

Abstract
BRCA1, a key factor in homologous recombination (HR) repair may also regulate base excision repair (BER). Targeting BRCA1‐BER deficient cells by blockade of ATM and DNA‐PKcs could be a promising strategy in breast cancer. We investigated BRCA1, XRCC1 and pol β protein expression in two cohorts (n = 1602 sporadic and n = 50 germ‐line BRCA1 mutated) and mRNA expression in two cohorts (n = 1952 and n = 249). Artificial neural network analysis for BRCA1‐DNA repair interacting genes was conducted in 249 tumours. Pre‐clinically, BRCA1 proficient and deficient cells were DNA repair expression profiled and evaluated for synthetic lethality using ATM and DNA‐PKcs inhibitors either alone or in combination with cisplatin. In human tumours, BRCA1 negativity was strongly associated with low XRCC1, and low pol β at mRNA and protein levels (p < 0.0001). In patients with BRCA1 negative tumours, low XRCC1 or low pol β expression was significantly associated with poor survival in univariate and multivariate analysis compared to high XRCC1 or high pol β expressing BRCA1 negative tumours (ps < 0.05). Pre‐clinically, BRCA1 negative cancer cells exhibit low mRNA and low protein expression of XRCC1 and pol β. BRCA1‐BER deficient cells were sensitive to ATM and DNA‐PKcs inhibitor treatment either alone or in combination with cisplatin and synthetic lethality was evidenced by DNA double strand breaks accumulation, cell cycle arrest and apoptosis. We conclude that XRCC1 and pol β expression status in BRCA1 negative tumours may have prognostic significance. BRCA1‐BER deficient cells could be targeted by ATM or DNA‐PKcs inhibitors for personalized therapy.
Keywords: BRCA1, Base excision repair, ATM, DNA‐PK, Small molecule inhibitors, Synthetic lethality, Cisplatin, Chemopotentiation
Highlights
BRCA1 may regulate base excision repair (BER).
BRCA1‐BER deficient cells may be sensitive to blockade of ATM and DNA‐PKcs.
We show that low XRCC1/low pol β have prognostic significance in BRCA1−/− tumours.
ATM or DNA‐PKcs inhibitors are synthetically lethal in BRCA1‐BER deficient cells.
Synthetic lethality is enhanced in combination with cisplatin.
1. Introduction
Breast Cancer Susceptibility Gene 1 (BRCA1) facilitates the efficient resolution of DNA double strand breaks (DSBs) through HR (Caestecker and Van de Walle, 2013; Huen et al., 2010). Cells lacking functional BRCA1 protein have impaired HR, and thus depend on the more error‐prone non‐homologous end joining (NHEJ) pathway leading to chromosomal instability that drive breast cancer development (Huen et al., 2010). In women, BRCA1 germ‐line mutation is associated with a 60%–70% lifetime risk of developing breast cancer (O'Donovan and Livingston, 2010). In the more common sporadic breast cancers, epigenetic silencing of the BRCA1 promoter has been reported in up to 11%–14% of tumours (Turner et al., 2004) and a dysfunctional BRCA pathway may also contribute to a BRCAness phenotype in about 25% of cancers (Turner et al., 2004), where breast cancers do not harbour germ‐line BRCA mutations but display similar phenotypes including HR deficiency. BER is critical for processing DNA damage caused by alkylation, oxidation, ring saturation, single strand breaks and base deamination. DNA polymerase β (pol β) and XRCC1 are key BER factors. PARP1 (poly [ADP‐ribose] polymerase 1) may play an essential role in single strand break repair (SSBR), a BER‐related pathway (Langelier and Pascal, 2013). The DNA repair intermediates generated during BER/SSBR, if unrepaired, may get converted to toxic double strand breaks (DSBs) (Dianov and Hubscher, 2013).
Emerging studies suggest a cross talk between BRCA1 and BER factors. BRCA1 mutated and basal‐like breast cancer cells were found to be sensitive to oxidative DNA damage induced by H2O2 treatment. The increased sensitivity was associated with defective BER as assessed by cell based BER assay in BRCA1 deficient cells (Alli et al., 2009). In a more recent study, BRCA1 deficient cells were sensitive to methyl methane sulfonate (alkylating agent) and functional interaction between pol β and BRCA1 was demonstrated in that study (Masaoka et al., 2013) implying a potential role for pol β in BRCA1 mediated DSB repair. In addition, BRCA1 has also been shown to be involved in the transcriptional regulation of BER factor such as OGG1, NTH1 and APE1 (Saha et al., 2010).
Synthetic lethality is a promising strategy for personalized cancer therapy. PARP [poly‐(ADP‐ribose) polymerase] inhibitors induce synthetic lethality in germ‐line BRCA1 deficient breast cancers and demonstrate clinical benefit in patients (Lord and Ashworth, 2008). Similarly, we have recently shown that APE1 inhibition is synthetically lethal in BRCA1 deficient breast cancer cells and in PTEN (and DSB repair) deficient melanoma cells. The data provides compelling reasons to investigate other potential synthetic lethal interactions targeting DNA repair for clinical application. Cells that are BRCA1 deficient as well as BER impaired may be reliant upon other back‐up repair pathways to maintain genomic integrity and survival. ATM and DNA‐PKcs play essential roles in the DNA damage response (DDR) and link DNA damage sensing to DDR effectors that regulate cell cycle progression and DNA repair (Shiloh and Ziv, 2013). ATM, a member of the phosphatidylinositol‐3‐kinase‐like protein kinase (PIKK) family, is a key sensor and transducer of DNA damage signalling during HR (Lee and Paull, 2007; Shiloh and Ziv, 2013). ATM recruitment at sites of DNA damage may be dependent upon functional BRCA1 in cells (Lee et al., 2010). DNA‐PKcs is another key member of the PIKK family and a critical component of NHEJ pathway required for repair of DSBs generated throughout the cell cycle (Hill and Lee, 2010). BRCA1 through a role in DNA end‐processing may also be involved in the regulation of NHEJ (Durant and Nickoloff, 2005).
Our hypothesis is that impaired BER in BRCA1 deficient tumours may influence prognosis. BRCA1‐BER deficient cells may be reliant upon ATM or DNA‐PKcs mediated back‐up pathways for cellular survival and could be targeted by synthetic lethality using inhibitors of ATM or DNA‐PKcs.
2. Materials and methods
2.1. Clinical study
2.1.1. BRCA1 and BER protein expression analysis in Nottingham Tenovus Primary Breast Carcinoma cohort
The study was performed in a consecutive series of 1650 patients with primary invasive breast carcinomas who were diagnosed between 1986 and 1999 and entered into the Nottingham Tenovus Primary Breast Carcinoma series described previously (Sultana et al., 2013). Supplemental Table S1 summarizes patient demographics. Supplemental treatment data 1 summarizes various adjuvant treatments received by patients in this cohort.
2.1.2. BRCA1 and BER protein expression analysis in germ‐line BRCA1 deficient breast cancer
The demographics of a cohort of 50 germ‐line BRCA1 mutated breast cancers confirmed by genetic testing is shown in Supplementary Table S6. All patients received surgery, adjuvant chemotherapy and radiotherapy according to our institutional policy (supplementary treatment data 1).
2.1.3. Tissue microarrays (TMAs) and immunohistochemistry (IHC)
Tumours were arrayed in tissue microarrays (TMAs) and immunohistochemically profiled for BRCA1, APE1, XRCC1, pol β, and other biological markers (Supplementary Table S2) as previously described (Sultana et al., 2013). Supplementary Table S2 summarizes immunohistochemistry protocols for the markers tested using the Bond Max automated staining machine and Leica Bond Refine Detection kit (DS9800) according to manufacturer instructions (Leica Microsystems). We have recently published optimisation and specificity of XRCC1 and pol β antibody used in the current study (Abdel‐Fatah et al., 2014b; Sultana et al., 2013). To validate the use of TMAs for immunophenotyping, full‐face sections of 40 cases were stained and protein expression levels of the different antibodies were compared. The concordance between TMAs and full‐face sections was excellent (k = 0.8). Positive and negative (by omission of the primary antibody and IgG‐matched serum) controls were included in each run.
2.1.4. Evaluation of immune staining
The tumour cores were evaluated by specialist pathologists blinded to the clinicopathological characteristics of patients, in two different settings. There was excellent intra and inter‐observer agreements (k > 0.8; Cohen's κ and multi‐rater κ tests, respectively). Whole field inspection of the core was scored and intensities of nuclear staining were grouped as follows: 0 = no staining, 1 = weak staining, 2 = moderate staining, 3 = strong staining. The percentage of each category was estimated (0–100%). H‐scores (range 0–300) were calculated by multiplying intensity of staining and percentage staining as previously described (Sultana et al., 2013). Supplementary Table S2 summarizes cut‐offs for individual markers.
2.1.5. Statistical analysis
Data analysis was performed using SPSS (SPSS, version 17 Chicago, IL). Where appropriate, Pearson's Chi‐square, Fisher's exact, Student's t and ANOVA one way tests were used. Cumulative survival probabilities were estimated using the Kaplan–Meier method, and differences between survival rates were tested for significance using the log‐rank test. Multivariate analysis for survival was performed using the Cox proportional hazard model. A p value <0.05 considered significant. For multiple comparisons, p values were adjusted according to Holm‐Bonferroni correction method.
2.1.6. Transcript levels in the METABRIC (molecular taxonomy of breast cancer international consortium) cohort
Investigation of the mRNA expression was performed in METABRIC cohort which refers to a set of 1980 breast cancer samples with a minimum of 5 years of clinical follow up where mRNA expression data was available (Curtis et al., 2012). Patient demographics are summarized in Supplementary Table S9. ER positive and/or lymph‐node negative patients did not receive adjuvant chemotherapy. ER negative and/or lymph‐node positive patients received adjuvant chemotherapy. All the samples were analysed as triplicates. A sliding window analysis was used to identify a cut‐off in gene expression values such that the resulting subgroups have significantly different survival courses.
2.1.7. Artificial neural network (ANN) analysis in Uppsala cohort
The demographics of the Uppsala cohort is summarized in Supplementary Table S10 and mRNA analysis has been described previously (Bergh et al., 1995). All microarray data are accessible at National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accession number: GSE4922) (Pawitan et al., 2005). All data were normalized using the global mean method (MAS5), and probe set signal intensities were natural log transformed and scaled by adjusting the mean signal to a target value of log 500. The expression levels of the BRCA1, for the probe 204531_s_at located on the HG‐U133A chip was utilized to generate the ANN based model as described previously (30) (Lancashire et al., 2010) (Lemetre et al., 2009). A non‐linear, ANN modelling based, data mining approach was utilised to identify the best gene probes for sample classification as described previously (30). 47,293 probes were screened for each sample in the test set (n = 249). The data mining algorithm comprised a three layer multilayer perception architecture modified with a feed forward back‐propagation algorithm and a sigmoidal transfer function, as previously described (Lancashire et al., 2010). The network momentum and learning rate were respectively set as 0.1 and 0.5. Two hidden nodes were utilised. The output node was coded as 0 if a case was low BRCA1 expression (<the median) and 1 if high BRCA1 expression (> median). Inputs were ranked in ascending order based on their classification error. The top 50 predictive genes identified were merged with 150 gene probes involved in the DNA repair process (Supplementary Table S11) and then applied to an ANN based network inference algorithm as described in earlier studies (Lemetre et al., 2009). This model predicted a weighted link (direction and magnitude) between each of the gene probe markers. This weighting was based on the non‐linear correlation between a source gene and a target gene in a multifactorial ANN model. This approach defines a linkage or interaction with a magnitude between every possible pair of genes in the set presented to the algorithm. The approach is data driven and unweighted by biological function. The 100 strongest interactions were then visualised as a map with Cytoscape (Smoot et al., 2011). In a second bioinformatics analysis step, we sought to obtain a robust ranking of genes that are differentially expressed between the mRNA BRCA1+ cases and the mRNA BRCA1− and have high predictive power, by applying an ensemble sample classification method within a leave‐one‐out cross‐validation scheme. For this purpose, the 249 patient samples were first grouped into 249 different training/test set partitions, using 248 samples for the training sets and the remaining sample as the test set. For each of the 248 training sets differentially expressed genes were selected independently with the “Empirical Bayes moderated t‐statistic” (Smyth, 2004) and used to train a machine learning model, which was evaluated based on the left‐out sample (a procedure known as “external cross‐validation”). To classify the left‐out sample, the prediction results of four algorithms (Support Vector Machine, Random Forest, kNN and Prediction Analysis for Microarrays, with all parameters being optimised by using a grid search within a nested cross‐validation) (Tibshirani et al., 2002) were combined to a majority‐vote ensemble classifier as to compensate for the inevitable inherent biases and variances that exists amongst each of these machine learning algorithms. In order to rank the genes based on the cross‐validation results, their frequency of occurrence in the list of significantly differentially expressed genes (p value <0.05) across different cross‐validation cycles was recorded, and genes received higher scores the more often they had been selected. All steps of the analysis were conducted using an in‐house web‐application for microarray analysis, available at www.arraymining.net.
2.2. Pre‐clinical study
2.2.1. Compounds and reagents
ATM inhibitors (KU55933 and KU60019) and DNA‐PKcs inhibitors (NU7441 and NU7026) were purchased from Tocris Bioscience, UK. The compounds were dissolved in 100% DMSO and stored at – 20 °C. Cisplatin was obtained from Nottingham University Hospitals.
2.2.2. Cell lines and culture media
BRCA1 deficient HeLa SilenciX® cells and control BRCA proficient HeLa SilenciX® cells were purchased from Tebu‐Bio (www.tebu‐bio.com). SilenciX cells were grown in DMEM medium (with l‐Glutamine 580 mg/L, 4500 mg/L D19 Glucose, with 110 mg/L Sodium Pyruvate) supplemented with 10% FBS, 1% penicillin/streptomycin and 125 μg/ml Hygromycin B. MDA‐MB‐436 (BRCA1 deficient human breast cancer cells) was grown in DMEM (Sigma, UK) and MCF7 (BRCA1 proficient human breast cancer cells) was grown in RPMI1640 (Sigma, UK). All media used to culture human cancer cell lines were supplemented with 10% FBS (PAA, UK) and 1% penicillin/streptomycin.
2.2.3. Clonogenic survival assay
200–500 hundred cells per well were seeded in six‐well plates. Cells were allowed to adhere for 4 h. Compounds (ATM inhibitors or DNA‐PKcs inhibitors) were added at the indicated concentrations. For cisplatin combination studies, cells were initially treated with cisplatin for 16 h and then gently washed twice with 1X phosphate buffered saline and incubated in fresh media with or without ATM or DNA‐PK inhibitors at indicated concentration. The plates were left in the incubator for 12–14 days. After incubation, the media was discarded, fixed (with methanol and acetic acid mixture) and stained with crystal violet and counted. Surviving Fraction = [No. of colonies formed/ (No. of cells seeded × Plating efficiency)] ×100. All clonogenic assays were done in triplicate.
2.2.4. Evaluation of drug interaction (combination index)
To investigate synergistic and additive activity, combination index was calculated as described previously (Berenbaum, 1981). If D (combination index) is <1 the effect of the combination is synergistic, whereas if D = 1 or D is >1 the effect is additive or antagonistic respectively.
2.2.5. γH2AX immunofluorescence microscopy
This assay was performed as described previously (Sultana et al., 2013). Briefly, cells were incubated in medium containing ATM inhibitor or DNA‐PKCS inhibitor for 48 h. For cisplatin combination studies, cells were initially treated with cisplatin for 16 h and then gently washed twice with 1X phosphate buffered saline and incubated in fresh media with or without ATM or DNA‐PK inhibitors at indicated concentration for 48 h.
2.2.6. Neutral COMET assay
COMET assay reagents were purchased from Trevigen and used according to the manufacturer's neutral COMET protocol (Gaithersburg, MD, USA). Briefly, 1 × 105 cells per well were seeded into a 6 well plate and treated with KU55933 (7.5 μM) or NU7441 (0.75 μM). After 48 h treatment cells were resuspended in 250 μl PBS. For COMET analysis 25 μl of cell suspension was mixed with 250 μl molten LMAgarose and then 75 μl of the mixture pipetted immediately onto COMET slides. Slides were allowed to set at 4 °C for 20 min and then immersed in lysis buffer for 60 min at 4 °C. Slides were then subjected to electrophoresis at 20 V for 60 min in chilled electrophoresis buffer (0.5 M Tris, 1.5 M sodium acetate). Following electrophoresis slides were incubated for 30 min in DNA precipitation solution (5 M ammonium acetate in 95% ethanol) at room temperature and subsequently fixed in 70% ethanol for 20 min at room temperature. Slides were left to dry overnight at 4 °C and then each sample stained with 50 μl Sybr green and scored using COMET ASSAY IV software (Perceptive Instruments Ltd, Bury St Edmonds, UK). Treatments were performed in triplicate and 50 cells counted for each.
2.2.7. Flow cytometric analyses (FACS)
Cells grown to sub‐confluence were exposed to ATM or DNA‐PKcs inhibitors either alone or in combination with cisplatin for 48 h and collected by trypsinization and centrifugation (1000 rpm for 5 min). FACS was performed as described previously (Sultana et al., 2013).
2.2.8. Annexin V flow cytometric analyses
Cells grown to sub‐confluence were exposed to ATM or DNA‐PKcs inhibitors either alone or in combination with cisplatin for 48 h and collected by trypsinization and centrifugation (1000 rpm for 5 min). The assay was performed as described previously (Sultana et al., 2013).
2.2.9. Quantitative real‐time PCR
RNA was extracted from cell lines using the RNeasy Mini Kit (Qiagen) and quantified using a microvolume spectrophotometer. cDNA synthesis was performed using the RT2 First Strand Kit (Qiagen). Primers used for RT‐PCR for BER genes are shown in Supplementary Table S9. Quantitative PCR was performed on an ABI prism 7700 (Applied Biosystems) using SYBR green detection (Applied Biosystems®, UK). The housekeeping gene GAPDH was used to standardise the samples.
2.2.10. RT² Profiler™ PCR array for global DNA repair expression analysis
To evaluate the expression of 84 DNA repair genes simultaneously, real‐time PCR was performed using the RT² Profiler™ PCR Array for global DNA Repair expression analysis in technical triplicates (ABI 7500 Fast Block Detection System; Applied Biosystems, Foster City, USA) and the data analysed as per manufacturer's recommendation. GAPDH was used for normalization of the data. A 2 fold change or greater change in expression was considered significant.
3. Results
3.1. Clinical studies
XRCC1, pol β, APE1 and SMUG1 are key BER proteins. Moreover, we have recently shown that XRCC1 (Sultana et al., 2013), pol β (Abdel‐Fatah et al., 2014b), APE1 (Abdel‐Fatah et al., 2014a) and SMUG1 (Abdel‐Fatah et al., 2013b) are promising biomarkers in breast cancer. To evaluate whether they also have prognostic and predictive significance based on BRCA1 status, we proceeded to immunohistochemical evaluation of XRCC1, pol β, APE1 and SMUG1 in BRCA1 positive and BRCA1 negative breast tumours.
3.1.1. BRCA1 negativity is associated with impaired XRCC1 and pol β protein expression in human sporadic breast cancers
A total of 1602 breast tumours were suitable for BRCA1 expression analysis. 1085/1602 (67.7%) of tumours were BRCA1 positive and 517/1602 (32.3%) were negative for BRCA1 expression (Figure 1A). As shown in Supplementary Table S3, BRCA1 negative tumours were highly significantly associated with low XRCC1 (p < 0.00001) and low pol β (p < 0.000001). In the BRCA1 negative cohort we then evaluated clinicopathological associations of XRCC1 and pol β protein expression (Figure 1A). The data for XRCC1 and pol β are summarized in Supplementary Tables S4 And S5 respectively. Although no significant associations were seen with stage, tumour grade, tumour types or pleomorphism, BRCA1 negative/pol β low tumours were more likely to be Bcl2 negative (p = 0.001) and BRCA1 negative/XRCC1 low tumours were more likely to be p53 negative (p = 0.015).
Figure 1.
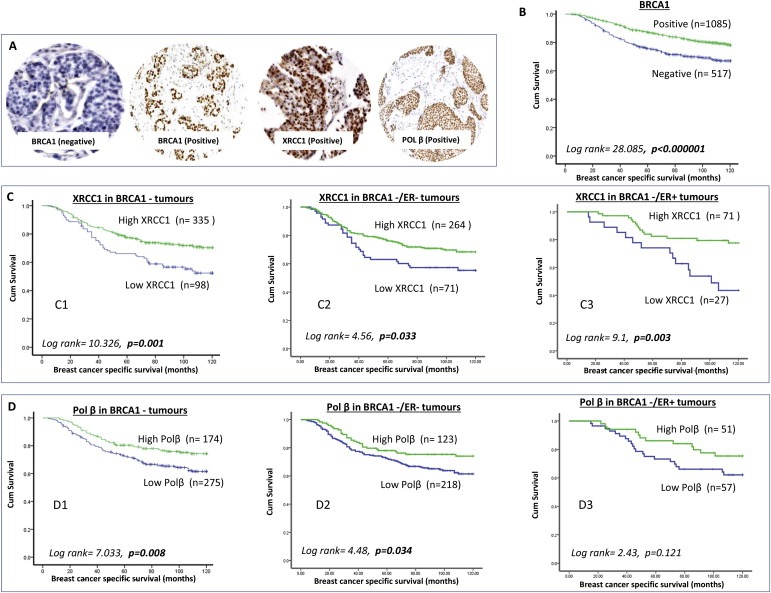
BRCA1 and BER protein expression in human breast cancer. A. Microphotographs of BRCA1 negative, BRCA1 positive, pol β positive and XRCC1 positive breast cancers. B. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients based on BRCA1 expression status. C1. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 negative tumours based on XRCC1 expression status. C2. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 negative/ER negative tumours based on XRCC1 expression status. C3. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 negative/ER positive tumours based on XRCC1 expression status. D1. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 negative tumours based on pol β expression status. D2. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 negative/ER negative tumours based on pol β expression status. D3. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 negative/ER positive tumours based on pol β expression status.
3.1.2. BRCA1 negative/low XRCC1 or BRCA1 negative/low pol β tumours are associated with poor breast cancer specific survival (BCSS)
BRCA1 negativity was significantly associated with poor BCSS compared to BRCA1 positive tumours (p < 0.000001) (Figure 1B) and is consistent with previous studies showing poor prognostic significance of BRCA1 silencing in sporadic breast tumours (Hsu et al., 2013; Wu et al., 2013). In the BRCA1 negative group we investigated the prognostic influence of XRCC1 and pol β. As shown in Figure 1C1, BRCA1 negative/low XRCC1 tumours had worse BCSS compared to BRCA1 negative/high XRCC1 tumours (p = 0.001). Similarly, BRCA1 negative/low pol β tumours had worse BCSS compared to BRCA1 negative/high pol β tumours (p = 0.008) (Figure 1D1). As BRCA1 negativity is likely to be associated with ER negative tumours we conducted further analysis. In the BRCA1 negative/ER negative subgroup, low XRCC1 or low pol β remains associated with poor survival (p = 0.033 and p = 0.034 respectively, Figure 1C2 and D2). In the BRCA1 negative/ER positive subgroup, similarly, low XRCC1 was associated with poor survival (p = 0.003, Figure 1C3) and although not significant there was trend with low pol β (p = 0.121, Figure 1D3). In multivariate cox regression analysis (Table 1), low XRCC1 (p = 0.005) and low pol β (p = 0.036) were independently associated with poor survival.
Table 1.
Multivariate analysis in BRCA1 negative sporadic breast cancers (Nottingham cohort).
| Variables | Multivariate model | ||
|---|---|---|---|
| Exp (B) | CI 95% | P value | |
| Pol beta expression | 0.810 | 0.666–0.986 | 0.036 |
| XRCC1 expression | 0.831 | 0.731–0.945 | 0.005 |
| Tumour stage | 3.088 | 2.414–3.950 | <0.0001 |
| ER receptor status | 0.522 | 0.328–0.833 | 0.006 |
| Chemotherapy status | 0.522 | 0.356–0.764 | 0.001 |
In the current study, APE1 and SMUG1 did not influence survival and was not associated with any clinicopathological parameters in BRCA1 negative breast tumours (data not shown).
3.1.3. XRCC1 and pol β expression in germ‐line BRCA1 mutated breast cancers
To investigate whether XRCC1 and pol β would also influence outcomes in germ‐line BRCA1 deficient breast cancer we investigated a cohort of 50 germ‐line BRCA1 mutated breast cancers. Demographics are summarised in Supplementary Table S6. No significant clinicopathological correlations were observed (Supplementary Tables S7 and S8). In this small exploratory cohort, low pol β (5/34 tumours) was significantly associated with poor survival (p = 0.007) in germ‐line BRCA1 mutated breast cancers (Figure 2A) compared to high pol β (29/34 tumours). Low XRCC1 expression did not influence survival in this cohort (Supplementary Figure S1A).
Figure 2.
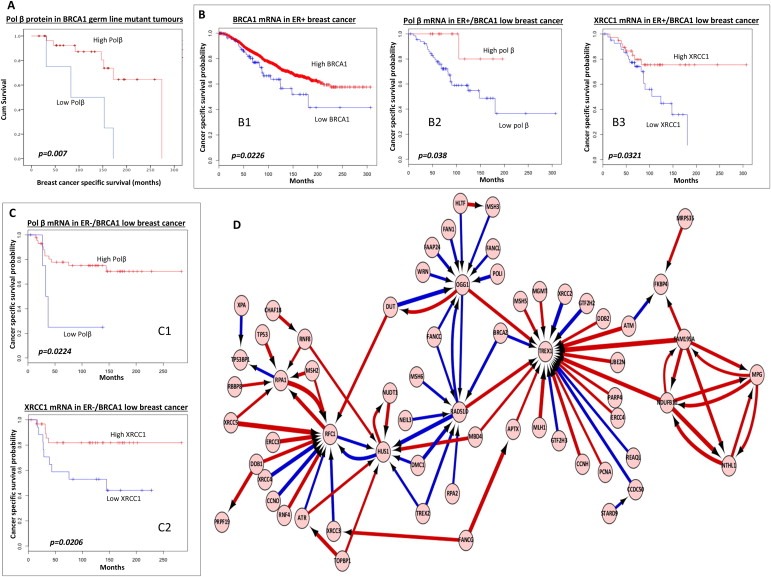
A. Kaplan–Meier curves showing overall survival in patients with germ‐line BRCA1mutated tumours based on pol β protein expression status. B1. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients based on BRCA1 mRNA expression status in ER+ breast cancer (METABRIC cohort). B2. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 mRNA low/ER+ who received adjuvant endocrine therapy based on pol β mRNA status (METABRIC cohort). B3. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 mRNA low/ER+ who received adjuvant endocrine therapy based on XRCC1 mRNA status (METABRIC cohort). C1. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 mRNA low/ER‐who received adjuvant chemotherapy based on pol β mRNA status (METABRIC cohort). C2. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 mRNA low/ER‐who received adjuvant chemotherapy based on XRCC1 mRNA status (METABRIC cohort). D. The neural network illustrates the top genes that interact with BRCA1 and other DNA repair genes. In addition, the artificial neural network also reveals how these top genes interact with each other. Top pair‐wise interactions for gene probe markers associated with BRCA1 expression and the DNA repair process in 249 breast cancers is shown here. Each gene probe is represented by a node and the interaction weight between them as an edge, the width being defined by the magnitude of the weight. Interactions are directed from a source gene to a target gene as indicated by arrows. Red interactions indicate an excitatory interaction and blue indicates an inhibitory interaction. Highly linked genes represent hubs that are indicated to be highly influential or highly regulated in the BRCA1‐DNA repair system. See supplementary data S12 for the biological functions of individual genes.
3.1.4. Low XRCC1 and low pol β transcript levels have prognostic significance in BRCA1 mRNA low sporadic breast cancers
To confirm whether the association between BRCA1 and BER also operated at the mRNA level we investigated the Metabric cohort (n = 1920, demographics summarized in Supplementary Table S9) and the Uppsala cohort (n = 249, demographics summarized in Supplementary Table S10) – cohorts where mRNA expression data was available. In ER+ tumours (n = 1485, Metabric), low BRCA1 (n = 81) was associated with poor survival compared to high BRCA1 mRNA expressing tumours (n = 1404) (p = 0.0226, Figure 2B1). In the low BRCA1/ER+ group, low pol β (n = 66) or low XRCC1 (n = 42) remains associated with poor survival (ps = 0.038 and 0.0321 respectively) compared to high pol β (n = 14) or high XRCC1 (n = 38) mRNA expressing tumours (Figure 2B2 and B3). In the ER‐tumours (n = 435, metabric), high BRCA1 (n = 385) was associated with poor survival compared to low BRCA1 mRNA expressing tumours (n = 50) (p = 0.0365, Supplementary Figure S2B). In the low BRCA1/ER‐group, low pol β (n = 5) or low XRCC1 (n = 17) remains associated with poor survival compared to high pol β (n = 43) or high XRCC1 (n = 31) mRNA expressing tumours (ps = 0.0224 and 0.0206 respectively) (Figure 2C2 and C3). In the Uppsala cohort, low pol β mRNA (36/175 tumours) was associated with poor survival in BRCA1 low mRNA breast cancers (p = 0.03, Supplementary Figure S1C) compared to high pol β mRNA tumours (139/175 tumours). XRCC1 mRNA expression levels did not influence survival in the Uppsala cohort (Supplementary Figure S1D).
3.1.5. Artificial neural network (ANN), ensemble classification and cross‐validation analysis for BRCA1 interacting DNA repair genes
The top 100 strongest are shown in Figure 2D. The biological functions of BRCA1 interaction genes are summarized in Supplementary Table S12. The predominant interactions with genes involved in BER, NER, HR, NHEJ, inter‐strand crosslink repair, MMR and transcription is not only consistent with the previously described functions of BRCA1 (Caestecker and Van de Walle, 2013; Huen et al., 2010; Silver and Livingston, 2012) but also reveals new BRCA1 interacting genes.
3.2. Pre‐clinical studies
The clinical and bioinformatics data presented above provides evidence that BRCA1 may influence the expression of multiple DNA repair genes. To provide additional pre‐clinical evidence we investigated the expression of DNA repair in BRCA1 deficient and proficient cancer cell lines.
3.2.1. BRCA1 deficient cancer cells exhibit impaired BER expression
BRCA1 deficient HeLa SilenciX cells, control BRCA1 proficient HeLa SilenciX cells, BRCA1 deficient MDA‐MB‐436 breast cancer cells and BRCA1 proficient MCF7 breast cancer cells were initially examined for the expression of BRCA1, XRCC1 and pol β proteins. BRCA1 deficiency was first confirmed at the protein level in BRCA1 deficient HeLa SilenciX and MDA‐MB‐436 cells compared to control HeLa SilenciX cells and MCF7 cells (Figure 3A1, A2, B1, B2). The relative expression of XRCC1 and pol β was also found to be low in BRCA1 deficient cells compared to BRCA1 proficient cells at the protein level (Figure 3A1, A2, B1, B2). Low mRNA expression of BRCA1, XRCC1 and pol β was confirmed by qRT‐PCR in MDA‐MB‐436 cells compared to MCF 7 cells and BRCA1 deficient HeLa SilenciX compared to control HeLa SilenciX cells (Figure 3A3, B3 respectively). The data is also summarized in Supplementary Tables S14 and S15. To provide additional evidence that low pol β and low XRCC1 expression confers phenotypic consequence, we investigated MMS sensitivity in MDA‐MB‐436 and MCF 7 cells. As shown in Supplementary Figure S3B, we found that MDA‐MB‐436 cells are sensitive to MMS. The data concurs with a recent study that showed a similar MMS sensitivity in BRCA1 deficient cells that was associated with impaired functional interaction between pol β and BRCA1 (Masaoka et al., 2013).
Figure 3.
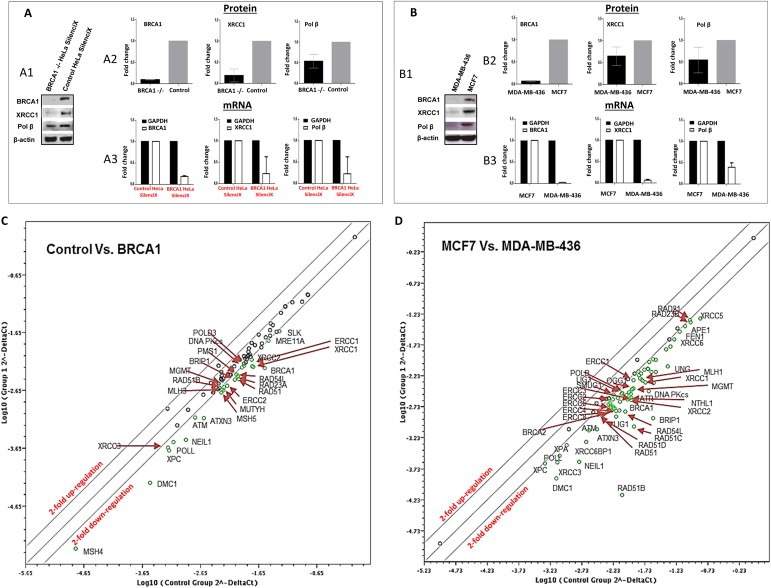
DNA repair expression in BRCA1 deficient and BRCA1 proficient cells A1. Representative Western blots of BRCA1, XRCC1 and pol β in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells. A2. Protein quantification in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells are shown here. The Figure shows fold change in BRCA1 deficient cells in comparison to BRCA1 proficient cells. A3. mRNA expression in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells are shown here. The Figure shows fold change in BRCA1 deficient cells in comparison to BRCA1 proficient cells. B1. Representative Western blots of BRCA1, XRCC1 and pol β in BRCA1 deficient MDA‐MB‐436 cells and BRCA1 proficient MCF7 cells. B2. Protein quantification in MDA‐MB‐436 cells and MCF7 cells are shown here. The Figure shows fold change in BRCA1 deficient cells in comparison to BRCA1 proficient cells. B3. mRNA expression in MDA‐MB‐436 cells and MCF7 cells are shown here. The Figure shows fold change in BRCA1 deficient cells in comparison to BRCA proficient cells. C. Scatter plots indicate up‐ and down‐regulation of DNA repair mRNA expression in BRCA1 deficient HeLa SilenciX cells compared to BRCA1 proficient HeLa SilenciX cells. D. Scatter plots indicate up‐ and down‐regulation of DNA repair mRNA expression in MDA‐MB‐436 cells compared to MCF7 cells are shown here. Green circles show genes that are two‐fold or more down‐regulated. See also Results section and Supplementary Tables S16 and S17.
3.2.2. BRCA1 deficient cells have deregulated gene expression of multiple DNA repair pathways
To investigate whether down‐regulation of DNA repair is restricted to pol β and XRCC1 or also includes additional DNA repair pathways, we profiled a panel of 84 DNA repair genes in BRCA1 deficient and BRCA1 proficient cells using the RT2 Profiler DNA Repair PCR array. All experiments were done in triplicates and DNA repair expression was compared between BRCA1 deficient and BRCA1 proficient cells [BRCA1 deficient HeLa SilenciX versus control BRCA1 proficient HeLa SilenciX cells and MDA‐MB‐436 versus MCF7 cells]. The data is summarized in Figure 3C (BRCA1 deficient and proficient HeLa SilenciX cells), Supplementary Table S16 (BRCA1 deficient and proficient HeLa SilenciX cells), Figure 3D (MDA‐MB‐436 and MCF7 cells), and Supplementary Table S17 (MDA‐MB‐436 and MCF7 cells). In MDA‐MB‐436 cells as well as in BRCA1 deficient HeLa SilenciX cells, we observed a consistent down‐regulation of several BER genes as well as genes involved in other pathways including base excision repair, nucleotide excision repair, homologous recombination, non‐homologous end joining, inter‐strand crosslink repair and mismatch repair.
3.2.3. BRCA1 deficient cancer cells are sensitive to ATM inhibitors either alone or in combination with cisplatin
KU55933 (2‐morpholin‐4‐yl‐6‐thianthren‐1‐yl‐pyran‐4‐one) is an ATP‐competitive potent ATM inhibitor with an IC50 of 13 nmol/L (Hickson et al., 2004). For additional validation we also tested KU60019 [(2R,6S‐rel)‐2,6‐Dimethyl‐N‐[5‐[6‐(4‐morpholinyl)‐4‐oxo‐4H‐pyran‐2‐yl]‐9H‐thioxanthen‐2‐yl‐4‐morpholineacetamide] another ATP‐competitive potent ATM inhibitor (Golding et al., 2009). Treatment with KU55933 resulted in reduced survival of BRCA1 deficient HeLa SilenciX cells compared to BRCA1 proficient HeLa SilenciX cells (Figure 4A1). Similarly, MDA‐MB‐436 cells were sensitive to KU55933 compared to MCF7 cells respectively (Figure 4B1). As an additional validation we investigated KU60019. As shown in Supplementary Figure S3A and S3E, BRCA1 deficient HeLa SilenciX cells and MDA‐MB‐436 cells were also sensitive to KU60019 compared to BRCA1 proficient cells. 3‐aminobenzamide, a PARP inhibitor (Shekh et al., 2014) was used as a positive control. As shown in Supplementary Figure S2A and S2B, BRCA1 deficient cells, as expected, are sensitive to PARP inhibitor.
Figure 4.
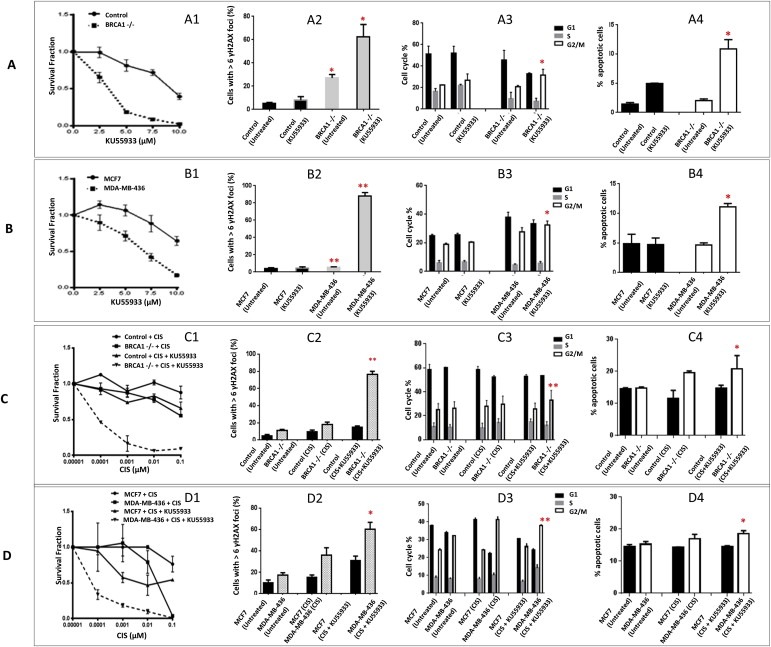
ATM inhibitors in BRCA1 deficient and BRCA1 proficient cells. A1. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. A2. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. A3. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. A4. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933.B1. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with KU55933. B2. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with KU55933. B3. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with KU55933. B4. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with KU55933. Inhibitors were added at the indicated concentrations (see Methods for details). C1. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with KU55933. C2. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with KU55933. C3. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with KU55933. C4. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with KU55933. D1. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with KU55933. D2. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with KU55933. D3. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with KU55933. D4. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with KU55933. *p < 0.05, **p < 0.01.
To provide mechanistic evidence that ATM inhibition leads to a synthetic lethality effect in BRCA1 deficient cells, we investigated the functional consequence of ATM inhibition in BRCA1 proficient and BRCA1 deficient cells. Double strand breaks (DSBs) induce phosphorylation of H2AX at serine 139 (γH2AX), and accumulation of γH2AX foci in the nucleus is a marker of DSBs. Therefore, γH2AX immunocytochemistry was performed in BRCA1 deficient HeLa SilenciX cells and MDA‐MB‐436 cells and compared to BRCA1 proficient control SilenciX or MCF7 cells (Supplementary Figure S3A). Nuclei containing more than six γH2AX foci were considered positive. Cells were treated with KU55933 (10 μM) for 48 h. The percentage of cells with more than six γH2AX foci was significantly higher in BRCA1 deficient cells in comparison to BRCA1 proficient cells (Figure 4A2, B2). Similar results were observed with KU60019 (Supplementary Figure S4B and S4F). The data provides evidence that BRCA1 deficient cells accumulate DSBs at an increased rate after treatment with an ATM inhibitor relative to BRCA1proficient cells. As an additional validation we performed neutral COMET assays after ATM or DNA‐PKcs inhibitor treatment in cells. As shown in Supplementary Figure S3C, BRCA1 deficient cells accumulated significantly more DSBs compared to BRCA1 proficient cells. Accumulation of DSBs may delay cell cycle progression. In BRCA1 deficient and BRCA1 proficient cells, cell cycle progression was monitored after 48 h of treatment with KU55933 (10 μM) (Supplementary Figure S4B). BRCA1 deficient cells were shown to be significantly arrested in G2/M phase of the cell cycle compared to BRCA1 proficient cells (Figure 4A3, B3). Similar results were observed with KU60019 (Supplementary Figure S5C and S5G). Accumulation of DSBs may result in eventual induction of apoptosis. Apoptosis detection by FITC‐annexin V flow cytometric analysis was therefore performed in cells treated with KU55933 (10 μM) for 48 h (Supplementary Figure S4C). The percentage of cells undergoing apoptosis following ATM inhibitor treatment was significantly higher in BRCA1 deficient cells in comparison to BRCA1 proficient cells (Figure 4A4, B4). Similar results were observed with KU60019 (Supplementary Figure S5D and S5H). The functional studies together provide evidence that ATM inhibition can induce synthetic lethality in BRCA1 deficient cells by causing accumulation of DSBs, G2M cell cycle arrest and induction of apoptosis. However the level of synthetic lethality effect seen with ATM inhibitor was modest compared that demonstrated previously using PARP inhibitors in BRCA1 deficient cells (Lord and Ashworth, 2008).
Cisplatin hypersensitivity has been well established in BRCA1 deficient cells (Tassone et al., 2009). We investigated whether low dose cisplatin could potentiate synthetic lethality induced by KU55933. Cells were treated with a combination of low dose cisplatin (0.00001 μM–0.1 μM) and KU55933 (5 μM). As shown in Figure 4C1 and D1, KU55933 treatment increased cytotoxicity of cisplatin in BRCA1 deficient HeLa SilenciX as well in MDA‐MB‐436 compared to BRCA1 proficient control SilenciX and MCF7 cells. The interaction was synergistic [combination index = 0.6 (BRCA1 deficient HeLa SilneciX) and 0.7 (MDA‐MB‐436), Supplementary Figure S7A]. In BRCA1 deficient cells treated with a combination of cisplatin and KU55933, the observed increased cytotoxicity was associated with accumulation of DSBs (Figure 4C2 and D2), G2/M cell cycle arrest (Figure 4C3 and D3) and increased apoptosis (Figure 4C4 and D4).
3.2.4. BRCA1 deficient cancer cells are sensitive to DNA‐PKcs inhibitors either alone or in combination with cisplatin
NU7441 (2‐N‐morpholino‐8‐dibenzothiophenyl‐chromen‐4‐one) is a potent and a specific inhibitor of DNA‐PKcs with an IC50 of 14 nmol/L for DNA‐PK inhibition (Tavecchio et al., 2012). NU7026 (2‐(morpholin‐4‐yl)‐benzo[h]chromen‐4‐one) is another DNA‐PKcs inhibitor (Nutley et al., 2005).
Treatment with NU7441 resulted in reduced survival of BRCA1 deficient HeLa SilenciX cells compared to BRCA1 deficient HeLa SilenciX cells (Figure 5A1). MDA‐MB‐436 cells were modestly sensitive to NU7441 compared to MCF7 cells respectively (Figure 5B1). As an additional validation we investigated NU7026. As shown in Supplementary Figure S6A and S6E, BRCA1 deficient HeLa SilenciX cells and MDA‐MB‐436 cells were also sensitive to NU7026 compared to BRCA1 proficient cells. To provide mechanistic evidence that DNA‐PKcs inhibition leads to a synthetic lethality effect in BRCA1 deficient cells, we investigated the functional consequence of DNA‐PKcs inhibition in BRCA1 proficient and BRCA1 deficient cells. Cells were treated with NU7441 (1.5 μM) for 48 h. The percentage of cells with more than six γH2AX foci was significantly higher in BRCA1 deficient cells in comparison to BRCA1 proficient cells (Figure 5A2, B2). Similar results were observed with NU7026 (Supplementary Figure S6B and S6F). In BRCA1 deficient and BRCA1 proficient cells, cell cycle progression was monitored after 48 h of treatment with NU7441 (1.5 μM). BRCA1 deficient cells were shown to be significantly arrested in G1 phase of the cell cycle compared to BRCA1 proficient cells (Figure 5A3, B3). Similar results were observed with NU7026 (Supplementary Figure S6C and S6G). Apoptosis detection by FITC‐annexin V flow cytometric analysis was therefore performed in cells treated with NU7441 (1.5 μM) for 48 h. The percentage of cells undergoing apoptosis following DNA‐PKcs inhibitor treatment was significantly higher in BRCA1 deficient cells in comparison to BRCA1 proficient cells (Figure 5A4, B4). Similar results were observed with NU7026 (Supplementary Figure S6D and S6H). The functional studies together provide evidence that DNA‐PKcs inhibition can induce synthetic lethality in BRCA1 deficient cells by causing accumulation of DSBs, G1 cell cycle arrest and induction of apoptosis. We then investigated whether low dose cisplatin could potentiate synthetic lethality induced by NU7441. Cells were treated with a combination of cisplatin (0.00001 μM–0.1 μM) and NU7441 (0.75 μM). As shown in Figure 5C1 and D1, NU7441 treatment substantially increased cytotoxicity of low dose cisplatin in BRCA1 deficient HeLa SilenciX as well as in MDA‐MB‐436 compared to BRCA1 proficient control SilenciX and MCF7 cells. The interaction was synergistic [combination index = 0.5 (BRCA1 deficient HeLa SilneciX) and 0.6 (MDA‐MB‐436), Supplementary Figure S7B]. Increased cytotoxicity was associated with accumulation of DSBs (Figure 5C2 and D2), G2/M cell cycle arrest (Figure 5C3 and D3) and increased apoptosis (Figure 5C4 and D4).
Figure 5.
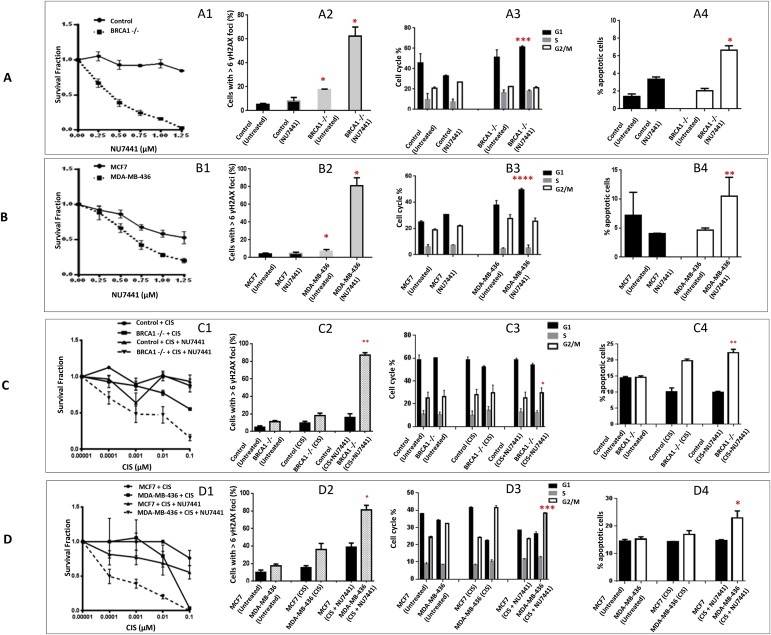
DNA‐PKcs inhibitors in BRCA1 deficient and BRCA1 proficient cells. A1. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7441. A2. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7441. A3. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7441. A4. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7441.B1. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with NU7441. B2. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with NU7441. B3. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with NU7441. B4. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with NU7441. Inhibitors were added at the indicated concentrations (see Methods for details). C1. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with NU7441. C2. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with NU7441. C3. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with NU7441. C4. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with cisplatin alone or in combination with NU7441. D1. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with NU7441. D2. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with NU7441. D3. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with NU7441. D4. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with cisplatin alone or in combination with NU7441. *p < 0.05, **p < 0.01.
4. Discussion
We have shown, for the first time, that BRCA1 negative tumours have significantly lower expression of XRCC1 and pol β, a feature that was also seen in BRCA1 deficient cancer cell lines. Patients with BRCA1 negative/low XRCC1 or BRCA1 negative/low pol β breast tumours also have worse breast cancer specific survival including in ER+ and ER‐sub‐groups. These new observations suggest that pol β/XRCC1 based sub‐stratification may refine prognostication in BRCA1 deficient phenotypes. In addition, poor prognostic significance of pol β in a small cohort of germ‐line BRCA1 mutated tumours is also consistent with a recent pre‐clinical study linking BRCA1 and pol β in cancer cell line models (Masaoka et al., 2013). Additionally, at the mRNA level we conducted neural network analysis and demonstrated that the predominant BRCA1 interactions were with genes involved in BER, NHEJ, NER and MMR inter‐strand crosslink repair and transcription. To compliment clinical observations we DNA repair profiled BRCA1 proficient and deficient cell lines and also demonstrated impairment in BER, NHEJ, NER and MMR. A surprising finding in the current study was that several genes involved in DDR were down‐regulated in BRCA1 deficient HeLa as well as in breast cancer cells as one would anticipate that this would reduce survival in the absence of exogenous DNA damage. Moreover, as inhibition of BER (via PARP inhibition) is synthetically lethal in association with BRCA defects, we expected BRCA deficient cells to have compensatory up‐regulation of BER pathway. In contrast we observed consistent down‐regulation of BER implying a complex regulation, including transcriptional regulation of BER by BRCA1. Although in BRCA deficient cells PARP1 may be up‐regulated as a compensatory mechanism, in our cell line models we actually observed low expression of PARP1 mRNA, PARP2 mRNA and PARP3 mRNA in BRCA1 deficient HeLa cells and low expression of PARP1 mRNA and PARP2 mRNA in MDA‐MB‐436 cells. Although these new observations are intriguing, mechanism for down‐regulation is currently unknown. However, we speculate that genomic instability in BRCA1 null cells may over a period of time, in the cell lines used in the current study, eventually lead to acquisition of new defects in other DNA repair pathways along with compensatory up‐regulation of pro‐survival pathways. In addition, recent studies implicating a role for BRCA1 in transcriptional regulation of BER (Saha et al., 2010) and NER(Hartman and Ford, 2002) suggests additional mechanisms may also operate for the observed genomic instability in BRCA1 deficient cells. The altered expression of XRCC1 and pol β shown here also concurs to a large extent with a recent study by De Summa et al. who investigated the expression of APE1, NTHL1, OGG1, PARP1, XRCC1 and miR17 in BRCA1/2 mutated and sporadic breast cancers (De Summa et al., 2014). Down‐regulation of XRCC1, APE1 and NTHL1 was evident in that study. Interestingly overexpression of PARP1 and miR17 were observed in BRCA1 mutated tumours implying that they could be investigated as biomarker of BRCAness phenotype. In contrast, we observed PARP1 down‐regulation in BRCA1 deficient HeLa as well as in MDA‐MB‐436 breast cancer cell line in our study. Larger studies in human breast tumours are required to confirm these findings.
Although from pre‐clinical studies we would expect DNA repair deficiency to be associated with chemosensitivity and improved survival in patient who received adjuvant chemotherapy, in the current clinical study we found that low BRCA1 or low BRCA1/low XRCC1 or low BRCA1/low pol β was associated with poor survival. Our recent studies of DNA repair in breast cancer suggest that the relationship may be complex. In previous studies we found that low XRCC1 (Sultana et al., 2013) and low DNA pol β (Abdel‐Fatah et al., 2014b) to be associated with poor survival. As BER deficiency could lead to accelerated accumulation of mutations resulting in a mutator phenotype (Bielas et al., 2006) that is associated with an aggressive biology, we speculate that the observed effect for XRCC1 and pol β may reflect this phenomenon. On the other hand, high SMUG1 (Abdel‐Fatah et al., 2013b) and high FEN1 (Abdel‐Fatah et al., 2014) was associated with poor survival in patients who have received adjuvant chemotherapy implying a DNA repair influenced predictive significance for SMUG1 and FEN1 in breast cancer.
We have demonstrated that a potential synthetic lethality relationship also exists between BRCA1 deficiency and blockade of ATM or DNA‐PKcs in cells. A model for synthetic lethality is shown in Supplementary Figure S8. We have concluded synthetic lethality for the following reasons: a) BRCA1 deficient cells have increased sensitivity to ATM or DNA‐PKcs inhibitors; b) upon ATM inhibitor treatment, BRCA1 deficient cells accumulate DSBs, exhibit G2/M cell cycle arrest and induction of apoptosis; and c) upon DNA‐PKcs inhibitor treatment, BRCA1 deficient cells accumulate DSBs, exhibit G1 cell cycle arrest and induction of apoptosis. Interestingly, BRCA1 deficient cells also exhibit low mRNA levels of ATM and DNA‐PKcs but are sensitive to ATM and DNA‐PKcs inhibitors. The data suggest a dose dependent effect where although ATM and DNA levels are low the cells are able to tolerate genomic instability and survive. However, with additional functional inhibition through small molecule inhibition, pronounced accumulation of DSBs could lead to a synthetic lethality effect. Although phosphorylation of H2AX is ATM and DNA‐Pkcs dependent, we observed accumulation of γH2AX foci in BRCA1 deficient cells incubated in medium containing ATM inhibitor or DNA‐PKcs inhibitor for 48 h. Whether reflects a timing issue are whether H2AX phosphorylation may be contributed by other factors such as activated ATR is unclear. To confirm DSB accumulation we also performed neutral COMET assay and have demonstrated DSB accumulation in BRCA1 deficient cells treated with ATM or DNA‐PKcs inhibitor. Moreover the effects on cell cycle progression, although significant, appear to be modest. In the current study, the magnitude of synthetic lethality seen in BRCA1 deficient cells treated with ATM or DNA‐PKcs inhibitor alone was not as pronounced as that demonstrated for PARP inhibitors (Lord and Ashworth, 2008), we found that low dose cisplatin combination significantly enhanced synthetic lethality. Of note, the dose of cisplatin used in our study was 1/10th the dose used in previous pre‐clinical studies investigating cisplatin sensitivity in vitro in BRCA1 deficient cells (Husain et al., 1998). At higher doses of cisplatin similar to the doses used by Husain et al., BRCA1 deficient cells remain highly sensitive to platinum therapy (data not shown). We speculate that low dose cisplatin treatment generates low levels of DSBs. In cells with proficient BRCA1 and BER, despite ATM or DNA‐PKcs blockade, DSBs may be rapidly repaired in back‐up DNA repair pathways and cells continue to survive. The back‐up repair could operate at multiple levels including the complex interactions/overlap between HR, NHEJ, alternative NHEJ (B‐NHEJ) (Chapman et al., 2012; Schipler and Iliakis, 2013), components of NER and ICL repair pathways. On the other hand, in cells with deficient BRCA1 and low BER, the associated pharmacological blockade with ATM or DNA‐PKcs inhibitors may lead to DSB accumulation which beyond a threshold, may severely compromise back‐up DNA repair machinery leading to DSB accumulation, cell cycle arrest and apoptosis. We have recently shown that XRCC1 deficient cells are cisplatin sensitive (Abdel‐Fatah et al., 2013a) and ATM or DNA‐PKcs inhibitors are also synthetically lethal in XRCC1 deficient cells (Sultana et al., 2013). Given the potential role of XRCC1 in B‐NHEJ (Mladenov and Iliakis, 2011) we speculate that BRCA1 deficient cells that have low XRCC1 could also have compromised B‐NHEJ in addition to BER resulting in increased genomic instability and enhanced synthetic lethality with cisplatin treatment seen in the current study. Moreover, ATM or DNA‐PKcs modulation has previously been shown to enhance cisplatin cytotoxicity (Dejmek et al., 2009; Yoshida et al., 2008). Taken together, the data suggests that cisplatin combination may be more successful than ATM or DNA‐PK monotherapy in BRCA1 negative tumours and would be consistent with a recent study showing enhancement of synthetic lethality with ABT‐888 (PARP inhibitor) in combination with platinum chemotherapy in BRCA deficient cells (Clark et al., 2012).
Conflicts of interest
The authors disclose no potential conflicts of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary Figure S1: A. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with germ‐line BRCA1 mutant tumours based on XRCC1 expression status. B. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with ER negative tumours based on BRCA1 mRNA expression status. C. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 low mRNA tumours based on pol β mRNA expression status. D. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 low mRNA tumours based on XRCC1 mRNA expression status. Supplementary Figure S2: A. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with 3‐aminobenzamide. B. Clonogenic survival assays in MCF 7 cells and MDA‐MB‐436 cells treated with 3‐aminobenzamide.Supplementary Figure S3: A. Survival assay in MCF‐7 and MDA‐MB‐436 cells treated with cisplatin. B. Survival assays in MCF‐7 and MDA‐MB‐436 cells treated with MMS. C. Neutral COMET assay in MCF‐7 and MDA‐MB‐436 cells treated with NU7441 or KU55933.Supplementary Figure S4: Functional analysis in cells (see Methods section for more details). A. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. B. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. C. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. Supplementary Figure S5: A. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. B. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. C. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. D. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. E. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with KU60019. F. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with KU60019. G. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with KU60019. H. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with KU60019. *p < 0.05, **p < 0.01. Supplementary Figure S6: A. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. B. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. C. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. D. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. E. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with NU7026. F. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with NU7026. G. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with NU7026. H. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with NU7026. *p < 0.05, **p < 0.01. Supplementary Figure S7: Combination index for synergism (see Results section for more details). A. ATM inhibitor (KU55933). B. DNA‐PKcs inhibitor (NU7441). Supplementary Figure S8: A model for synthetic lethality in BRCA1 deficient cells using ATM or DNA‐PKcs inhibitors either alone or in combination with cisplatin chemotherapy is shown here. See Discussion section for details.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.08.001.
Albarakati Nada, Abdel-Fatah Tarek M.A., Doherty Rachel, Russell Roslin, Agarwal Devika, Moseley Paul, Perry Christina, Arora Arvind, Alsubhi Nouf, Seedhouse Claire, Rakha Emad A., Green Andrew, Ball Graham, Chan Stephen, Caldas Carlos, Ellis Ian O., Madhusudan Srinivasan, (2015), Targeting BRCA1-BER deficient breast cancer by ATM or DNA‐PKcs blockade either alone or in combination with cisplatin for personalized therapy, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.08.001.
References
- Abdel-Fatah, T. , Sultana, R. , Abbotts, R. , Hawkes, C. , Seedhouse, C. , Chan, S. , Madhusudan, S. , 2013. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int. J. Cancer J. Int. du Cancer. 132, 2778–2786. [DOI] [PubMed] [Google Scholar]
- Abdel-Fatah, T.M. , Albarakati, N. , Bowell, L. , Agarwal, D. , Moseley, P. , Hawkes, C. , Ball, G. , Chan, S. , Ellis, I.O. , Madhusudan, S. , 2013. Single-strand selective monofunctional uracil-DNA glycosylase (SMUG1) deficiency is linked to aggressive breast cancer and predicts response to adjuvant therapy. Breast Cancer Res. Treat. 142, 515–527. [DOI] [PubMed] [Google Scholar]
- Abdel-Fatah, T.M. , Perry, C. , Moseley, P. , Johnson, K. , Arora, A. , Chan, S. , Ellis, I.O. , Madhusudan, S. , 2014. Clinicopathological significance of human apurinic/apyrimidinic endonuclease 1 (APE1) expression in oestrogen-receptor-positive breast cancer. Breast Cancer Res. Treat. 143, 411–421. [DOI] [PubMed] [Google Scholar]
- Abdel-Fatah, T.M. , Russell, R. , Agarwal, D. , Moseley, P. , Abayomi, M.A. , Perry, C. , Albarakati, N. , Ball, G. , Chan, S. , Caldas, C. , Ellis, I.O. , Madhusudan, S. , 2014. DNA polymerase beta deficiency is linked to aggressive breast cancer: a comprehensive analysis of gene copy number, mRNA and protein expression in multiple cohorts. Mol. Oncol. 8, 520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Fatah, T.M. , Russell, R. , Albarakati, N. , Maloney, D.J. , Dorjsuren, D. , Rueda, O.M. , Moseley, P. , Mohan, V. , Sun, H. , Abbotts, R. , Mukherjee, A. , Agarwal, D. , Illuzzi, J.L. , Jadhav, A. , Simeonov, A. , Ball, G. , Chan, S. , Caldas, C. , Ellis, I.O. , Wilson, D.M. , Madhusudan, S. , 2014. Genomic and protein expression analysis reveals flap endonuclease 1 (FEN1) as a key biomarker in breast and ovarian cancer. Mol. Oncol. 10.1016/j.molonc.2014.04.009 pii: S1574-7891(14)00092-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alli, E. , Sharma, V.B. , Sunderesakumar, P. , Ford, J.M. , 2009. Defective repair of oxidative dna damage in triple-negative breast cancer confers sensitivity to inhibition of poly(ADP-ribose) polymerase. Cancer Res. 69, 3589–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum, M.C. , 1981. Criteria for analyzing interactions between biologically active agents. Adv. Cancer Res. 35, 269–335. [DOI] [PubMed] [Google Scholar]
- Bergh, J. , Norberg, T. , Sjogren, S. , Lindgren, A. , Holmberg, L. , 1995. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Bielas, J.H. , Loeb, K.R. , Rubin, B.P. , True, L.D. , Loeb, L.A. , 2006. Human cancers express a mutator phenotype. Proc. Natl. Acad. Sci. U S A. 103, 18238–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caestecker, K.W. , Van de Walle, G.R. , 2013. The role of BRCA1 in DNA double-strand repair: past and present. Exp. Cell Res. 319, 575–587. [DOI] [PubMed] [Google Scholar]
- Chapman, J.R. , Taylor, M.R. , Boulton, S.J. , 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 47, 497–510. [DOI] [PubMed] [Google Scholar]
- Clark, C.C. , Weitzel, J.N. , O'Connor, T.R. , 2012. Enhancement of synthetic lethality via combinations of ABT-888, a PARP inhibitor, and carboplatin in vitro and in vivo using BRCA1 and BRCA2 isogenic models. Mol. Cancer Ther. 11, 1948–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, C. , Shah, S.P. , Chin, S.F. , Turashvili, G. , Rueda, O.M. , Dunning, M.J. , Speed, D. , Lynch, A.G. , Samarajiwa, S. , Yuan, Y. , Graf, S. , Ha, G. , Haffari, G. , Bashashati, A. , Russell, R. , McKinney, S. , Langerod, A. , Green, A. , Provenzano, E. , Wishart, G. , Pinder, S. , Watson, P. , Markowetz, F. , Murphy, L. , Ellis, I. , Purushotham, A. , Borresen-Dale, A.L. , Brenton, J.D. , Tavare, S. , Caldas, C. , Aparicio, S. , 2012. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Summa, S. , Pinto, R. , Pilato, B. , Sambiasi, D. , Porcelli, L. , Guida, G. , Mattioli, E. , Paradiso, A. , Merla, G. , Micale, L. , De Nittis, P. , Tommasi, S. , 2014. Expression of base excision repair key factors and miR17 in familial and sporadic breast cancer. Cell Death Dis. 5, e1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejmek, J. , Iglehart, J.D. , Lazaro, J.B. , 2009. DNA-dependent protein kinase (DNA-PK)-dependent cisplatin-induced loss of nucleolar facilitator of chromatin transcription (FACT) and regulation of cisplatin sensitivity by DNA-PK and FACT. Mol. Cancer Res. 7, 581–591. [DOI] [PubMed] [Google Scholar]
- Dianov, G.L. , Hubscher, U. , 2013. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 41, 3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant, S.T. , Nickoloff, J.A. , 2005. Good timing in the cell cycle for precise DNA repair by BRCA1. Cell Cycle. 4, 1216–1222. [DOI] [PubMed] [Google Scholar]
- Golding, S.E. , Rosenberg, E. , Valerie, N. , Hussaini, I. , Frigerio, M. , Cockcroft, X.F. , Chong, W.Y. , Hummersone, M. , Rigoreau, L. , Menear, K.A. , O'Connor, M.J. , Povirk, L.F. , van Meter, T. , Valerie, K. , 2009. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 8, 2894–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, A.R. , Ford, J.M. , 2002. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat. Genet. 32, 180–184. [DOI] [PubMed] [Google Scholar]
- Hickson, I. , Zhao, Y. , Richardson, C.J. , Green, S.J. , Martin, N.M. , Orr, A.I. , Reaper, P.M. , Jackson, S.P. , Curtin, N.J. , Smith, G.C. , 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64, 9152–9159. [DOI] [PubMed] [Google Scholar]
- Hill, R. , Lee, P.W. , 2010. The DNA-dependent protein kinase (DNA-PK): more than just a case of making ends meet?. Cell Cycle. 9, 3460–3469. [DOI] [PubMed] [Google Scholar]
- Hsu, N.C. , Huang, Y.F. , Yokoyama, K.K. , Chu, P.Y. , Chen, F.M. , Hou, M.F. , 2013. Methylation of BRCA1 promoter region is associated with unfavorable prognosis in women with early-stage breast cancer. PloS One. 8, e56256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen, M.S. , Sy, S.M. , Chen, J. , 2010. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 11, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain, A. , He, G. , Venkatraman, E.S. , Spriggs, D.R. , 1998. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum(II). Cancer Res. 58, 1120–1123. [PubMed] [Google Scholar]
- Lancashire, L.J. , Powe, D.G. , Reis-Filho, J.S. , Rakha, E. , Lemetre, C. , Weigelt, B. , Abdel-Fatah, T.M. , Green, A.R. , Mukta, R. , Blamey, R. , Paish, E.C. , Rees, R.C. , Ellis, I.O. , Ball, G.R. , 2010. A validated gene expression profile for detecting clinical outcome in breast cancer using artificial neural networks. Breast Cancer Res. Treat. 120, 83–93. [DOI] [PubMed] [Google Scholar]
- Langelier, M.F. , Pascal, J.M. , 2013. PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr. Opin. Struct. Biol. 23, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.H. , Goodarzi, A.A. , Jeggo, P.A. , Paull, T.T. , 2010. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 29, 574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.H. , Paull, T.T. , 2007. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 26, 7741–7748. [DOI] [PubMed] [Google Scholar]
- Lemetre, C. , 2009. Artificial neural network based algorithm for biomolecular interaction modeling Bio-Inspired Systems: Computational and Ambient Intellegence Lecture Notes in Computer Science. 5517, 877–885. [Google Scholar]
- Lord, C.J. , Ashworth, A. , 2008. Targeted therapy for cancer using PARP inhibitors. Curr. Opin. Pharmacol. 8, 363–369. [DOI] [PubMed] [Google Scholar]
- Masaoka, A. , Gassman, N.R. , Horton, J.K. , Kedar, P.S. , Witt, K.L. , Hobbs, C.A. , Kissling, G.E. , Tano, K. , Asagoshi, K. , Wilson, S.H. , 2013. Interaction between DNA polymerase beta and BRCA1. PloS One. 8, e66801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladenov, E. , Iliakis, G. , 2011. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat. Res. 711, 61–72. [DOI] [PubMed] [Google Scholar]
- Nutley, B.P. , Smith, N.F. , Hayes, A. , Kelland, L.R. , Brunton, L. , Golding, B.T. , Smith, G.C. , Martin, N.M. , Workman, P. , Raynaud, F.I. , 2005. Preclinical pharmacokinetics and metabolism of a novel prototype DNA-PK inhibitor NU7026. Br. J. Cancer. 93, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan, P.J. , Livingston, D.M. , 2010. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 31, 961–967. [DOI] [PubMed] [Google Scholar]
- Pawitan, Y. , Bjohle, J. , Amler, L. , Borg, A.L. , Egyhazi, S. , Hall, P. , Han, X. , Holmberg, L. , Huang, F. , Klaar, S. , Liu, E.T. , Miller, L. , Nordgren, H. , Ploner, A. , Sandelin, K. , Shaw, P.M. , Smeds, J. , Skoog, L. , Wedren, S. , Bergh, J. , 2005. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 7, R953–R964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha, T. , Rih, J.K. , Roy, R. , Ballal, R. , Rosen, E.M. , 2010. Transcriptional regulation of the base excision repair pathway by BRCA1. J. Biol. Chem. 285, 19092–19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipler, A. , Iliakis, G. , 2013. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 41, 7589–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekh, K. , Khan, S. , Jena, G. , Kansara, B.R. , Kushwaha, S. , 2014. 3-Aminobenzamide–a PARP inhibitor enhances the sensitivity of peripheral blood micronucleus and comet assays in mice. Toxicol. Mech. Methods. 24, 332–341. [DOI] [PubMed] [Google Scholar]
- Shiloh, Y. , Ziv, Y. , 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210. [PubMed] [Google Scholar]
- Silver, D.P. , Livingston, D.M. , 2012. Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot, M.E. , Ono, K. , Ruscheinski, J. , Wang, P.L. , Ideker, T. , 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, G.K. , 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- Sultana, R. , Abdel-Fatah, T. , Abbotts, R. , Hawkes, C. , Albarakati, N. , Seedhouse, C. , Ball, G. , Chan, S. , Rakha, E.A. , Ellis, I.O. , Madhusudan, S. , 2013. Targeting XRCC1 deficiency in breast cancer for personalized therapy. Cancer Res. 73, 1621–1634. [DOI] [PubMed] [Google Scholar]
- Tassone, P. , Di Martino, M.T. , Ventura, M. , Pietragalla, A. , Cucinotto, I. , Calimeri, T. , Bulotta, A. , Neri, P. , Caraglia, M. , Tagliaferri, P. , 2009. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol. Ther. 8, 648–653. [DOI] [PubMed] [Google Scholar]
- Tavecchio, M. , Munck, J.M. , Cano, C. , Newell, D.R. , Curtin, N.J. , 2012. Further characterisation of the cellular activity of the DNA-PK inhibitor, NU7441, reveals potential cross-talk with homologous recombination. Cancer Chemother. Pharmacol. 69, 155–164. [DOI] [PubMed] [Google Scholar]
- Tibshirani, R. , Hastie, T. , Narasimhan, B. , Chu, G. , 2002. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. U S A. 99, 6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, N. , Tutt, A. , Ashworth, A. , 2004. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer. 4, 814–819. [DOI] [PubMed] [Google Scholar]
- Wu, L. , Wang, F. , Xu, R. , Zhang, S. , Peng, X. , Feng, Y. , Wang, J. , Lu, C. , 2013. Promoter methylation of BRCA1 in the prognosis of breast cancer: a meta-analysis. Breast Cancer Res. Treat. 142, 619–627. [DOI] [PubMed] [Google Scholar]
- Yoshida, K. , Ozaki, T. , Furuya, K. , Nakanishi, M. , Kikuchi, H. , Yamamoto, H. , Ono, S. , Koda, T. , Omura, K. , Nakagawara, A. , 2008. ATM-dependent nuclear accumulation of IKK-alpha plays an important role in the regulation of p73-mediated apoptosis in response to cisplatin. Oncogene. 27, 1183–1188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary Figure S1: A. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with germ‐line BRCA1 mutant tumours based on XRCC1 expression status. B. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with ER negative tumours based on BRCA1 mRNA expression status. C. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 low mRNA tumours based on pol β mRNA expression status. D. Kaplan–Meier curves showing breast cancer specific survival (BCSS) in patients with BRCA1 low mRNA tumours based on XRCC1 mRNA expression status. Supplementary Figure S2: A. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with 3‐aminobenzamide. B. Clonogenic survival assays in MCF 7 cells and MDA‐MB‐436 cells treated with 3‐aminobenzamide.Supplementary Figure S3: A. Survival assay in MCF‐7 and MDA‐MB‐436 cells treated with cisplatin. B. Survival assays in MCF‐7 and MDA‐MB‐436 cells treated with MMS. C. Neutral COMET assay in MCF‐7 and MDA‐MB‐436 cells treated with NU7441 or KU55933.Supplementary Figure S4: Functional analysis in cells (see Methods section for more details). A. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. B. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. C. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU55933. Supplementary Figure S5: A. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. B. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. C. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. D. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with KU60019. E. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with KU60019. F. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with KU60019. G. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with KU60019. H. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with KU60019. *p < 0.05, **p < 0.01. Supplementary Figure S6: A. Clonogenic survival assays in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. B. ↠H2AX immunohistochemistry in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. C. FACS analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. D. Annexin V flow cytometric analysis in BRCA1 deficient HeLa SilenciX cells and control BRCA1 proficient HeLa SilenciX cells treated with NU7026. E. Clonogenic survival assays in MDA‐MB‐436 and MCF7 cells treated with NU7026. F. ↠H2AX immunohistochemistry in MDA‐MB‐436 and MCF7 cells treated with NU7026. G. FACS analysis in MDA‐MB‐436 and MCF7 cells treated with NU7026. H. Annexin V flow cytometric analysis in MDA‐MB‐436 and MCF7 cells treated with NU7026. *p < 0.05, **p < 0.01. Supplementary Figure S7: Combination index for synergism (see Results section for more details). A. ATM inhibitor (KU55933). B. DNA‐PKcs inhibitor (NU7441). Supplementary Figure S8: A model for synthetic lethality in BRCA1 deficient cells using ATM or DNA‐PKcs inhibitors either alone or in combination with cisplatin chemotherapy is shown here. See Discussion section for details.