

Abstract
Introduction
Various studies have identified aberrantly expressed miRNAs in breast cancer and demonstrated an association between distinct miRNAs and malignant progression as well as metastasis. Even though tumor‐associated macrophages (TAM) are known mediators of these processes, little is known regarding their miRNA expression upon education by malignant cells in vivo.
Methods
We profiled miRNA and mRNA expression of in vitro tumor‐educated macrophages (TEM) by indirectly co‐culturing with estrogen‐receptor‐positive (ER+) MCF‐7 breast cancer cells. The prognostic power of the resulting miRNA list was investigated in primary breast cancer datasets and compared to other signatures. Furthermore, miRNA expression levels were correlated to mRNA expression of macrophage markers and the impact on prognosis was assessed.
Results
Through the evaluation of the group effects between differentially‐expressed miRNAs and their target mRNAs in TEM, the power of detecting regulated miRNAs was greatly increased. The resulting list of 96 miRNAs predicts disease‐free survival (DFS) in external datasets of ER+ breast cancer patients and performs well in comparison with other miRNA signatures. Clustering with the predefined miRNA list revealed a significant difference in survival between the two resulting patient groups. Furthermore, an optimized miRNA list, based on correlations with macrophages markers, proved even more capable at identifying patient clusters significantly differing in DFS.
Conclusions
In vitro profiling of TEM and subsequent bioinformatic verification identified miRNAs with a high prognostic power for DFS when transferred into the clinical setting of primary breast cancer. The resulting miRNAs not only verify previously established findings but also lead to new prognostic markers. Furthermore, our data suggest that TAM contribute to the total miRNA expression profile of ER + breast cancers.
Keywords: miRNA, Tumor‐associated macrophages, Breast cancer
Highlights
miRNA and mRNA were measured in macrophages exposed to ER + breast cancer cells.
Regulated miRNAs were detected by analyzing group effects of mRNA targets.
The resulting miRNA list has good prognostic value for DFS in ER + breast cancer.
Correlation of miRNAs to macrophage markers improved identification of clusters differing in DFS.
Abbreviations
- ER
estrogen receptor
- ERBB2/HER2
human epidermal growth factor 2
- miRNA
micro-RNA
- TAM
tumor-associated macrophages
- TEM
tumor-educated macrophages
- PR
progesterone receptor
- miRNA
micro RNA
1. Introduction
The heterogeneity of breast cancer poses serious clinical challenges. Thus, different approaches to stratifying patients with respect to their prognosis have been developed. In clinical practice, the management of breast cancer is based upon established criteria such as tumor size, the presence of distant metastases, histologic type and grade, the expression of hormone (i.e. estrogen and progesterone) receptors (ER and PR), and human epidermal growth factor 2 (ERBB2/HER2) receptor. These parameters are complemented by gene expression studies, which resulted in the identification of molecular subtypes (Perou et al., 2000; Smid et al., 2008; Sorlie et al., 2001) such as luminal A, luminal B, normal‐like, basal‐like, and HER2‐enriched. This classification has great clinical relevance, as the different subtypes have distinct prognoses and patterns of metastasis (Smid et al., 2008). In ER‐positive (ER+) patients, distinct gene expression signatures have been identified to predict clinical outcome with a greater degree of accuracy (Arranz et al., 2012). Subsequently, simplified qRT‐PCR based approaches of molecular subtyping have been developed to allow a transfer into routine clinical decision making (Parker et al., 2009).
Recently, the field of clinicogenomics has been further expanded through the use of microRNA analyses. MicroRNAs (miRNAs) are small, non‐coding RNA molecules of 20–25 nucleotides in length, which can regulate target mRNAs on the transcriptional and posttranscriptional level (Du and Zamore, 2005). The latter mechanism is predominant in the mammalian system (Guo et al., 2010). miRNAs are present at abnormal levels in human tumors with pathogenetic consequences through the action as tumor suppressors or oncogenes (Nicoloso et al., 2009; Tian et al., 2013). As such, the expression of miRNAs has been implicated in various aspects of breast cancer progression, such as tumor development, drug resistance, and metastasis (Volinia et al., 2012; Tian et al., 2013). Several paradigmatic studies (Camps et al., 2008; Rothe et al., 2011; Volinia et al., 2012) revealed an up‐regulation of miR‐210 during invasive transition and correlation with tumor aggressiveness and poor prognosis. Subsequently, Buffa et al. validated the previously established prognostic relevance of miR‐210 expression and explored regulatory effects on target genes integrating miRNA and mRNA global expression profiles (Buffa et al., 2011). Furthermore, these authors identified a list of miRNAs independently associated with prognosis in breast cancer. On the contrary, a study investigating another cohort of similar patients found no single miRNA profile to predict outcome (Lyng et al., 2012).
Most gene expression and miRNA studies rely on the analysis of whole tumor specimens comprised of malignant epithelial cells and the surrounding tissue. Thus, these data reflect alterations within both cancer and stromal cells. Even though the stromal compartment is highly tumor‐supportive, only limited gene expression data from the tumor stroma of clinical samples exist (Allinen et al., 2004; Finak et al., 2008; Singer et al., 2008). This transformed benign tumor compartment contains large populations of myoepithelial cells, fibroblasts, endothelial cells, and immune cells. Among the latter, tumor‐associated macrophages (TAM) play a pivotal role in facilitating tumor progression and metastasis (Qian and Pollard, 2010; Quail and Joyce, 2013). Several clinical studies have demonstrated a correlation between the rate of macrophage infiltration of the primary tumor and tumor cell infiltration, vascularity, and ultimately patient prognosis (Eiro et al., 2012; Leek et al., 1996; Medrek et al., 2012). Furthermore, gene expression studies of murine TAM have proven to predict poor prognosis and reduced survival if applied to clinical datasets (Ojalvo et al., 2010; Zabuawala et al., 2010). However, little is known on the involvement of miRNA expression in TAM‐mediated tumor progression. While miRNAs regulate macrophage differentiation and response to external stimuli, i.e. alternative M2‐type activation, the implications for TAM function are about to be elucidated (Squadrito et al., 2013). So far, down‐regulation of miR‐155 targeting cytokine production (He et al., 2009) and up‐regulation of miR‐511‐3p as part of a negative feedback loop (Squadrito et al., 2012) have been described in TAM.
Assuming that in vitro tumor‐educated macrophages (TEM) constitute a model for TAMs in vivo, we hypothesized that miRNA expression data from TEM can be used to determine unfavorable clinical subsets in breast cancer. Thus, we obtained miRNA and mRNA expression data from macrophages co‐cultured in vitro with breast cancer cells. These data were filtered by testing for their meaningful relation, i.e. diametrically opposed miRNA and mRNA expression levels (Artmann et al., 2012). The resulting miRNA list was then analyzed further to determine its discriminative power within datasets from breast cancer patients (for an overview see Figure 1). This enabled us to identify miRNA classifiers distinguishing clinical subgroups with distinctively different prognoses. These clinical subgroups were finally further refined when correlations between miRNAs and macrophage marker expression levels were taken into consideration.
Figure 1.
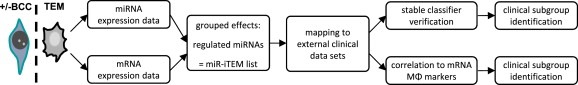
Experimental and bioinformatics workflow of this study. After co‐culture with breast cancer cells (BCC), the miRNA and target set mRNA expression levels of tumor‐educated macrophages (TEM) were analyzed separately and the p‐values obtained were combined. The resulting miR‐iTEM list was mapped to external clinical datasets of breast cancer primaries and tested for prognostic power. In parallel, correlation to macrophage markers was determined. Prognostic relevance was revealed by supervised clustering and subsequent Kaplan–Meier analysis of DFS.
2. Material and methods
2.1. Cell lines, cell culture and co‐culture with human macrophages
The ER+ and PR+ human breast cancer cell line MCF‐7 was purchased from DSMZ and maintained in RPMI‐1640 medium (PAA Laboratories Inc., Cölbe, Germany) supplemented with 10% fetal calf serum (FCS, Invitrogen, Karlsruhe, Germany). Human macrophages were derived from mononuclear peripheral blood cells and were collected with approval of the local ethics committee in Göttingen.
Briefly, monocytes were obtained from buffy coats of healthy donors through double‐density‐gradient isolation. Macrophages were differentiated by culturing monocytes in fluorinated ethylene propylene‐coated cell culture bags (CellGenix, Freiburg, Germany) in the presence of M‐CSF (Immunotools, Friesoythe, Germany) for 7 days. Differentiation into mature macrophages was assessed by flow cytometry verification of CD11b, CD11c, CD14 and CD45 (Beckman Coulter, Krefeld, Germany) expression and negativity for CD209 (BioLegend, Fell, Germany). For indirect co‐culture experiments, macrophages were seeded in hanging cell culture inserts (0.4 μm pore size, PET; Millipore, Billerica, MA, USA) and co‐cultured with MCF‐7 cells at a ratio of 2:1 under normoxia for 24 h. All co‐culture experiments were performed with at least 3 biological replicates, i.e. with different passages of MCF‐7 cells and macrophages derived from different donors.
2.2. RNA extraction
Total RNA for array experiments was isolated using TRIZOL reagent according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). RNA integrity for each sample was confirmed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Samples used for microarray experiments exhibited a RIN number greater than 7.5. Each RNA sample was then split into two aliquots that were either processed for the miRNA or the mRNA microarray.
2.3. Gene expression and miRNA microarray studies
Global gene expression analyses utilized the “Human GE 4 × 44K v2 Microarray Kit” (Agilent, Böblingen, Germany) and the QuickAmp Labeling Kit Cy3 One‐Color (Agilent) as well as the RNA Spike‐In Kit (Agilent). 500 ng of total RNA were used as starting material for the synthesis of cDNA. Following in‐vitro transcription, the quality and quantity of labeled RNA was determined using the NanoDrop D‐1000 UV‐VIS Spectrophotometer (Peqlab, Erlangen, Germany). 1.65 μg of each labeled sample were fragmented and applied onto “Human GE 4 × 44K v2” (#G4845A, Agilent) microarrays. Hybridizations were performed for 17 h at 10 rpm and 65 °C in a hybridization oven (Agilent). Washing and array processing were completed according to the manufacturer's recommendations. Cy3 intensities were detected by one‐color scanning using the G2505B Agilent DNA microarray scanner at 5 μm resolution. Intensity data were extracted using the Feature Extraction application (version 9.5, Agilent).
For human miRNA microarray analyses, 200 ng of total RNA were labeled using the miRNA Complete Labeling and Hyb Kit (Agilent). Labeled samples were hybridized to “Human miRNA 8 × 15K V3” microarrays (#G4470C, Agilent), based on miRBase (release 12.1, (Griffiths‐Jones et al., 2008)). Hybridizations were performed for 20 h at 10 rpm and 65 °C in a hybridization oven (Agilent). After washing and array processing according to the manufacturer's protocol, Cy3 intensities were detected by one color scanning at 3 μm resolution.
All miRNA and mRNA expression data have been submitted to the NCBI Gene Expression Omnibus (GEO) data repository (Barrett and Edgar, 2006), SuperSeries GSE55024.
2.4. Detection of differentially‐expressed miRNA
All expression data were log‐transformed and quantile‐normalized. We utilized the R‐package ‘miRtest’ (Artmann et al., 2012) to detect miRNAs that were both differentially regulated and had an effect on their target mRNA set. Matching of miRNA to mRNA data was obtained from microCosm (Griffiths‐Jones et al., 2008). Gene identifiers were mapped using the biomaRt R‐package (Durinck et al., 2005). Group effects of mRNA target gene sets were determined using the gene set test ‘Romer’ (Majewski et al., 2010) available in the ‘miRtest’ R‐package. The resulting lists of p‐values from miRNA and set‐wise testing were combined by means of Stouffer's inverse normal method (Marot and Mayer, 2009; Stouffer, 1949). Possible confounding effects of macrophage donors and microarray types were adjusted for in the design matrix.
2.5. miRNA qRT‐PCR
2 μg of total RNA was used as starting material for reverse transcription. SYBR based quantification of mature miR‐210 was performed with the miScript system and the appropriate primer assay (both Qiagen, Hilden, Germany) on an ABI 7900HT instrument (Applied Biosystems, Foster City, CA, USA). RNU6B miRNA levels were used to normalize between different samples. Relative miR210 expression was analyzed using RQ Manager software (Applied Biosystems). Significance was tested using the paired two‐sided Student's T‐test.
2.6. External microarray datasets
Two primary breast cancer miRNA expression datasets were retrieved from GEO. All data were downloaded as raw expression data, log‐transformed, and quantile‐normalized. For all samples, annotations of the ER status and information on disease‐free survival were available. In both breast cancer datasets, all ER+ patients included in our analysis underwent adjuvant antihormonal tamoxifen treatment. Both published datasets were originally collected with approval of the respective local ethics committees.
The first dataset (GSE22216) contained 210 whole tumor samples from patients treated in Oxford, UK and the corresponding miRNA expression levels for 735 miRNAs (Buffa et al., 2011). No information on the tumor cell content within the samples was provided. For all 210 samples within the GSE22216 dataset, mRNA expression levels for 24332 mRNAs were available via GEO (GSE22219). For the sake of readability, the datasets will be called “Oxford miRNA” and “mRNA set”, respectively. The authors reported different miRNA signatures for ER+ and ER− patients. As macrophages were co‐cultured with hormone‐receptor‐positive MCF‐7 cells, we split the dataset into two subsets and only performed the analysis on the 128 ER+ patients. Within this cohort the authors identified a set of miRNAs prognostic for distant relapse‐free survival (DRFS). For clarity, this list will be referred to as “miR‐Oxford ER+”.
The second dataset (GSE37405) comprised three datasets (GPL14149, GPL13703 and GPL15462) (Lyng et al., 2012) of only ER+ patients who underwent surgery in Odense, Denmark. Thus, this dataset will herein after be titled “Odense miRNA”. The authors included only samples which exhibited >50% tumor cells. Due to the amount of miRNA expression data missing in the GPL14149 subset, only GPL13703 and GPL15462 were analyzed. Together these two sets comprised 113 samples with expression levels for 761 miRNAs. No prognostic miRNA profile could be detected reliably in the original study.
For both these datasets disease‐free survival (DFS) as provided by the authors was used as primary end point. All probes which were not annotated to any miRNA were discarded. For probes annotated with the same miRNA, the median of their expression was taken.
2.7. Statistical evaluation of prognostic power of miRNA sets
All analyses were performed using the free statistical software R version 3.0.0 (R Development Core Team, 2013). In cases of multiple hypothesis testing P‐values were adjusted for multiplicity using the method established by Benjamini and Hochberg (Benjamini and Hochberg, 1995) using the R function p.adjust. A false discovery rate (FDR, q‐value) up to 5% was considered significant.
On both external datasets, three miRNA lists were evaluated with respect to their prognostic power: 1) the list of all miRNAs present on the chip, 2) the set of differentially‐regulated miRNAs in TEM (for details see Results) and 3) the miR‐Oxford ER+ list. The logarithmic gene expression levels were assessed for their potential to predict disease‐free survival (DFS) based on the Cox proportional hazards model (Andersen and Gill, 1982). Analysis of survival data was performed using the R package survival. CoxBoost (Binder and Schumacher, 2009), an algorithm and R package for sparse Cox modeling, was applied to all three sets for feature selection. For purposes of comparison, an additional null model was included into the analysis. This null model was calculated by permuting the survival information. In order to avoid empty feature sets in the null model, which occurs if CoxBoost does not detect any informative features, non‐sparse Cox modeling was applied for the null model estimation. To keep the feature set small enough for non‐sparse Cox modeling, the ten least informative features (tested in univariate Cox regression), were used in this permutation.
Stable features selected by CoxBoost were determined by an internal 10‐fold cross‐validation. The features that were selected in at least 50% of the internal folds were used as variables in the final Cox regression model.
To assess the predictive performance of the different miRNA sets the time‐dependent Brier score was calculated (Brier, 1950). In our setting this score represents differences between the patient's disease status and the predicted value at each time point. The Brier score is expressed in numbers between 0 and 1 – a lower number indicates an improved ability to predict DFS. Calculation of the Brier score was performed in a 10‐fold cross‐validation with the R package survAUC. The resulting Brier score curves were tested for significant difference using a sign‐rank test on the integrated Brier scores as proposed by Wiel et al. (Wiel et al., 2009).
Correlation between miRNA expression levels and the mRNA expression levels of macrophage markers was determined using Pearson's correlation test in the Oxford miRNA and mRNA breast cancer datasets (i.e. GSE22216 and GSE22219). The mean of the normalized expression levels of CD14 and CD68 (IDs: GI_4557416 and GI_4557434) was used as a macrophage marker. To establish a macrophage‐related list, miRNAs were included according to their correlations (FDR < 0.05).
Breast cancer samples were clustered within the obtained lists according to their miRNA expression profile by means of hierarchical clustering. Individual results of the cluster validation methods were combined by rank aggregation (Pihur and Datta, 2009) using the R package clValid (Brock et al., 2011). The resulting clusters were subsequently subjected to a Kaplan–Meier analysis of disease‐free survival (DFS). These Kaplan–Meier curves were then compared using a log rank test. Hazard ratios (HR) and 95% confidence intervals (CI) were computed using the Cox proportional hazard model using the function coxph from the R package survival.
3. Results
3.1. miRNA profiling in TEM through miRNA and mRNA group effects
Our aim was to identify differentially‐expressed miRNAs by comparing naive macrophages with tumor‐educated macrophages (TEM). We therefore performed both miRNA arrays and mRNA arrays from the same macrophage preparations with and without the addition of tumor cells. In this combined dataset we identified differentially‐expressed miRNAs through the test statistic miRtest that evaluates not only the individual miRNAs but also their predicted target genes for consistent differential expression.
Out of the 738 miRNAs present on the array, 642 had annotated gene sets that allowed the application of the miRtest method. Thus, group effects were determined individually for each of these 642 miRNAs and combined with their mRNA target gene sets. Through this approach, 96 miRNAs revealed both a differential expression and consistently regulated target mRNA sets in TEM. This panel of miRNAs will be referred to as miRNAs in TEM – miR‐iTEM. Combined p‐ and FDR‐values for these miRNAs are given in Supplementary Table 1. An assessment of the direction of the respective fold changes is shown in Supplementary Table 2.
Interestingly, miR‐210 was among the members of the miR‐iTEM list, which has previously been shown to be associated with poor patient outcome (Camps et al., 2008; Rothe et al., 2011; Volinia et al., 2012).
We thus aimed to verify its increased expression levels by qRT‐PCR in independent samples of TEM. In line with the array data these experiments showed a significant increase of miR‐210 expression (Supplementary Figure 1).
3.2. Mapping of miRNA sets to external breast cancer datasets
The retrieved miR‐iTEM list of 96 miRNAs was mapped to the two external microarray datasets. While for the Oxford miRNA dataset thirteen out of the 96 miRNAs could not be mapped, mapping was successful in all cases for the Odense dataset (Supplementary Table 1). To increase mapping efficiency and retain as much information as possible we allowed for mapping to the complementary strand if no exact match was found.
The miR‐Oxford ER+ list contained four miRNAs, of these only three were found in the Odense dataset because miR‐135a was not present (Table 1) on the array used in the original study.
Table 1.
Stable feature selection of miR‐iTEM in breast cancer datasets. List of stably selected miRNAs from the different miRNA sets (miR‐iTEM list, miR‐Oxford ER + list, all miRNAs list) and their corresponding inclusion frequencies (in % of all cross validation runs) for the two external breast cancer datasets. “‐“ indicates that this miRNA was not present in the respective list of miRNAs.
| Oxford dataset (GSE22216) | Odense dataset (GSE37405) | ||||||
|---|---|---|---|---|---|---|---|
| miRNA | miR‐iTEM | miR‐Oxford ER+ | All miRNAs | miRNA | miR‐iTEM | miR‐Oxford ER+ | All miRNAs |
| hsa‐miR‐767‐3p | 90 | 100 | 50 | hsa‐miR‐767‐3p | 0 | 100 | 0 |
| hsa‐miR‐210 | 100 | – | 20 | hsa‐miR‐941 | 100 | – | 90 |
| hsa‐miR‐486‐5p | 90 | – | 30 | hsa‐miR‐548c‐5p | 100 | – | 90 |
| hsa‐miR‐941 | 100 | – | 60 | hsa‐miR‐30a* | 70 | – | 20 |
| hsa‐miR‐769‐3p | – | 100 | 50 | hsa‐miR‐769‐3p | – | 100 | 0 |
| hsa‐miR‐181d | 60 | – | 0 | hsa‐miR‐128 | – | 70 | 0 |
| hsa‐miR‐135a | – | 100 | 30 | ||||
| hsa‐miR‐125a‐5p | 60 | – | 0 | ||||
| hsa‐miR‐128a:9.1 | – | 100 | 0 | ||||
| hsa‐miR‐296‐5p | 70 | – | 0 | ||||
| hsa‐miR‐578 | 60 | – | 0 | ||||
| hsa‐miR‐493 | 50 | – | 0 | ||||
| HS_285 | – | – | 50 | ||||
3.3. Robustness of miR‐iTEM as a stratifier in clinical breast cancer samples
To validate the usefulness of the miR‐iTEM list obtained in vitro in a clinical setting, we investigated its prognostic power for DFS in external datasets of primary breast cancers. Through this approach, we aimed to clarify whether our miR‐iTEM list is beneficial in comparison with already established prognostic miRNA panels. Hence, we trained prognostic models for DFS and compared the prediction accuracy of the miR‐iTEM list to that of the miR‐Oxford ER+ list. It is noteworthy that there was a low overlap between the two lists – only miR767‐3p was present in both. To prove that the miR‐iTEM list is suitable to predict DFS, we also included for each dataset the list of all miRNAs identified in the corresponding experiment and a null model into our calculations.
The estimated error, i.e. Brier score, for survival prediction was calculated for the Oxford dataset (Figure 2a). This statistical measure determines the accuracy of prediction – the lower the Brier score the better the accuracy. As was to be expected, the miR‐Oxford ER+ list demonstrated the lowest prediction error in the dataset in which it was generated (p‐value of comparison to null model 0.10). Surprisingly, the list of all miRNAs provided only marginal improvements compared to the null model. However, the miR‐iTEM list exhibited a low prediction error compared to both the null model and the list of all miRNAs (p‐value of comparison to null mode 0.14). Its performance was only slightly worse than the miR‐Oxford ER+ list (p‐value 0.24). This highlights the validity of the miR‐iTEM list even when transferred to clinical in vivo data.
Figure 2.
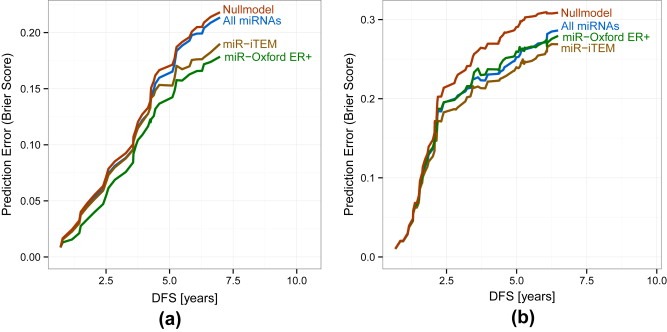
Low estimated error of the miR‐iTEM list for survival prediction in breast cancer datasets. Brier score for DFS prediction at different time points of the different miRNA sets (green: miR‐iTEM; brown: miR‐Oxford ER+; blue: all miRNAs; red: null model). Results for the Oxford dataset are illustrated in (a) and the Odense data set in (b). Lower curves indicate fewer prediction errors.
In order to perform a further unbiased analysis to validate our findings and compare the performance of the miR‐iTEM to the miR‐Oxford ER+ list, we tested their impact on DFS in an independent dataset. Thus, we calculated the Brier scores in the Odense dataset. The miR‐Oxford ER+ and complete miRNA lists perform comparably and show distinct improvement over the null model, whereas both were outperformed by the miR‐iTEM list from the macrophages (Figure 2b). Only the miR‐iTEM list yields a significantly reduced prediction error (p‐value 0.3) while the miR‐Oxford ER+ stays not significant (p‐value 0.10).
3.4. Identification of clinically relevant subgroups by miR‐iTEM
Having shown that miR‐iTEM is suitable as a stratifier in external datasets of breast cancer patients, we were interested as to whether this would translate into any prognostic ability. Thus we performed cluster analysis based on the expression profile of the miR‐iTEM list in the Oxford and Odense breast cancer datasets to define clinically relevant subgroups. In both ER+, tamoxifen‐treated patient cohorts clustering defined two distinct patient sets (Figure 3a and b) as the optimal number of clusters. More strikingly, these two clusters separate clearly in their DFS (Oxford: p = 0.0341, HR = 1.8, 95% CI [1.04 to 3.11]), Odense: p = 0.00654, HR = 2.33, 95% CI [1.24 to 4.36] (Figure 3c and d) as confirmed by the log rank test.
Figure 3.
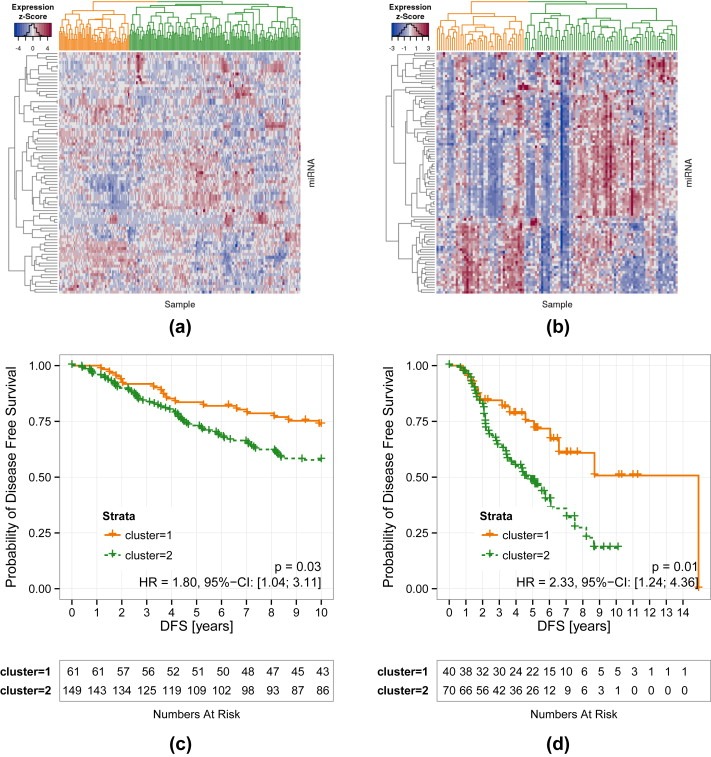
Supervised clustering according to miR‐iTEM distinguishes DFS. Heatmap of the two separate clusters (given in orange and green in both datasets) based on the miRNA expression values of the miR‐iTEM list in the (a) Oxford and (b) Odense breast cancer datasets. Corresponding Kaplan–Meier graphs of DFS are depicted in (c) and (d), p‐values were determined by log rank test.
3.5. Stable feature selection within miR‐iTEM
Our aim was to condense the information contained within the miR‐iTEM list into a smaller subset. To achieve this goal, we performed stable feature selection, i.e. determination of the most informative miRNAs. To this end, sparse Cox models (see Methods) were used and a miRNA was defined as stable if chosen in at least 50% of the training steps of cross validation runs. The resulting stable miRNAs from the different lists are given in Supplementary Table 1. Unsurprisingly, all of the four miRNAs from the miR‐Oxford ER+ list were selected consistently in the original Oxford miRNA dataset. From the miR‐iTEM list nine miRNAs were considered stable. The overlap of both lists comprised only one miRNA (miR‐767‐3p), which was stably selected from both lists. In the Odense miRNA dataset, three out of the four available miR‐Oxford ER+ miRNAs were chosen, while from the miR‐iTEM list three miRNAs were picked. Among these only miR‐941 was congruently selected from our miR‐iTEM list as a stable feature in both external datasets.
3.6. Correlation of miR‐iTEM with mRNA expression of macrophage markers
As stable feature selection did not yield a subset applicable to all external datasets, we hoped to extract the most useful miRNAs from miR‐iTEM by regarding the data from a more biological point of view. We therefore evaluated whether the individual miR‐iTEMs are associated to the extent of TAM infiltration in primary breast cancers. We thus correlated miRNA expression levels of the miR‐iTEM members to mRNA expression levels of macrophage markers, i.e. CD68 and CD14, in the Oxford mRNA dataset. In order to uncover relations within the complex tumor tissue, even low correlation coefficients were considered meaningful as long as the FDR was <0.05. Out of the original 96 miR‐iTEM members only 23 – which will be referred to as the miR‐iTEMMΦ set – significantly correlate with CD14 and CD68 (Table 2, Figure 4).
Table 2.
Correlations of miR‐iTEM list to macrophage markers. Significant correlations between members of the miR‐iTEMMΦ list and the macrophage markers CD68 and CD14.
| miRNA | rho | FDR |
|---|---|---|
| hsa‐miR‐150 | 0.22 | <0.001 |
| hsa‐miR‐30a* | −0.31 | <0.001 |
| hsa‐miR‐342‐3p | −0.2 | 0.001 |
| hsa‐miR‐148b | −0.27 | 0.001 |
| hsa‐miR‐941 | 0.26 | 0.002 |
| hsa‐miR‐138 | 0.26 | 0.003 |
| hsa‐miR‐194 | −0.25 | 0.004 |
| hsa‐miR‐766 | 0.24 | 0.004 |
| hsa‐miR‐185 | 0.24 | 0.004 |
| hsa‐miR‐125a‐5p | −0.23 | 0.006 |
| hsa‐miR‐483‐3p | −0.23 | 0.006 |
| hsa‐miR‐578 | −0.23 | 0.006 |
| hsa‐miR‐550* | 0.22 | 0.008 |
| hsa‐miR‐184 | −0.21 | 0.011 |
| hsa‐miR‐151:9.1 | −0.21 | 0.011 |
| hsa‐miR‐18a* | 0.21 | 0.011 |
| hsa‐miR‐645 | −0.21 | 0.014 |
| hsa‐miR‐602 | −0.2 | 0.014 |
| hsa‐miR‐181d | −0.2 | 0.015 |
| hsa‐miR‐579 | 0.19 | 0.025 |
| hsa‐miR‐130a | 0.18 | 0.035 |
| hsa‐miR‐767‐3p | 0.18 | 0.035 |
| hsa‐miR‐384 | −0.18 | 0.04 |
Figure 4.
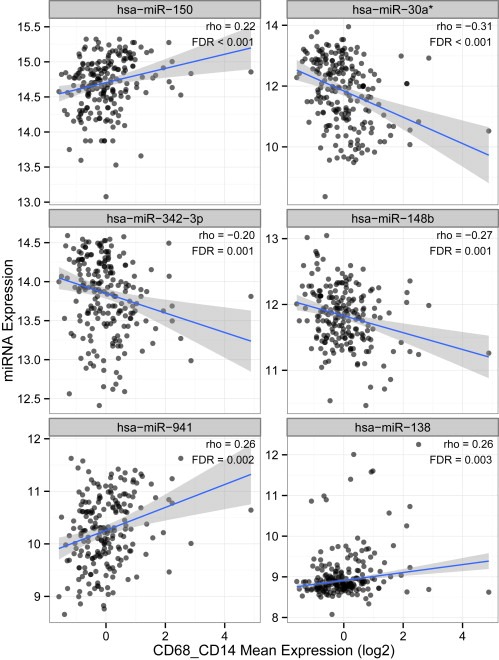
Correlations between miR‐iTEM and macrophage markers in breast cancer primaries. Correlations of the macrophage markers CD14 and CD68 with the exemplary miR‐iTEM members hsa−miR−150, hsa−miR−30a*, hsa−miR−342−3p, and hsa−miR−148b in breast cancer samples as determined by Pearson's correlation test. Correlation coefficient (rho) and p‐values are given.
3.7. Improved prognostic subgroups based on miR‐iTEMMΦ
To verify whether the smaller miR‐iTEMMΦ set still contains the same information as its precursor we performed once again supervised clustering of the external datasets. Remarkably, using the miR‐iTEMMΦ list leads to the identification of two clusters in both external datasets (Figure 5, Supplementary Figure 2) which once again exhibit a significant difference in DFS (Oxford: p = 0.00141, HR = 2.24, 95% CI [1.35 to 3.71]), Odense: p = 0.00423, HR = 2,48, 95% CI [1.30 to 4.73]. While these clusters significantly overlap with the previously described patient groups their discriminative ability is increased in comparison to the miR‐iTEM clusters. This suggests that the miR‐iTEMMΦ carries additional prognostic power.
Figure 5.
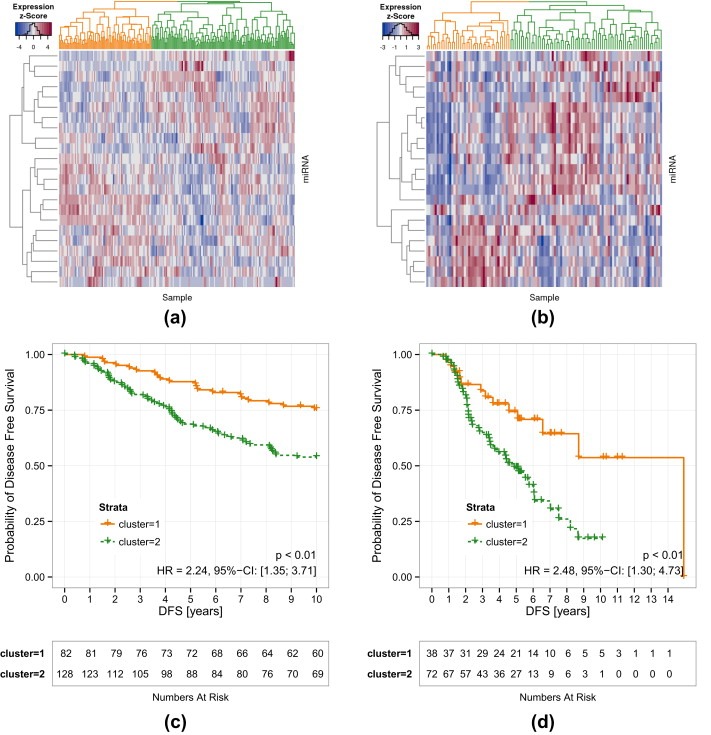
Increased prognostic value of the miR‐iTEMMΦ list. Cluster analyses based on miR‐iTEMMΦ miRNA expression values lead to two separate clusters (orange and green) in both breast cancer datasets: (a) Oxford, (b) Odense. DFS analyses are depicted in (c) and (d) respectively.
4. Discussion
In our study, miRNAs that were regulated in human TEM in vitro proved to be prognostic if transferred to datasets of breast cancer primaries. Analyzing miRNA expression in TEM can be challenging, as the regulatory effects are small and thus can be difficult to separate from the biological background. As a consequence, the induction of an M2 phenotype in TAM often relies on an induction through external stimuli such as IL‐4 or TGF‐β treatment (Graff et al., 2012; Yang et al., 2011) or the use of a murine model (Squadrito et al., 2012). Even under these stringent conditions there appears to be only little detectable regulation of miRNA expression. This can be attributed to increased basal miRNA abundance in macrophages relative to their monocytic predecessors. Consequently, polarizing conditions result in smaller fold‐changes in miRNA expression in macrophages than in activated monocytes (Graff et al., 2012). However, the induction of a tumor‐supportive phenotype through the co‐culture of tumor cells with macrophages is robust and has been validated previously (Hagemann et al., 2005; Pukrop et al., 2006). Our approach of combining the expression levels of miRNAs and their respective target genes greatly increased the power of detecting differentially‐expressed miRNAs (Artmann et al., 2012). This method mimics the physiologic functions of miRNAs, since their effects are orchestrated through the combined regulation of multiple target genes. On the whole, this allowed us to use a syngeneic human co‐culture system.
By testing for these aforementioned grouped effects, we identified 96 miRNAs, whose expression is significantly regulated in macrophages upon co‐culture with ER+ MCF‐7 cells. This approach differs from the work of other groups, which generate their signatures from whole tissue samples. The observation that our miR‐iTEM panel carries prognostic information even if applied to clinical data of ER+ breast cancer primaries highlights the validity of our procedure. Moreover, in a second independent dataset, the miR‐iTEM list's prediction error for DFS was even lower than that of the miRNA list obtained by Buffa et al. (Buffa et al., 2011). Interestingly, the predictive ability of the miR‐iTEM list unfolds its greatest effect in the long term. This mirrors the clinical course of ER positive breast cancers, which greatly benefit from adjuvant chemotherapy and endocrine therapy but exhibit a great risk for late recurrence (Esserman et al., 2011). This risk persists even under adjuvant endocrine therapy as in the two external cohorts studied in our manuscript (Cuzick et al., 2010).
Nonetheless, the miR‐iTEM list is still quite extensive – containing approximately one eighth of all the miRNAs tested. The information contained within these 96 miRNAs appears to depend on the context. This is highlighted by the fact that the stable feature selection results in two distinct panels of 13 and 6 miRNAs for the Oxford and Odense datasets, respectively. There is little overlap of the stably selected features between these two independent datasets– only miR‐941 is chosen in both lists. This miRNA is restricted to humans and has been shown to target hedgehog and insulin signaling. Both pathways have been implicated in cell differentiation and stemness (Hu et al., 2012), suggesting a possible involvement in cancer. Further studies of miR‐941 expression and more importantly localization in tumor samples by e.g. in‐situ hybridization are warranted. On a side note, this result greatly reinforces the use of a human system, since it would not have been detectable in a murine model.
Beside its low prediction error, the mi‐iTEM list also carries a substantial prognostic value as illustrated by its ability to identify patient cohorts which differ significantly in their DFS. In order to improve our miRNA list further, we examined its correlation to macrophage markers. While macrophage infiltration has already been demonstrated as being prognostic (Eiro et al., 2012; Leek et al., 1996; Medrek et al., 2012), there are only sparse data on miRNAs in breast‐cancer‐associated macrophages. Excitingly, most of the miRNAs chosen in the stable feature selection were also present in the miR‐iTEMMΦ list – once again miR‐941 was among the results. Another interesting finding is miR‐150, which has been linked so far to natural killer cell maturation (Bezman et al., 2011).
The correlation coefficients observed by us are rather low, which may be attributed to the complex nature of the tumor tissue. However, as our approach includes a priori knowledge, i.e. the miR‐iTEM list, we are confident that we identified TAM‐enriched rather than tumor‐cell‐intrinsic prognostic miRNAs. One may hypothesize that instead of mirroring macrophage infiltration, these miRNAs represent a specifically polarized subset of TAMs, i.e. of a pro‐metastatic vs. pro‐angiogenic phenotype (Laoui et al., 2011). This would fit our observation of both positive and negative correlations. Furthermore, the low values of the correlation coefficients could be accounted for by a directed, horizontal miRNA transfer between TAMs and breast cancer cells through microvesicles (Yang et al., 2011). This can result in a dynamic exchange of miRNAs between different cellular populations of the tumor tissue. Along these lines we found that miR‐210 which has previously been implicated in breast cancer cell proliferation, migration and invasion in vitro and is associated with poor patient outcome (Camps et al., 2008; Rothe et al., 2011; Volinia et al., 2012), was also significantly upregulated in TEM. It remains to be seen whether this is an actual increase of miR expression in the TEM or due to possible exchange between the different cell populations.
Overall, the narrowed‐down miR‐iTEMMΦ subset results in even more defined clusters with distinct DFS in two independent datasets.
5. Conclusions
In summary, prognostic miRNAs in ER+ tumors were identified through the integration of miRNA and mRNA expression data from breast cancer TEM. These miRNAs partially correlated with the expression levels of macrophage markers and have proved able to distinguish patient clusters with distinct DFS. Thus, even though it originates from in vitro TEM data, our approach can be reasonably applied to the study of whole tumor sample miRNA expression.
Authors' contributions
AB designed the experiments, analyzed data and wrote the manuscript. AL and SA analyzed the data. KM performed the co‐culture experiments. GS‐R provided miRNA arrays and tools. TP and CB revised the manuscript. TB guided the data analysis and revised the manuscript. FK participated in the research design, data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Supporting information
The following are the supplementary data related to this article:
Supplementary Table 1: miR‐iTEM list and its mapping to external breast cancer datasets. The miR‐iTEM list containing 96 miRNAs (corresponding miRBase accessions shown in column 2) was mapped to both external datasets. While a tick mark indicates successful mapping, discrepancies are listed. Columns 5 – 7 contain the q‐values for the differentially‐expressed miRNAs, mRNAs, and their combination in TEM in comparison with naive macrophages.
Supplementary Table 2: miRNA and target set directional fold changes. Color coded table illustrating the direction of the respective fold changes as determined by the formula: −log10 (min(pup;pdown)) × sign, where sign = −1; if pdown < pup or =1; if pup < pdown. The background color corresponds to value = −log10 (min(pup;pdown)) × sign.
Supplementary Figure 1:Validation of increased miR‐210 expression in TEM. Relative quantification of miR‐210 expression by qRT‐PCR verified increased expression in TEM (n = 5, mean fold change in TEM = 2.193, p = 0.01375 as determined by paired two‐sided Student's T‐test).
Supplementary Figure 2: Differential miR‐iTEMMΦ expression between patient clusters. Box plots (whiskers equal 1.5× IQR, dots represent outliers) illustrating the differential expression of miR‐iTEMMΦ members between the patient clusters shown in Figure 5. Color coding of the clusters and order of the miRNAs are identical to the heatmap.
Acknowledgments
The authors would like to thank Matthias Schulz and Meike Schaffrinski for their excellent technical assistance and express their gratitude to Andrew Entwistle for his critical review of the manuscript. This work was supported by the German Research Foundation (DFG) research group 942 and by the German Ministry of Education and Research (BMBF) projects BreastSys (0315396) and MetastaSys (0316173).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.07.023.
Bleckmann Annalen, Leha Andreas, Artmann Stephan, Menck Kerstin, Salinas-Riester Gabriela, Binder Claudia, Pukrop Tobias, Beissbarth Tim, Klemm Florian, (2015), Integrated miRNA and mRNA profiling of tumor-educated macrophages identifies prognostic subgroups in estrogen receptor-positive breast cancer, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.07.023.
Contributor Information
Annalen Bleckmann, Email: a.bleckmann@med.uni-goettingen.de.
Andreas Leha, Email: Andreas.Leha@med.uni-goettingen.de.
Stephan Artmann, Email: artmanns@student.ethz.ch.
Kerstin Menck, Email: Kerstin.Menck@med.uni-goettingen.de.
Gabriela Salinas-Riester, Email: gsalina@gwdg.de.
Claudia Binder, Email: claudia.binder@med.uni-goettingen.de.
Tobias Pukrop, Email: tobias.pukrop@med.uni-goettingen.de.
Tim Beissbarth, Email: Tim.Beissbarth@med.uni-goettingen.de.
Florian Klemm, Email: florian.klemm@med.uni-goettingen.de.
References
- Allinen, M. , Beroukhim, R. , Cai, L. , Brennan, C. , Lahti-Domenici, J. , Huang, H. , Porter, D. , Hu, M. , Chin, L. , Richardson, A. , Schnitt, S. , Sellers, W.R. , Polyak, K. , 2004. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 6, 17–32. 10.1016/j.ccr.2004.06.010 [DOI] [PubMed] [Google Scholar]
- Andersen, P.K. , Gill, R.D. , 1982. Cox's regression model for counting processes: a large sample study. Ann. Stat. 10, 1100–1120. 10.1214/aos/1176345976 [Google Scholar]
- Arranz, E.E. , Vara, J.A. , Gamez-Pozo, A. , Zamora, P. , 2012. Gene signatures in breast cancer: current and future uses. Transl. Oncol. 5, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artmann, S. , Jung, K. , Bleckmann, A. , Beissbarth, T. , 2012. Detection of simultaneous group effects in microRNA expression and related target gene sets. PLoS One. 7, e38365 10.1371/journal.pone.0038365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, T. , Edgar, R. , 2006. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods Enzym. 411, 352–369. 10.1016/S0076-6879(06)11019-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , Hochberg, Y. , 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B (Methodological). 57, 289–300. 10.2307/2346101 [Google Scholar]
- Bezman, N.A. , Chakraborty, T. , Bender, T. , Lanier, L.L. , 2011. miR-150 regulates the development of NK and iNKT cells. J. Exp. Med. 208, 2717–2731. 10.1084/jem.20111386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder, H. , Schumacher, M. , 2009. Incorporating pathway information into boosting estimation of high-dimensional risk prediction models. BMC Bioinform. 10, 18 10.1186/1471-2105-10-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier, G.W. , 1950. Verification of forecasts expressed in terms of probability. Mon. Weather Rev. 78, (1–3) 10.1175/1520-0493(1950)078<0001:VOFEIT>2.0.CO;2 [Google Scholar]
- Brock, G. , Pihur, V. , Datta, S. , Datta, S. , 2011. clValid, an R package for cluster validation. J. Stat. Softw. 25, 1–22. Brock Al March 2008 [Google Scholar]
- Buffa, F.M. , Camps, C. , Winchester, L. , Snell, C.E. , Gee, H.E. , Sheldon, H. , Taylor, M. , Harris, A.L. , Ragoussis, J. , 2011. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast Cancer. Cancer Res. 71, 5635–5645. 10.1158/0008-5472.CAN-11-0489 [DOI] [PubMed] [Google Scholar]
- Camps, C. , Buffa, F.M. , Colella, S. , Moore, J. , Sotiriou, C. , Sheldon, H. , Harris, A.L. , Gleadle, J.M. , Ragoussis, J. , 2008. hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin. Cancer Res. 14, 1340–1348. 10.1158/1078-0432.CCR-07-1755 [DOI] [PubMed] [Google Scholar]
- Cuzick, J. , Sestak, I. , Baum, M. , Buzdar, A. , Howell, A. , Dowsett, M. , Forbes, J.F. , 2010. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 11, 1135–1141. 10.1016/S1470-2045(10)70257-6 [DOI] [PubMed] [Google Scholar]
- Du, T. , Zamore, P.D. , 2005. microPrimer: the biogenesis and function of microRNA. Development. 132, 4645–4652. 10.1242/dev.02070 [DOI] [PubMed] [Google Scholar]
- Durinck, S. , Moreau, Y. , Kasprzyk, A. , Davis, S. , De Moor, B. , Brazma, A. , Huber, W. , 2005. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 21, 3439–3440. 10.1093/bioinformatics/bti525 [DOI] [PubMed] [Google Scholar]
- Eiro, N. , Pidal, I. , Fernandez-Garcia, B. , Junquera, S. , Lamelas, M.L. , del Casar, J.M. , Gonzalez, L.O. , Lopez-Muniz, A. , Vizoso, F.J. , 2012. Impact of CD68/(CD3 + CD20) ratio at the invasive front of primary tumors on distant metastasis development in breast cancer. PLoS One. 7, e52796 10.1371/journal.pone.0052796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esserman, L.J. , Moore, D.H. , Tsing, P.J. , Chu, P.W. , Yau, C. , Ozanne, E. , Chung, R.E. , Tandon, V.J. , Park, J.W. , Baehner, F.L. , Kreps, S. , Tutt, A.N.J. , Gillett, C.E. , Benz, C.C. , 2011. Biologic markers determine both the risk and the timing of recurrence in breast cancer. Breast Cancer Res. Treat. 129, 607–616. 10.1007/s10549-011-1564-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak, G. , Bertos, N. , Pepin, F. , Sadekova, S. , Souleimanova, M. , Zhao, H. , Chen, H. , Omeroglu, G. , Meterissian, S. , Omeroglu, A. , Hallett, M. , Park, M. , 2008. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 14, 518–527. 10.1038/nm1764 [DOI] [PubMed] [Google Scholar]
- Graff, J.W. , Dickson, A.M. , Clay, G. , McCaffrey, A.P. , Wilson, M.E. , 2012. Identifying functional microRNAs in macrophages with polarized phenotypes. J. Biol. Chem. 287, 21816–21825. 10.1074/jbc.M111.327031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S. , Saini, H.K. , van Dongen, S. , Enright, A.J. , 2008. miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158. 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, H. , Ingolia, N.T. , Weissman, J.S. , Bartel, D.P. , 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 466, 835–840. 10.1038/nature09267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann, T. , Wilson, J. , Kulbe, H. , Li, N.F. , Leinster, D.A. , Charles, K. , Klemm, F. , Pukrop, T. , Binder, C. , Balkwill, F.R. , 2005. Macrophages induce invasiveness of epithelial cancer cells via NF-κB and JNK. J. Immunol. 175, 1197–1205. [DOI] [PubMed] [Google Scholar]
- He, M. , Xu, Z. , Ding, T. , Kuang, D.M. , Zheng, L. , 2009. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol. Immunol. 6, 343–352. 10.1038/cmi.2009.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H.Y. , He, L. , Fominykh, K. , Yan, Z. , Guo, S. , Zhang, X. , Taylor, M.S. , Tang, L. , Li, J. , Liu, J. , Wang, W. , Yu, H. , Khaitovich, P. , 2012. Evolution of the human-specific microRNA miR-941. Nat. Commun. 3, 1145 10.1038/ncomms2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoui, D. , Movahedi, K. , Van Overmeire, E. , Van den Bossche, J. , Schouppe, E. , Mommer, C. , Nikolaou, A. , Morias, Y. , De Baetselier, P. , Van Ginderachter, J.A. , 2011. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int. J. Dev. Biol. 55, 861–867. 10.1387/ijdb.113371dl [DOI] [PubMed] [Google Scholar]
- Leek, R.D. , Lewis, C.E. , Whitehouse, R. , Greenall, M. , Clarke, J. , Harris, A.L. , 1996. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 56, 4625–4629. [PubMed] [Google Scholar]
- Lyng, M.B. , Laenkholm, A.V. , Sokilde, R. , Gravgaard, K.H. , Litman, T. , Ditzel, H.J. , 2012. Global microRNA expression profiling of high-risk ER+ breast cancers from patients receiving adjuvant tamoxifen mono-therapy: a DBCG study. PLoS One. 7, e36170 10.1371/journal.pone.0036170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski, I.J. , Ritchie, M.E. , Phipson, B. , Corbin, J. , Pakusch, M. , Ebert, A. , Busslinger, M. , Koseki, H. , Hu, Y. , Smyth, G.K. , Alexander, W.S. , Hilton, D.J. , Blewitt, M.E. , 2010. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood. 116, 731–739. 10.1182/blood-2009-12-260760 [DOI] [PubMed] [Google Scholar]
- Marot, G. , Mayer, C.D. , 2009. Sequential analysis for microarray data based on sensitivity and meta-analysis. Stat. Appl. Genet. Mol. Biol. 8, 10.2202/1544-6115.1368 Article 3 [DOI] [PubMed] [Google Scholar]
- Medrek, C. , Ponten, F. , Jirstrom, K. , Leandersson, K. , 2012. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 12, 306 10.1186/1471-2407-12-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoloso, M.S. , Spizzo, R. , Shimizu, M. , Rossi, S. , Calin, G.A. , 2009. MicroRNAs–the micro steering wheel of tumour metastases. Nat. Rev. Cancer. 9, 293–302. 10.1038/nrc2619 [DOI] [PubMed] [Google Scholar]
- Ojalvo, L.S. , Whittaker, C.A. , Condeelis, J.S. , Pollard, J.W. , 2010. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J. Immunol. 184, 702–712. 10.4049/jimmunol.0902360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, J.S. , Mullins, M. , Cheang, M.C. , Leung, S. , Voduc, D. , Vickery, T. , Davies, S. , Fauron, C. , He, X. , Hu, Z. , Quackenbush, J.F. , Stijleman, I.J. , Palazzo, J. , Marron, J.S. , Nobel, A.B. , Mardis, E. , Nielsen, T.O. , Ellis, M.J. , Perou, C.M. , Bernard, P.S. , 2009. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167. 10.1200/JCO.2008.18.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou, C.M. , Sorlie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Rees, C.A. , Pollack, J.R. , Ross, D.T. , Johnsen, H. , Akslen, L.A. , Fluge, O. , Pergamenschikov, A. , Williams, C. , Zhu, S.X. , Lonning, P.E. , Borresen-Dale, A.L. , Brown, P.O. , Botstein, D. , 2000. Molecular portraits of human breast tumours. Nature. 406, 747–752. 10.1038/35021093 [DOI] [PubMed] [Google Scholar]
- Pihur, V. , Datta, S. , 2009. RankAggreg, an R package for weighted rank aggregation. BMC Bioinform. 10, 62 10.1186/1471-2105-10-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukrop, T. , Klemm, F. , Hagemann, T. , Gradl, D. , Schulz, M. , Siemes, S. , Trümper, L. , Binder, C. , 2006. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl. Acad. Sci.U. S. A. 103, 5454–5459. 10.1073/pnas.0509703103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, B.Z. , Pollard, J.W. , 2010. Macrophage diversity enhances tumor progression and metastasis. Cell. 141, 39–51. 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail, D.F. , Joyce, J.A. , 2013. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2013. R: A Language and Environment for Statistical Computing R Foundation for Statistical Computing; Vienna, Austria: [Google Scholar]
- Rothe, F. , Ignatiadis, M. , Chaboteaux, C. , Haibe-Kains, B. , Kheddoumi, N. , Majjaj, S. , Badran, B. , Fayyad-Kazan, H. , Desmedt, C. , Harris, A.L. , Piccart, M. , Sotiriou, C. , 2011. Global microRNA expression profiling identifies MiR-210 associated with tumor proliferation, invasion and poor clinical outcome in breast cancer. PLoS One. 6, e20980 10.1371/journal.pone.0020980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, C.F. , Gschwantler-Kaulich, D. , Fink-Retter, A. , Haas, C. , Hudelist, G. , Czerwenka, K. , Kubista, E. , 2008. Differential gene expression profile in breast cancer-derived stromal fibroblasts. Breast Cancer Res. Treat. 110, 273–281. 10.1007/s10549-007-9725-2 [DOI] [PubMed] [Google Scholar]
- Smid, M. , Wang, Y. , Zhang, Y. , Sieuwerts, A.M. , Yu, J. , Klijn, J.G. , Foekens, J.A. , Martens, J.W. , 2008. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 68, 3108–3114. 10.1158/0008-5472.CAN-07-5644 [DOI] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , Hastie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Thorsen, T. , Quist, H. , Matese, J.C. , Brown, P.O. , Botstein, D. , Lonning, P.E. , Borresen-Dale, A.L. , 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U. S. A. 98, 10869–10874. 10.1073/pnas.191367098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squadrito, M.L. , Etzrodt, M. , De Palma, M. , Pittet, M.J. , 2013. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 34, 350–359. 10.1016/j.it.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squadrito, M.L. , Pucci, F. , Magri, L. , Moi, D. , Gilfillan, G.D. , Ranghetti, A. , Casazza, A. , Mazzone, M. , Lyle, R. , Naldini, L. , De Palma, M. , 2012. miR-511-3p modulates genetic programs of tumor-associated macrophages. Cell Rep. 1, 141–154. 10.1016/j.celrep.2011.12.005 [DOI] [PubMed] [Google Scholar]
- Stouffer, S.A. , 1949. The American Soldier, Studies in Social Psychology in World War II Princeton University Press; Princeton: [Google Scholar]
- Tian, W. , Chen, J. , He, H. , Deng, Y. , 2013. MicroRNAs and drug resistance of breast cancer: basic evidence and clinical applications. Clin. Transl. Oncol. 15, 335–342. 10.1007/s12094-012-0929-5 [DOI] [PubMed] [Google Scholar]
- Volinia, S. , Galasso, M. , Sana, M.E. , Wise, T.F. , Palatini, J. , Huebner, K. , Croce, C.M. , 2012. Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc. Natl. Acad. Sci. U. S. A. 109, 3024–3029. 10.1073/pnas.1200010109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiel, M.A. van de , Berkhof, J. , Wieringen, W.N. van , 2009. Testing the prediction error difference between 2 predictors. Biostatistics. 10, 550–560. 10.1093/biostatistics/kxp011 [DOI] [PubMed] [Google Scholar]
- Yang, M. , Chen, J. , Su, F. , Yu, B. , Lin, L. , Liu, Y. , Huang, J.D. , Song, E. , 2011. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer. 10, 117 10.1186/1476-4598-10-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabuawala, T. , Taffany, D.A. , Sharma, S.M. , Merchant, A. , Adair, B. , Srinivasan, R. , Rosol, T.J. , Fernandez, S. , Huang, K. , Leone, G. , Ostrowski, M.C. , 2010. An ets2-driven transcriptional program in tumor-associated macrophages promotes tumor metastasis. Cancer Res. 70, 1323–1333. 10.1158/0008-5472.CAN-09-1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary Table 1: miR‐iTEM list and its mapping to external breast cancer datasets. The miR‐iTEM list containing 96 miRNAs (corresponding miRBase accessions shown in column 2) was mapped to both external datasets. While a tick mark indicates successful mapping, discrepancies are listed. Columns 5 – 7 contain the q‐values for the differentially‐expressed miRNAs, mRNAs, and their combination in TEM in comparison with naive macrophages.
Supplementary Table 2: miRNA and target set directional fold changes. Color coded table illustrating the direction of the respective fold changes as determined by the formula: −log10 (min(pup;pdown)) × sign, where sign = −1; if pdown < pup or =1; if pup < pdown. The background color corresponds to value = −log10 (min(pup;pdown)) × sign.
Supplementary Figure 1:Validation of increased miR‐210 expression in TEM. Relative quantification of miR‐210 expression by qRT‐PCR verified increased expression in TEM (n = 5, mean fold change in TEM = 2.193, p = 0.01375 as determined by paired two‐sided Student's T‐test).
Supplementary Figure 2: Differential miR‐iTEMMΦ expression between patient clusters. Box plots (whiskers equal 1.5× IQR, dots represent outliers) illustrating the differential expression of miR‐iTEMMΦ members between the patient clusters shown in Figure 5. Color coding of the clusters and order of the miRNAs are identical to the heatmap.