

Abstract
Novel combinations aiming at maximizing the efficacy of bortezomib are highly valued in the clinic. Therefore the current study investigated the outcomes of combining bortezomib with dipyridamole, a well‐known antiplatelet. The co‐treatment exerted a synergistic lethality in a panel of human leukemia/lymphoma cell lines of different origin. Mechanistically, dipyridamole did not modulate the proteasome inhibitory activity of bortezomib. However, dipyridamole triggered an endoplasmic reticulum (ER) stress, and co‐treatment with bortezomib resulted in higher levels of ER stress than either monotherapies. Relieving ER stress with the protein translation inhibitor, cycloheximide suppressed cell death. Moreover, the enhanced ER stress by the co‐treatment was associated with an aggravation of reactive oxygen species (ROS) generation and glutathione (GSH) depletion. Replenishing GSH pools significantly scavenged ROS and rescued the cells. Importantly, the cytotoxicity of the co‐treatment was executed mainly via the mitochondrial apoptotic pathway with an efficient suppression of the key anti‐apoptotic regulators, Mcl‐1, Bcl‐xl, Bcl‐2 and XIAP, driving the independence of the co‐treatment‐induced apoptosis of a single apoptotic trigger. Furthermore, the intrinsic potential of bortezomib to inhibit important pro‐survival pathways was enhanced by dipyridamole in a GSH/ROS‐dependent manner. Interestingly, dipyridamole abrogated JAK2 phosphorylation indirectly and selectively in cancer cells, and the co‐treatment‐induced cytotoxicity was preserved in a model of stromal‐mediated chemoresistance. In nude mice, the antitumor activity of the co‐treatment surpassed that of bortezomib monotherapy despite that synergy was lacking. In summary, findings of the present study provided a preclinical rationale which warrants further clinical evaluation of bortezomib/dipyridamole novel combination in hematologic malignancies.
Keywords: Hematologic malignancies, Dipyridamole, Bortezomib, GSH/ROS, ER stress, Pro‐survival pathways, JAK2
Highlights
Bortezomib/dipyridamole synergistically induced apoptosis.
Bortezomib/dipyridamole‐induced apoptosis is multifaceted & cooperatively executed.
Bortezomib/dipyridamole enhanced proapoptotic & prosurvival pathways targeting.
Dipyridamole abrogated JAK2 phosphorylation indirectly & selectively in cancer cells.
Bortezomib/dipyridamole is superior to bortezomib monotherapy in vivo.
1. Introduction
Bortezomib is the first proteasome inhibitor to be used clinically for the treatment of hematologic malignancies. Bortezomib reversibly inhibits the chymotrypsin‐like activity of the proteasomes through a complex formation with the active site of the threonine hydroxyl group in the β5‐subunit, resulting in the accumulation of misfolded proteins. The ensuing proteotoxic conditions eventually impair the balance between the pro‐apoptotic and anti‐apoptotic signaling pathways and trigger cell death (McConkey and Zhu, 2008; Reddy and Czuczman, 2010).
Although initial promising results were obtained with bortezomib monotherapy of a number of hematologic malignancies, certain subsets of patients had a partial response. Moreover, relapse is still a major challenge and a poorly understood drug resistance remains a crucial clinical problem in the majority of treated patients. Different bortezomib resistance mechanisms have been reported including overexpression or point mutations in proteasome β5 subunit, impaired pro‐apoptotic protein accumulation, increased protein chaperone expression, and activation of a number of pro‐survival pathways (McConkey and Zhu, 2008; Horton et al., 2007). Therefore, combining bortezomib with chemotherapeutic agents is an important therapeutic modality aiming at boosting the efficacy and/or overcoming the resistance of hematologic malignancies (Reddy and Czuczman, 2010; Chen et al., 2011).
Dipyridamole has been in clinical use for decades as an anti‐platelet agent. Apart from this anti‐platelet activity, dipyridamole has shown a good potential to exert antitumor activity and/or enhance the cytotoxicity of a number of antitumor agents like TRAIL, cisplatin, methotrexate and 5‐fluorouracil (Kennedy et al., 1986; Sato et al., 1993; Rodrigues et al., 2004; Goda et al., 2008; Spano et al., 2013). In the present study we investigated the potential of dipyridamole to enhance bortezomib cytotoxicity against human malignant cell lines of different hematologic origin, and provided insights into the underlying mechanisms.
2. Materials and methods
2.1. Chemicals
Bortezomib (LC laboratoreis) was dissolved in dimethyl sulfoxide (DMSO) (Sigma–Aldrich), aliquoted, and stored at −80 °C for no more than 3 weeks. Dipyridamole (Sigma–Aldrich) was freshly dissolved in DMSO just before addition to cell cultures. N‐acetyl‐l‐cysteine (NAC) (Sigma–Aldrich) was freshly dissolved in RPMI 1640 medium, neutralized, and filter sterilized immediately prior to addition to cell cultures. A cell‐permeable form of reduced glutathione (glutathione monoethyl ester, Sigma–Aldrich) was dissolved in water, aliquoted, and stored at −80 °C. Dibutyryl cAMP (Sigma–Aldrich) and dibutyryl cGMP (Enzo Life Sciences Inc.) were freshly dissolved in water. The pan caspase inhibitor z‐VAD‐fmk (R&D Systems), and cycloheximide (Sigma–Aldrich) were dissolved in DMSO, aliquoted and stored at −20 °C. Imatinib (Santa Cruz Biotechnology), monochlorobimane (Sigma–Aldrich) and fluorogenic proteasome substrates: Suc‐LLVY‐AMC, Z‐ARR‐AMC, and Z‐LLE‐AMC (Calbiochem) were dissolved in DMSO, aliquoted, and stored at −80 °C. Glutathione‐S‐transferase (Sigma–Aldrich) was dissolved in PBS, aliquoted and stored at −20 °C.
2.2. Cell lines and culture conditions
Human p53‐mutant cell lines of different hematologic origin (ATCC): K562 (chronic myelogenous leukemia), HL60 (acute promyelocytic leukemia), MOLT4 (acute lymphoblastic leukemia), Jurkat and FADD−/−Jurkat (acute T cell leukemia), HEL92.1.7 (erythroleukemia) and U937 (histiocytic lymphoma) were cultured in RPMI 1640 medium (Hyclone) supplemented with 10% FBS (Hyclone), l‐glutamine (2 mmol/L), penicillin (100 units/mL), and streptomycin (100 μg/mL) (Gibco, Life Technologies Corp.) and maintained at 37 °C in a humidified atmosphere of 5% CO2.
2.3. Assessment of cytotoxicity via flow cytometry‐based propidium iodide staining
Cytotoxicity was evaluated based on the selective incorporation of propidium iodide (PI) into dead cells which lost the integrity of their plasma membrane (Moore et al., 1998) using FACSCaliburflow cytometer and CellQuest software package (BD Biosciences). A representative of 3 independent experiments (triplicates in each) was shown. Differences were considered statistically significant at p < 0.05 using Student's t test.
2.4. Assessment of the combination index (CI)
A quantitative assessment of non‐constant ratio combinations (fixed‐dose combinations) of bortezomib with dipyridamole in vitro was carried out using the Compusyn software (ComboSyn Inc.) that utilizes a median‐effect mathematical algorithm to calculate the combination index (CI) which expresses pharmacologic drug interactions based on the method of Chou and Talalay (Chou and Talalay, 1984). Values of the CI less than 1 reflect synergistic interactions, whereas CI values equal to 1 indicate additive effects. Drug antagonism is indicated by CI values more than 1.
2.5. FACS analysis of the intracellular reactive oxygen species (ROS)
Intracellular ROS generation in K562 cells was assessed using CM‐H2DCFDA (Molecular Probes) according to the manufacturer's instructions using FACSCalibur flow cytometer and CellQuest software package (BD Biosciences). A representative of 3 independent experiments (triplicates in each) was shown. Differences were considered statistically significant at p < 0.05 using Student's t test.
2.6. Analysis of mitochondrial membrane potential (MMP) using TMRE
The drop in MMP was analyzed using TMRE (Molecular Probes) as described previously (Goda et al., 2013) on FACSCalibur flow cytometer and CellQuest software package (BD Biosciences). A representative of 3 independent experiments (triplicates in each) was shown. Differences were considered statistically significant at p < 0.05 using Student's t test.
2.7. Immunoblotting
Protein isolation and immunoblotting were performed as previously described (Goda et al., 2008). The primary antibodies used were: anti‐Bcl‐xl, anti‐Bcl‐2, anti‐NOXA, anti‐XIAP, anti‐Akt (Santa Cruz Biotechnology); anti‐cleaved caspase‐3, anti‐PARP, anti‐Bid, anti‐Mcl‐1, anti‐GRP78, anti‐CHOP, anti‐phsopho Bcr‐Abl (Tyr245), anti‐phsopho Bcr‐Abl (Tyr412), anti‐phospho Akt (Ser473), anti‐phospho mTOR (Ser2448), anti‐phospho mTOR (Ser2481), anti‐mTOR, anti‐phospho ERK, anti‐ERK, anti‐phospho JAK2 (Tyr1007/1008), anti‐phospho JAK3, anti‐phospho Tyk2, anti‐JAK2, anti‐JAK3, anti‐Tyk2, anti‐c‐IAP‐1, anti‐survivin (Cell Signaling Technologies); anti‐caspase‐8, anti‐caspase‐9 (BD PharMingen); anti‐polyubiquitinylated proteins (clone FK1, Millipore); anti‐β‐actin (Sigma–Aldrich). Secondary antibodies used were: HRP‐anti‐rabbit IgG, HRP‐ anti‐mouse IgG (Cell Signaling Technologies). Relative band intensities were quantified using ImageJ software (http://imagej.nih.gov/ij/), and were indicated underneath relevant blots.
2.8. Assessment of different proteasomes activities
Chymotrypsin‐like, trypsin‐like and caspase‐like activities of the proteasomes were assessed via their respective fluorogenic substrates: Suc‐LLVY‐AMC, Z‐ARR‐AMC, and Z‐LLE‐AMC using a commercially available assay kit (Cayman Chemical) according to the manufacturer's instructions. Fluorescence values were acquired using Victor X2 multilabel plate reader (Perkin Elmer) and were normalized to the protein concentration of each sample. A representative of 3 independent experiments (triplicates in each) was shown. Differences were considered statistically significant at p < 0.05 using Student's t test.
2.9. Direct assay of intracellular reduced glutathione (GSH) levels using monochlorobimane (MCB)
A methodology based on the fluorescence detection of GST‐catalyzed conjugates of GSH to MCB (Young et al., 1994) was applied to de‐proteinized cell lysates prepared using ultra‐0.5, ultracel‐3 centrifugal filter units (Amicon). Fluorescence values were acquired using Victor X2 multilabel plate reader (PerkinElmer) and were normalized to the protein concentration of each sample that was assayed before de‐proteinization. A representative of 3 independent experiments (triplicates in each) was shown. Differences were considered statistically significant at p < 0.05 using Student's t test.
2.10. Generation of HS‐5 stromal conditioned medium (CM)
The bone marrow stromal cells HS‐5 (ATCC) were used to generate CM using RPMI 1640 medium as described previously (Traer et al., 2012).
2.11. Knockdown of gene expression via small interfering RNA (siRNA)
Commercially available CHOP siRNA (SMARTpool ON‐TARGETplus Human DDIT3 siRNA) was purchased from Dharmacon. Previously reported sequences of siRNA for NOXA and Bid (Bai et al., 2008; Liu et al., 2011) were synthesized by Cosmogenetech Co. Ltd. and transfected into K562 cells using HiPerFect Transfection Reagent (QIAGEN) according to the manufacturer instructions. Knockdown efficiency was assessed by immunoblotting.
2.12. In vivo evaluation of the efficacy of bortezomib/dipyridamole combination against K562 xenograft in nude mice
In vivo studies on nude mice were conducted according to the guidelines and following the approval of the KRIBB‐IACUC (Korea Research Institute of Bioscience & Biotechnology‐Institutional Animal Care and Use Committee). Athymic male BALB/c nude (nu/nu) mice (8 weeks old) (Charles River‐Orient Bio Inc., Republic of Korea) were acclimatized for one week before the beginning of the study in climate‐controlled conditions with 12‐h dark/light cycles and free access to food and water. For tumor implantation, K562 cells were diluted with BD matrigel basement membrane matrix (1:1). Each mouse was injected with 107 cells/mouse subcutaneously in the left flank. When tumors reached about 150 mm3, mice were randomly assigned to 4 groups (5 mice/group): control group, dipyridamole group (70 mg/kg administered in on–off cycles of two days on‐one day off), bortezomib group (0.8 mg/kg administered every third day on the dipyridamole‐off day), and bortezomib/dipyridamole group (0.8 mg/kg bortezomib and 70 mg/kg dipyridamole). A total of 6 divided doses of bortezomib were administered in PBS. Dipyridamole was injected as a solution in DMSO/PBS/Polyethylene glycol (PEG) vehicle. Both the control and bortezomib groups received the same DMSO/PBS/PEG vehicle on dipyridamole injection days throughout the whole study. Drug treatments were administered once daily by intraperitoneal route. Tumor volumes were measured every 3–4 days using a caliber and applying the formula: [(long axis) X (squared short axis) X 0.54].
3. Results
3.1. Generalized nature of the enhancement of bortezomib cytotoxicity by dipyridamole in human leukemia/lymphoma cells
To investigate the in vitro outcome of combining bortezomib with dipyridamole, a subclinical concentration of bortezomib (Horton et al., 2007; Attar et al., 2008) was combined for 48 h with 5, 10, 15 and 20 μM concentrations of dipyridamole which flank the clinically achievable serum concentrations (Willson et al., 1988; Budd et al., 1990). Cytotoxicity assessment showed that bortezomib monotherapy induced about a 30% cell death, whereas dipyridamole monotherapy did not show any appreciable cytotoxicity at the tested concentrations (Figure 1A). Interestingly, combining bortezomib with dipyridamole showed concentration‐dependent increments in the cytotoxicity of K562 cells, where the magnitude of cell death was almost doubled compared to bortezomib monotherapy (Figure 1A). Combination index (CI) analysis revealed the synergistic nature of bortezomib/dipyridamole combination in K562 cells (Table 1). Likewise, there was a concentration‐dependent increase in the cytotoxicity of K562 cells when a single concentration of dipyridamole (20 μM) was combined with escalating concentrations of bortezomib (5, 7.5, 10, 12.5, 15 nM), where the concentration of bortezomib that induces 50% cytotoxicity of the cells (EC50) was reduced from 15 nM to approximately 6.25 nM (Figure 1B), and the CI values for all combinations were 0.48, 0.43, 0.49, 0.59 and 0.65, respectively, reflecting synergistic interactions.
Figure 1.
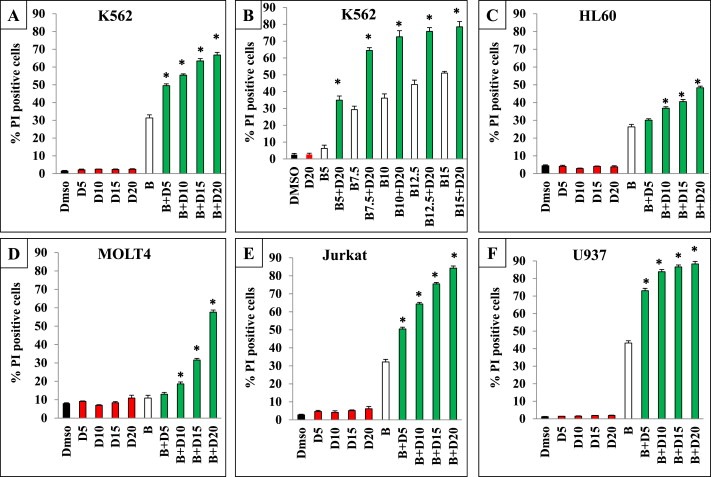
In vitro enhancement of cell death by bortezomib/dipyridamole co‐treatment (A and C through F): FACS analysis of the cytotoxicity of leukemia/lymphoma cells of different origin treated with bortezomib (K562: 7.5 nM; HL60: 3 nM; MOLT4: 1.5 nM; Jurkat: 4 nM; U937: 4 nM) for 30 min followed by co‐treatment with different concentrations of dipyridamole (5, 10, 15, 20 μM) for 48 h (B) FACS analysis of the cytotoxicity of K562 cells treated with different concentrations of bortezomib (5, 7.5, 10, 12.5 15 nM) for 30 min followed by co‐treatment with a single concentration of dipyridamole (20 μM) for 48 h. Abbreviations: “B”: bortezomib; “D”: dipyridamole. Asterisk indicates a statistically significant difference at p < 0.05.
Table 1.
The combination index values of different dose levels of dipyridamole in combination with bortezomib (K562:7.5 nM; HL60:3 nM; MOLT4:1.5 nM; Jurkat:4 nM; U937:4 nM) on different leukemia/lymphoma cell lines. Abbreviations: “B”: bortezomib; “D”: dipyridamole.
| B + D5 | B + D10 | B + D15 | B + D20 | |
|---|---|---|---|---|
| K562 | 0.7 | 0.65 | 0.57 | 0.53 |
| HL60 | 0.93 | 0.84 | 0.79 | 0.72 |
| MOLT4 | 0.98 | 0.93 | 0.86 | 0.77 |
| Jurkat | 0.73 | 0.58 | 0.47 | 0.37 |
| U937 | 0.61 | 0.49 | 0.45 | 0.42 |
To test whether the enhancement of bortezomib cytotoxicity by dipyridamole is of a generalized nature, a panel of human leukemia/lymphoma cells were treated with a single subclinical concentration of bortezomib and/or the same range of dipyridamole concentrations. FACS analysis showed dose‐dependent and manifold increments in bortezomib cytotoxicity upon combination with dipyridamole in HL60, MOLT4, Jurkat and U937 cells (Figure 1C through 1F). Moreover, combination index values of bortezomib with different concentrations of dipyridamole in these cell lines were less than 1, reflecting synergistic interactions (Table 1).
3.2. Dipyridamole acted downstream of the proteasome inhibition step by bortezomib through aggravation of ER stress induction, GSH depletion and ROS generation, to enhance the cytotoxicity of bortezomib independently of the protein kinase A (PKA) or protein kinase G (PKG) pathways
To gain mechanistic insights into the observed enhancement of bortezomib antitumor activity upon combination with dipyridamole, we investigated sequential steps along bortezomib signaling cascade to identify those targeted by co‐treatment with dipyridamole. Firstly, the influence of dipyridamole on the proteasome inhibitory potential of bortezomib was investigated at an early time point of drug treatments. The three different types of proteasomes activities, chymotrypsin‐like, trypsin‐like and caspase‐like activities were assessed in K562 cells following bortezomib and/or dipyridamole treatment for 3.5 h. Results showed that chymotrypsin‐like activity, which is the main target of bortezomib, was almost equally inhibited by bortezomib monotherapy and the combination treatment (Figure 2A). A similar result but with a lower magnitude was observed for the caspase‐like activity, whereas the trypsin‐like activity was not targeted by bortezomib monotherapy or the combination treatment (Figure 2A). Furthermore, immunoblotting of the polyubiquitinated proteins in K562 cells did not show any difference between bortezomib mono‐ or co‐treatments following 6 h of drugs exposure (Figure 2B). These results suggested that proteasome inhibition and accumulation of polyubiquitinated proteins by bortezomib were not targeted by dipyridamole.
Figure 2.
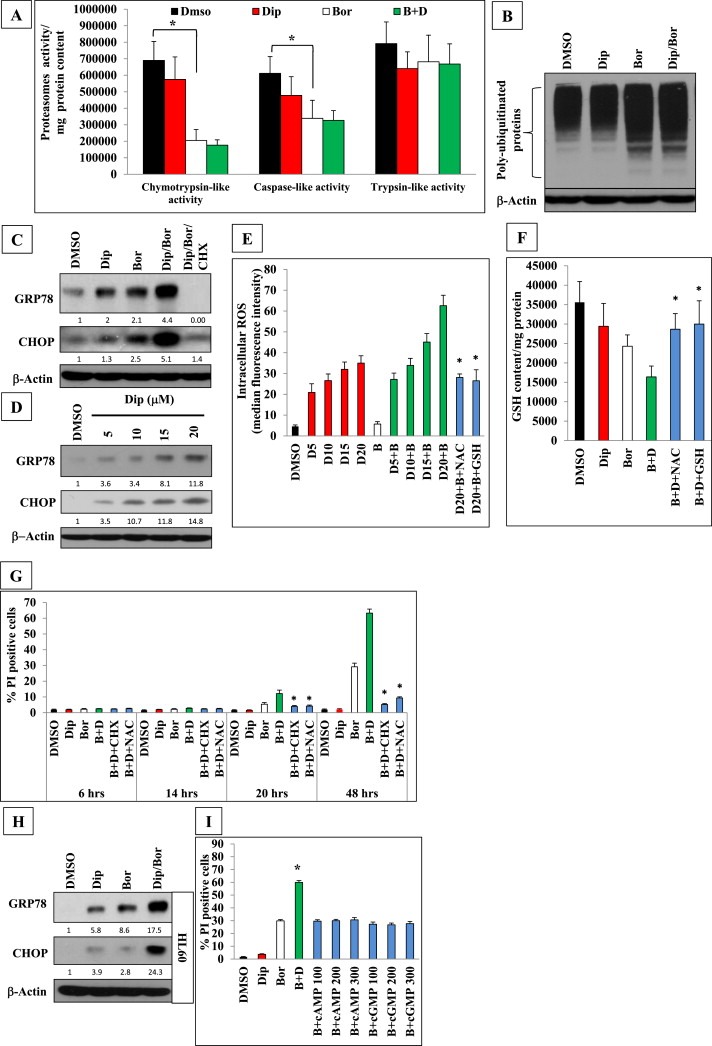
Dipyridamole enhanced bortezomib‐induced ER stress, ROS generation and GSH depletion downstream of the proteasome inhibition step and independently of the PKA or PKG. (A) Different proteasomes activities were assessed in K562 cells following bortezomib (7.5 nM) and/or dipyridamole (20 μM) co‐treatment for 3.5 h. (B) Immunoblot analysis of polyubiquitinated proteins in K562 cells treated with bortezomib (7.5 nM) and/or dipyridamole (20 μM) for 6 h. (C) Immunoblot analysis of ER stress markers in K562 cells following 6‐h of bortezomib (7.5 nM) and/or dipyridamole (20 μM) treatments. Cycloheximide (CHX) pre‐treatment (2 μg/mL) was performed for 2 h.(D) Immunoblotting of ER stress markers in K562 cells following 48‐h treatment with different dose levels of dipyridamole. (E) FACS analysis of ROS generation in K562 cells pretreated with NAC (10 mM) or cell‐permeable form of GSH (2.5 mM) for 2 h followed by 6‐h treatment with different concentrations of dipyridamole alone or in combination with bortezomib (7.5 nM). (F) Fluorimetric assay of intracellular GSH content in K562 cells. Bortezomib (7.5 nM) was applied for 30 min followed by dipyridamole (20 μM) co‐treatment for 6 h. NAC (10 mM) or cell‐permeable form of GSH (2.5 mM) were applied for 2 h before bortezomib treatment. (G) FACS analysis of the cytotoxicity of K562 cells pretreated with NAC (10 mM) or cycloheximide (CHX) (2 μg/mL) for 2 h then bortezomib (7.5 nM) was applied for 30 min followed by dipyridamole (20 μM) co‐treatment for 6, 14, 20, 48 h. (H) Immunoblot analysis of ER stress markers in HL60 cells following 6‐h co‐treatment with bortezomib (3 nM)/dipyridamole (20 μM). (I) FACS analysis of cell death of K562 cells treated with bortezomib (7.5 nM) for 30 min followed by co‐treatment with either dipyridamole (20 μM) or with escalating concentrations (100, 200, 300 μM) of the cell‐permeable long‐lasting dibutyryl cAMP or dibutyryl cGMP, respectively, for 48 h. Abbreviations: “Bor”or”B”: bortezomib; “Dip”or“D”: dipyridamole. Asterisk indicates a statistically significant difference at p < 0.05.
The inhibition of proteasomes and increased accumulation of misfolded proteins following bortezomib treatment eventually leads to the induction of endoplasmic reticulum (ER) stress (McConkey and Zhu, 2008; Reddy and Czuczman, 2010). Therefore, we assessed the impact of dipyridamole on the cellular levels of ER stress in K562 cells following6 hours of bortezomib treatment. Immunoblot analysis showed that dipyridamole and bortezomib monotherapies induced ER stress evidenced by the upregulation of GRP78 and CHOP, (Figure 2C). Cells received the co‐treatment experienced even much higher levels of ER stress that were abrogated by pre‐treatment with the protein translation inhibitor cycloheximide (Figure 2C). This novel ER stress‐inducing potential of dipyridamole was sustained since it could be maintained dose‐dependently up to 48 h (Figure 2D).
Given the well‐known cross‐talk between ER stress and the cell redox status, (Higa and Chevet, 2012) we investigated the possible modulation of intracellular ROS following bortezomib and/or dipyridamole. Intriguingly, K562 cells treated with different concentrations of dipyridamole for 6 h showed concentration‐dependent manifold increments in ROS generation which ranged from 4‐ to 7‐folds compared to the control level. However, bortezomib monotherapy weakly induced ROS by about 20% of the control level (Figure 2E). Of note, bortezomib/dipyridamole combination showed higher and concentration‐dependent increments in ROS generation which ranged from 6‐ to 13‐folds compared to the control level (Figure 2E). Pretreating the cells with NAC or a cell‐permeable form of GSH comparably scavenged about 55% of the ROS generated by bortezomib/dipyridamole co‐treatment (Figure 2E).Importantly, other scavengers like EUK132 or edaravone failed to affect the co‐treatment‐induced ROS generation (data not shown), suggesting that the depletion of GSH pools played an important role into ROS generation. To further validate this hypothesis, the intracellular levels of GSH were assessed in K562 cells following6 hours of bortezomib and/or dipyridamole treatment. Results showed 17% and 31% reductions in GSH basal levels by monotherapy with either dipyridamole or bortezomib, respectively, whereas the combination treatment brought about a 54% depletion of GSH pools (Figure 2F). Of note, pretreatment with NAC or the cell‐permeable form of GSH significantly restored the intracellular GSH pool in bortezomib/dipyridamole co‐treated cells to less than 20% of the basal GSH levels (Figure 2F). Taken together, these findings indicated that co‐treated K562 cells experienced profound ER stress, GSH depletion and ROS generation after 6 h of treatment, the time point at which cell death was undetectable (Figure 2G). However, the cytotoxicity was apparent after 20 h of treatment and followed an accelerated kinetics thereafter, and could be effectively inhibited by NAC or cycloheximide pretreatments (Figure 2G).
To gain insights whether this finding could be extended to another cell line of different origin, HL60 cells were treated with bortezomib and/or dipyridamole, and the level of ER stress was assessed after 6 h via immunoblotting. Results showed that co‐treated cells experienced higher levels of ER stress as compared to dipyridamole or bortezomib monotherapies (Figure 2H), corroborating findings obtained with K562 cells.
Moreover, since dipyridamole has previously been shown to activate signaling through PKA or PKG via the inhibition of cAMP‐ or cGMP‐phosphodiesterases, respectively (Salzman et al., 1972; Clarke et al., 1994), we assessed the probable involvement of these two kinases in modulating bortezomib cytotoxicity against K562 cells through the use of escalating concentrations of the cell‐permeable and long‐acting analogues of cAMP or cGMP. FACS analysis showed that bortezomib cytotoxicity was not enhanced by co‐treatment with either dibutyryl cAMP or dibutyryl cGMP even when they were applied at concentrations up to 300 μM (Figure 2I), suggesting that PKA and PKG signaling pathways are dispensable for enhancing bortezomib cytotoxicity.
3.3. Dipyridamole/bortezomib co‐treatment induced a GSH/ROS‐dependent caspase‐mediated mitochondrial apoptosis which involved multiple apoptotic modulators
Next, the pattern of cell death induced by bortezomib/dipyridamole co‐treatment was characterized. Pre‐treatment of K562 cells with the pan caspase inhibitor z‐VAD‐fmk almost completely abrogated the cytotoxicity of the co‐treatment (Figure 3A), indicating a caspase‐mediated apoptosis. Given this, we analyzed the relative contribution of the intrinsic and extrinsic apoptotic pathways in mediating the cytotoxicity of bortezomib/dipyridamole co‐treatment. To address this, FADD−/− Jurkat cells were used as a model cell line because of its resistance to death receptor‐initiated cell death. The lack of FADD expression in this cell line was confirmed by immunoblotting (Figure 3B). Both wild‐type and FADD−/− Jurkat cell lines were treated with bortezomib/dipyridamole combination, and cytotoxicity was assessed. Results showed a 22% reduction in the cytotoxicity of the combination treatment in FADD−/−Jurkat cells as compared to the parental cell line (Figure 3B), indicating that the extrinsic apoptotic pathway played a relatively minor role as compared to the intrinsic apoptotic pathway which largely regulated the cytotoxicity of bortezomib/dipyridamole co‐treatment.
Figure 3.
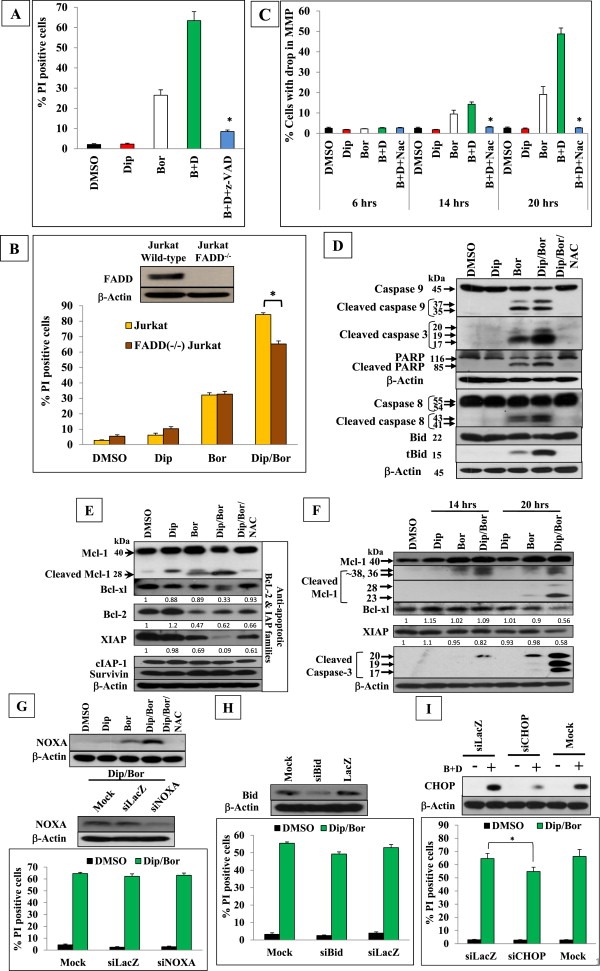
GSH/ROS‐dependent caspase‐mediated mitochondrial apoptosis induction by bortezomib/dipyridamole combination with the involvement of multiple apoptotic triggers in K562 cells. (A) FACS analysis of cell death of K562 cells pre‐treated with z‐VAD (40 μM) for 2 h, then with bortezomib (7.5 nM) for 30 min followed by co‐treatment with dipyridamole (20 μM) for 48 h (B) FACS analysis of cell death of Jurkat cells and FADD−/‐ Jurkat cells treated with bortezomib (4 nM) for 30 min followed by co‐treatment with dipyridamole (20 μM) for 48 h. The expression level of FADD in both cell lines was confirmed by immunoblotting. (C) FACS analysis of the drop in MMP in K562 cells pre‐treated with NAC (10 mM) for 2 h, then with bortezomib (7.5 nM) for 30 min followed by co‐treatment with dipyridamole (20 μM) for 6, 14, 20 h. (D) Immunoblot analysis of the effect of NAC on caspase‐8, ‐9, ‐3, PARP and Bid cleavage in K562 cells pre‐treated with NAC (10 mM) for 2 h, then with bortezomib (7.5 nM) for 30 min followed by co‐treatment with dipyridamole (20 μM) for 48 h. (E) Immunoblot analysis of Mcl‐1, Bcl‐xl, Bcl‐2, XIAP,c‐IAP‐1, and survivin in K562 cells pre‐treated with NAC (10 mM) for 2 h, then with bortezomib (7.5 nM) for 30 min followed by dipyridamole (20 μM) for 48 h. (F) Time‐course immunoblotting of Mcl‐1, Bcl‐xl, XIAP and cleaved caspase‐3 following bortezomib (7.5 nM) and/or dipyridamole (20 μM) treatments for 14 and 20 h (G) upper panel: Immunoblot analysis of NOXA in K562 cells pre‐treated with NAC (10 mM) for 2 h, then with bortezomib (7.5 nM) for 30 min followed by dipyridamole (20 μM) for 48 h (G lower panel, H and I) FACS analysis of the impact of siRNA‐mediated knockdown of NOXA, Bid and CHOP, respectively, on the cytotoxicity of K562 cells following bortezomib (7.5 nM)/dipyridamole(20 μM) 48‐h co‐treatment. Knockdown efficiency was evaluated by immunoblotting. Abbreviations: “Bor”or”B”: bortezomib; “Dip”or“D”: dipyridamole. Asterisk indicates a statistically significant difference at p < 0.05.
Since marked replenishment of the intracellular GSH and ROS scavenging could rescue K562 cells from the lethality of co‐treatment (Figures 2E and 2G), we investigated the GSH/ROS dependency of the observed apoptotic cell death. To this end, the MMP was assessed in K562 cells following bortezomib/dipyridamole co‐treatment in the presence or absence of NAC, and in a time‐course manner. FACS analysis showed that after 6 h of drug treatments, there was not any detectable drop in the MMP (Figure 3C), however after 14 h there was a differential loss of MMP in bortezomib mono‐ or co‐treated cells, which preceded the induction of cell death (Figure 2G, 14‐h′ time point), and markedly increased in intensity after 20 h where the co‐treatment induced about 2‐folds increase in the percentage of cells with dissipated MMP as compared to bortezomib monotherapy (Figure 3C). Importantly, NAC pretreatment could overcome such a drop in MMP by the co‐treatment (Figure 3C), confirming that GSH depletion and ROS generation acted upstream and mediated the apoptotic cell death. Further, having demonstrated the caspase‐dependency of the co‐treatment‐induced apoptosis (Figure 3A), we aimed at providing a direct evidence for the role of GSH/ROS in controlling caspases in K562 cells treated with bortezomib and/or dipyridamole in the presence or absence of NAC. Immunoblotting showed that NAC pretreatment efficiently abrogated the co‐treatment‐induced cleavage of caspase‐9, ‐8, ‐3 and their downstream targets PARP and Bid (Figure 3D). These findings confirmed that caspases processing was regulated by GSH depletion and ROS generation.
Having shown that the mitochondrial pathway largely mediated bortezomib/dipyridamole‐induced apoptosis, it was important to investigate which apoptototic trigger(s) could be involved. To this end, we firstly analyzed whether the co‐treatment may modulate the expression levels of the anti‐apoptotic members of the Bcl‐2 and IAP protein families, since they sequester and neutralize several pro‐apoptotic binding partners (Chipuk et al., 2010). Immunoblotting of K562 cell lysates harvested after 48 h of treatment showed that bortezomib monotherapy upregulated the anti‐apoptotic Mcl‐1 protein and weakly increased its truncation to the 28‐kDa fragment which has a pro‐apoptotic function (Le Gouill et al., 2004). Interestingly, the combination treatment not only downregulated Mcl‐1 total protein levels but also strongly enriched the abundance of the pro‐apoptotic 28‐kDa fragment (Figure 3E). In addition, bortezomib/dipyridamole combination efficiently downregulated the expression level of the Bcl‐xl, Bcl‐2 as well as the potent endogenous caspase inhibitor XIAP (Eckelman et al., 2006) (Figure 3E). On the other hand, the expression levels of c‐IAP‐1 and survivin were not downregulated by the co‐treatment (Figure 3E). Of note, NAC pre‐treatment markedly normalized all the observed changes in the expression levels of Mcl‐1, Bcl‐xl, Bcl‐2 and XIAP, which further supported the key role played by the aggravated GSH depletion and ROS generation by bortezomib/dipyridamole co‐treatment (Figure 3E). Kinetic analysis revealed that at early time points of drug exposure, Mcl‐1 total levels were not downregulated by the co‐treatment (Figure 3F), however the 36 and 38 kDa fragments were observed with a higher abundance after 14 h of the co‐treatment, whereas after 20 h there was a marked increase in the co‐treatment‐induced Mcl‐1 cleavage, generating fragments that are either pro‐apoptotic (28 kDa) or non‐functional (23 kDa) (Le Gouill et al., 2004) (Figure 3F). This increased cleavage may account for the downregulation of the total Mcl‐1 levels observed after 48 h of the co‐treatment (Figure 3E). Furthermore, the downregulation of XIAP was detectable as early as 14 h and intensified thereafter, while the suppression of Bcl‐xl was clearly evident after 20 h of treatment (Figure 3F). These findings indicated that the modulation of these anti‐apoptotic regulators followed a kinetics that matched the drop in MMP (Figure 3C), caspase‐3 cleavage (Figure 3F) and the ensuing cell death (Figure 2G), suggesting that these anti‐apoptotic regulators were closely associated with the induction of cell death.
The aforementioned findings suggested that the downregulation of Mcl‐1, Bcl‐xl, Bcl‐2 and XIAP by the co‐treatment could have de‐repressed their multiple pro‐apoptotic binding partners (Chipuk et al., 2010), accordingly multiple triggers would be expected to activate the mitochondrial pathway. To verify this hypothesis, we investigated the relative contribution of two pro‐apoptotic Bcl‐2 family members, NOXA and Bid that were previously reported to mediate bortezomib‐induced apoptosis in some cell lines (Pérez‐Galán et al., 2006; Unterkircher et al., 2011) whose origin is different from the cells being tested in the current study. Immunoblotting showed that bortezomib/dipyridamoleco‐treatment was more efficient than bortezomib monotherapy in the induction of NOXA in K562 cells in a GSH/ROS dependent manner (Figure 3G upper panel). However, knockdown of NOXA did not rescue K562 cells from the lethality of the co‐treatment (Figure 3G lower panel). Similarly, Bid knockdown failed to abrogate the cytotoxicity of the co‐treatment (Figure 3H). These findings were not surprising since there was no combined modulations of the three anti‐apoptotic Bcl‐2 family members, Mcl‐1, Bcl‐xl and Bcl‐2as well as XIAP in the previous reports where interference with NOXA or Bid was protective (Pérez‐Galán et al., 2006; Unterkircher et al., 2011). Furthermore, knockdown of CHOP, a mediator of the pro‐apoptotic arm of the ER stress which eventually engages the mitochondrial pathway, (Higa and Chevet, 2012) conferred about 14% protection against bortezomib/dipyridamole‐induced cell death (Figure 3I). Taken together, these findings suggested that the anticipated de‐repression of several pro‐apoptotic Bcl‐2 family members driven by the downregulation of their anti‐apoptotic binding partners, Mcl‐1, Bcl‐xl and Bcl‐2, could have masked the individual contribution of NOXA, Bid or CHOP towards apoptosis induction by the co‐treatment, which strongly supported the hypothesis that mitochondrial apoptosis induction by bortezomib/dipyridamole combination is multi‐faceted and cooperatively executed via a number of pro‐apoptotic triggers.
3.4. Dipyridamole enhanced bortezomib potential to target pro‐survival signaling pathways in a GSH/ROS‐dependent manner
The observed expression changes in Mcl‐1, Bcl‐xl, Bcl‐2 and XIAP following bortezomib/dipyridamole co‐treatment of K562 cells suggested a probable modulation of the pro‐survival signaling pathways which keep the apoptotic circuitry under a tight control (McCubrey et al., 2008). Given that the Bcr‐Abl, Akt/mTOR, and ERK pathways are well‐known to act individually and in concert to suppress apoptosis, (McCubrey et al., 2008; Cilloni and Saglio, 2012) and that a crosstalk exists among these pro‐survival signaling pathways and the ER stress signaling which can be negatively regulated by the Bcr‐Abl, (Piwocka et al., 2006) and likewise, the ER stress can suppress the mTOR signaling, (Appenzeller‐Herzog and Hall, 2012) we therefore profiled the potential expression changes in the Bcr‐Abl, Akt/mTOR, and ERK pathways in K562 cells following bortezomib/dipyridamole co‐treatment. Immunoblotting showed that the expression of the Bcr‐Abl, and Akt/mTOR (both the phosphorylated and total levels) were more suppressed by bortezomib/dipyridamole combination as compared to that induced by bortezomib monotherapy (Figure 4A). On the other hand, only the phosphorylated form of ERK was almost completely downregulated by the co‐treatment (Figure 4A). Further, the inhibition of these pro‐survival pathways by the co‐treatment occurred in a GSH/ROS‐dependent manner since pre‐treatment with NAC could normalize the observed changes (Figure 4A). Of note, the extent of suppression of the pro‐survival pathways by bortezomib (7.5 nM) when combined with dipyridamole (20 μM) (Figure 4A) could approximately mimic the inhibition level obtained with bortezomib (20 nM) monotherapy (Figure 4B), indicating that dipyridamole increased the multi‐targeting potential of bortezomib by about 2‐folds. Importantly, kinetic analysis revealed that the modulation of the expression of Bcr‐Abl, mTOR, ERK was evident after 14 h of the co‐treatment, with more suppressions after 20 h (Figure 4C). This is in concert with the expression kinetics of CHOP induction (Figure 4C), anti‐apoptotic regulators and cleaved caspase‐3 (Figure 3F), and cell death (Figure 2G). Of note, Akt phosphorylation was strongly induced early during bortezomib monotherapy, however this induction was of lower intensity and shorter duration with the co‐treatment (Figure 4C). Collectively, these time‐course investigations in K562 cells shed some light on the molecular events that accompanied and may account for the enhanced cell death by bortezomib/dipyridamole combination.
Figure 4.
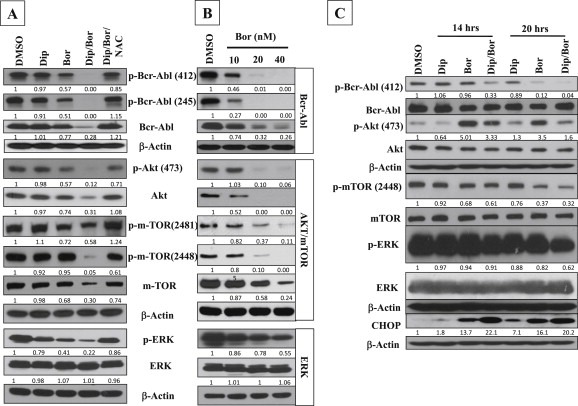
Enhanced multi‐targeting potential of bortezomib/dipyridamole combination. (A) Immunoblot analysis of signaling components of the Bcr‐Abl, Akt/mTOR and ERK pathways in K562 cells. NAC (10 mM) was applied for 2 h followed by bortezomib (7.5 nM) for 30 min then dipyridamole (20 μM) was added for additional 48 h.(B) Bortezomib different dose levels (10, 20, 40 nM) were applied to K562 cells for 48 h and different signaling components of the Bcr‐Abl, Akt/mTOR and ERK pathways were analyzed by immunoblotting. (C) Time‐course immunoblotting of signaling components of the Bcr‐Abl, Akt/mTOR and ERK pathways as well as CHOP in K562 cells following bortezomib (7.5 nM) and/or dipyridamole (20 μM) treatments for 14 and 20 h. Abbreviations: “Bor”: bortezomib; “Dip”: dipyridamole.
3.5. Downregulation of JAK family members by bortezomib/dipyridamole combination and its potential impact on the stromal‐mediated chemoresistance
The JAK family of tyrosine kinases is another important pro‐survival pathway which regulate the apoptotic signals in hematologic malignancies (McCubrey et al., 2008). Therefore, we investigated the influence of bortezomib and/or dipyridamole treatments on the expression levels of JAK family members in K562 cells. Immunoblotting showed that bortezomib monotherapy upregulated the JAK2 activating phosphorylationon tyrosine 1007/1008 residues (Figure 5A), most likely due to the regulation of JAK2 by the ubiquitin/proteasome pathway (Ungureanu et al., 2002). Interestingly, dipyridamole effectively abolished the basal as well as bortezomib‐induced JAK2 phosphorylation (Figure 5A). The total levels of JAK2 were unaffected by dipyridamole monotherapy but were markedly downregulated by bortezomib and almost completely abolished by the co‐treatment (Figure 5A). Of note, the suppression of JAK2 phosphorylation by dipyridamole/bortezomib combination was weakly recovered by NAC even though the total level of JAK2 was markedly recovered (Figure 5A), suggesting a differential regulation by GSH/ROS. With regards to other JAK family members, the combination treatment was more efficient than bortezomib monotherapy in downregulating the expression levels of both the phosphorylated and total forms of Tyk2 and JAK3, while dipyridamole lacked any remarkable effect on these two JAK family members (Figure 5A). The phosphorylation of JAK1 could not be detected in K562 cells (data not shown). Of note, NAC pretreatment recovered both the phosphorylated and total forms of Tyk2, however its effects on JAK3 were similar to those observed with JAK2 suggesting a similar regulatory mechanism (Figure 5A). Moreover, time‐course immunoblot analysis showed that the suppression of JAK family members by the co‐treatment (Figure 5B) was closely associated with other pro‐survival pathways (Figure 4C) and the anti‐apoptotic regulators (Figure 3F), suggesting its potential role in regulating the co‐treatment‐induced cytotoxicity. Of note, the novel inhibitory effect of dipyridamole monotherapy on the JAK2 activating phosphorylation on tyrosine 1007/1008 residues could be detected as early as 14 h of treatment (Figure 5B) and was sustained dose‐dependently up to 48 h without any detectable changes in the phosphorylation status of either JAK3 or Tyk2 (Figure 5C), suggesting a selectivity in dipyridamole action.
Figure 5.
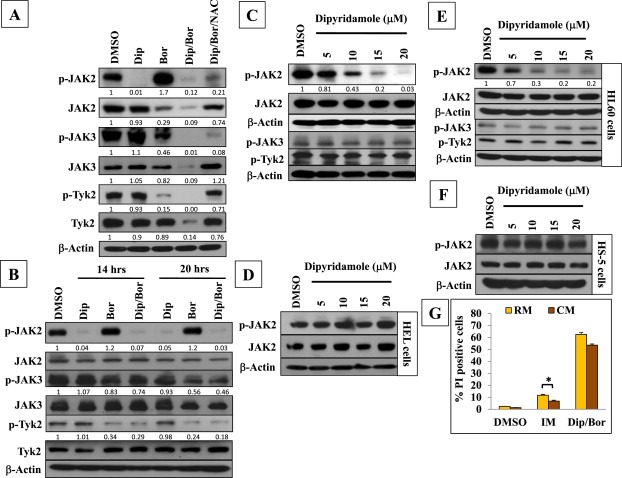
Efficient suppression of different JAK family members by bortezomib/dipyridamole co‐treatment and the lack of marked impact of the stromal‐mediated chemoresistance. (A) Immunoblot analysis of JAK family members in K562 cells. NAC (10 mM) was applied for 2 h followed by bortezomib (7.5 nM)/dipyridamole (20 μM) 48‐h co‐treatment. (B) Time‐course immunoblotting of JAK family members in K562 cells following bortezomib (7.5 nM) and/or dipyridamole (20 μM) treatments for 14 and 20 h. (C) Immunoblot analysis of JAK family members in K562 cells following dipyridamole different dose levels treatment for 48 h. (D) Immunoblot analysis of JAK2 in HEL cells following dipyridamole different dose levels treatment for 48 h. (E) Immunoblot analysis of JAK family members in HL60 cells following dipyridamole different dose levels treatment for 48 h. (F) Immunoblot analysis of JAK2 in HS‐5 cells following dipyridamole different dose levels treatment for 48 h (G) FACS analysis comparing the effects of RM and HS‐5 CM on the cytotoxicity of bortezomib (7.5 nM)/dipyridamole (20 μM) co‐treatment treatment for 48 h. Imatinib (1 μM for 48 h) was included as a positive control. Asterisk indicates a statistically significant difference at p < 0.05.Abbreviations: “Bor”: bortezomib; “Dip”: dipyridamole.
To gain some insights into the mechanism(s) underlying the suppression of JAK2 phosphorylation by dipyridamole, HEL92.1.7 erythroleukemia cell line was used which has a JAK2 point mutation (V617F) (Zhao et al., 2012) that renders JAK2 constitutively phosphorylated independently of the cytokine/growth factor receptors (Reuther, 2008). Immunoblotting showed that treatment of HEL92.1.7 cells with dipyridamole failed to inhibit JAK2 phosphorylation (Figure 5D), suggesting that the abrogation of JAK2 phosphorylation by dipyridamole occurred indirectly through interference with the signaling events that regulate cytokine/growth factor receptor‐dependent phosphorylation.
Moreover, the potential of dipyridamole to suppress basal JAK2 phosphorylation was investigated in another leukemia cell line, HL60 cells. Immunoblotting showed that dipyridamole downregulated JAK2 phosphorylation dose‐dependently and even more prominently than that observed with K562 cells, whereas no changes could be detected in the expression levels of phosphorylated Tyk2 or JAK3 (Figure 5E). Further, in view of the crucial roles of JAK2 in controlling the erythroid/myeloid lineage differentiation during normal hematopoiesis (Neubauer et al., 1998), we investigated whether dipyridamole may influence JAK2 phosphorylation in HS‐5 stromal cell line. Immunoblotting showed that treatment of HS‐5 stromal cells with dipyridamole failed to affect JAK2 phosphorylation (Figure 5F). Collectively, these results indicated the dipyridamole selectively abrogated JAK2 phosphorylation in cancer cells but not in stromal cells.
Given the role of JAK family in mediating the survival signals within the cytokine‐rich bone marrow microenvironment which drive resistance to chemotherapy (Traer et al., 2012), we tested whether the abrogation of JAK signaling by the co‐treatment would be of value in ameliorating the impact of the stromal‐mediated chemoresistance. Firstly, to validate the model, K562 cells were treated with imatinib in either routine medium (RM) or HS‐5‐conditioned medium (CM) to validate the proper generation of the CM, and the cytotoxicity was assessed by FACS. Results showed that CM induced about 50% reduction in imatinib cytotoxicity (Figure 5G). Next, we evaluated the cytotoxicity of bortezomib/dipyridamole combination against K562 cells in both RM and CM. FACS analysis showed that the values of co‐treatment‐induced cytotoxicity in CM and RM were about 53% and 62%, respectively, (i.e., 15% reduction in cytotoxicity) (Figure 5G), indicating that the efficacy of bortezomib/dipyridamole combination is weekly affected by the stromal‐mediated chemoresistance.
3.6. In vivo efficacy of bortezomib/dipyridamole co‐treatment against K562 xenografts in nude mice
Next, we aimed at evaluating the in vivo efficacy of bortezomib/dipyridamole co‐treatment against xenografted K562 cells. To this end, we conducted preliminary experiments to determine the appropriate dosing regimens of bortezomib and dipyridamole in combination. Dipyridamole dose of 70 mg/kg was selected which did not affect nude mice survival when combined with bortezomib (0.8 mg/kg) throughout the study. The duration of drug treatments was set to 17 consecutive days during which a total of 6 divided doses of bortezomib (0.8 mg/kg) were administered. Longer treatment periods were disapproved because nude mice receiving more than 6 divided doses of bortezomib monotherapy showed a compromise in survival rates (data not shown). Next, nude mice were engrafted with K562 cells and received bortezomib and/or dipyridamole treatments or the control vehicle, and the tumor volumes were monitored in a time‐course manner. Results showed that at the end of treatment period, the average tumor volume in the bortezomib‐treated group was 2717 ± 1025 mm3 (mean ± SD) as compared to 3129 ± 1268 mm3 in the control group (Figure 6) with an average reduction in tumor volume by 13%, indicating that bortezomib monotherapy was weakly effective in retarding solid tumor growth. Interestingly, in the dipyridamole‐treated group the average tumor volume was 1795 ± 290 mm3, which means that dipyridamole was much more effective than bortezomib monotherapy in reducing tumor volume by 42% (Figure 6). This observed in vivo antitumor activity of dipyridamole on K562 cells is in line with results obtained from other cancer cell lines of different origin (Spano et al., 2013). Furthermore, mice receiving bortezomib/dipyridamole combination had about a 48% reduction in their xenografts with an average tumor volume of 1630 ± 581 mm3 (Figure 6). Findings of the current in vivo study even though were not synergistic in nature unlike the results obtained in vitro with the leukemia/lymphoma cells growing in suspension form which is the main way of disease progression, however the superior efficacy of bortezomib/dipyridamole co‐treatment driven by dipyridamole was demonstrated as compared to the weak antitumor activity of bortezomib monotherapy against engrafted K562 tumors.
Figure 6.
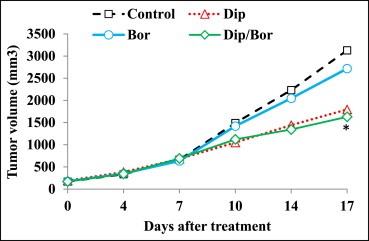
In vivo efficacy of bortezomib/dipyridamole co‐treatment against K562 xenografts. Time‐course assessment of the tumor volumes in nude mice harboring K562 xenografts following treatment with dipyridamole (70 mg/kg) and/or bortezomib (0.8 mg/kg). Asterisk indicates a statistically significant difference at p < 0.05 as compared to bortezomib monotherapy. Abbreviations: “Bor”: bortezomib; “Dip”: dipyridamole.
4. Discussion
Bortezomib monotherapy of hematologic malignancies is frequently undermined by partial response, dose‐limiting toxicities and drug resistance, which necessitate drug combinations to improve therapeutic outcomes. According to the database of the “ClinicalTrials.gov” (www.clinicaltrials.gov) there are over 300 interventional clinical trials that have evaluated/currently evaluating different bortezomib‐based combinations in the management of a variety of malignant blood disorders. In the current study, it has been shown for the first time that the antitumor activity of bortezomib against leukemia/lymphoma cells could be markedly improved upon combination with the clinically achievable serum concentrations of dipyridamole. Of note, the lack of the contribution of cAMP‐ or cGMP‐dependent signaling towards the enhancement of bortezomib cytotoxicity by dipyridamole suggested an alternative molecular target of dipyridamole, the identity of which is yet to be uncovered.
Based on the data presented herein, several clinically relevant conclusions could be drawn. Firstly, bortezomib/dipyridamole combination was able to overcome a number of clinically relevant resistance factors, Mcl‐1, Bcl‐xl, Bcl‐2, XIAP, FADD‐deficiency, Bcr‐Abl, Akt/mTOR and ERK which mediate chemotherapy failure (McCubrey et al., 2008; Testa and Riccioni, 2007). Secondly, the efficacy of the combination treatment was independent of the p53 tumor suppressor signaling given that all cell lines tested were p53‐mutant. This is of value in bypassing leukemia drug resistance driven by p53 mutations (Wattel et al., 1994). Thirdly, the antitumor activity of the bortezomib/dipyridamole combination was largely unaffected in a model of cytokine‐rich bone marrow microenvironment, at least in part, due to the strong suppression of different JAK family members by the co‐treatment. Fourthly, the independence of bortezomib/dipyridamole‐induced apoptosis of a single trigger could be of value in overcoming the sporadic inactivating mutations of the Bcl‐2 family (Packham, 1998). Lastly, the enhanced efficacy of subclinical concentrations of bortezomib upon combination with dipyridamole should enable bortezomib dosage reductions without undermining the clinical outcomes, which may allow for possible treatment cost cuts given the relatively high annual expenses of bortezomib therapy (Durie et al., 2013).
Unlike the in vitro findings, there was a lack of in vivo synergy between bortezomib and dipyridamole in the current study. This may be attributed to the notion that all in vitro experiments were conducted under normoxia conditions, while it is generally well established that solid tumors present hypoxic environments which constitute a selective pressure for the development of acquired drug resistance (Hockel and Vaupel, 2001). Hypoxia was previously shown to select for bortezomib‐resistant K562 cells (Tanturli et al., 2011) which may explain the weak in vivo antitumor activity of bortezomib monotherapy in the current study. Moreover, it is very likely that hypoxic tumor cell adaptation could have negatively influenced different signaling pathways that were exploited by bortezomib/dipyridamole co‐treatment, thereby undermining synergistic interactions. Importantly, under these in vivo hypoxic conditions dipyridamole alone or in combination with bortezomib could exert a significant antitumor activity which is at least 3 folds that of bortezomib monotherapy.
In summary, the current study introduced a mechanism‐based novel therapeutic modality which warrants further clinical investigation of bortezomib/dipyridamole combination in hematologic malignancies.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgment
This work was supported by the World Class Institute (WCI 2009‐002), Global R&D Center (GRDC) and KRIBB Research Initiative Program funded by the Ministry of Science, ICT and Future Planning (MSIP), Republic of Korea.
Goda Ahmed E., Erikson Raymond L., Sakai Toshiyuki, Ahn Jong-Seog, Kim Bo-Yeon, (2015), Preclinical evaluation of bortezomib/dipyridamole novel combination as a potential therapeutic modality for hematologic malignancies, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.08.010.
Contributor Information
Ahmed E. Goda, Email: AHMEDELSAYEDGODA@pharm.tanta.edu.eg
Bo-Yeon Kim, Email: bykim@kribb.re.kr.
References
- Appenzeller-Herzog, C. , Hall, M.N. , 2012. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 22, 274–282. [DOI] [PubMed] [Google Scholar]
- Attar, E.C. , De Angelo, D.J. , Supko, J.G. , D'Amato, F. , Zahrieh, D. , Sirulnik, A. , Wadleigh, M. , Ballen, K.K. , McAfee, S. , Miller, K.B. , Levine, J. , Galinsky, I. , Trehu, E.G. , Schenkein, D. , Neuberg, D. , Stone, R.M. , Amrein, P.C. , 2008. Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clin. Cancer Res. 14, 1446–1454. [DOI] [PubMed] [Google Scholar]
- Bai, Y. , Ahmad, U. , Wang, Y. , Li, J.H. , Choy, J.C. , Kim, R.W. , Kirkiles-Smith, N. , Maher, S.E. , Karras, J.G. , Bennett, C.F. , Bothwell, A.L. , Pober, J.S. , Tellides, G. , 2008. Interferon-gamma induces X-linked inhibitor of apoptosis-associated factor-1 and Noxa expression and potentiates human vascular smooth muscle cell apoptosis by STAT3 activation. J. Biol. Chem. 283, 6832–6842. [DOI] [PubMed] [Google Scholar]
- Budd, G.T. , Jayaraj, A. , Grabowski, D. , Adelstein, D. , Bauer, L. , Boyett, J. , Bukowski, R. , Murthy, S. , Weick, J. , 1990. Phase I trial of dipyridamole with 5-fluorouracil and folinic acid. Cancer Res. 50, 7206–7211. [PubMed] [Google Scholar]
- Chen, D. , Frezza, M. , Schmitt, S. , Kanwar, J. , Dou, Q.P. , 2011. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets. 11, 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk, J.E. , Moldoveanu, T. , Llambi, F. , Parsons, M.J. , Green, D.R. , 2010. The Bcl-2 family reunion. Mol. Cell. 37, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T.C. , Talalay, P. , 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme. Regul. 22, 27–55. [DOI] [PubMed] [Google Scholar]
- Cilloni, D. , Saglio, G. , 2012. Molecular pathways: BCR-ABL. Clin. Cancer Res. 18, 930–937. [DOI] [PubMed] [Google Scholar]
- Clarke, W.R. , Uezono, S. , Chambers, A. , Doepfner, P. , 1994. The type III phosphodiesterase inhibitor milrinone and type V PDE inhibitor dipyridamole individually and synergistically reduce elevated pulmonary vascular resistance. Pulm. Pharmacol. 7, 81–89. [DOI] [PubMed] [Google Scholar]
- Durie, B. , Binder, G. , Pashos, C. , Khan, Z. , Hussein, M. , Borrello, I. , 2013. Total cost comparison in relapsed/refractory multiple myeloma. J. Med. Econ. 16, 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckelman, B.P. , Salvesen, G.S. , Scott, F.L. , 2006. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 7, 988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda, A.E. , Yoshida, T. , Horinaka, M. , Yasuda, T. , Shiraishi, T. , Wakada, M. , Sakai, T. , 2008. Mechanisms of enhancement of TRAIL tumoricidal activity against human cancer cells of different origin by dipyridamole. Oncogene. 27, 3435–3445. [DOI] [PubMed] [Google Scholar]
- Goda, A.E. , Koyama, M. , Sowa, Y. , Elokely, K.M. , Yoshida, T. , Kim, B.Y. , Sakai, T. , 2013. Molecular mechanisms of the antitumor activity of SB225002: a novel microtubule inhibitor. Biochem. Pharmacol. 85, 1741–1752. [DOI] [PubMed] [Google Scholar]
- Higa, A. , Chevet, E. , 2012. Redox signaling loops in the unfolded protein response. Cell. Signal. 24, 1548–1555. [DOI] [PubMed] [Google Scholar]
- Hockel, M. , Vaupel, P. , 2001. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 93, 266–276. [DOI] [PubMed] [Google Scholar]
- Horton, T.M. , Pati, D. , Plon, S.E. , Thompson, P.A. , Bomgaars, L.R. , Adamson, P.C. , Ingle, A.M. , Wright, J. , Brockman, A.H. , Paton, M. , Blaney, S.M. , 2007. A phase 1 study of the proteasome inhibitor bortezomib in pediatric patients with refractory leukemia: a Children's Oncology Group study. Clin. Cancer Res. 13, 1516–1522. [DOI] [PubMed] [Google Scholar]
- Kennedy, D.G. , Van den Berg, H.W. , Clarke, R. , Murphy, R.F. , 1986. Enhancement of methotrexate cytotoxicity towards the MDA.MB.436 human breast cancer cell line by dipyridamole. The role of methotrexate polyglutamates. Biochem. Pharmacol. 35, 3053–3056. [DOI] [PubMed] [Google Scholar]
- Le Gouill, S. , Podar, K. , Harousseau, J.L. , Anderson, K.C. , 2004. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle. 3, 1259–1262. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Vaithiyalingam, S. , Shi, Q. , Chazin, W.J. , Zinkel, S.S. , 2011. BID binds to replication protein A and stimulates ATR function following replicative stress. Mol. Cell Biol. 31, 4298–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey, D.J. , Zhu, K. , 2008. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug. Resist. Updat. 11, 164–179. [DOI] [PubMed] [Google Scholar]
- McCubrey, J.A. , Steelman, L.S. , Abrams, S.L. , Bertrand, F.E. , Ludwig, D.E. , Bäsecke, J. , Libra, M. , Stivala, F. , Milella, M. , Tafuri, A. , Lunghi, P. , Bonati, A. , Martelli, A.M. , 2008. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 22, 708–722. [DOI] [PubMed] [Google Scholar]
- Moore, A. , Donahue, C.J. , Bauer, K.D. , Mather, J.P. , 1998. Simultaneous measurement of cell cycle and apoptotic cell death. Methods Cell Biol. 57, 265–278. [DOI] [PubMed] [Google Scholar]
- Neubauer, H. , Cumano, A. , Müller, M. , Wu, H. , Huffstadt, U. , Pfeffer, K. , 1998. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 93, 397–409. [DOI] [PubMed] [Google Scholar]
- Packham, G. , 1998. Mutation of Bcl-2 family proteins in Cancer. Apoptosis. 3, 75–82. [DOI] [PubMed] [Google Scholar]
- Pérez-Galán, P. , Roué, G. , Villamor, N. , Montserrat, E. , Campo, E. , Colomer, D. , 2006. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 107, 257–264. [DOI] [PubMed] [Google Scholar]
- Piwocka, K. , Vejda, S. , Cotter, T.G. , O'Sullivan, G.C. , McKenna, S.L. , 2006. Bcr-Abl reduces endoplasmic reticulum releasable calcium levels by a Bcl-2-independent mechanism and inhibits calcium-dependent apoptotic signaling. Blood. 107, 4003–4010. [DOI] [PubMed] [Google Scholar]
- Reddy, N. , Czuczman, M.S. , 2010. Enhancing activity and overcoming chemoresistance in hematologic malignancies with bortezomib: preclinical mechanistic studies. Ann. Oncol. 21, 1756–1764. [DOI] [PubMed] [Google Scholar]
- Reuther, G.W. , 2008. JAK2 activation in myeloproliferative neoplasms: a potential role for heterodimeric receptors. Cell Cycle. 7, 714–719. [DOI] [PubMed] [Google Scholar]
- Rodrigues, M. , Barbosa, F. , Perussi, J.R. , 2004. Dipyridamole increases the cytotoxicity of cisplatin in human larynx cancer cells in vitro . Braz. J. Med. Biol. Res. 37, 591–599. [DOI] [PubMed] [Google Scholar]
- Salzman, E.W. , Kensler, P.C. , Levine, L. , 1972. Cyclic 3'5'-adenosine monophosphate in human blood platelets: regulatory role of cyclic AMP in platelet function. Ann. N. Y. Acad. Sci. 201, 61–71. [DOI] [PubMed] [Google Scholar]
- Sato, S. , Kohno, K. , Hidaka, K. , Hisatsugu, T. , Kuwano, M. , Komiyama, S. , 1993. Differentially potentiating effects by dipyridamole on cytotoxicity of 5-fluorouracil against three human maxillary cancer cell lines derived from a single tumor. Anticancer. Drug. Des. 8, 289–297. [PubMed] [Google Scholar]
- Spano, D. , Marshall, J.C. , Marino, N. , De Martino, D. , Romano, A. , Scoppettuolo, M.N. , Bello, A.M. , Di Dato, V. , Navas, L. , De Vita, G. , Medaglia, C. , Steeg, P.S. , Zollo, M. , 2013. Dipyridamole prevents triple-negative breast-cancer progression. Clin. Exp. Metastasis. 30, 47–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanturli, M. , Giuntoli, S. , Barbetti, V. , Rovida, E. , Dello Sbarba, P. , 2011. Hypoxia selects bortezomib-resistant stem cells of chronic myeloid leukemia. PLoS One. 6, e17008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa, U. , Riccioni, R. , 2007. Deregulation of apoptosis in acute myeloid leukemia. Haematologica. 92, 81–94. [DOI] [PubMed] [Google Scholar]
- Traer, E. , MacKenzie, R. , Snead, J. , Agarwal, A. , Eiring, A.M. , O'Hare, T. , Druker, B.J. , Deininger, M.W. , 2012. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 26, 1140–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungureanu, D. , Saharinen, P. , Junttila, I. , Hilton, D.J. , Silvennoinen, O. , 2002. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol. Cell Biol. 22, 3316–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterkircher, T. , Cristofanon, S. , Vellanki, S.H. , Nonnenmacher, L. , Karpel-Massler, G. , Wirtz, C.R. , Debatin, K.M. , Fulda, S. , 2011. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin. Cancer Res. 17, 4019–4030. [DOI] [PubMed] [Google Scholar]
- Wattel, E. , Preudhomme, C. , Hecquet, B. , Vanrumbeke, M. , Quesnel, B. , Dervite, I. , Morel, P. , Fenaux, P. , 1994. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 84, 3148–3157. [PubMed] [Google Scholar]
- Willson, J.K. , Fischer, P.H. , Tutsch, K. , Alberti, D. , Simon, K. , Hamilton, R.D. , Bruggink, J. , Koeller, J.M. , Tormey, D.C. , Earhart, R.H. , Ranhsoky, A. , Trump, D.L. , 1988. Phase I clinical trial of a combination of dipyridamole and acivicin based upon inhibition of nucleoside salvage. Cancer Res. 48, 5585–5590. [PubMed] [Google Scholar]
- Young, P.R. , ConnorsWhite, A.L. , Dzido, G.A. , 1994. Kinetic analysis of the intracellular conjugation of monochlorobimane by IC-21 murine macrophage glutathione-S-transferase. Biochim. Biophys. Acta. 1201, 461–465. [DOI] [PubMed] [Google Scholar]
- Zhao, W. , Du, Y. , Ho, W.T. , Fu, X. , Zhao, Z.J. , 2012. JAK2V617F and p53 mutations coexist in erythroleukemia and megakaryoblastic leukemic cell lines. Exp. Hematol. Oncol. 1, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]