treatment with UV (20 J/m2) or 3 h continuous exposure to 4NQO (2.5 μM) (A). MCF7 cells were harvested 1 h after treatment with UV (20 J/m2) or 2 h continuous exposure to 4NQO (2.5 μM) (B). Data are (i) immunofluorescence images and quantitation (ii) pChk1Ser345 and (iii) γH2AX immunofluorescence data where each point represents data from a single cell with at least 100 cells counted per treatment. Significance is given by: ns, not significant, ***, p < 0.001.
Figure 3.
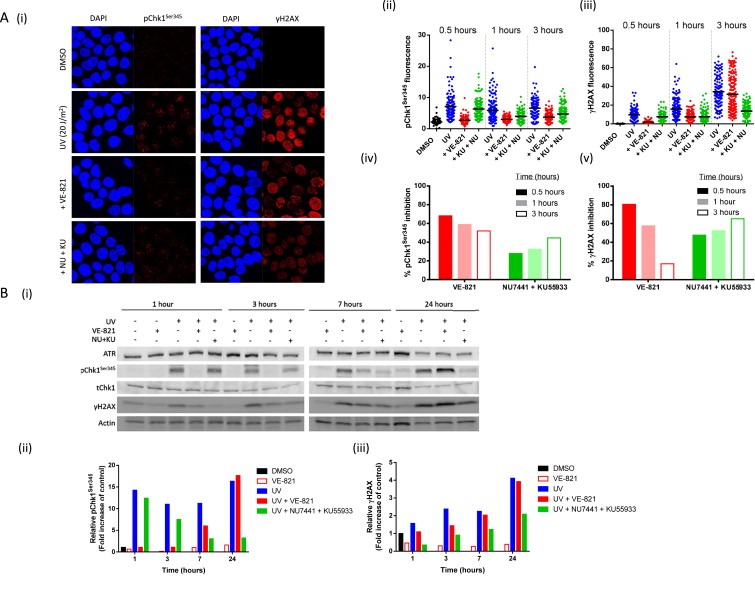
Specificity of UV‐induced Chk1 Ser345 and H2AX phosphorylation for ATR, ATM and DNA‐PK over time. Analysis of UV‐induced pChk1Ser345 and γH2AX by immunofluorescence microscopy (A) and western blotting (B) with or without VE‐821 or NU7441 and KU55933. MCF7 cells were treated for 15 min with either VE‐821 (10 μM) or NU7441 and KU55933 (1 μM and 10 μM, respectively) before being exposed to UV (20 J/m2). Cells (in the presence/absence of inhibitor(s)) were incubated for a further hour and either fixed for immunofluorescence (A) or lysed for western blotting (B). A) Immunofluorescence data: images (i) and quantitation of pChk1Ser345 (ii) or γH2AX (iii) where each point represents data from a single cell with at least 100 cells counted in each group. These were used to calculate the % inhibition of Chk1Ser345 (iv) or H2AX (v) phosphorylation by kinase inhibitors. B) Western blot: images (i) and quantitation of pChk1Ser345 (ii) or γH2AX (iii) by densitometry.
There is some evidence that ATR activation by UV is not limited to S phase (Vrouwe et al., 2011), and our data showing that most cells were positive for phosphorylation biomarkers supports this hypothesis. Chemical inhibitors of cell cycle progression or nutrient deprivation have been criticized for not properly synchronizing cells as well as imposing cell stress and cytotoxicity (Cooper, 2003). Therefore, to confirm ATR activation in non S‐phase cells, we used an elutriation system to separate K562 cells into G1/early S and late S/G2 fractions. EdU labeling was used to mark cells undergoing DNA synthesis. HU‐induced pChk1Ser345 and γH2AX only in the fraction of cells that were also EdU positive. In contrast, UV‐induced pChk1Ser345 and γH2AX in almost all the cells (Figure S5). Both HU‐ and UV‐induced pChk1Ser345 and γH2AX signals were significantly inhibited by VE‐821 (Figure S5).
3.5. Immunofluorescence detection of biomarkers in human PBMCs
Having confirmed that both pChk1Ser345 and γH2AX were induced by UV in G1 and G2 phase K562 cells we investigated ATR‐dependent phosphorylation in non‐replicating PBMCs. UV‐induced pChk1Ser345 (confirming the Western blot data, Figure S3) and γH2AX signals in PBMCs were evident by immunofluorescence microscopy, and were inhibited by VE‐821 following a 1 h exposure (Figure 5A). Similarly, 3 h exposure to 4NQO‐induced both Chk1 and H2AX phosphorylation, which was inhibited by VE‐821. As expected, HU did not induce pChk1Ser345 or γH2AX in PBMCs because HU only induces ATR activity in proliferating cells.
Figure 5.
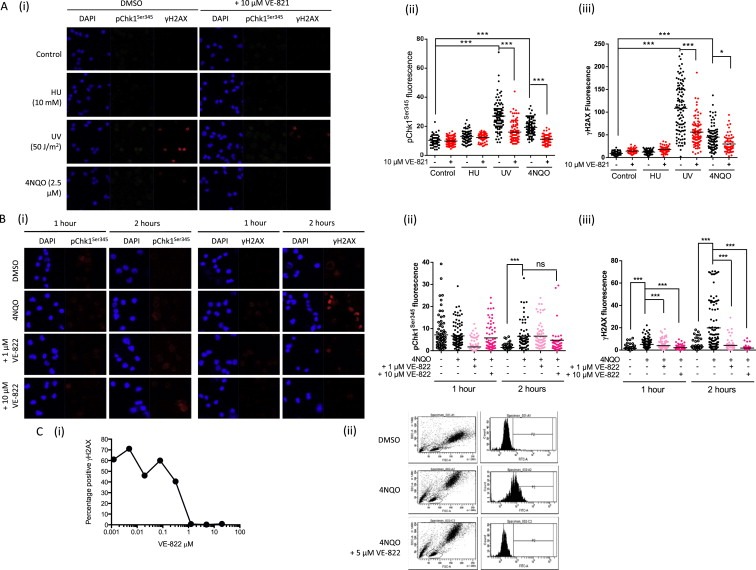
UV induced ATR activity in human PBMCs. Analysis of pChk1Ser345 and γH2AX by immunofluorescence microscopy following exposure of isolated PBMCs (A) to HU (10 mM, 1 h), UV (20 J/m2 followed by 1 h incubation) or 3 h 4NQO (2.5 μM) treatment or in PBMCs isolated from whole blood (B) that had been treated with 240 μM 4NQO for 1 or 2 h with or without 1 or 10 μM VE‐822 prior to isolation of the PBMCs. Data are (i) immunofluorescence images and (ii) quantitation of the fluorescence of pChk1Ser345 and γH2AX in each cell with over 100 cells being counted in each group. Each experiment was repeated at least twice. Representative pictures and quantitation results from one experiment were presented here. (C) Measurement of γH2AX positive PBMCs following exposure of whole blood to 62 μM 4NQO in the presence of increasing concentrations of VE‐822 by flow cytometry (i) and representative data in the presence of 5 μM VE‐822 (ii). Significance is given by: ns, not significant, *p < 0.05; ***p < 0.001.
Since VE‐821 inhibition of ATR was rapidly reversible (Figure S2) and inhibition may be lost during PBMC purification we treated whole blood with UV at a high dose (40 J/m2) 1 h before PBMC purification. This caused a modest but significant increase in pChk1Ser345 fluorescence that was inhibited by VE‐821 (Figure S6). There was a much greater increase in γH2AX and VE‐821 reduced the mean fluorescence.
Whilst exposure of whole blood to UV does induce pChk1Ser345 and γH2AX, the use of UV in a clinical setting is somewhat impractical. We therefore optimized the concentration of 4NQO for biomarker induction by treating whole blood with varying concentrations of 4NQO. γH2AX fluorescence increased with increasing 4NQO, however consistent induction of pChk1Ser345 was not observed in these studies.
To assess the potential for 4NQO‐induced H2AX and Chk1Ser345 phosphorylation as a clinically relevant biomarker in whole blood we used VE‐822, an ATR inhibitor that is in Phase I clinical studies. Knowing that ATR‐dependent H2AX phosphorylation is time‐dependent (Figure 4A and Supplementary Figure S4), whole blood was treated with 4NQO (240 μM) for both 1 and 2 h 4NQO‐induced at least a 3‐ and 13‐fold increase in γH2AX fluorescence at 1 and 2 h respectively in two independent experiments, and this was inhibited in a concentration‐dependent manner by VE‐822. At 2 h 10 μM VE‐822 inhibited γH2AX by >90% (Figure 5B).
Immunofluorescence for γH2AX, while successful, is labour intensive and low throughput. To investigate if biomarker identification could be achieved in a higher throughput format we measured the induction of γH2AX formation following 4NQO exposure, and its inhibition by VE‐822, by flow cytometry. Initial studies with isolated PBMCs demonstrated that exposure to just 1.5 μM 4NQO for 1 h induced H2AX phosphorylation in 35% of the cells and this was reduced with increasing concentrations of VE‐822, the calculated IC50 for VE‐822 was 20 nM (Figure S8A). This is consistent with the previously reported cellular IC50 for inhibition of ATR by VE‐822 (Fokas et al., 2012). There was no increase in γH2AX in PBMCs following exposure of whole blood to 1.5 μM 4 NQO for 1 h but raising the concentration to 62 μM increased the proportion of γH2AX positive cells by 60% (Figure S8B). This 4NQO‐induced H2AX phosphorylation was inhibited by co‐exposure to increasing concentrations of VE‐822 (Figure 5C) and the IC50 of VE‐822 in whole blood was 600 nM, i.e. 30× higher than in isolated PBMCs. This difference is most likely explained by large fractions of the drug being bound to proteins in whole blood.
4. Discussion
Chk1 is the primary target of ATR and phosphorylation at Ser317 and Ser345 have been widely used as biomarkers for ATR activity (Cliby et al., 2002; Nishida et al., 2009; Kawasumi et al., 2011). Here we confirmed that HU‐ and UV‐induced pChk1Ser345 was dependent on ATR at early time points by treating MCF7 cells with siATR, shATR and the ATR‐specific inhibitor VE‐821. Chemosensitisation by VE‐821 has previously been shown to be associated with the inhibition of Chk1Ser345 phosphorylation, validating it as a potential biomarker (reviewed in (Fokas et al., 2014)). However, by 24 h ATR inhibition had no impact on pChk1Ser345. Similarly, VE‐821 inhibited HU‐, 4NQO‐ and UV‐induced pChk1Ser345 in K562 cells at early time points. The loss of ATR‐dependent phosphorylation of Chk1 at Ser345 is attributed to an increased role for ATM and DNA‐PK (Chanoux et al., 2009). The timing of the measurement may be critical and cell‐dependent in replicating cells, for example, VE‐821 failed to inhibit gemcitabine‐induced pChk1Ser345 inOVCAR‐8, SKOV‐3 and MiaPaCa cells when measured at 4 h (Huntoon et al., 2013) and a specific ATR inhibitor (ATRi) inhibited HU‐induced pChk1Ser345 in U2‐OS cells at 40 min but not 80 min (Toledo et al., 2013).
The phosphorylation of thousands of H2AX molecules into individual foci, detectable by immunofluorescence microscopy, is one of the earliest events after DNA damage and has been used as a biomarker for DNA double strand breaks and stalled replication forks (Redon et al., 2010). Although H2AX can be phosphorylated by a number of enzymes such as ATM and DNA‐PK, replication stress induced by HU and UV‐induced DNA damage has been shown to lead to an ATR‐dependent increase in γH2AX (Ward and Chen, 2001; Ward et al., 2003). Here we confirmed that UV and 4NQO induced H2AX phosphorylation is ATR‐dependent at early time points with a steady decrease in inhibition by VE‐821 between 0.5 and 3 h. However, phosphorylation of H2AX was increasingly inhibited by NU7441 and KU55933 with time. We believe the highly transient nature of ATR dependence of γH2AX in cycling cells may be attributed to DNA lesions colliding with replication forks, which in turn triggers the activation of ATM and DNA‐PK phosphorylation of H2AX (Chanoux et al., 2009). Again the time‐dependence may be cell‐type specific, for example in Ovcar‐8 cells VE‐821 increased cisplatin and topotecan‐induced H2AX phosphorylation after 4 h exposure (Huntoon et al., 2013).
It has been proposed that UV‐induced H2AX phosphorylation is not dependent on the induction of replication stress and occurs in all phases of the cell cycle and in non‐cycling cells, and that the kinase responsible is ATR (Matsumoto et al., 2007; Stiff et al., 2008; Vrouwe et al., 2011). Here we confirmed that UV exposure induced γH2AX and pChk1Ser345 in all phases of the cell cycle in K562 cells and exposure to 10 μM VE‐821 inhibited both UV‐ and 4NQO‐ induced pChk1Ser345 by 60–75% in K562 and MCF7 cells. This suggests that it should be possible to measure ATR activity in cells from non‐cycling tissues such as un‐stimulated PBMCs and that this may be exploitable for the development of clinically applicable biomarkers. In this study, we found ATR‐mediated phosphorylation of Chk1 and H2AX in isolated PBMCs could be activated by UV irradiation and the UV mimetic 4NQO, although the ATR activity in these terminally differentiated PBMC cells was weak. It had previously been reported that ATR protein could not be detected in un‐stimulated PBMCs by western blotting, but this may be due to the sensitivity of western blotting itself (Jones et al., 2004). The γH2AX signal in PBMCs was much stronger than pChk1Ser345 in all of our studies, potentially making it a better biomarker for ATR activity and its inhibition.
We found that ATR inhibition by VE‐821 was rapidly reversed on removal of the drug, suggesting that inhibition would be lost during the preparation of the PBMCs from patients' blood prior to ex vivo UV or 4NQO induction of ATR, precluding their use as a surrogate biomarker. To avoid losing inhibition whole blood was treated with UV or 4NQO prior to isolation of the PBMCs. Whilst UV‐induced pChk1Ser345 and γH2AX, ex vivo stimulation of patient samples with UV is impractical to perform in the clinic. We therefore favour the use of 4NQO to induce ATR activity in whole blood. In order to validate this method for clinical application, we used VE‐822, an analog of VE‐821 that is in clinical trials. A consistent induction of γH2AX was observed by 4NQO that was fully inhibited by VE‐822. This was evident up to 2 h after treatment with VE‐822, which contrasts with the observations from cycling MCF7 cells where ATR‐dependent phosphorylation of H2AX was observed for <1 h. In whole blood 4NQO does not appear to induce Chk1Ser345 phosphorylation consistently in healthy volunteer samples making it a less attractive event for clinical biomarker studies. Finally, we demonstrated inhibition of 4NQO‐induced H2AX phosphorylation in whole blood using flow cytometry. This provides a higher throughput analytical method that is better suited to clinical application.
In conclusion, our studies in two different human cell lines, one from a solid tumour and the other from a haematological malignancy, in which ATR activity has been reduced by genetic means and with small molecule inhibitors, demonstrate that pChk1Ser345 and γH2AX after UV or 4NQO can be used as biomarkers for ATR activity. Importantly, we demonstrate that ATR‐mediated phosphorylation is detectable in non‐cycling PBMCs meaning that they can be used as a surrogate tissue for clinical evaluation of ATR inhibitors. With both pChk1Ser345 and γH2AX we show ATR dependence at early time points in cycling cells, however phosphorylation becomes increasingly dependent on other DNA damage‐activated kinases over time. This was most evident with γH2AX where ATR dependence was lost after 1 h. In contrast, in response to UV or 4NQO, ATR is activated in isolated non‐cycling PBMCs, and under these conditions we showed γH2AX was ATR‐dependent for at least 2 h. Timing of the measurements is therefore critical and this may explain some of the variable and contradictory data previously reported. Since the process of isolating PBMCs is likely to wash out any ATR inhibitor from a patient's blood, we developed a method whereby a blood sample is first treated with 4NQO and then the PBMCs isolated and markers of ATR activity analysed by flow cytometry. With currently available antibodies we did not observe a consistent pChk1Ser345 signal from whole blood. However, γH2AX was robustly observed and could be blocked by spiking the blood with an ATR inhibitor. We suggest that this biomarker method could be an important tool to help define the optimal biological dose of an ATR inhibitor in the clinic. This may be critical for the successful development for such molecularly targeted agents where the maximum tolerated dose may not be informative.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Acknowledgements
We gratefully acknowledge support from the Medical Research Council UK (G1000400: for FKM) and Cancer Research UK (for TC).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.09.012.
Chen Tao, Middleton Fiona K., Falcon Susanna, Reaper Philip M., Pollard John R., Curtin Nicola J., (2015), Development of pharmacodynamic biomarkers for ATR inhibitors, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.09.012.
References
- Chanoux, R.A. , Yin, B. , Urtishak, K.A. , Asare, A. , Bassing, C.H. , Brown, E.J. , 2009. ATR and H2AX cooperate in maintaining genome stability under replication stress. J. Biol. Chem. 284, (9) 5994–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T. , Stephens, P.A. , Middleton, F.K. , Curtin, N.J. , 2012. Targeting the S and G2 checkpoint to treat cancer. Drug Discov. Today. 17, (5–6) 194–202. [DOI] [PubMed] [Google Scholar]
- Cliby, W.A. , Lewis, K.A. , Lilly, K.K. , Kaufmann, S.H. , 2002. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J. Biol. Chem. 277, (2) 1599–1606. [DOI] [PubMed] [Google Scholar]
- Cliby, W.A. , Roberts, C.J. , Cimprich, K.A. , Stringer, C.M. , Lamb, J.R. , Schreiber, S.L. , Friend, S.H. , 1998. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 17, (1) 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, S. , 2003. Rethinking synchronization of mammalian cells for cell cycle analysis. Cell Mol. Life Sci. 60, (6) 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, H.A. , Marietta, P.M. , Van Den Berg, C.L. , 2003. UV-induced apoptosis is mediated independent of caspase-9 in MCF-7 cells: a model for cytochrome c resistance. J. Biol. Chem. 278, (46) 45793–45800. [DOI] [PubMed] [Google Scholar]
- Fokas, E. , Prevo, R. , Hammond, E.M. , Brunner, T.B. , McKenna, W.G. , Muschel, R.J. , 2014. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 40, (1) 109–117. [DOI] [PubMed] [Google Scholar]
- Fokas, E. , Prevo, R. , Pollard, J.R. , Reaper, P.M. , Charlton, P.A. , Cornelissen, B. , Vallis, K.A. , Hammond, E.M. , Olcina, M.M. , Gillies McKenna, W. , Muschel, R.J. , Brunner, T.B. , 2012. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 3, e441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote, K.M. , Blades, K. , Cronin, A. , Fillery, S. , Guichard, S.S. , Hassall, L. , Hickson, I. , Jacq, X. , Jewsbury, P.J. , McGuire, T.M. , Nissink, J.W. , Odedra, R. , Page, K. , Perkins, P. , Suleman, A. , Tam, K. , Thommes, P. , Broadhurst, R. , Wood, C. , 2013. Discovery of 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-y l}-1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 56, (5) 2125–2138. [DOI] [PubMed] [Google Scholar]
- Gagou, M.E. , Zuazua-Villar, P. , Meuth, M. , 2010. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol. Biol. Cell. 21, (5) 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntoon, C.J. , Flatten, K.S. , Wahner Hendrickson, A.E. , Huehls, A.M. , Sutor, S.L. , Kaufmann, S.H. , Karnitz, L.M. , 2013. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 73, (12) 3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacq, X. , Smith, L. , Brown, E. , Hughes, A. , Odedra, R. , Heathcote, D. , Barnes, J. , Powell, S. , Maguire, S. , Pearson, V. , Boros, J. , Caie, P. , Thommes, P.A. , Nissink, W. , Foote, K. , Jewsbury, P.J. , Guichard, S.M. , 2012. AACR 103rd Annual Meeting. Chicago, IL, Mar 31–Apr 4. [Google Scholar]
- Jones, C.D. , Blades, K. , Foote, K. , Guichard, S.M. , Jewsbury, P.J. , McGuire, T.M. , Nissink, J.W. , Odedra, R. , Tam, K. , Thommes, P. , Turner, P. , Wilkinson, G. , Wood, C. , Yates, J.W. , 2013. AACR Annual Meeting. Boston, MA, USA, April 15, 2013. [Google Scholar]
- Jones, G.G. , Reaper, P.M. , Pettitt, A.R. , Sherrington, P.D. , 2004. The ATR-p53 pathway is suppressed in noncycling normal and malignant lymphocytes. Oncogene. 23, (10) 1911–1921. [DOI] [PubMed] [Google Scholar]
- Kawasumi, M. , Lemos, B. , Bradner, J.E. , Thibodeau, R. , Kim, Y.S. , Schmidt, M. , Higgins, E. , Koo, S.W. , Angle-Zahn, A. , Chen, A. , Levine, D. , Nguyen, L. , Heffernan, T.P. , Longo, I. , Mandinova, A. , Lu, Y.P. , Conney, A.H. , Nghiem, P. , 2011. Protection from UV-induced skin carcinogenesis by genetic inhibition of the ataxia telangiectasia and Rad3-related (ATR) kinase. Proc. Natl. Acad. Sci. U S A. 108, (33) 13716–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague, J. , 2004. G1 cell-cycle control and cancer. Nature. 432, (7015) 298–306. [DOI] [PubMed] [Google Scholar]
- Matsumoto, M. , Yaginuma, K. , Igarashi, A. , Imura, M. , Hasegawa, M. , Iwabuchi, K. , Date, T. , Mori, T. , Ishizaki, K. , Yamashita, K. , Inobe, M. , Matsunaga, T. , 2007. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J. Cell Sci. 120, (Pt 6) 1104–1112. [DOI] [PubMed] [Google Scholar]
- Nghiem, P. , Park, P.K. , Kim, Y. , Vaziri, C. , Schreiber, S.L. , 2001. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. U S A. 98, (16) 9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida, H. , Tatewaki, N. , Nakajima, Y. , Magara, T. , Ko, K.M. , Hamamori, Y. , Konishi, T. , 2009. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 37, (17) 5678–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peasland, A. , Wang, L.Z. , Rowling, E. , Kyle, S. , Chen, T. , Hopkins, A. , Cliby, W.A. , Sarkaria, J. , Beale, G. , Edmondson, R.J. , Curtin, N.J. , 2011. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer. 105, (3) 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires, I.M. , Olcina, M.M. , Anbalagan, S. , Pollard, J.R. , Reaper, P.M. , Charlton, P.A. , McKenna, W.G. , Hammond, E.M. , 2012. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer. 107, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevo, R. , Fokas, E. , Reaper, P.M. , Charlton, P.A. , Pollard, J.R. , McKenna, W.G. , Muschel, R.J. , Brunner, T.B. , 2012. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 13, (11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaper, P.M. , Griffiths, M.R. , Long, J.M. , Charrier, J.D. , MacCormick, S. , Charlton, P.A. , Golec, J.M.C. , Pollard, J.R. , 2011. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 7, 428–430. [DOI] [PubMed] [Google Scholar]
- Redon, C.E. , Nakamura, A.J. , Zhang, Y.W. , Ji, J.J. , Bonner, W.M. , Kinders, R.J. , Parchment, R.E. , Doroshow, J.H. , Pommier, Y. , 2010. Histone {gamma}H2AX and Poly(ADP-Ribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res. 16, (18) 4532–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. , Tho, L.M. , Xu, N. , Gillespie, D.A. , 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112. [DOI] [PubMed] [Google Scholar]
- Stiff, T. , Cerosaletti, K. , Concannon, P. , O'Driscoll, M. , Jeggo, P.A. , 2008. Replication independent ATR signalling leads to G2/M arrest requiring Nbs1, 53BP1 and MDC1. Hum. Mol. Genet. 17, (20) 3247–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana, R. , Abdel-Fatah, T. , Perry, C. , Moseley, P. , Albarakti, N. , Mohan, V. , Seedhouse, C. , Chan, S. , Madhusudan, S. , 2013. Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS One. 8, (2) e57098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo, L.I. , Altmeyer, M. , Rask, M.B. , Lukas, C. , Larsen, D.H. , Povlsen, L.K. , Bekker-Jensen, S. , Mailand, N. , Bartek, J. , Lukas, J. , 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 155, (5) 1088–1103. [DOI] [PubMed] [Google Scholar]
- Toledo, L.I. , Murga, M. , Zur, R. , Soria, R. , Rodriguez, A. , Martinez, S. , Oyarzabal, J. , Pastor, J. , Bischoff, J.R. , Fernandez-Capetillo, O. , 2011. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 18, (6) 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrouwe, M.G. , Pines, A. , Overmeer, R.M. , Hanada, K. , Mullenders, L.H. , 2011. UV-induced photolesions elicit ATR-kinase-dependent signaling in non-cycling cells through nucleotide excision repair-dependent and -independent pathways. J. Cell Sci. 124, (Pt 3) 435–446. [DOI] [PubMed] [Google Scholar]
- Ward, I.M. , Chen, J. , 2001. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 276, (51) 47759–47762. [DOI] [PubMed] [Google Scholar]
- Ward, I.M. , Minn, K. , Jorda, K.G. , Chen, J. , 2003. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 278, (22) 19579–19582. [DOI] [PubMed] [Google Scholar]
- Zhao, H. , Piwnica-Worms, H. , 2001. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 21, (13) 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data