

Abstract
Basal cell carcinomas (BCCs) and a subset of medulloblastomas are characterized by loss‐of‐function mutations in the tumor suppressor gene, PTCH1. PTCH1 normally functions by repressing the activity of the Smoothened (SMO) receptor. Inactivating PTCH1 mutations result in constitutive Hedgehog pathway activity through uncontrolled SMO signaling. Targeting this pathway with vismodegib, a novel SMO inhibitor, results in impressive tumor regression in patients harboring genetic defects in this pathway. However, a secondary mutation in SMO has been reported in medulloblastoma patients following relapse on vismodegib to date. This mutation preserves pathway activity, but appears to confer resistance by interfering with drug binding.
Here we report for the first time on the molecular mechanisms of resistance to vismodegib in two BCC cases. The first case, showing progression after 2 months of continuous vismodegib (primary resistance), exhibited the new SMO G497W mutation. The second case, showing a complete clinical response after 5 months of treatment and a subsequent progression after 11 months on vismodegib (secondary resistance), exhibited a PTCH1 nonsense mutation in both the pre‐ and the post‐treatment specimens, and the SMO D473Y mutation in the post‐treatment specimens only. In silico analysis demonstrated that SMOG497W undergoes a conformational rearrangement resulting in a partial obstruction of the protein drug entry site, whereas the SMO D473Y mutation induces a direct effect on the binding site geometry leading to a total disruption of a stabilizing hydrogen bond network. Thus, the G497W and D473Y SMO mutations may represent two different mechanisms leading to primary and secondary resistance to vismodegib, respectively.
Keywords: Basal cell carcinoma, Hedgehog pathway, PTCH1, SMO, Vismodegib, Primary resistance, Secondary resistance
1. Introduction
Basal cell carcinoma (BCC) is the most frequent skin cancer with a recognized increasing incidence in the last 30 years. (Flohil et al., 2013) Treatment of BCC mainly consists of local approaches, surgery being the most effective one, followed by radiotherapy, photodynamic therapy, and imiquimod as alternative therapeutic strategies. (Rubin et al., 2005) Systemic therapy for metastatic and locally advanced BCCs not amenable to curative surgery or radiotherapy has been recently added to the therapeutic arsenal, with the approval by FDA and EMA of vismodegib, a small molecule inhibitor of the Hedgehog (Hh) pathway. Indeed, BCC pathogenesis is based on an inappropriate activation of the Hh pathway. The Hh ligand binds the transmembrane receptor Patched Homolog 1 (PTCH1) that in turn prevents the inhibition of the transmembrane Smoothened (SMO) receptor. SMO signaling results in the activation of GLI family zinc finger transcription factors and their target genes such as Cyclin D1. PTCH1 inactivating mutations, characterizing >90% of BCCs, and activating SMO mutations, even if less frequent, (Reifenberger et al., 2005) can act as ligand‐independent oncogenic drivers of this disease. (Rubin et al., 2005) Vismodegib binds and inhibits SMO, thus preventing systemic activation of the forward signaling. Also, the significant antitumor activity of vismodegib in BCC strongly suggests the addiction of this tumor to SMO activity. (Von Hoff et al., 2009).
Results of the pivotal phase II trial with vismodegib in metastatic or locally advanced BCCs showed a response rate of 30% and 43% in the two settings of disease, respectively. (Sekulic et al., 2012) Median duration of response was 7.6 months.
Acquired resistance to vismodegib has been reported in medulloblastomas where the drug, after an initial dramatic response, rapidly turned into resistance. (Yauch et al., 2009) This happened through the acquisition of the SMO D473H mutation which, according to the authors, prevents vismodegib from binding without altering the ability of the receptor to activate the downstream Hh pathway. (Yauch et al., 2009) To our knowledge, however, no evidence of molecular mechanisms associated with primary or acquired resistance to vismodegib in BCCs has been reported to date. Accordingly, in this study we present and discuss for the first time clinical, molecular and in silico evidence of primary and acquired SMO mutation‐mediated resistance to vismodegib in BCCs.
2. Materials and methods
2.1. PTCH1 and SMO mutational analysis
Formalin‐fixed paraffin‐embedded (FFPE) tumor specimens of pre‐treatment liver metastasis, primary tumor and node metastasis (first case), and of both pre‐treatment primary tumor and recurrence arisen on vismodegib regimen (second case) were reviewed and subjected to micro‐dissection under microscopy control. Genomic DNA was extracted using the Qiamp FFPE DNA kit (Qiagen, Chatsworth, CA), following manufacturer's instructions.
Exons 1‐23 of PTCH1 and 8‐10 of SMO were amplified by PCR; all PCR primer sequences are available upon request. The PCR products were subjected to direct sequencing using an ABI Prism 3500 DX Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), and then evaluated by means of the ChromasPro software.
2.2. In silico experiments
The Amber ff03 force field (Duan et al., 2003) was used to parametrize all protein structures. The atomic partial charges for vismodegib were obtained using the RESP procedure (Bayly et al., 1993), and the electrostatic potentials were produced by single‐point quantum mechanical calculations at the Hartree‐Fock level with a 6‐31G* basis set, using the Merz‐Singh‐Kollman van der Waals parameters. (Singh and Kollman, 1984; Besler et al., 1990) Eventual missing force field parameters for the inhibitor molecule were generated using the Antechamber tool (Wang et al., 2006) of Amber 12 (Case et al., 2012) and the general AMBER force field (GAFF) (Wang et al., 2004) for rational drug design.
The structures of each SMO proteins in complex with vismodegib were obtained exploited a well‐validated procedure reported in details in our previous works. (Bozzi et al., 2013; Laurini et al., 2013; Conca et al., 2013; Dileo et al., 2011; Pierotti et al., 2011) However, since SMO is a transmembrane receptor protein, each protein/drug complex was inserted into a lipid membrane structure prior to solvation in an explicit water environment. To the purpose, the CHARMM‐GUI Lipid Builder web application was employed. (Woolf and Roux, 1996; Jo et al., 2007) The lipidic bilayer consisted in a 2:2 proportion of 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC) and 1‐palmitoyl‐2‐oleoyl‐sn‐ glycero‐3‐phosphoethanolamine (POPE). The two sides (top and bottom) of the membrane were constituted by 125 and 115 lipid molecules, respectively. The entire system was parameterized using the lipid11 library (Skjevik et al., 2012) of the ff03 force field.
Each system was then allowed to relax in a box of TIP3P water molecules. (Jorgensen et al., 1983) The solvated systems were minimized with a gradual decrease in the position restraints of the protein atoms. Finally, to achieve electroneutrality, a suitable number of neutralizing ions were added; further, the solution ionic strength was adjusted to the physiological value of 0.15 M by adding the required amounts of Na+ and Cl− ions. After energy minimization of the added ions for 1500 steps, the entire water/ion box was equilibrated via 2ns constant volume/constant temperature (NVT) molecular dynamics (MD) simulations. Further unfavorable interactions within the structures were relieved by imposing progressively smaller positional restraints on the protein/inhibitor complex (from 25 to 0 kcal/(mol Å2) for a total of 5 ns of MD simulations. Each hydrated complex system was then gradually heated to 310 K in three intervals, allowing a 2 ns interval per each 100 K, and then equilibrated for 5 ns at 310 K. Finally, starting from each MD equilibrated system 50 ns of data collection runs were carried out to estimate the corresponding free energy of binding.
The production MD simulations were performed in the constant pressure/constant temperature (NPT) ensemble at T = 310 K using the Berendsen et al. algorithm (Berendsen et al., 1984) with an integration time step of 2 fs, and the applications of the Shake algorithm. (Ryckaert et al., 1977) Long‐range nonbonded van der Waals interactions were truncated by using a dual cutoff of 9 and 13 Å, respectively, where energies and forces due to interactions between 9 and 13 Å were updated every 20‐time steps. The particle mesh Ewald method (Toukmaji et al., 2000) was used to treat the long‐range electrostatics. For the calculation of the binding free energy between SMO and vismodegib in water, a total of 50,000 snapshots were saved during the MD data collection period described above.
The free energies of binding ΔGbind for the wild‐type (WT) and mutant SMO/vismodegib complexes were obtained using an extensively validated procedure based on the Molecular Mechanics/Poisson–Boltzmann Surface Area (MM‐PBSA) methodology. (Bozzi et al., 2013; Laurini et al., 2013; Conca et al., 2013; Dileo et al., 2011; Pierotti et al., 2011; Gibbons et al., 2014) Accordingly, ΔGbind values were calculated for equilibrated structures extracted from the corresponding MD trajectories. The average value of ΔGbind was estimated as the sum of different energetic contributions, corresponding to the average molecular mechanics energies (ΔEMM = ΔEele + ΔEvdW), the average solvation free energy (ΔGsolv = ΔGsolv,pol + ΔGsolv,nonpol), and the entropic contribution (−TΔS). The molecular mechanics energies (ΔEMM) were evaluated from a single 50 ns MD trajectory of each receptor/inhibitor complex. The solvation energies were obtained solving the Poisson–Boltzmann equation for the polar part (ΔGsolv,pol) plus a non‐polar contribution (ΔGsolv,nonpol), proportional to the solvent‐accessible surface area and including the entropy cost of creating a solute‐size cavity in the solvent. Finally, the variation of entropy upon binding was evaluated by utilizing the quasi‐harmonic approach. (Andricioaei and Karplus, 2011).
A per residue binding free energy decomposition was performed exploiting the MD trajectory of each complex. This analysis was carried out using the MM/GBSA approach, (Tsui and Case, 2000; Onufriev et al., 2000) and was based on the same snapshots used in the binding free energy calculation.
The steered molecular dynamics (SMD) simulations (Isralewitz et al., 2001) required to pull off the inhibitor from its binding site were performed using snapshots randomly taken from the MD equilibrated runs as initial structures. The center of mass of vismodegib was chosen as the acting point for the pulling force. Harmonic constraints were applied to the Cα atoms of all helices in the SMO, while the exponent appearing in the harmonic constraint energy functions was set to the value of 2. The force constant and velocity used in all SMD simulations were 4 kcal mol−1 Å−2 (277.9 pN Å−1) and 0.0001 Å timestep−1 (0.030 Å ps−1), respectively. With these settings, 3.0 nm were covered in 1000 ps of SMD simulation. The same parameters were exploited to simulate the entry process of vismodegib into the SMO binding pocket.
The entire MM/PBSA computational procedure was optimized by integrating AMBER 12 in modeFRONTIER, (http://www.esteco.com/home/mode_frontier/mode_frontier.htm) a multidisciplinary and multi‐objective optimization and design environment, and run on the EURORA supercomputer (CINECA, Bologna, Italy).
3. Results
Two patients enrolled in a phase II trial with vismodegib for metastatic and locally advanced BCC (ClinicalTrials.gov Identifier: NCT01367665) are reported and discussed in this work as examples of primary and acquired resistance to the vismodegib, respectively.
3.1. Patient 1: a case of vismodeginb primary resistance in BCCs
The first patient was an 82‐years old woman presenting with a BCC metastatic to liver, lung and bones. After histological confirmation of liver metastasis, she received radiation therapy to lumbar (L2‐L4) and cervical (C3) secondary lesions and she started vismodegib 150 mg/die. After 2 months of continuous treatment, CT scans showed disease progression at all sites (primary resistance).
3.2. Patient 2: a case of vismodegib acquired resistance in BCCs
The second patient was a 78‐years old man presenting with a large (12 cm), ulcerated lesion of the supra‐scapular skin on which a diagnosis of BCC was rendered (Figure S1, left panel). Because a surgical approach would have led to substantial morbidity, this patient was started on a 150 mg/day vismodegib regimen. The lesion dramatically reduced after the first month, and a complete clinical response was obtained 5 months later (Figure S1, right panel). However, after 11 months on vismodegib, two subcutaneous nodules appeared in the area of the previous lesion. Both lesions were surgically removed and the histology was consistent with recurrent BCC suggesting the development of an acquired resistance.
3.3. PTCH1 is wild type in vismodegib primary resistance and mutated in acquired resistance to SMO in BCC
To explore possible mechanisms of Hh pathway activation in these two BCC cases considered, we first performed PTCH1 sequencing. In the pre‐treatment BCC liver metastasis of the case showing primary resistance (patient 1), no PTCH1 mutation was observed (Figure 1A). By contrast, the pre‐treatment primary biopsy of the case showing acquired resistance (patient 2) revealed the PTCH1 nonsense mutation CAG > TAG creating a premature STOP codon (pQ84) in the exon 2 (Figure 1B). This PTCH1 mutation predicts translation to a heavily truncated protein, causing loss of function of the protein and preventing inhibition of SMO. As expected, the same PTCH1 mutation was also observed in the recurrence sample taken during vismodegib (Figure 1B).
Figure 1.

Pre‐treatment BCC liver metastasis of the first case (patient 1, primary resistance) showed PTCH1 wild type gene (A) and the SMO G49W mutation (C). Pre‐treatment primary tumor and BCC recurrence of the second case (patient 2, acquired resistance) carried the nonsense Q84Stop PTCH1 mutation (B) while the SMO D473Y mutation was observed only in the recurrence BCC sample (D).
3.4. Two distinct missense SMO mutations characterize primary and secondary vismodegib resistance in BCCs
Next, we evaluated the SMO mutational status. In the case showing primary resistance (patient 1), the analysis of pre‐treatment liver metastasis revealed the SMO missense mutation GGG > TGG at exon 9, leading to the amino acid substitution G497W in the corresponding protein (Figure 1C). Primary tumor and node metastasis also exhibited the same SMO missense mutation GGG > TGG at exon 9. By contrast, in the pre‐treatment primary BCC of the second case (acquired resistance, patient 2), no SMO mutations were detected (Figure 1D, left panel); however the sample obtained from the recurrence on vismodegib showed the missense mutation GAC > TAC at exon 8, leading to the single amino acid substitution D473Y (Figure 1D, right panel) confirming the acquired resistance to the inhibitor.
3.5. SMOG497W: a distal mutation that obstructs vismodegib entry to SMO binding site leading to primary BCC resistance
To gain mechanistic insight regarding the resistance posed by the mutant SMO proteins we performed a thorough in silico analysis of the wild type and both clinically relevant SMO mutant alleles (Figure 2).
Figure 2.

Cross section of the 3D structure of the SMO receptor embedded in a 1‐hexadecanoyl‐2‐[(9Z)‐octadecenoyl]‐sn‐glycero‐3‐phosphocholine (POPC)/1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphoethanolamine (POPE) (2:2) membrane model. Water is shown as a light cyan surface, while Na+ and Cl− ions are visible as green and purple spheres, respectively. Lipids are portrayed as ball‐and‐sticks, the polar heads of POPC and POPE depicted in white and cyan, respectively, while the corresponding hydrophobic tails are colored green and salmon, respectively. The membrane solvent accessible surface area is highlighted in transparent forest green. The SMO receptor protein is shown as a red ribbon, the inhibitor binding region being evidenced by a yellow sphere.
As shown in Table 1, the MM/PBSA‐derived IC50 for the SMOG497W/vismodegib complex is 69 nM, a value slightly higher than the experimental/calculated IC50 derived for the wild‐type (WT) receptor (3 and 2.5 nM, respectively). This result indicates only a moderate direct effect of the mutation G497W on the protein affinity for the inhibitor, in keeping with a distal position of G497W with respect to the protein drug binding site.
Table 1.
In silico estimation of free energy of binding (ΔGbind) for wild type (WT), SMOD473H and SMOG497W mutant receptors in complex with vismodegib. Errors are given in parenthesis as standard errors of the mean (SEM).
| SMO complex | ΔGbind (kcal/mol) | a ΔΔGbind (kcal/mol) | b IC50,calc (kcal/mol) | IC50,exp (kcal/mol) |
|---|---|---|---|---|
| WT | −11.75 (0.11) | – | 2.5 | 3 |
| D473H | −9.27 (0.10) | −2.48 | 159 | – |
| G497W | −9.78 (0.10) | −1.97 | 69 | – |
ΔΔGbind = ΔGbind,WT − ΔGbind,mutant. By definition, a negative value of ΔΔGbind indicates that the WT/vismodegib complex is favored with respect to the mutated one, and vice versa.
ΔGbind and the concentration of ligand that inhibits the kinase activity by 50% (i.e., IC50) are related by the following fundamental equation: ΔGbind = ‐RTln 1/IC50. Thus, once ΔGbind for a given kinase/inhibitor couple is estimated by MM‐PBSA simulations, the relative IC50 value is also known by virtue of this relationship.
However, in the presence of the mutant residue the entire protein region undergoes a conformational rearrangement, ultimately resulting in a partially obstructed drug entry site. Our simulations of the binding process of vismodegib to SMO indeed shows that the vismodegib link with the protein binding pocket is substantially hindered in the presence of the G497W mutation (Figure 3A–B and movies).
Figure 3.
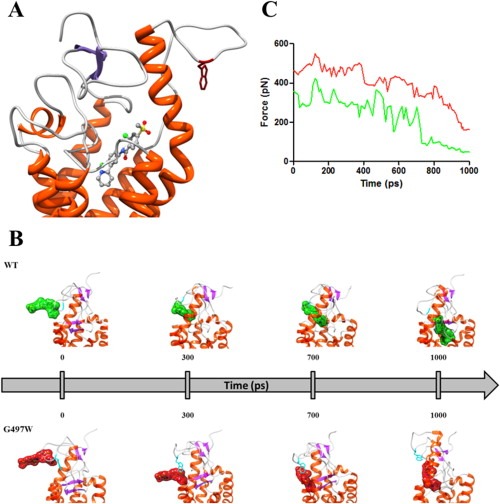
(A) Zoomed view of the SMOG497W binding site in complex with vismodegib. The receptor is shown as a secondary‐structure colored ribbon (orange, α‐helices; purple, β‐sheets; gray, coils). Vismodegib is portrayed as atom‐colored sticks‐and‐balls (red, O; blue, N; green, Cl; S, sulfur; gray, C). Residue W497 is evidenced as dark red sticks. Hydrogen atoms, water molecules, ions and counterions are omitted for clarity. (B) SMD snapshots of vismodegib entering the receptor binding pocket. Vismodegib is highlighted by its green/red van der Waals surface. Hydrogen atoms, ions, counterions and water molecules are omitted for clarity. (C) Rupture force vs. time during the entry process of vismodegib within the WT (green) and SMOG497W (red) binding site.
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.molonc.2014.09.003
The following are the Supplementary data related to this article:
This fundamentally implies that, in time, the effective vismodegib concentration within the SMOG497W binding site is significantly lower than in the case of the WT receptor. Accordingly to our simulation, other molecular mechanisms are likely contributing to the resistance of SMOG497W to vismodegib. A direct comparison of the WT/mutated protein structures in the area surrounding position 497 revealed that, in the presence of the tryptophan mutant residue, the entire region undergoes a conformational rearrangement, thus resulting in a narrowing of the protein drug entry site (Figure 3A). For this reason, in the presence of the G497W mutation vismodegib might be less able to reach the protein binding pocket and, hence, less effective in its inhibitory activity. Further, as we can see from Figure 3B and 3C, not only quite a higher force (and hence energy) is required to vismodegib to reach its binding site in the presence of the mutant residue (Figure 3C) but also, and perhaps even more interestingly, contrarily to the WT case, for SMOG497W the drug has not yet reached its correct orientation within the protein binding pocket after the same time lag (Figure 3B and movies). In practical terms this fundamentally might imply that, in time, the effective vismodegib concentration within the SMOG497W binding site is significantly lower than in the case of the WT receptor and this, is our opinion, constitutes a novel and effective mechanism of drug primary resistance.
3.6. SMOD473Y: a binding site mutation that directly interferes with vismodegib binding and leads to secondary BCC resistance
When considering vismodegib in complex with SMOD473Y, the alternative mutant isoform of SMO detected in the patient specimen with acquired BCC resistance, the calculated IC50 value is 159 nM. This translates into an almost two orders of magnitude decrease in protein affinity to vismodegib with respect to the WT receptor (Table 1), clearly revealing a direct effect of the mutated residue on vismodegib binding. Notably, D473 is involved with other two residues (R400 and H470) in a pivotal network of hydrogen bonds that keeps the SMO binding pocket in the proper shape and stabilizes vismodegib binding (Figure 4A–B). The aromatic side chain of the 473Y residue induces a considerable effect on the binding site geometry and leads to the total disruption of the stabilizing hydrogen bond network; hence, the inhibitor is shifted from its optimal position (Figure 4B). In details, in the presence of the D473Y mutation, a considerable effect on the binding site geometry is induced, required for the proper accommodation of the residue aromatic side chain, ultimately leading to the total disruption of the stabilizing hydrogen bond network. The inhibitor is hence shifted from its optimal position, the only surviving interaction with the receptor being an intermitted hydrogen bond with Y394.
Figure 4.
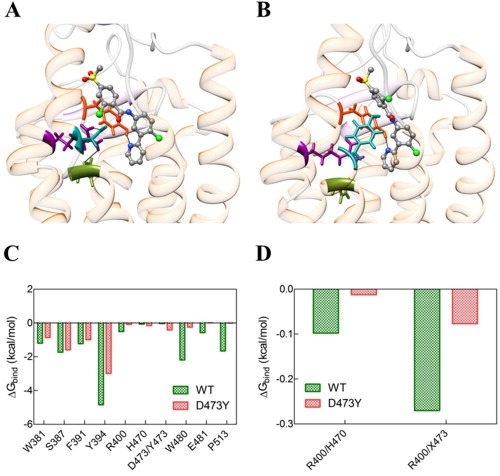
(A) Zoomed views of the wild type (WT) and (B) SMOD473Y mutant binding sites in complex with vismodegib. In both panels, the receptor secondary structure is outlined as a semi‐transparent ribbon (orange, α‐helices; purple, β‐sheets; gray, coils). Vismodegib is portrayed as atom‐colored sticks‐and‐balls (red, O; blue, N; green, Cl; S, sulfur; gray, C). The triad of residues involved in the hydrogen‐bond network are highlighted colored sticks: R300, dark magenta; H470, olive drab; D/Y473, dark cyan. Y394 is also shown as dark red sticks. H‐bonds are evidenced as black lines. (C) Comparison of vismodegib binding energy contributions from WT (green) and D473Y (red) SMO residues. (D) Comparison between hydrogen bond network stabilization energies for SMO residues belonging to the WT (green) and SMOD473Y triad residues in the relevant vismodegib complexes. X denotes either D or Y residue at position 473.
These evidences are substantiated by the corresponding per residue energy deconvolution of the free energy of binding, as shown in Figure 4C–D. As we see, all receptor residues mainly involved in vismodegib binding undergo a severe reduction in the stabilizing contribution to inhibitor binding in the presence of the mutation (Figure 4C). Analogously, upon disruption of the hydrogen bond network among the SMO triad residues, the relevant, favorable contribution to receptor/inhibitor binding plummets drastically (Figure 4D).
4. Discussion
This study presents for the first time clinical, molecular and in silico evidence of primary and acquired SMO mutation‐mediated resistance to vismodegib in BCC. Indeed, the true‐cut based assessment of the SMO G497W missense mutation in the liver pre‐treatment metastasis of the first patient exhibiting a progression of disease after two months on vismodegib provides a good example of primary resistance to the drug. To our knowledge, this SMO mutation has never been previously described. However, its location in the most frequently mutated SMO region in BCC (exons 8–10) where other SMO mutations have already shown a constitutive ligand‐independent signal transduction, argues in favor of its oncogenic role. (Xie et al., 1998) Furthermore, since the lack of a PTCH1 inactivating mutation reinforced this assumption, we sought to explore the effects exerted by this mutation on the inhibitor binding to the mutated SMO protein. By in silico experiments we demonstrated that in the presence of the W497 residue the entire protein region undergoes a conformational rearrangement, which ultimately results in a partial obstruction of the protein drug entry site. This obstruction, leading to a significant decreasing of the effective vismodegib concentration within the SMOG497W binding site (lower than that in the case of the WT receptor), gives a mechanistic explanation for a novel effective drug resistance. In this respect, the presence of such a SMO mutation in BCC candidate to vismodegib should definitely be assessed in a greater number of tumors with primary resistance to the inhibitor. This would eventually allow defining SMOG497W as a possible biomarker for drug resistance, ultimately resulting in the avoidance of unnecessary toxicity effects and costs limitation to non‐responding patients.
Concerning the second patient, the presence of the PTCH1 inactivating mutation in the pre‐treatment primary lesion along with absence of SMO mutation could explain the complete clinical response obtained after 6 months of continuous vismodegib treatment. Indeed, loss of PTCH1 function by inactivating mutations relieves normal SMO inhibition leading to the activation of Hh targets genes. By contrast, the secondary drug resistance observed after 11 months is likely to be ascribable to the appearance of the missense SMO D473Y substitution in the recurrence sample. Notably, another amino acid substitution of the same SMO aspartic acid (D473H) was designated as the mechanism of vismodegib resistance in medulloblastoma. (Yauch et al., 2009) More recently, the detrimental role of D473 in SMO function has been found out by an elegant in vitro study where, by replacing this aspartic acid residue with every other amino acid, all mutants were resistant to vismodegib. (Dijkgraaf et al., 2011) In keeping with these findings, our analysis of the structural effects of the SMO D473Y clearly revealed a direct and significant effect on the binding site geometry. The inhibitor is shifted from its optimal position because the aromatic side chain of the 473Y residue causes the total disruption of the stabilizing hydrogen bond network involving the D473 with other two residues (R400 and H470). As for G497W, this SMO point mutation should also be confirmed in a greater series of BCCs in order to definitely claim its role in secondary resistance to vismodegib. As a general remark, we acknowledge that the presented evidences could be considered only reasonably correlated to the resistance phenotype, and indeed our analysis requires further confirmation through functional analysis. Under this perspective, in vitro and in vivo experiments are under way in our laboratories to definitively assess the role of these two SMO mutations as possible biomarkers for vismodegib resistance.
Similarly to medulloblastoma, our data further raise the issue of overcoming the resistance due to mutation of the drug target. Under this perspective, efforts aimed at identifying second‐generation drugs showing potent activity against SMO mutants are mandatory. To this goal, a panel of compounds has already been screened in vitro. Several promising antagonists able to inhibit tumor growth mediated by drug‐resistant SMOs were selected in murine allograft model of medulloblastoma, (Dijkgraaf et al., 2011) that could be tested also in BCC.
Another strategy worth pursuing is the inhibition of the Hh pathway through other mechanisms, the activity of itraconazole or arsenic oxide in blocking vismodegib‐resistant BCC both in vitro and in vivo being prime examples. (Kim et al., 2013, 2014).
A further alternative approach for facing vismodegib resistance could consist in exploring other pathways interacting with Hh signaling in BCC and leading to an SMO‐independent stimulation of Gli1 such as EGFR, (Eberl et al., 2012) the atypical protein kinase C ι/λ, (Atwood et al., 2013) the protein kinase A (Makinodan and Marneros, 2012) and, similarly to medulloblastoma, the PI3K pathway. (Dijkgraaf et al., 2011; Buonamici et al., 2010).
Finally, a clinical point to be stressed is that our two cases of vismodegib resistance occurred in patients who had not received previous radiation therapy. We cannot exclude that Hh inhibitor resistance outbreak could be higher in patients having received a radiation insult, as well as it is possible that alternative therapeutic schedules may alter resistance mechanism development. (Buonamici et al., 2010).
In this report we discussed two different SMO mutations representing examples of primary or secondary resistance respectively to vismodegib in two distinct BCC cases. The screening for the reported SMO mutations and the search for new therapeutic strategies to overcome related resistance mechanisms represent a priority in the treatment of patients with advanced BCCs.
Conflict of interest
The authors declare they have no potential conflicts of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Acknowledgment
We would like to acknowledge Roche‐Genentech for reviewing the manuscript for scientific accuracy. Part of this work was carried out in the framework of the HPC‐Europa 2 projects INSIDER and SYMBIOSI (CINECA Supercomputing Center, Bologna, Italy), funded by the European Commission – DG Research in the 7th Framework Program (Grant agreement n° 228398), granted to E. Laurini and S. Pricl.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.09.003.
Pricl Sabrina, Cortelazzi Barbara, Dal Col Valentina, Marson Domenico, Laurini Erik, Fermeglia Maurizio, Licitra Lisa, Pilotti Silvana, Bossi Paolo, Perrone Federica, (2015), Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.09.003.
Contributor Information
Sabrina Pricl, Email: sabrina.pricl@di3.units.it.
Federica Perrone, Email: federica.perrone@istitutotumori.mi.it.
References
- Andricioaei, I. , Karplus, M. , 2011. On the calculation of entropy from covariance matrices of the atomic fluctuations. J. Chem. Phys. 115, 6289–6292. [Google Scholar]
- Atwood, S.X. , Li, M. , Lee, A. , Tang, J.Y. , Oro, A.E. , 2013. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature. 494, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly, C.I. , Cieplak, P. , Cornell, W.D. , Kollman, P.A. , 1993. A well-behaved electrostatic potential based method using charge restraints for determining atom-centered charges: the RESP model. J. Phys. Chem. 97, 10269–10280. [Google Scholar]
- Berendsen, H.J.C. , Postma, J.P.M. , Van Gunsteren, W.F. , DiNola, A. , Haak, J.R. , 1984. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Besler, B.H. , Merz, K.M. , Kollman, P.A. , 1990. Atomic charges derived from semiempirical methods. J. Comput. Chem. 11, 431–439. [Google Scholar]
- Bozzi, F. , Conca, E. , Laurini, E. , Posocco, P. , Lo Sardo, A. , Jocollè, G. , Sanfilippo, R. , Gronchi, A. , Perrone, F. , Tamborini, E. , Pelosi, G. , Pierotti, M.A. , Maestro, R. , Pricl, S. , Pilotti, S. , 2013. In vitro and in silico studies of MDM2/MDMX isoforms predict Nutlin-3A sensitivity in well/de-differentiated liposarcomas. Lab. Invest. 93, 1232–1240. [DOI] [PubMed] [Google Scholar]
- Buonamici, S. , Williams, J. , Morrissey, M. , Wang, A. , Guo, R. , Vattay, A. , Hsiao, K. , Yuan, J. , Green, J. , Ospina, B. , Yu, Q. , Ostrom, L. , Fordjour, P. , Anderson, D.L. , Monahan, J.E. , Kelleher, J.F. , Peukert, S. , Pan, S. , Wu, X. , Maira, S.M. , García-Echeverría, C. , Briggs, K.J. , Watkins, D.N. , Yao, Y.M. , Lengauer, C. , Warmuth, M. , Sellers, W.R. , Dorsch, M. , 2010. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2, 51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D.A. , Darden, T.A. , Cheatham, T.E. , 2012. AMBER 12 University of California; San Francisco: [Google Scholar]
- Conca, E. , Miranda, C. , Dal Col, V. , Fumagalli, E. , Pelosi, G. , Mazzoni, M. , Fermeglia, M. , Laurini, E. , Pierotti, M.A. , Pilotti, S. , Greco, A. , Pricl, S. , Tamborini, E. , 2013. Are two better than one? A novel double-mutant KIT in GIST that responds to Imatinib. Mol. Oncol. 7, 756–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkgraaf, G.J. , Alicke, B. , Weinmann, L. , Januario, T. , West, K. , Modrusan, Z. , Burdick, D. , Goldsmith, R. , Robarge, K. , Sutherlin, D. , Scales, S.J. , Gould, S.E. , Yauch, R.L. , de Sauvage, F.J. , 2011. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71, 435–444. [DOI] [PubMed] [Google Scholar]
- Dileo, P. , Pricl, S. , Tamborini, E. , Negri, T. , Stacchiotti, S. , Gronchi, A. , Posocco, P. , Laurini, E. , Coco, P. , Fumagalli, E. , Casali, P.G. , Pilotti, S. , 2011. Imatinib response in two GIST patients carrying two hitherto functionally uncharacterized PDGFRA mutations: an imaging, biochemical and molecular modeling study. Int. J. Cancer. 128, 983–990. [DOI] [PubMed] [Google Scholar]
- Duan, Y. , Wu, C. , Chowdhury, S. , Lee, M.C. , Xiong, G. , Zhang, W. , Yang, R. , Cieplak, P. , Luo, R. , Lee, T. , 2003. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- Eberl, M. , Klingler, S. , Mangelberger, D. , Loipetzberger, A. , Damhofer, H. , Zoidl, K. , Schnidar, H. , Hache, H. , Bauer, H.C. , Solca, F. , Hauser-Kronberger, C. , Ermilov, A.N. , Verhaegen, M.E. , Bichakjian, C.K. , Dlugosz, A.A. , Nietfeld, W. , Sibilia, M. , Lehrach, H. , Wierling, C. , Aberger, F. , 2012. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumor-initiating pancreatic cancer cells. EMBO Mol. Med. 4, 218–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohil, S.C. , Seubring, I. , van Rossum, M.M. , Coebergh, J.W. , de Vries, E. , Nijsten, T. , 2013. Trends in Basal cell carcinoma incidence rates: a 37-year Dutch observational study. J. Invest. Dermatol. 133, 913–918. [DOI] [PubMed] [Google Scholar]
- Gibbons, D.L. , Pricl, S. , Posocco, P. , Laurini, E. , Fermeglia, M. , Sun, H. , Talpaz, M. , Donato, N. , Quintás-Cardama, A. , 2014. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc. Natl. Acad. Sci. 111, 3550–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isralewitz, B. , Gao, M. , Schulten, K. , 2001. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11, 224–230. [DOI] [PubMed] [Google Scholar]
- Jo, S. , Kim, T. , Im, W. , 2007. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One. 2, (9) e880 10.1371/journal.pone.0000880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, W.L. , Chandrasekhar, J. , Madura, J.D. , Impey, R.W. , Klein, M.L. , 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Kim, J. , Aftab, B.T. , Tang, J.Y. , Kim, D. , Lee, A.H. , Rezaee, M. , Kim, J. , Chen, B. , King, E.M. , Borodovsky, A. , Riggins, G.J. , Epstein, E.H. , Beachy, P.A. , Rudin, C.M. , 2013. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 23, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.J. , Kim, J. , Spaunhurst, K. , Montoya, J. , Khodosh, R. , Chandra, K. , Fu, T. , Gilliam, A. , Molgo, M. , Beachy, P.A. , Tang, J.Y. , 2014. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 32, 745–751. [DOI] [PubMed] [Google Scholar]
- Laurini, E. , Posocco, P. , Fermeglia, M. , Gibbons, D.L. , Quintás-Cardama, A. , Pricl, S. , 2013. Through the open door: Preferential binding of dasatinib to the active form of BCR-ABL unveiled by in silico experiments. Mol. Oncol. 7, 968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan, E. , Marneros, A.G. , 2012. Protein kinase A activation inhibits oncogenic Sonic hedghog signalling and suppresses basal cel carcinoma of the skin. Exp. Dermatol. 21, 847–852. [DOI] [PubMed] [Google Scholar]
- Onufriev, A. , Bashford, D. , Case, D.A. , 2000. Modification of the generalized born model suitable for macromolecules. J. Phys. Chem. B. 104, 3712–3720. [Google Scholar]
- Pierotti, M.A. , Tamborini, E. , Negri, T. , Pricl, S. , Pilotti, S. , 2011. Targeted therapy in GIST: in silico modeling for prediction of resistance. Nat. Rev. Clin. Oncol. 8, 161–170. [DOI] [PubMed] [Google Scholar]
- Reifenberger, J. , Wolter, M. , Knobbe, C.B. , Kohler, B. , Schonicke, A. , Scharwachter, C. , Kumar, K. , Blaschke, B. , Ruzicka, T. , Reifenberger, G. , 2005. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 152, 43–51. [DOI] [PubMed] [Google Scholar]
- Rubin, A.I. , Chen, E.H. , Ratner, D. , 2005. Basal-cell carcinoma. New Engl. J. Med. 353, 2262–2269. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J.P. , Ciccotti, G. , Berendsen, H.J.C. , 1977. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Sekulic, A. , Migden, M.R. , Oro, A.E. , Dirix, L. , Lewis, K.D. , Hainsworth, J.D. , Solomon, J.A. , Yoo, S. , Arron, S.T. , Friedlander, P.A. , Marmur, E. , Rudin, C.M. , Chang, A.L.S. , Low, J.A. , Mackey, H.M. , Yauch, R.L. , Graham, R.A. , Reddy, J.C. , Hauschild, A. , 2012. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. New Engl. J. Med. 366, 2171–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, U.C. , Kollman, P.A. , 1984. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 5, 129–145. [Google Scholar]
- Skjevik, A.A. , Madej, B.D. , Walker, R.C. , Teigen, K. , 2012. LIPID11: a modular framework for lipid simulations using amber. J. Phys. Chem. B. 116, 11124–11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toukmaji, A. , Sagui, C. , Board, J. , Darden, T. , 2000. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 113, 10913–10927. [Google Scholar]
- Tsui, V. , Case, D.A. , 2000. Theory and applications of the Generalized Born solvation model in macromolecular simulations. Biopolymers. 56, 75–291. [DOI] [PubMed] [Google Scholar]
- Von Hoff, D.D. , LoRusso, P.M. , Rudin, C.M. , Reddy, J.C. , Yauch, R.L. , Tibes, R. , Weiss, G.J. , Borad, M.J. , Hann, C.L. , Brahmer, J.R. , Mackey, H.M. , Lum, B.L. , Darbonne, W.C. , Marsters, J.C. , de Sauvage, F.J. , Low, J.A. , 2009. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. New Engl. J. Med. 361, 1164–1172. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Wolf, R.M. , Caldwell, J.W. , Kollman, P.A. , Case, D.A. , 2004. Development and testing of a general AMBER force field. J. Comput. Chem. 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Wang, W. , Kollman, P.A. , Case, D.A. , 2006. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Model. 25, 247–260. [DOI] [PubMed] [Google Scholar]
- Woolf, T.B. , Roux, B. , 1996. Structure, energetics, and dynamics of lipid-protein interactions: a molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins. 24, 92–114. [DOI] [PubMed] [Google Scholar]
- Xie, J. , Murone, M. , Luoh, S.M. , Ryan, A. , Gu, Q. , Zhang, C. , Bonifas, J.M. , Lam, C.W. , Hynes, M. , Goddard, A. , Rosenthal, A. , Epstein, E.H. , de Sauvage, F.J. , 1998. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 391, 90–92. [DOI] [PubMed] [Google Scholar]
- Yauch, R.L. , Dijkgraaf, G.J. , Alicke, B. , Januario, T. , Ahn, C.P. , Holcomb, T. , Pujara, K. , Stinson, J. , Callahan, C.A. , Tang, T. , Bazan, J.F. , Kan, Z. , Seshagiri, S. , Hann, C.L. , Gould, S.E. , Low, J.A. , Rudin, C.M. , de Sauvage, F.J. , 2009. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 326, 572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data