

Abstract
PARP inhibitors are mostly effective as anticancer drugs in association with DNA damaging agents. We have previously shown that the oncolytic adenovirus dl922‐947 induces extensive DNA damage, therefore we hypothesized a synergistic antitumoral effect of the PARP inhibitor olaparib in association with dl922‐947. Anaplastic thyroid carcinoma was chosen as model since it is a particularly aggressive tumor and, because of its localized growth, it is suitable for intratumoral treatment with oncolytic viruses.
Here, we show that dl922‐947 infection induces PARP activation, and we confirm in vitro and in vivo that PARP inhibition increases dl922‐947 replication and oncolytic activity. In vitro, the combination with olaparib exacerbates the appearance of cell death markers, such as Annexin V positivity, caspase 3 cleavage, cytochrome C release and propidium iodide permeability. In vivo, we also observed a better viral distribution upon PARP inhibition. Changes in CD31 levels suggest a direct effect of olaparib on tumor vascularization and on the viral distribution within the tumor mass. The observation that PARP inhibition enhances the effects of dl922‐947 is highly promising not only for the treatment of anaplastic thyroid carcinoma but, in general, for the treatment of other tumors that could benefit from the use of oncolytic viruses.
Keywords: Anaplastic thyroid carcinoma, Oncolytic virus, PARP inhibitors
Highlights
dl922‐947 infection induces PARP hyperactivation in anaplastic thyroid carcinoma cells.
PARP inhibitor olaparib has a synergistic effect on dl922‐947 cytotoxicity both in vitro and in vivo.
Olaparib improves viral distribution in the tumor mass.
1. Introduction
In recent years, the cure rate of neoplastic lesions has been improved by the availability of novel therapeutic agents such as small molecules (Levitzki and Mishani, 2006) and humanized antibodies (Weiner et al., 2010). Despite these advances, some lesions still represent a medical challenge: glioblastomas (Furnari et al., 2007), pancreatic carcinomas (Jemal et al., 2010) and anaplastic thyroid carcinoma (ATC) (Smallridge et al., 2009; Taccaliti et al., 2012) are among them.
ATC is one of the deadliest human solid tumors: although it counts for less than 3% of all thyroid cancers, ATC contributes up to 14–50% of the annual mortality associated with thyroid tumors. At the time of first clinical observation, patients present a rapidly enlarging mass, with compressive symptoms and invasion of surrounding structures. Death is often caused by tracheal obstruction, usually within 6 months from diagnosis (Nagaiah et al., 2011). The treatment of ATC consists of surgery combined with radiation and chemotherapy. This multimodality treatment confers only a short term palliative and survival benefit. The failure of all known ATC treatment is the main reason of the continuing search for novel strategies. Several novel approaches have been tested for the treatment of ATC, including the use of a new class of pharmacological agents targeting angiogenesis and vascularization, gene therapy (to restore p53 or of the human sodium iodine symporter), suicide therapy and oncolytic viruses (Elisei, 2012).
Oncolytic viruses (OVs) are particularly promising, since several preclinical studies have demonstrated the effectiveness of virotherapy, alone or in combination with rationally‐designed molecularly‐targeted drugs, for the treatment of ATC (Hallden and Portella, 2012).
OVs are replication competent viral mutants able to complete their life cycle selectively in tumor cells. Viruses from different families (adenoviruses, herpes simplex virus and poxviruses) are under consideration for cancer gene therapy, and have confirmed their safety with varying degrees of success in clinical trials (Hallden and Portella, 2012; Russell et al., 2012). Among them, adenoviruses have been extensively studied. H101, a modified version of dl1520, the first oncolytic virus described (Heise et al., 1997), has been approved in China for the treatment of head and neck carcinoma in combination with cisplatin (Garber, 2006). However, the clinical efficacy of dl1520 and related adenoviruses is limited, since their replication is impaired (O'Shea et al., 2004).
dl922‐947 is a second generation adenoviral mutant bearing a 24‐bp deletion in E1A‐Conserved Region 2 (Heise et al., 2000a). This region binds to and inactivates pRb protein, dissociating the pRb‐E2F complex and driving S phase entry and viral replication. Since the mutant E1A protein encoded by dl922‐947 is unable to bind pRb, the virus cannot trigger S phase entry in normal cells, but can still replicate in cells with an aberrant G1‐S checkpoint, a defect observed in over 90% human cancers (Sherr and McCormick, 2002). Superior cell killing potency of dl922‐947 compared to dl1520 has been observed in vitro and in vivo in human cancer cells of different origin such as pancreas (Bhattacharyya et al., 2011), prostate (Radhakrishnan et al., 2010), ovaries (Lockley et al., 2006), thyroid (Libertini et al., 2008) and brain (Botta et al., 2012, 2010).
Oncolytic adenoviruses can interact synergistically with different type of drugs (mitoxantrone, etoposide, gemcitabine, cisplatin, taxanes, irinotecan and others) (Cheong et al., 2008; Heise et al., 2000b; Radhakrishnan et al., 2010; Raki et al., 2005); however, the mechanisms underlying these synergistic effects are still unclear. OVs infection exerts multiple effects in host neoplastic cells, converging toward cell cycle control and inactivation of cell death pathways. The host cell response, aimed to extend cell survival and prevent viral replication, further complicates the scenario.
DNA damage response (DDR) represents a barrier against viruses, which processes viral genetic material as damaged DNA to restrict viral infection. Viruses have evolved strategies to counteract this process; many of these include either degradation or mislocalization of key players involved in DNA damage response (Lilley et al., 2007; Nichols et al., 2009; Touchefeu et al., 2011). In anaplastic thyroid carcinoma cells, dl922‐947 triggers an inefficient DDR: although ATM and Chk1 kinases are activated, a reduction of the MRN complex components and the accumulation of γH2AX are observed, indicating persistence of DNA damage (Passaro et al., 2013). A specific competitive ATP‐inhibitor of ATM (KU55933) increased the replication and the oncolytic activity of dl922‐947, confirming that the DDR acts as an intrinsic cellular defense against the virus. This observation is also supported in ovarian cancer cells, where the inhibition of ATR or Chk1 increased dl922‐947 cytotoxicity (Connell et al., 2011).
Different DNA repair pathways protect the integrity of the genome. Poly (ADP‐ribose) polymerase 1 and 2 (PARP‐1 and PARP‐2) are key mediators in base excision repair (BER) pathway, interacting with BER factors and recruiting them to damaged sites. These effects are mainly mediated by PARP‐1. Once activated, PARP‐1 rapidly catalyzes the transfer of ADP‐ribose moieties from NAD+ to the acceptor protein (PARylation), including histones, DNA repair proteins, transcription factors and chromatin modulators. PARylation introduces flexibility into the damaged region of DNA, loosening the chromatin and allowing access to the other proteins of the repair complex. In addition to its role in BER, PARP‐1 is involved in several other DNA damage repair processes, including the repair of DNA double strand breaks (Ame et al., 2004).
During viral infections, cells are pushed toward S phase (Davy and Doorbar, 2007) and it is known that an unscheduled DNA synthesis induces the accumulation of single strand breaks (SSBs) as replication‐associated DNA lesions (Kunkel, 2004; Smith et al., 2010). We hypothesized that dl922‐947 infection, driving neoplastic cells toward S phase, induces the accumulation SSBs and subsequent PARP‐1 activation. A robust protein PARylation in anaplastic thyroid carcinoma cells infected with dl922‐947 confirmed our hypothesis.
PARP‐1 deficiency leads to the persistence of the SSBs, converted to Double Strand Breaks (DSBs) that are eventually repaired by the error free homologous recombination pathway at the replication fork. Cells defective in homologous recombination are highly sensitive to PARP‐1 inhibition due to the persistence of multiple unrepaired DSBs, in line with the concept that two genes are synthetically lethal if loss of function of either gene alone is compatible with cell survival, whereas inactivation of both genes causes cell death. Studies have confirmed that PARP inhibitors selectively kill BRCA2‐deficient tumor cells increasing genomic instability and cell death; furthermore, PARP inhibitors sensitize tumor cells to DNA damaging agents such as temozolomide and ionizing radiation. We have decided to exploit this approach in anaplastic thyroid carcinoma cells where dl922‐947 infection induces single and double strand breaks, simulating the effects of a typical DNA damaging drug (Passaro et al., 2013). Therefore, to increase the efficacy of the viral treatment, we have combined dl922‐947 with a specific PARP inhibitor. Our study shows that this combined treatment greatly increased DNA damage accumulation and cell death.
2. Material and methods
2.1. Cells, adenoviruses and drugs
Human thyroid anaplastic carcinoma cell lines BHT101‐5, FRO and Cal62 have been described and authenticated as shown elsewhere (Schweppe et al., 2008). dl922‐947, non‐replicating reporter adenovirus AdGFP (ΔE1) and non‐replicating adenovirus dl312 (ΔE1A, ΔE3B) were expanded in the human embryonic kidney cell line HEK‐293, purified and stored as previously reported (Libertini et al., 2007). For in vitro study, olaparib (Selleckchem, Houston, TX, USA) was dissolved in DMSO to a final concentration of 10 mM. For in vivo study, olaparib was solubilized in DMSO and diluted to 5 mg/mL with PBS containing 10% 2‐hydroxy‐propyl‐betacyclodextrin. zVAD‐fmk (Tocris Bioscience, Bristol, UK) was dissolved in DMSO to a final concentration of 10 mM. BrdU was dissolved in PBS to a final concentration of 10 mM. Digitonin was dissolved in absolute ethanol to a final concentration of 2 mg/mL 2‐hydroxy‐propyl‐betacyclodextrin, BrdU and digitonin were purchased from Sigma–Aldrich, St. Louis, MO, USA. MNNG (1‐Methyl‐3‐nitro‐1‐nitrosoguanidine) was purchased from Carbosinth (Berkshire, UK) and solubilized in DMSO to a final concentration of 10 mM. Drugs were stored at −20 °C.
2.2. FACS analysis
For all FACS experiments, cells were seeded in 100 mm cell culture dish at density of 4 × 105 cells/dish and treated as indicated.
2.2.1. BrdU/PI staining
FRO, BHT101‐5 and Cal62 cells were infected with dl922‐947 for 24 h (1 pfu/cell for BHT101‐5, 5 pfu/cells for the other cell lines). Thirty minutes before harvesting, cells were labeled with 10 μM BrdU. Cells were fixed in ice‐cold 70% ethanol in PBS. After an overnight incubation at −20 °C, cells were washed once with PBS and incubated for 15′ at room temperature with 2N HCl to denature DNA. Cells were then washed with PBT (0.5% BSA, 0.1% Tween20 in PBS) and resuspended in PBT containing anti‐BrdU antibody (M0744, 1:40, Dako, Carpinteria, CA, USA). After 30′, cells were washed twice with PBT, and then resuspended in PBT containing Alexa488 anti‐mouse (#A11001, 1:100, Invitrogen, Grand Island, NY, USA) in the dark for 30′. Cells were washed twice with PBS, resuspended in Propidium Iodide (PI) 0.015 M (Sigma–Aldrich) in PBS for 20′ and analyzed for the emission in FL1 and FL3 channels.
2.2.2. γH2AX/PI staining and cell cycle
Procedure as described in Passaro et al., 2013 (Passaro et al., 2013) was used. To remove artifacts ‐doublets and aggregates‐ from the analysis, an electronic doublet discrimination was performed using the area and width of the fluorescence (FL3) pulse.
2.2.3. AdGFP infection
FRO and BHT101‐5 cells were infected with AdGFP (25 pfu/cell) and harvested 48 h later. Pellet was resuspended in PBS and emission in the FL1 channel analyzed.
2.2.4. Annexin V/PI staining
Attached and detached FRO and BHT101‐5 cells were collected and washed with Annexin V binding buffer (BioLegend, San Diego, CA, USA). Pellet was resuspended in Annexin V binding buffer containing FITC‐conjugated Annexin V (#640906, 1:40, BioLegend). After 15′ incubation at room temperature, PI to a final concentration of 0.0015 M was added to each sample just before analysis.
2.2.5. Cytochrome C release from mitochondria
Procedure described by Waterhouse and Trapani (Waterhouse and Trapani, 2003) was used. Briefly, attached and detached FRO and BHT101‐5 cells were harvested and treated with digitonin (50 μg/mL in 150 mM KCl and 1 mM EDTA in PBS) for 10′ on ice. The selective permeabilization of the plasma membrane with digitonin allows virtually all of the cytoplasmic cytochrome C to diffuse out of the cell. Permeabilized cells were fixed in 4% paraformaldehyde in PBS for 20′ on ice, washed 3× in PBS and incubated in blocking buffer (0.5% w/v saponin, 1% w/v BSA in PBS) for 20′ at room temperature. Cells were then incubated for 2 h at room temperature with anti‐Cytochrome C antibody (#556432, 1:200, BD, Franklin Lakes, NJ, USA) in blocking buffer, washed 3× and incubated for 30′ at room temperature with anti‐mouse Alexa488 in blocking buffer at dark. Stained cells were washed twice with PBS, resuspended in PBS and analyzed by flow cytometry detecting Alexa488 fluorescence in FL1 channel.
All samples were acquired by a BD LSRFortessa (BD Biosciences, San Jose, CA, USA) and analyzed using BD FACSDiva Software.
2.3. Protein extraction and western blot analysis
After the indicated treatments, attached and detached cells were harvested and lysates prepared as already described (Botta et al., 2012). 50 μg of lysate proteins were probed with the following antibodies: caspase 3 (#Ab13585, 1:500, Abcam, Cambridge, UK), p‐H2AX s139 (#05‐636, 1:1000, Millipore, Billerica, MA, USA), PAR (#4336‐BPC‐100, 1:1000, Trevigen, Gaithersburg, MD, USA), PARP (#9542, 1:1000, Cell Signaling, Boston, MA, USA), Lamin A/C (1:2000, Cell Signaling, Boston, MA, USA), AIF (D39D2, 1:1000, Cell Signaling, Boston, MA, USA), α‐tubulin (#T9026, 1:5000, Sigma–Aldrich, St. Louis, MO, USA).
Cellular fractionation was performed using NE‐PER™ Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer's instruction.
2.4. In vitro and in vivo evaluation of dl922‐947 replication and activity
For all the in vitro experiments, 104 cells/well were seeded in 12‐well plates and 24 h later infected with viruses (5 and 1 pfu/cell for FRO and BHT101‐5 cells, respectively). At 48 hpi (hours post infection), cells and media were separately collected. To release the virus, the two fractions (cells and media) were disrupted by 3 freeze–thaw cycles, centrifuged at 1000 g for 5′ and supernatants were collected. Viral replication (Real‐Time PCR (Passaro et al., 2013)) and viral activity (determination of 50% tissue culture infective dose, TCID50, (Wang et al., 2003)) were analyzed in both culture media (released virus) and supernatants (cellular virus).
For the in vivo experiments, tumors from treated mice (infected only or infected plus olaparib) were excised at day 27. To analyze in vivo viral activity, tumor samples were pulverized in liquid nitrogen and resuspended in 1 mL of serum‐free DMEM. Samples underwent 3 freeze–thaw cycles followed by a centrifugation at 1000 g for 5′; supernatant were then collected and a TCID50 assay performed. To analyze viral replication, total DNA was extracted from 25 mg of sample using phenol/chloroform protocol. Viral titre was then quantified by Real‐Time PCR as previously described (Passaro et al., 2013).
2.5. Immunofluorescence staining
FRO cells were seeded on cover slips in 24‐well dishes and infected with 5 pfu/cell of dl922‐947. At indicated times, cells were collected and fixed with 4% paraformaldehyde for 15′ at room temperature, washed and permeabilized with 0.1% Triton X‐100 in PBS. Cells were then blocked with blocking buffer (0.5% BSA in PBS) for 30′ and then incubated overnight at 4 °C with primary antibodies diluted in blocking buffer (p‐H2AX s139, Millipore #05‐636, 1:250; AIF, Cell Signaling D39D2, 1:400). After washing three times with 0.1% Triton X‐100 in PBS, cells were incubated with Alexa488 anti‐mouse and Alexa594 anti‐rabbit (Invitrogen #A11001 and #A11012 respectively, 1:100). Fixed and stained cells were mounted using Vectashield‐mounting medium with DAPI (H‐1200, Vector Laboratories, Burlingame, CA, USA). Images were acquired with a Nikon A1R confocal microscope (Nikon, Melville, NY, USA), using a 60× oil objective and processed with NIS element software (Nikon, Melville, NY, USA).
2.6. Viability assay and analysis of the combined drug effect
FRO, BHT101‐5 and Cal62 cells were seeded in 96‐well plates at densities of 500 cells/well and allowed to attach for 24 h. Cells were exposed to graded concentrations (six replicates) of olaparib or dl922‐947 either alone or in combination for 6 days. At the end of the treatment, cells were fixed with 50% v/v TCA and stained with 0.4% w/v sulforhodamine B in 1% v/v acetic acid as already described (Libertini et al., 2008; Vichai and Kirtikara, 2006). The percentages of surviving cells after treatment were calculated by assuming that the number of surviving untreated cells is 100%. The concentration of each agent or of the combined treatment required to kill 50% of cells (median lethal dose, LD50) was determined by a dose‐response curve using GraphPad prism (GraphPad Software). Drug synergy was determined by the isobologram and combination‐index methods as previously described (Tallarida, 2001). Isobologram was obtained by plotting the individual drug doses required to generate that LD50 on their respective x‐ and y‐axes.
Combination‐index (CI) method is a mathematical and quantitative representation of a two‐drug pharmacologic interaction. Using data from the growth inhibitory experiments and computerized software, CI values were generated. A CI of 1 indicates an additive effect between two agents, whereas a CI < 1 or CI > 1 indicates synergism or antagonism, respectively.
2.7. Tumorigenicity assay and in vivo viral distribution
FRO cells in exponential phase were prepared at a concentration of 4 × 107cells/ml in DMEM medium. CD‐I athymic mice were obtained from Charles River (Wilmington, MA, USA), all experiments were carried out with 6‐week‐old females. To initiate tumor xenografts, 0.2 mL of cell suspension was injected into the right flank of 60 animals. Tumor diameters were measured with calipers and tumor volume (V) was calculated by the formula for a rotational ellipsoid: V = A·B2/2 (A, axial diameter; B, rotational diameter). Mice weights were monitored weekly.
Twenty days post‐injection mice with similar tumor size were randomized into four groups (15 animals/group): untreated, treated with olaparib, treated with dl922–947, or treated with both. Olaparib (50 mg/kg) was administered intraperitoneally three consecutive days a week. A low viral dose (2 × 106 pfu/cell) was administered two times a week (the day before and the day after olaparib administration) by intratumoral injection to avoid any first‐pass effect (Libertini et al., 2011, 2007, 2008). Olaparib vehicle was administered to the control groups. Statistical analysis was done by ANOVA (analysis of variance) and the Bonferroni post hoc test, using commercial software (Prism 4; GraphPad Software). Differences in the rate of tumor growth in mice were assessed for each time point of the observation period.
Mice were maintained at the Dipartimento di Biologia e Patologia Cellulare e Molecolare‐Animal Facility. All animal experiments were conducted in accordance with accepted standards of animal care and in accordance with the Italian regulations for the welfare of animals used in studies of experimental neoplasia. Study was approved by our institutional committee on animal care.
In vivo viral distribution were evaluated as previously described (Libertini et al., 2008).
2.8. RNA isolation and Real‐Time PCR
Tumors excised at day 27 were homogenized and total RNA was extracted using Trizol reagent (Invitrogen, Carlsberg, CA, USA) according to the manufacturer's instructions. 1 μg of total RNA was reverse‐transcribed using Superscript III Reverse Transcriptase (Invitrogen, Carlsberg, CA, USA). Real‐Time PCR was carried out using a CFX96 Real‐Time System (Biorad, Hercules, CA, USA). Reactions were run in triplicate in three independent experiments.
Specific primers for CD31 were used to measure its expression by Real‐Time PCR: 5'‐ATT GCA GTG GTT ATC ATC GGA GTG‐3' and 5'‐CTC GTT GTT GGA GTT CAG AAG TGG‐3'. Expression data were normalized to the geometric mean of housekeeping gene GAPDH to control the variability in expression levels and were analyzed using the 2−ΔΔCT method described by Livak and Schmittgen (Livak and Schmittgen, 2001). Specific primers for GAPDH were used: 5'‐CGC TCT CTG CTC CTC CTG TTC‐3' and 5'‐TTG ACT CCG ACC TTC ACC TTC C‐3'.
2.9. Immunohistochemistry and microvessel density measurement
Formalin fixed, paraffin embedded 4 μm sections of tumors were processed as already described (Esposito et al., 2009). Antigen retrieval was performed in citrate buffer pH 6 and primary antibodies were left ON at 4 °C. GFP antibody (AM1009a, 1:100) was purchased by Abgent, CD31 (rb10333, 1:30) from Thermo Scientific. To calculate the tumor microvessel density (MVD), CD31‐positive area and total tumor area per field were measured using ImageJ software. MVD was then determined as a percentage of CD31‐positive area per field. Three randomly selected areas from three different tumors were analyzed.
3. Results
3.1. dl922‐947 infection induces S phase and PARP activation
Adenoviruses are known to induce S phase to push viral genome production by the host cell machinery (Davy and Doorbar, 2007). To verify whether this mechanism is shared by dl922‐947, we monitored S phase induction in infected cells. Anaplastic Thyroid Carcinoma (ATC) cell lines FRO, BHT101‐5 and Cal62 were infected with dl922‐947; BrdU incorporation was evaluated by FACS analysis 24 h after infection (hpi). As shown in Figure 1A, dl922‐947 infection increased by ∼15% the number of cells in S phase in all cell lines analyzed, indicating that dl922‐947 infection drives DNA synthesis.
Figure 1.
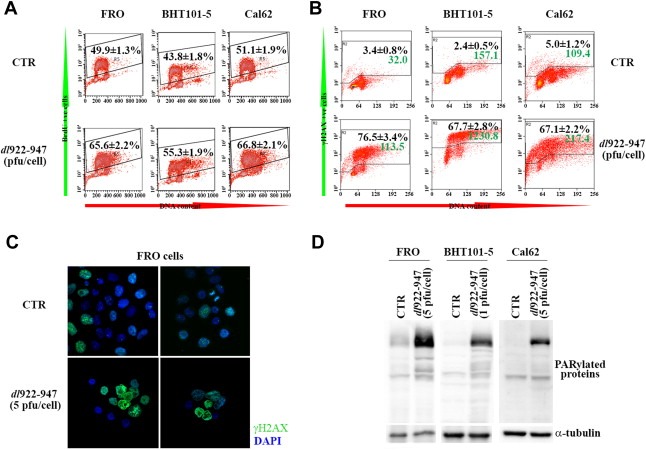
dl922‐947 infection induces S phase, γH2AX accumulation and PARP activation. A) FRO, BHT101‐5 and Cal62 cells were infected or not (CTR) with dl922‐947 (5, 1, 5 pfu/cell respectively) for 24 h and pulse‐labeled with BrdU for 30 min before harvesting. FACS analysis of samples stained for BrdU and, to quantify the amount of DNA, for propidium iodide was performed. Numbers represent the % of BrdU+ve cells ± sd (n = 3 for each experiment, repeated 3 times). B) FRO, BHT101‐5 and Cal62 cells were infected or not (CTR) with dl922‐947 (10, 5, 10 pfu/cell respectively) for 24 h and stained for γH2AX and propidium iodide. Black numbers represent the % of γH2AX+ve cells ± sd, green numbers represent the median fluorescence in FL1 channel (n = 3 for each experiment, repeated 3 times). C) Immunofluorescence for γH2AX on FRO cells infected for 24 h with dl922‐947. D) Protein lysates from cells treated as in Figure 1A were analyzed by Western blot with an anti‐PAR antibody. α‐tubulin used as loading control. Data are representative of three independent experiments.
DNA synthesis can cause single strand break (SSB) as replication‐associated DNA lesions (Kunkel, 2004; Smith et al., 2010). SSBs are repaired by the SSB repair machinery, which requires PARP activity. To evaluate SSB accumulation, we assessed the positivity of infected cells for γH2AX (histone H2AX phosphorylated on serine 139). To assess PARP activation, we analyzed by western blot the presence of PARylated proteins. Both FACS analysis and IF confirmed γH2AX activation in infected cells (Figure 1B and C), while Western Blot analysis indicated a robust PARylation with respect to uninfected controls (Figure 1D).
Interestingly, infection with the non‐replicating adenovirus dl312 neither activated PARP nor induced γH2AX (Supplementary Figure 1), suggesting that the observed effects are specifically due to dl922‐947 replication.
3.2. PARP inhibition has a synergistic effect on dl922‐947 cytotoxicity
Since PARP activation is required for SSB repair and SSB repair antagonizes viral replication, we wondered whether PARP inhibition could facilitate viral replication and viral cytotoxicity. To test this hypothesis, we used olaparib, a competitive inhibitor of NAD+ in the PARP catalytic site (Peralta‐Leal et al., 2009). ATC cells were treated with olaparib in combination with dl922‐947 infection. PARylated protein levels were evaluated by western blot 24 hpi, showing that olaparib treatment was sufficient to revert dl922‐947 induced PARP activation (Figure 2A). We have, then, evaluated γH2AX levels to understand whether olaparib, combined with the functional block of DSB repair mediated by dl922‐947, induces the accumulation of an unrepaired DNA (Figure 2B). The combined treatment led to a robust increase in the median fluorescence for γH2AX with respect to dl922‐947 infection alone (∼100% and 50% increase in FRO and BHT101‐5 cells, respectively). Interestingly, olaparib treatment alone did not induce γH2AX accumulation. To explore the effect of PARP inhibition on the oncolytic activity of dl922‐947, ATC cells were infected and treated with olaparib. Combination‐index showed a potent, statistically significant, synergy of cell killing in both cell lines, at all combinations used (Figure 2C).
Figure 2.
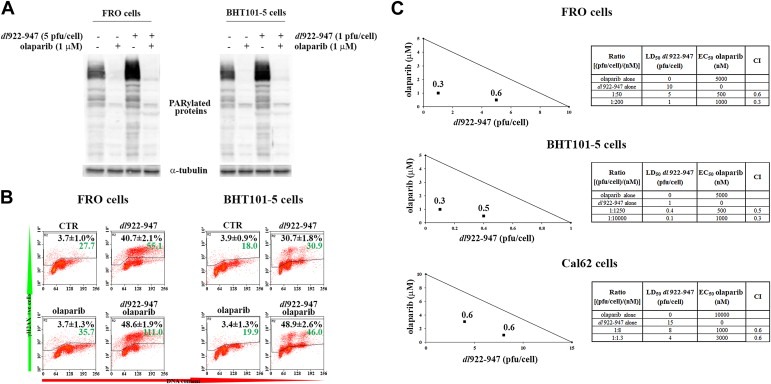
Olaparib increases dl922‐947 induced γH2AX accumulation and cell death. A) FRO and BHT101‐5 cells were treated with olaparib (1 μM) and infected with dl922‐947 for 24 h at indicated pfu/cell. Expression of PARylated proteins was analyzed by Western blot using an anti‐PAR antibody. α‐tubulin was used as a loading control. Results shown are representative of three independent experiments. B) Cells treated as described in Figure 2A were collected, fixed, stained for γH2AX and propidium iodide, and analyzed by FACS. Black numbers represent the % of γH2AX+ve cells ± sd, green numbers represent the median fluorescence of γH2AX staining (n = 3 for each experiment, repeated 3 times). C) FRO and BHT101‐5 cells were treated with different concentrations of dl922‐947 and olaparib. Cell viability was determined 7 days post infection by sulforhodamine B assay. Dose‐response curve and LD50 or EC50 values were generated and used to construct isobolograms and calculate combination‐indexes (CI) for each ratio. The shown line connects the LD50 values of the virus and EC50 of the drug, both as a single agent. A ratio producing a CI < 1 is considered synergistic, CI = 1 additive, CI > 1 antagonistic. A synergistic effect was observed in all used combination (CI < 1). Tables show olaparib EC50 and dl922‐947 LD50 for each cell line, the ratio between the dose of the virus and the dose of the drug, and the CI for the combinations plotted on the graph.
3.3. dl922‐947 replication, but not entry, is affected by PARP inhibition
To verify whether olaparib enhanced oncolytic activity of dl922‐947 by increasing viral entry into target cells, infection efficiency was evaluated (Figure 3A). FRO and BHT101‐5 were treated with olaparib (1 μM) and infected with a non‐replicating reporter adenovirus transducing GFP (AdGFP, 25 pfu/cell). After 48 h from infection, GFP emission, indicative of viral entry, was evaluated by cytofluorimetric analysis. The treatment with olaparib did not induce any modification in GFP emission after AdGFP infection, suggesting that the drug is not acting on viral entry.
Figure 3.
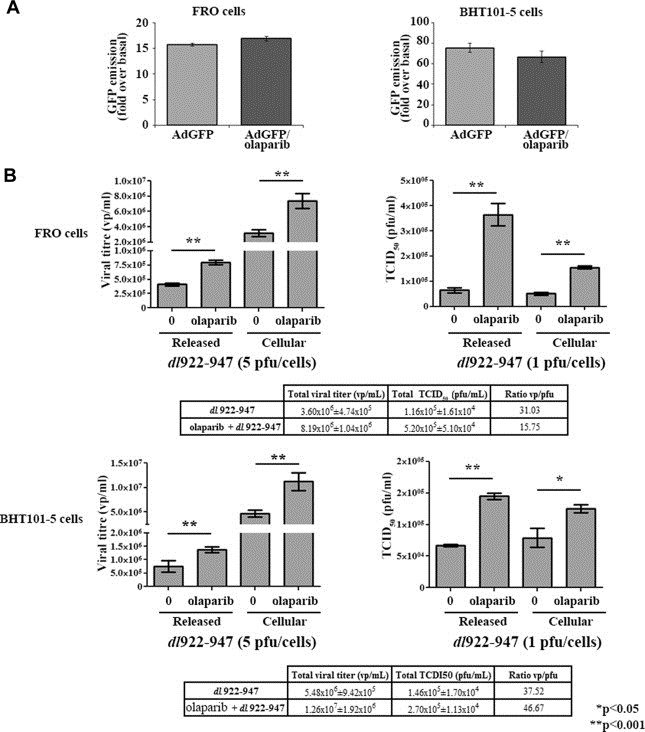
Viral replication, but not viral entry, is affected by olaparib. A) FRO and BHT101‐5 were treated with olaparib (1 μM) and infected with 25 pfu/cell of AdGFP. GFP average emission was analyzed by FACS in freshly collected, unfixed samples. The histograms show the fold change in GFP emission compared to uninfected cells. Data represent the mean of three independent experiments. B) FRO and BHT101‐5 cells were infected with dl922‐947 (5 or 1 pfu/cell, respectively) in presence or not of olaparib (1 μM). After 48 h, supernatant and adherent cells were collected separately to evaluate, respectively, released and cellular viral particles. Viral DNA was then extracted and used to quantify viral titre by Real‐Time PCR (on the left). Viral activity of both released and cellular virus was evaluated by TCID50 assay (on the right). Significant (*p < 0.05) differences were observed in olaparib treated cells compared to untreated cells. The data represent the mean of three different experiments. Tables show total viral titre (cellular + released) and total TCID50 (cellular + released). The ratio vp/pfu was also shown in table. vp/mL = viral particles/mL, pfu/mL = plaque forming units/mL.
To quantify viral replication, ATC cells were infected with dl922‐947 and treated or not with olaparib (1 μM). 48 h later, cells and supernatant were collected and viral DNA extracted: Real‐Time PCR showed, in both cell lines, a statistically significant drug induced increase of cellular and released viral titre (Figure 3B, left). A similar trend was observed when a TCID50 analysis was performed to assess viral activity (Figure 3B, right).
We evaluated also the effects of PARP activation on viral replication and γH2AX accumulation. FRO and BHT101‐5 cells were pre‐treated for 15′ with MNNG, a strong PARP activator, and then infected with dl922‐947. Cells were collected 24 hpi to evaluate PARP activation by western blot and γH2AX induction by FACS analysis (see Supplementary Figure 2a–b). Viral replication was assessed 48 hpi (see Supplementary Figure 2c). MNNG treatment significantly decreased both viral titre and γH2AX accumulation.
3.4. PARP inhibition increases the polyploidy and the percentage of cells in sub‐G1 phase
To evaluate changes in cell cycle, FRO and BHT101‐5 cell were treated with olaparib and infected with dl922‐947 (5 or 1 pfu/cell, respectively). 72 h later cells were stained with Propidium Iodide and cell cycle profile evaluated by FACS analysis (Figure 4A). A highly significant accumulation of polyploidy cells was observed in FRO cells undergoing the combined treatment. This effect was paralleled by a significant increase in the percentage of cells in sub‐G1 phase. Similar results were obtained on BHT101‐5 cells, although a higher percentage of polyploidy cells and a lower percentage of cells in sub‐G1 phase were observed. In accordance with the hypothesis that polyploidy can result from mitotic defects leading to cell death, the differences between FRO and BHT101‐5 cells could be explained by a different timing in the escape from mitotic block and in cell death pathways activation. Olaparib treatment alone did not modify the cell cycle profile in FRO and BHT101‐5 cells (data not shown).
Figure 4.
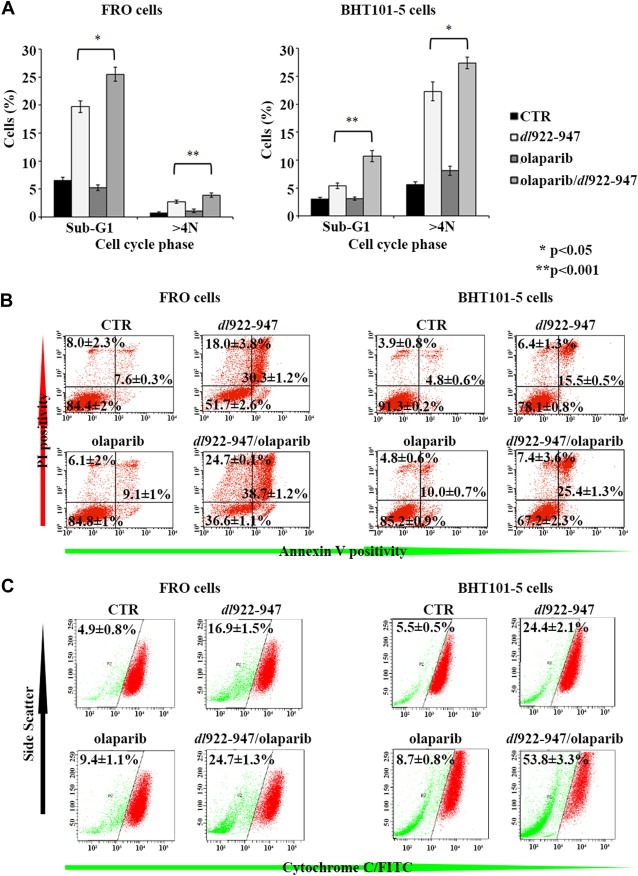
PARP inhibition increases cell death induced by dl922‐947. A) FRO (left panel) and BHT101‐5 (right panel) were infected with dl922‐947 (5 or 1 pfu/cell respectively) for 72 h in the presence or not of olaparib (1 μM). Fixed cells were stained with propidium iodide and cell cycle distribution analyzed by FACS. The percentage of cells in sub‐G1 phase and polyploid cells was significantly (*p < 0.05) or highly significantly (**p < 0.001) increased by the combined treatment with respect to dl922‐947 infection. Bars represent sd; data are the mean of three different experiments. B) FRO (left panel) and BHT101‐5 (right panel) cells were treated as in Figure 4A. After 72 h, cells were collected and stained with Annexin V. Propidium iodide was added just before analysis to identify cells with damaged cellular membrane. Numbers on the right quadrants represent the average percentage of Annexin V positive cells; numbers on the lower left quadrant and on the upper left quadrant represent the average percentage of living and necrotic cells respectively ± sd (n = 3, experiment repeated 3 times). C) FRO and BHT101‐5 cells were treated as in Figure 4A. After 72 h, cytochrome C release from mitochondria was assessed by FACS analysis. Numbers represent the percentage of cytochrome C negative cells ± sd of three independent experiments.
3.5. Viral infection and cell death
It has been demonstrated that dl922‐947 infection induces in ATC and in other cancer cells a programmed cell death that partially resembles apoptosis. It is also known that, when dl922‐947 is used in combination with chemotherapeutic drugs, alternative cell death pathways could be activated. Therefore, we decided to analyze some apoptotic markers in ATC cells treated with dl922‐947 and olaparib.
First, phosphatidylserine exposition and plasma membrane permeabilization were evaluated by staining with Annexin V and PI, respectively (Figure 4B). At 72 hpi, in both cell lines, an increase in Annexin V staining was observed in the combined treatment with respect to single treatments.
Second, we analyzed the release of pro‐apoptotic protein cytochrome C from the mitochondrial inner membrane space to the cytoplasm. Selective permeabilisation of the mitochondrial outer membrane is an integral event in apoptosis (Martinou and Green, 2001): in samples stained for cytochrome C and analyzed by FACS, cells with intact mitochondria result strongly positive, while a lower fluorescence is indicative of higher release of cytochrome C to the cytosol. As shown in Figure 4C, dl922‐947 infection increased the percentage of cytochrome C low cells in both cell lines. This effect was amplified by the combination with olaparib (16.9% vs 24.7 in FRO cells; 24.4 vs 53.8 in BHT101‐5 cells). Olaparib treatment only slightly increased basal cytochrome C release.
Third, levels of pro‐ and cleaved caspase 3 were analyzed by western blot (Figure 5A). A decrease of pro‐caspase 3, paralleled by a concomitant slight increase of the cleaved form, suggested the activation of caspase 3 in cells undergoing the combined treatment. To further evaluate the role of caspases in dl922‐947 induced cell death, ATC cells were treated with the pan‐caspase inhibitor zVAD‐fmk (Figure 5B). zVAD‐fmk induced only a slight increase in cell survival, indicating that also caspase independent cell death pathways are involved in dl922‐947 induced cell death.
Figure 5.
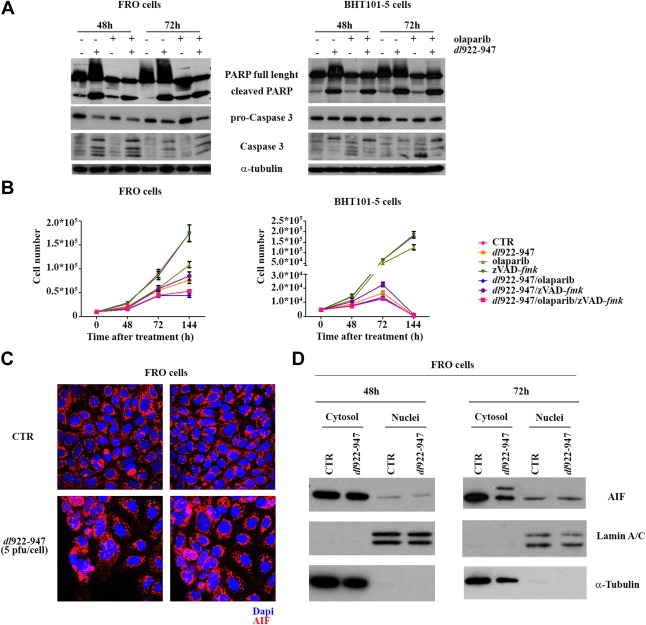
dl922‐947 activates an apoptotic‐like cell death. A) FRO (left panel) and BHT101‐5 cells (right panel) were infected with dl922‐947 (5 and 1 pfu/cell respectively), in the presence or not of olaparib (1 μM). After 48 and 72 h, PARP and caspase 3 cleavage was analysed by Western blot, using α‐tubulin as a loading control. B) FRO and BHT101‐5 cells were plated in 12‐well plates and infected with dl922‐947 (5 and 1 pfu/cell respectively) in the presence or not of zVAD‐fmk (20 μM) and olaparib (1 μM). Cell number was evaluated in the following days. The data represent the mean of three independent experiments. C) FRO cells were infected for 48 h with 5 pfu/cell of dl922‐947. Immunofluorescence shows AIF (in red), 60× magnification. D) FRO cells were infected for 48 and 72 h with 5 pfu/cell of dl922‐947. Cellular fractionation was performed to isolate nuclear and cytosolic fraction. Lysates were probed with AIF antibody, as well as Lamin A/C and α‐tubulin antibodies to verify the purity of the preparation. The extra band of 67 kDa observed 72 hpi likely represents an accumulation of AIF precursor form.
Parthanatos is a type of apoptosis induced by PARP hyperactivation, therefore we wondered whether parthanatos could be the cell death mechanism responsible for dl922‐947 effects. Apoptosis‐Inducing Factor (AIF) is a key mediator of parthanatos: upon PARP‐1 activation, AIF translocates from the mitochondria to the nucleus. We analyzed AIF translocation by both immunofluorescence and western blotting after cellular fractionation. As shown in Figure 5C and D, dl922‐947 infection did not modify AIF localization, excluding parthanatos as main effector of dl922‐947 induced cell death.
3.6. PARP inhibitor olaparib increases dl922‐947 oncolytic activity in vivo
To validate the therapeutic potential of dl922‐947 and olaparib in vivo, the effects of the combined treatment on ATC tumor xenografts were analyzed. Athymic mice were inoculated subcutaneously with FRO cells. Mice were then randomized into four groups (15 animals/group) with similar average tumor size (>100 mm3). In accordance to previous reports (Kortmann et al., 2011), animals were injected i.p. with olaparib at the dose of 50 mg/kg three consecutive day/week. dl922‐947 (2 × 106 pfu) was administered two times per week by intratumoral injection to avoid first‐pass effect. A low viral dose (2 × 106 pfu) was chosen to enable a better visualization of the effects of the combined treatment versus dl922‐947 infection alone.
As shown in Figure 6A, a significant reduction of tumor growth was achieved in the combined treatment group compared to single treatments and untreated groups. At day 13, the difference between the combined treatment group vs the single treatments and vs the control group become statistically significant (*p < 0.05). From day 17, the difference was highly significant (*p < 0.001).
Figure 6.
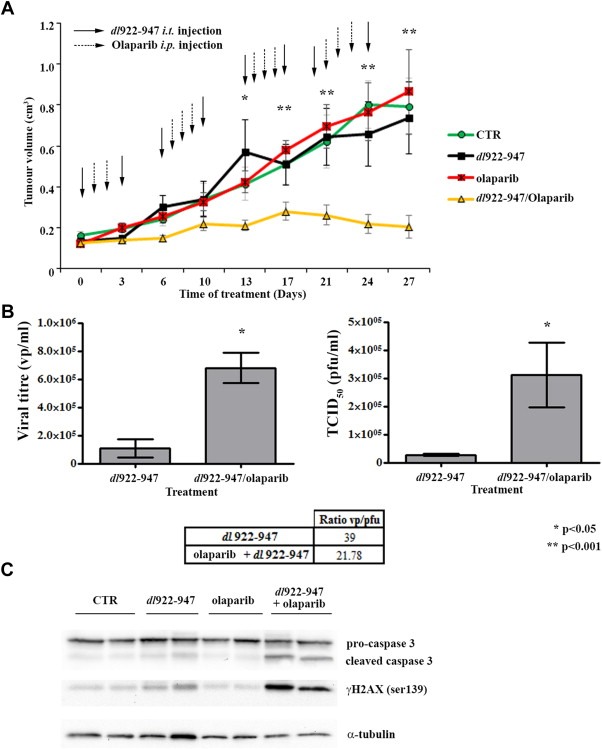
Olaparib increases the oncolytic activity of dl922‐947 in vivo. A) Animals were injected i.p. with olaparib (dashed line arrows) three consecutive days per week; dl922‐947 (solid line arrows) was administered two times per week (the day before and the day after olaparib treatment) by intratumoral injection. Tumor diameters were measured with calipers every third day, mice weights were monitored weekly. At day 13, the difference between the combined treatment group vs the single treatments and vs the control group become statistically significant (*p < 0.05). From day 17, the difference was highly significant (*p < 0.001). B) At day 27, total DNA was extracted from tumors (4/group) treated as in Figure 6A and used to quantify viral titre by Real‐Time PCR (on the left). Viral activity was evaluated by TCID50 assay (on the right). Table shows the ratio vp/pfu. Bars represent standard deviation. C) Western blot analysis of lysates obtained from tumors treated as in Figure 6A. At day 27 lysates from two different animals/group were probed with anti caspase 3 and γH2AX antibodies; a‐tubulin was used as loading control.
Animal weight was monitored weekly and did not show any significant differences between all the groups (see Supplementary Figure 3).
Real‐Time PCR analysis of viral genome equivalent copies showed a significant higher viral DNA content (*p < 0.05) in tumors receiving the combined treatment (Figure 6B). Western blot analysis showed increased γH2AX levels and caspase 3 activation in the combined treatment group compared to single treatment groups (Figure 6C).
3.7. PARP inhibitor olaparib improves viral distribution in the tumor mass
PARP inhibitors negatively regulate tumor angiogenesis in experimental models (Rajesh et al., 2006), and we have previously shown that the treatment with the antiangiogenic antibody bevacizumab improved the distribution of the reporter virus AdGFP in ATC xenografts (Libertini et al., 2008).
We wondered whether a similar effect on viral distribution could be exerted by olaparib. To this aim, 10 animals were injected s.c. with FRO cells and, after 25 days, when tumors were clearly visible, 5 mice were treated i.p. with olaparib (50 mg/kg) for three consecutive days. At day four, both treated and untreated animals received within the tumor mass the non‐replicating adenovirus AdGFP. Mice were sacrificed 48 hpi and tumors excised. AdGFP distribution was evaluated by immunohistochemistry for GFP, showing that olaparib treatment clearly ameliorates viral distribution in vivo (Figure 7A).
Figure 7.
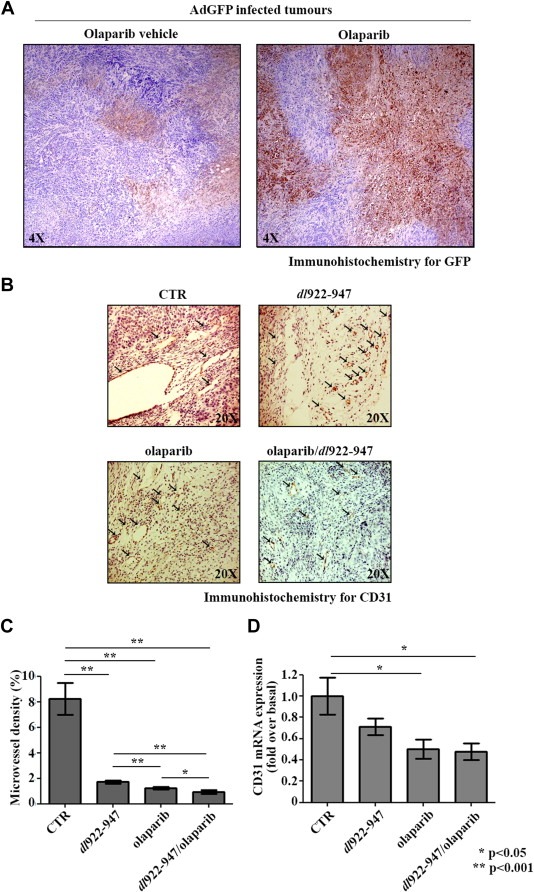
Olaparib improves viral distribution. A) Animals bearing FRO xenografts were divided into two groups with similar tumor size. Animals received olaparib or olaparib vehicle i.p. for 3 consecutive days. AdGFP virus was injected intratumourally in both groups at day 4. After 2 days, tumors were collected. Viral distribution was evaluated using an anti‐GFP antibody. B) Histological analysis of CD31 expression from tumor samples excised at day 27 treated as in Figure 6A. Arrows in figure indicate CD31‐positive cells (blood vessels). C) Quantification of data represented in Figure 7B. Microvessel density was determined as a percentage of CD31‐positive area on total tumor area per field. Three randomly selected areas from three different tumors for each group were analysed. D) Real‐Time PCR analysis of CD31 mRNA levels from tumors excised at day 27 (4 for each group) treated as in Figure 6A. Bars represent the standard deviation.
In order to assess the effect of olaparib on tumor angiogenesis, we have also analyzed the expression of CD31, a marker constitutively expressed on the surface of endothelial cells. As shown by immunohistochemistry (Figure 7B and C) and Real‐Time PCR (Figure 7D), CD31 levels were significantly reduced by olaparib, either alone or in combination with dl922‐947.
4. Discussion
Anaplastic thyroid carcinoma is one of the deadliest human malignancies, characterized by an uncontrollable local growth, rapidly leading to tracheal obstruction, asphyxia and death. Surgical resection, followed by irradiation and chemotherapy, only provides a temporary palliative benefit, leaving the patients with a survival expectancy of only six months after the diagnosis (Smallridge et al., 2009; Taccaliti et al., 2012). More effective treatments are, therefore, desperately required.
Clinical studies have proved oncolytic viruses safe, well–tolerated and effective against localized lesions, supporting their use for the treatment of ATC (Hallden and Portella, 2012).
It is known that an increased DNA synthesis could induce single strand breaks (SSBs) (Kunkel, 2004), which activate base excision repair pathway, followed by PARP‐1 activation, protein PARylation and SSBs repair (Ame et al., 2004). Viral infection pushes cell toward S phase (Davy and Doorbar, 2007), therefore we have evaluated whether dl922‐947 infection could lead to PARP activation. Our data show a robust protein PARylation in response to the infection.
Inhibition of PARP induces accumulation of large numbers of unrepaired SSBs, causing the collapse of replication forks during S phase and the subsequent generation of double strand breaks (DSBs). In cells homozygous for a defect in homologous recombination, the DSBs cannot be repaired, leading to a synthetic lethality effect (McCabe et al., 2006). This effect has been exploited to successfully treat with PARP inhibitors BRCA‐deficient mammary carcinoma patients or tumors previously treated with DNA damaging drugs. In this latter case, PARP inhibitors sensitize tumor cells to DNA damaging agents, increasing their therapeutic index (Farmer et al., 2005).
dl922‐947 infection alone induces DNA damage (Connell et al., 2011; Passaro et al., 2013), therefore we wondered whether the use of a PARP inhibitor (olaparib) could increase DNA damage accumulation. Our data show that olaparib treatment increases dl922‐947 induced γH2AX positivity.
γH2AX formation has historically been associated with the induction of DSBs, however γH2AX has also been shown after replication stalling and SSBs accumulation (Ewald et al., 2007; Ward and Chen, 2001). In the latter case, the phosphorylation of H2AX is likely dependent on ATR kinase rather than ATM (Burdak‐Rothkamm et al., 2007; Ward and Chen, 2001). We previously demonstrated ATR activation upon dl922‐947 infection and an ATM‐independent H2AX phosphorylation. We also observed that γH2AX was predominantly accumulated in replicative and post‐replicative phases, suggesting a correlation with viral induced replicative stress (Passaro et al., 2013). Therefore the assessment of γH2AX likely represented the best marker to evaluate the replication‐associated DNA lesions produced after dl922‐947 infection.
Accumulation of DNA damage is a potent stimulus for cell death, indeed γH2AX levels correlate with cell death in response to DNA damaging agents (Cook et al., 2009). Accordingly, olaparib treatment increased dl922‐947 cell killing effect. Isobolograms analysis showed a potent synergistic effect at all concentrations used, confirming the hypothesis that the block of the SSB repair by a PARP inhibitor enhances the effect of dl922‐947. It is worth noting that olaparib had no effect on DNA damage accumulation and only a modest effect on cell viability.
The role of viral replication on the efficacy of oncolysis is controversial: in some studies, the efficacy of oncolytic virotherapy have been correlated with the efficacy of viral replication (Touchefeu et al., 2011), while we have previously reported an enhancement of cell death in the absence of increased replication (Botta et al., 2012; Libertini et al., 2011). Here, we showed a slight, albeit significant increase, in viral genome copies in infected ATC cells treated with olaparib.
In order to better understand the observed synergistic effect, we have evaluated the cell cycle profile and some markers of cells death in cells undergoing both treatments.
The combined treatment induced a significant increase in cells with >4N content (polyploidy) and significantly increased the percentage of cells in sub‐G1 fraction. Sub‐G1 phase indicates the presence of DNA fragmentation occurring either during necrosis or apoptosis. Likely, cells in sub‐G1 fraction arise from fragmented polyploid cells.
The association with olaparib augmented the percentage of viral induced Annexin V positive cells and caspase 3 cleavage. In previous studies, the release of Cytochrome C has not been analyzed, here, we also showed the activation of this other apoptotic marker by dl922‐947 infection.
However, despite the presence of several apoptotic markers, the treatment with caspase inhibitor zVAD‐fmk did not induce statistically significant changes in cell survival, Annexin V positivity and cytochrome C release (not shown), confirming the involvement of non‐apoptotic pathways in dl922‐947 induced cell death.
Recently, a PARP‐mediated cell death has been described (parthanatos). This death causes phosphatidylserine flipping onto the outer plasma membrane, dissipation of mitochondrial membrane potential, chromatin condensation and large DNA fragmentation (Delettre et al., 2006; Susin et al., 1999; Yu et al., 2002). However, parthanatos differs from apoptosis, since it does not cause apoptotic body formation or small scale DNA fragmentation and cannot be rescued by zVAD‐fmk. We have evaluated the activation of parthanatos in ATC cells infected with dl922‐947. Lack of AIF translocation leads us to exclude the involvement of parthanatos in viral induced cell death.
To validate the potential therapeutic use of olaparib in association with dl922‐947, we analyzed the effects of the combined treatment on ATC xenograft tumors, by injecting FRO cells in athymic mice. Starting from day 13, we observed that the combined treatment olaparib/dl922‐947 significantly reduced tumor growth compared to single treatment group and control group.
Although xenograft tumors do not completely reproduce the features of thyroid carcinomas in orthotopic models, our data demonstrate the efficacy of the combination in vivo.
In addition, we have demonstrated that the combined treatment increased viral replication, γH2AX levels and Caspase 3 activation. Overall, these data clearly demonstrate the efficacy of olaparib/dl922‐947 combination in ATC both in vitro and in vivo.
A number of reports have shown an antiangiogenic effect of PARP inhibitors. PARP inhibitors have been efficiently used in vitro (Peralta‐Leal et al., 2009) to inhibit vascular endothelial growth factor (VEGF)‐induced proliferation, migration and tube formation in tumor models. Furthermore, it has been shown that PARP modulates the expression of hypoxia inducible factor HIF. In olaparib treated tumors we observed, by Real‐Time PCR and immunohistochemistry, a significant decrease in CD31 levels, indicating a reduction in tumor vessel formation, probably due to the anti‐angiogenic effects of PARP inhibitor. In line with this observation, we have previously reported that the normalization of tumor vasculature by the anti‐angiogenic antibody bevacizumab improves viral distribution and oncolytic activity (Libertini et al., 2008). The analysis of AdGFP distribution in olaparib treated tumors showed a more diffuse and intense signal. Although these data are not conclusive, they suggest that olaparib treatment likely enhances the effects of dl922‐947 also by improving viral diffusion. Interestingly, tumors infected with dl922‐947 alone showed a significant reduction in microvessel density and a trend toward a decrease in CD31 mRNA expression. These results are in line with previous data indicating an innate antiangiogenic effect of oncolytic viruses (Angarita et al., 2013). Further studies are required to specifically analyze the effects of dl922‐947 on tumor angiogenesis in ATC.
Multiple PARP inhibitors that may have an anticancer potential have been identified. PARP inhibitors could be used either as a single agent in HR‐deficient cells or to sensitize tumors to chemotherapy or radiotherapy, improving the therapeutic index of these approaches. The use of PARP inhibitors for the treatment of a variety of cancers is currently in phase I and II clinical trials, supporting the great clinical potential of this class of drugs. However, optimization of the doses is crucial to mitigate the increased toxicity in the combined treatments. Our data indicate that the selective inhibition of PARP‐1 increases the oncolytic activity of the mutant adenovirus dl922‐947 without inducing any significant weight loss compared to the control and the virus alone group.
In conclusion, data presented here show that the combined treatment olaparib/dl922‐947 is highly effective against anaplastic thyroid carcinoma cells and tumor xenografts, and could represent a novel therapeutic option for the treatment of this dismal disease. Furthermore, dl922‐947 has been proved to be effective in glioblastoma and pancreatic carcinoma models, therefore the association with olaparib could be also effective against other aggressive and not curable lesions.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
We thank Salvatore Sequino for his excellent technical assistance.
This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC). Silvana Libertini was a Marie Curie fellow.
1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.07.022.
Passaro Carmela, Volpe Massimiliano, Botta Ginevra, Scamardella Eloise, Perruolo Giuseppe, Gillespie David, Libertini Silvana, Portella Giuseppe, (2015), PARP inhibitor olaparib increases the oncolytic activity of dl922-947 in in vitro and in vivo model of anaplastic thyroid carcinoma, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.07.022.
Contributor Information
Silvana Libertini, Email: silvanalibertini@yahoo.it.
Giuseppe Portella, Email: portella@unina.it.
References
- Ame, J.C. , Spenlehauer, C. , de Murcia, G. , 2004. The PARP superfamily. Bioessays. 26, 882–893. [DOI] [PubMed] [Google Scholar]
- Angarita, F.A. , Acuna, S.A. , Ottolino-Perry, K. , Zerhouni, S. , McCart, J.A. , 2013. Mounting a strategic offense: fighting tumor vasculature with oncolytic viruses. Trends Mol. Med. 19, 378–392. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya, M. , Francis, J. , Eddouadi, A. , Lemoine, N.R. , Hallden, G. , 2011. An oncolytic adenovirus defective in pRb-binding (dl922-947) can efficiently eliminate pancreatic cancer cells and tumors in vivo in combination with 5-FU or gemcitabine. Cancer Gene Ther. 18, 734–743. [DOI] [PubMed] [Google Scholar]
- Botta, G. , Passaro, C. , Libertini, S. , Abagnale, A. , Barbato, S. , Maione, A.S. , Hallden, G. , Beguinot, F. , Formisano, P. , Portella, G. , 2012. Inhibition of autophagy enhances the effects of E1A-defective oncolytic adenovirus dl922-947 against glioma cells in vitro and in vivo. Hum. Gene Ther. 23, 623–634. [DOI] [PubMed] [Google Scholar]
- Botta, G. , Perruolo, G. , Libertini, S. , Cassese, A. , Abagnale, A. , Beguinot, F. , Formisano, P. , Portella, G. , 2010. PED/PEA-15 modulates coxsackievirus-adenovirus receptor expression and adenoviral infectivity via ERK-mediated signals in glioma cells. Hum. Gene Ther. 21, 1067–1076. [DOI] [PubMed] [Google Scholar]
- Burdak-Rothkamm, S. , Short, S.C. , Folkard, M. , Rothkamm, K. , Prise, K.M. , 2007. ATR-dependent radiation-induced gamma H2AX foci in bystander primary human astrocytes and glioma cells. Oncogene. 26, 993–1002. [DOI] [PubMed] [Google Scholar]
- Cheong, S.C. , Wang, Y. , Meng, J.H. , Hill, R. , Sweeney, K. , Kirn, D. , Lemoine, N.R. , Hallden, G. , 2008. E1A-expressing adenoviral E3B mutants act synergistically with chemotherapeutics in immunocompetent tumor models. Cancer Gene Ther. 15, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell, C.M. , Shibata, A. , Tookman, L.A. , Archibald, K.M. , Flak, M.B. , Pirlo, K.J. , Lockley, M. , Wheatley, S.P. , McNeish, I.A. , 2011. Genomic DNA damage and ATR-Chk1 signaling determine oncolytic adenoviral efficacy in human ovarian cancer cells. J. Clin. Invest. 121, 1283–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, P.J. , Ju, B.G. , Telese, F. , Wang, X. , Glass, C.K. , Rosenfeld, M.G. , 2009. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 458, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy, C. , Doorbar, J. , 2007. G2/M cell cycle arrest in the life cycle of viruses. Virology. 368, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre, C. , Yuste, V.J. , Moubarak, R.S. , Bras, M. , Lesbordes-Brion, J.C. , Petres, S. , Bellalou, J. , Susin, S.A. , 2006. AIFsh, a novel apoptosis-inducing factor (AIF) pro-apoptotic isoform with potential pathological relevance in human cancer. J. Biol. Chem. 281, 6413–6427. [DOI] [PubMed] [Google Scholar]
- Elisei, R. , 2012. Anaplastic thyroid cancer therapy: dream or reality?. Endocrine. 42, 468–470. [DOI] [PubMed] [Google Scholar]
- Esposito, F. , Libertini, S. , Franco, R. , Abagnale, A. , Marra, L. , Portella, G. , Chieffi, P. , 2009. Aurora B expression in post-puberal testicular germ cell tumours. J. Cell Physiol. 221, 435–439. [DOI] [PubMed] [Google Scholar]
- Ewald, B. , Sampath, D. , Plunkett, W. , 2007. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol. Cancer Ther. 6, 1239–1248. [DOI] [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C.J. , Tutt, A.N. , Johnson, D.A. , Richardson, T.B. , Santarosa, M. , Dillon, K.J. , Hickson, I. , Knights, C. , Martin, N.M. , Jackson, S.P. , Smith, G.C. , Ashworth, A. , 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Furnari, F.B. , Fenton, T. , Bachoo, R.M. , Mukasa, A. , Stommel, J.M. , Stegh, A. , Hahn, W.C. , Ligon, K.L. , Louis, D.N. , Brennan, C. , Chin, L. , DePinho, R.A. , Cavenee, W.K. , 2007. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 21, 2683–2710. [DOI] [PubMed] [Google Scholar]
- Garber, K. , 2006. China approves world's first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 98, 298–300. [DOI] [PubMed] [Google Scholar]
- Hallden, G. , Portella, G. , 2012. Oncolytic virotherapy with modified adenoviruses and novel therapeutic targets. Expert Opin. Ther. Targets. 16, 945–958. [DOI] [PubMed] [Google Scholar]
- Heise, C. , Hermiston, T. , Johnson, L. , Brooks, G. , Sampson-Johannes, A. , Williams, A. , Hawkins, L. , Kirn, D. , 2000. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 6, 1134–1139. [DOI] [PubMed] [Google Scholar]
- Heise, C. , Lemmon, M. , Kirn, D. , 2000. Efficacy with a replication-selective adenovirus plus cisplatin-based chemotherapy: dependence on sequencing but not p53 functional status or route of administration. Clin. Cancer Res. 6, 4908–4914. [PubMed] [Google Scholar]
- Heise, C. , Sampson-Johannes, A. , Williams, A. , McCormick, F. , Von Hoff, D.D. , Kirn, D.H. , 1997. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat. Med. 3, 639–645. [DOI] [PubMed] [Google Scholar]
- Jemal, A. , Siegel, R. , Xu, J. , Ward, E. , 2010. Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300. [DOI] [PubMed] [Google Scholar]
- Kortmann, U. , McAlpine, J.N. , Xue, H. , Guan, J. , Ha, G. , Tully, S. , Shafait, S. , Lau, A. , Cranston, A.N. , O'Connor, M.J. , Huntsman, D.G. , Wang, Y. , Gilks, C.B. , 2011. Tumor growth inhibition by olaparib in BRCA2 germline-mutated patient-derived ovarian cancer tissue xenografts. Clin. Cancer Res. 17, 783–791. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A. , 2004. DNA replication fidelity. J. Biol. Chem. 279, 16895–16898. [DOI] [PubMed] [Google Scholar]
- Levitzki, A. , Mishani, E. , 2006. Tyrphostins and other tyrosine kinase inhibitors. Annu. Rev. Biochem. 75, 93–109. [DOI] [PubMed] [Google Scholar]
- Libertini, S. , Abagnale, A. , Passaro, C. , Botta, G. , Barbato, S. , Chieffi, P. , Portella, G. , 2011. AZD1152 negatively affects the growth of anaplastic thyroid carcinoma cells and enhances the effects of oncolytic virus dl922-947. Endocr. Relat. Cancer. 18, 129–141. [DOI] [PubMed] [Google Scholar]
- Libertini, S. , Iacuzzo, I. , Ferraro, A. , Vitale, M. , Bifulco, M. , Fusco, A. , Portella, G. , 2007. Lovastatin enhances the replication of the oncolytic adenovirus dl1520 and its antineoplastic activity against anaplastic thyroid carcinoma cells. Endocrinology. 148, 5186–5194. [DOI] [PubMed] [Google Scholar]
- Libertini, S. , Iacuzzo, I. , Perruolo, G. , Scala, S. , Ierano, C. , Franco, R. , Hallden, G. , Portella, G. , 2008. Bevacizumab increases viral distribution in human anaplastic thyroid carcinoma xenografts and enhances the effects of E1A-defective adenovirus dl922-947. Clin. Cancer Res. 14, 6505–6514. [DOI] [PubMed] [Google Scholar]
- Lilley, C.E. , Schwartz, R.A. , Weitzman, M.D. , 2007. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 15, 119–126. [DOI] [PubMed] [Google Scholar]
- Livak, K.J. , Schmittgen, T.D. , 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lockley, M. , Fernandez, M. , Wang, Y. , Li, N.F. , Conroy, S. , Lemoine, N. , McNeish, I. , 2006. Activity of the adenoviral E1A deletion mutant dl922-947 in ovarian cancer: comparison with E1A wild-type viruses, bioluminescence monitoring, and intraperitoneal delivery in icodextrin. Cancer Res. 66, 989–998. [DOI] [PubMed] [Google Scholar]
- Martinou, J.C. , Green, D.R. , 2001. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2, 63–67. [DOI] [PubMed] [Google Scholar]
- McCabe, N. , Turner, N.C. , Lord, C.J. , Kluzek, K. , Bialkowska, A. , Swift, S. , Giavara, S. , O'Connor, M.J. , Tutt, A.N. , Zdzienicka, M.Z. , Smith, G.C. , Ashworth, A. , 2006. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 66, 8109–8115. [DOI] [PubMed] [Google Scholar]
- Nagaiah, G. , Hossain, A. , Mooney, C.J. , Parmentier, J. , Remick, S.C. , 2011. Anaplastic thyroid cancer: a review of epidemiology, pathogenesis, and treatment. J. Oncol. 2011, 542358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, G.J. , Schaack, J. , Ornelles, D.A. , 2009. Widespread phosphorylation of histone H2AX by species C adenovirus infection requires viral DNA replication. J. Virol. 83, 5987–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea, C.C. , Johnson, L. , Bagus, B. , Choi, S. , Nicholas, C. , Shen, A. , Boyle, L. , Pandey, K. , Soria, C. , Kunich, J. , Shen, Y. , Habets, G. , Ginzinger, D. , McCormick, F. , 2004. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 6, 611–623. [DOI] [PubMed] [Google Scholar]
- Passaro, C. , Abagnale, A. , Libertini, S. , Volpe, M. , Botta, G. , Cella, L. , Pacelli, R. , Hallden, G. , Gillespie, D. , Portella, G. , 2013. Ionizing radiation enhances dl922-947-mediated cell death of anaplastic thyroid carcinoma cells. Endocr. Relat. Cancer. 20, 633–647. [DOI] [PubMed] [Google Scholar]
- Peralta-Leal, A. , Rodriguez-Vargas, J.M. , Aguilar-Quesada, R. , Rodriguez, M.I. , Linares, J.L. , de Almodovar, M.R. , Oliver, F.J. , 2009. PARP inhibitors: new partners in the therapy of cancer and inflammatory diseases. Free Radic. Biol. Med. 47, 13–26. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan, S. , Miranda, E. , Ekblad, M. , Holford, A. , Pizarro, M.T. , Lemoine, N.R. , Hallden, G. , 2010. Efficacy of oncolytic mutants targeting pRb and p53 pathways is synergistically enhanced when combined with cytotoxic drugs in prostate cancer cells and tumor xenografts. Hum. Gene Ther. 21, 1311–1325. [DOI] [PubMed] [Google Scholar]
- Rajesh, M. , Mukhopadhyay, P. , Batkai, S. , Godlewski, G. , Hasko, G. , Liaudet, L. , Pacher, P. , 2006. Pharmacological inhibition of poly (ADP-ribose) polymerase inhibits angiogenesis. Biochem. Biophys. Res. Commun. 350, 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raki, M. , Kanerva, A. , Ristimaki, A. , Desmond, R.A. , Chen, D.T. , Ranki, T. , Sarkioja, M. , Kangasniemi, L. , Hemminki, A. , 2005. Combination of gemcitabine and Ad5/3-Delta24, a tropism modified conditionally replicating adenovirus, for the treatment of ovarian cancer. Gene Ther. 12, 1198–1205. [DOI] [PubMed] [Google Scholar]
- Russell, S.J. , Peng, K.W. , Bell, J.C. , 2012. Oncolytic virotherapy. Nature Biotechnol. 30, 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe, R.E. , Klopper, J.P. , Korch, C. , Pugazhenthi, U. , Benezra, M. , Knauf, J.A. , Fagin, J.A. , Marlow, L.A. , Copland, J.A. , Smallridge, R.C. , Haugen, B.R. , 2008. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J. Clin. Endocrinol. Metab. 93, 4331–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr, C.J. , McCormick, F. , 2002. The RB and p53 pathways in cancer. Cancer Cell. 2, 103–112. [DOI] [PubMed] [Google Scholar]
- Smallridge, R.C. , Marlow, L.A. , Copland, J.A. , 2009. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr. Relat. Cancer. 16, 17–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. , Tho, L.M. , Xu, N. , Gillespie, D.A. , 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112. [DOI] [PubMed] [Google Scholar]
- Susin, S.A. , Lorenzo, H.K. , Zamzami, N. , Marzo, I. , Snow, B.E. , Brothers, G.M. , Mangion, J. , Jacotot, E. , Costantini, P. , Loeffler, M. , Larochette, N. , Goodlett, D.R. , Aebersold, R. , Siderovski, D.P. , Penninger, J.M. , Kroemer, G. , 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 397, 441–446. [DOI] [PubMed] [Google Scholar]
- Taccaliti, A. , Silvetti, F. , Palmonella, G. , Boscaro, M. , 2012. Anaplastic thyroid carcinoma. Front. Endocrinol. 3, 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida, R.J. , 2001. Drug synergism: its detection and applications. J. Pharmacol. Exp. Ther. 298, 865–872. [PubMed] [Google Scholar]
- Touchefeu, Y. , Vassaux, G. , Harrington, K.J. , 2011. Oncolytic viruses in radiation oncology. Radiother. Oncol. 99, 262–270. [DOI] [PubMed] [Google Scholar]
- Vichai, V. , Kirtikara, K. , 2006. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Hallden, G. , Hill, R. , Anand, A. , Liu, T.C. , Francis, J. , Brooks, G. , Lemoine, N. , Kirn, D. , 2003. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 21, 1328–1335. [DOI] [PubMed] [Google Scholar]
- Ward, I.M. , Chen, J. , 2001. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 276, 47759–47762. [DOI] [PubMed] [Google Scholar]
- Waterhouse, N.J. , Trapani, J.A. , 2003. A new quantitative assay for cytochrome c release in apoptotic cells. Cell Death Differ. 10, 853–855. [DOI] [PubMed] [Google Scholar]
- Weiner, L.M. , Surana, R. , Wang, S. , 2010. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 10, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, S.W. , Wang, H. , Poitras, M.F. , Coombs, C. , Bowers, W.J. , Federoff, H.J. , Poirier, G.G. , Dawson, T.M. , Dawson, V.L. , 2002. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 297, 259–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data