

Abstract
Identification of the primary tumor site in patients with metastatic cancer is clinically important, but remains a challenge. Hence, efforts have been made towards establishing new diagnostic tools. Molecular profiling is a promising diagnostic approach, but tissue heterogeneity and inadequacy may negatively affect the accuracy and usability of molecular classifiers. We have developed and validated a microRNA‐based classifier, which predicts the primary tumor site of liver biopsies, containing a limited number of tumor cells. Concurrently we explored the influence of surrounding normal tissue on classification. MicroRNA profiling was performed using quantitative Real‐Time PCR on formalin‐fixed paraffin‐embedded samples. 278 primary tumors and liver metastases, representing nine primary tumor classes, as well as normal liver samples were used as a training set. A statistical model was applied to adjust for normal liver tissue contamination. Performance was estimated by cross‐validation, followed by independent validation on 55 liver core biopsies with a tumor content as low as 10%. A microRNA classifier developed, using the statistical contamination model, showed an overall classification accuracy of 74.5% upon independent validation. Two‐thirds of the samples were classified with high‐confidence, with an accuracy of 92% on high‐confidence predictions. A classifier trained without adjusting for liver tissue contamination, showed a classification accuracy of 38.2%. Our results indicate that surrounding normal tissue from the biopsy site may critically influence molecular classification. A significant improvement in classification accuracy was obtained when the influence of normal tissue was limited by application of a statistical contamination model.
Keywords: microRNA, Classification, Liver biopsy, Metastases, Surrounding tissue, Tissue contamination
Highlights
Metastatic core biopsies contain a mixture of malignant‐ and non‐malignant cells.
We explore the impact of non‐malignant cells on tissue of origin classification.
Non‐malignant cells significantly hamper correct tissue of origin classification.
A statistical model adjusts for the signal provided by non‐malignant cells.
Applying this model to a microRNA tissue of origin test improves classification.
Abbreviations
- PRIM classifier
primary tumor based classifier
- CCM classifier
contamination model based classifier
- CCM + CB classifier
contamination model and liver core biopsy based classifier
1. Introduction
Current cancer treatment strategies are based on the anatomical site of the primary tumor. Therefore, a correct diagnosis of the primary tumor site remains an essential first step in disease management. Since more specific treatment regimens have emerged for many solid tumors, correct primary tumor site identification has become increasingly important.
Despite improvements in imaging techniques and the use of immunohistochemical (IHC) markers, cancer patients presenting with metastatic disease at the time of diagnosis still represent a diagnostic challenge and in 3–5% the primary tumor site remains undetectable (Pavlidis et al., 2012). As a result, these patients may be subjected to a time‐consuming and expensive diagnostic work‐up, resulting in treatment delay or even a suboptimal or incorrect treatment strategy.
In recent years, effort has been made towards establishing new supplementary diagnostic tools for primary tumor site identification. Molecular profiling is a promising diagnostic approach, which has the potential to provide an objective classification of uncertain or unknown metastatic cancers and render the diagnostic work‐up of cancer patients more time‐ and cost‐effective.
For the majority of patients with metastatic cancer, classification of the primary tumor site relies on formalin‐fixed and paraffin‐embedded (FFPE) core biopsies from metastatic lesions. Standard specimen sampling methods result in heterogeneous specimens, consisting of varying amounts of malignant cells and normal tissue (Cheng et al., 2013). A molecular classifier for primary tumor site identification in patients with metastatic disease must therefore be compatible with FFPE biopsy specimens, representing metastatic tissue with limited tumor content. Furthermore, the possible influence on classification by normal tissue contamination must be considered. Essentially, the classifier performance must be assessed on representative samples for which the classifier is intended to perform.
Several molecular classifiers, based on either messenger RNA (mRNA) or microRNA (miRNA) analysis, have been developed for primary tumor site identification. These classifiers show promising cross‐validation and independent validation results. However, validation is often performed on a sample set predominantly constituted by primary tumors (Ma et al., 2006; Meiri et al., 2012; Pillai et al., 2011; Ramaswamy et al., 2001; Su et al., 2001; Talantov et al., 2006). Primary tumors and their corresponding metastases may exhibit significant molecular differences due to altered biology or diversity in specimen sampling, which may influence classification accuracy. Such an influence may potentially be overlooked if metastatic samples represent a small part of the total validation set. Additionally, it is not well established to which extend contamination by non‐malignant tissue in the specimens affects molecular classification.
The primary objective of this study was to develop a classifier able to identify the primary tumor site of FFPE liver core biopsies. Additionally, the classifier should be easy to apply in the daily clinic. Hence, the classifier should be able to perform on limited tumor tissue without the need for prior microdissection. We used miRNA, which is a class of small (21–24 nucleotides) non‐coding RNA molecules (Finnegan et al., 2013), since these are highly stable in FFPE tissue (Hall et al., 2012). The biopsy site was limited to a single organ in order to explore the influence of surrounding (“contaminating”) non‐malignant tissue on primary tumor site classification. A statistical contamination model was incorporated to allow classification of core biopsies even in the presence of normal liver tissue (Vincent et al., 2014). Furthermore we explored if the miRNA profile of metastases provides additional information necessary for correct classification, when compared to primary tumors.
2. Materials and methods
2.1. Clinical samples
Tissue samples from 338 patients, corresponding to one of the following ten predefined assay classes, were obtained from tissue archives at the Department of Pathology, Copenhagen University Hospital, Denmark: Lung cancer, breast cancer, gastric/cardia cancer (GC), colorectal cancer (CRC), bladder cancer, pancreatic cancer, hepatocellular carcinoma (HCC), cholangiocarcinoma (CCA), squamous cell cancers (SCC) of different origin, and non‐malignant (“normal”) liver tissue (Table 1).
Table 1.
Selected characteristics of samples included in classifier training and validation. The tissue of origin, histology and number (no.) of samples used for classifier training (TR) and independent validation (V) are listed. Normal liver was subdivided into reactive and cirrhotic liver, but was regarded as one class. Squamous cell carcinoma was regarded as one class of mixed population. Resection, primary tumor and normal liver resections; Biopsy, liver core biopsies consisting of liver metastases, primary liver cancer and normal liver.
| Tissue of origin | Histology | Resection no. (TR) | Biopsy no. (TR) | Biopsy no. (V) |
|---|---|---|---|---|
| Bladder | Urothelial carcinoma | 17 | 2 | 5 |
| Breast | Invasive ductal, lobular, medullar | 17 | 7 | 5 |
| Billiary tract | Adenocarcinoma | 20 | 4 | 5 |
| Colorectal | Adenocarcinoma, mucinous adenocarcinoma | 20 | 12 | 5 |
| Gastric/cardia | Adenocarcinoma, signet ring cell carcinoma | 18 | 12 | 5 |
| Liver | Hepatocellular carcinoma | 17 | 3 | 5 |
| Normal liver | Reactive | 20 | 7 | 5 |
| Normal liver | Cirrhotic | 17 | 8 | 5 |
| Lung | Adenocarcinoma, Mixed type, Large cell | 17 | 2 | 5 |
| Pancreas | Adenocarcinoma | 20 | 10 | 5 |
| Cervix, Lung, Anal, Esophagus, Head and Neck | Squamous cell carcinoma | 16 | 12 | 5 |
| Total | 199 | 79 | 55 |
The study was conducted according to national guidelines.
The selection of primary tumor classes in the present study was made to encompass: (i) primary tumors that often metastasize to the liver, (ii) primary tumors difficult to diagnose with conventional diagnostic methods and (iii) common primary tumors for which an effective systemic treatment is available, making a correct tumor classification clinically important.
When selecting samples, the following issues were considered: (i) a single confident reference diagnosis was required. The reference diagnosis was established based on the original pathology report, clinical data, radiological findings or, when available, autopsy reports. Primary tumors were assigned a differentiation grade, according to international guidelines. In addition, all samples were independently reviewed by an expert pathologist to confirm the reference diagnosis. Metastatic samples with several primary tumor sites suggested by histopathology were included, if one of those suggestions was in agreement with the clinical and radiological findings; (ii) the training set should include the most common histological subtypes and represent a varied spectrum of dedifferentiation; (iii) each patient could only be represented by one sample, hence primary tumor samples and metastatic samples were unmatched.
Samples were formalin‐fixed and paraffin‐embedded (FFPE) tissue specimens archived in the time period 2000‐2012. The sample set consisted of 199 surgical resections (162 primary tumors and 37 normal liver samples) and 134 liver core biopsies (109 primary liver cancers and liver metastases of known origin, and 25 normal liver samples). Normal liver samples, defined as liver samples without tumor tissue, were obtained from large surgical liver resections for colorectal metastases or from explanted livers. These samples were subdivided into: (i) liver samples containing mild reactive changes due to the presence of a tumor in the proximity and (ii) cirrhotic liver. Cirrhosis was included in order to differentiate non‐neoplastic fibrosis from the desmoplastic stromal reaction of metastatic lesions. Characteristics of the samples are shown in Table 1.
All samples were assigned an estimated tumor percentage by an expert pathologist. The percentage of tumor tissue in resected samples (primary tumors) was defined as the relative amount of tumor cells. In core biopsies, the tumor tissue content was defined as the relative area of combined tumor cells and desmoplastic stroma. The tumor percentage was estimated from a hematoxylin and eosin‐stained section.
From each surgical resection, one 10‐μm section was cut. To obtain tumor specific miRNA expression profiles, primary tumor samples were microdissected using the Arcturus XT Microdissection System (Applied Biosystems, Foster City, CA) to ensure a tumor cell content of ≥60%. The influence of non‐malignant cells was limited by excluding samples with ≥50% fibrosis, hemorrhage or necrosis (arbitrary cut‐off).
Two sections of 5‐μm were cut from each liver core biopsy. No microdissection was performed on these samples. The only requirement was a minimum of 10% tumor tissue without further limitations, regarding fibrosis, hemorrhage or necrosis.
2.2. RNA extraction
Total RNA was extracted from FFPE tissue using a combination of ReCover All Total Nucleic Acid Isolation Kit (Ambion, Austin, Tx) and RNAqueous Micro Kit (Ambion). Briefly, the microdissected sections were deparaffinized by first adding 1 ml 100% xylene and subsequently 1 ml 100% ethanol. The later RNA extraction steps were similar for all dissected and non‐dissected samples. The tissue was digested using 100 μl digestion buffer and 4 μl ProteinaseK (ReCover All) at 50 °C for 15 min and 80 °C for 15 min according to the manufacturer's instructions. RNA was subsequently purified on columns and eluted in 15 μl elution solution (RNAqueous) according to the manufacturer's protocol. Total RNA yield and quality was evaluated using Nanodrop ND‐1000 spectrophotometer (NanoDrop Technologies, Wilmington).
2.3. miRNA quantitative real‐time PCR profiling
miRNA profiling was performed using TaqMan low density array (TLDA) cards, human MicroRNA array A (Applied Biosystems) according to the manufacturer's instructions. A detailed description of material and methods used for miRNA profiling is provided as Supplementary Data.
Successful analysis was performed for 333 samples (98.5%).
The PCR data has been deposited at the Gene Expression Omnibus (GEO), accession number GSE51429.
2.4. Study design
Samples were initially split into a training set, consisting of the 199 surgical resections and 79 core biopsies (2‐12 biopsies in each class), and a validation set consisting of 55 liver core biopsies (5 samples in each class).
Following miRNA expression profiling, a stepwise development of three different classifiers was performed, as illustrated in Figure 1. First, a primary tumor based classifier (PRIM classifier) was developed. This classifier was trained exclusively on normalized miRNA expression data from primary tumor‐ and normal liver resections. Second, a contamination model based classifier (CCM classifier) was developed, using the same sample set as for PRIM classifier training. However, for the CCM classifier a contamination model was applied before classifier training, to adjust for normal liver contamination. The statistical contamination model (Vincent et al., 2014) uses the original training samples to produce a set of computationally constructed samples mimicking liver core biopsies. The computationally constructed samples constituted the CCM classifier training set. Third, a contamination model and liver core biopsy based classifier (CCM + CB classifier) was developed. This classifier was trained on the same sample set as the CCM classifier, together with the 79 liver core biopsies.
Figure 1.
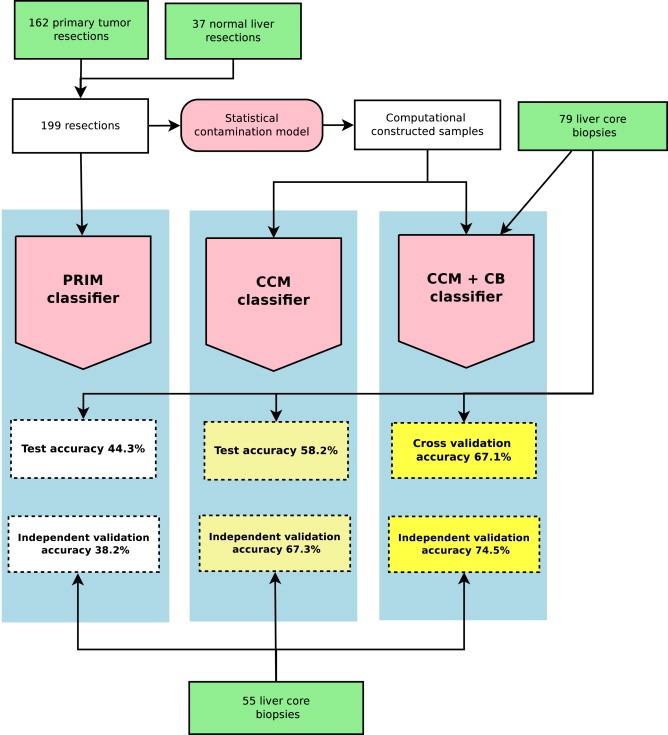
Development and validation of three different miRNA classifiers for primary tumor site classification of liver core biopsies. The PRIM classifier was trained on 162 primary tumor resections and 37 normal liver resections. Primary tumor resections represented the following 9 tumor classes: bladder, breast, biliary tract, colorectal, gastric/cardia, liver, pancreatic, lung and a class of mixed squamous tumors. Each class was represented by 16–20 samples. Normal liver resections consisted of 20 reactive liver resections and 17 cirrhotic liver resections. The CCM classifier was trained by applying a statistical contamination model to the same 162 primary tumor resections and 37 normal liver resections before classifier training, resulting in a set of computationally constructed samples mimicking liver core biopsies. The CCM + CB classifier was trained on the same computationally constructed samples together with 79 liver core biopsies. Core biopsies consisted of 57 metastatic samples, 7 primary liver cancer samples and 15 normal liver samples. An initial cross‐validation/test was performed for each of the three classifiers, using the 79 liver core biopsies. Finally, an independent validation was performed for each of the three classifiers using 55 liver core biopsies, consisting of 35 metastatic samples, 10 primary liver samples and 10 normal liver samples.
2.5. Data preparation, sample construction and normalization
TaqMan array controls and miRNAs not expressed in primary tumor and liver samples were removed.
Using the 199 training samples (primary tumors and normal liver resections), a new data set was constructed, by applying a statistical contamination model (Vincent et al., 2014). The constructed samples were a mixture of miRNA expressions from primary tumor and normal liver samples according to:
where α denotes the tumor percentage. The tumor percentage was taken to follow a beta distribution with first shape parameter equal to four and second shape parameter equal to three. Due to non‐linearity of the PCR amplification, the model was not applied on the observed scale but on a suitable transformed scale (Vincent et al., 2014). The contamination model uses random sampling with replacement of the primary tumor and the normal liver samples in the training data set, as well as random sampling of α to construct miRNA expression profiles mimicking liver core biopsies.
All samples were normalized by centering and scaling the individual samples to mean 0 and variance 1.
2.6. Multinomial logistic regression model training and validation
The multinomial group lasso method (Vincent et al., 2014) was used to train three different multinomial logistic regression models (Hosmer et al., 2013) for classification, reflecting the three different training data sets. In our set‐up, the multinomial logistic regression model is a model of the probability of the 10 assay classes given the observed 377 miRNA expression measurements from each sample. The log‐probability of each class is, up to a constant, a weighted sum of the miRNA expressions. The model provides an estimate of the class probability and not just a classification. As a consequence of the multinomial group lasso method, the weights for some miRNAs will be 0 for all classes. The method thus automatically selects those miRNAs that are most relevant for classification. Standardization of miRNA expressions across samples was done internally in the training algorithm to avoid that difference in scale could influence the miRNA selection.
The multinomial group lasso method produces a sequence of 100 models, each consisting of different combinations of miRNAs. To select a final model, an additional model selection procedure was performed. For the PRIM and CCM classifiers, both exclusively trained on primary tumor and normal liver resections, the 79 liver core biopsies were used as a test set. The model with the best classification performance, measured by negative log‐likelihood, was selected as the final classifier. For the CCM + CB classifier, the 79 liver core biopsies were included in the training set and the negative log‐likelihood was estimated by cross‐validation.
An unbiased assessment of the PRIM and CCM classification accuracies was obtained by cross‐validating the entire training and model selection procedure. Since the 79 liver core biopsies were included in the CCM + CB training data, the classification accuracy of the CCM + CB classifier was obtained by nested cross‐validation. To strengthen the performance assessment of each of the three classifiers (PRIM, CCM and CCM + CB); an independent validation was performed, using the 55 liver core biopsy validation set.
3. Results
3.1. MicroRNA classifier based exclusively on primary tumors misclassifies core biopsies
To investigate whether a miRNA profile obtained exclusively from primary tumor and normal liver resections was able to classify the primary tumor site of liver core biopsies, predominantly consisting of metastases, a classifier based on 55 miRNA expression profiles (PRIM classifier) was developed only using the 199 primary tumor and normal liver resections as a training set. The PRIM classifier showed a 90% overall accuracy upon 10‐fold cross validation (Supplementary Figure S1). When applied to the 79‐core biopsy test set, the accuracy dropped to 44.3% (Table 2) with a pronounced difference in classification accuracy across the different assay classes. The PRIM classifier performed well on core biopsies consisting of normal liver, but generally poor on metastases from non‐liver derived primary tumors. Liver metastases from colorectal cancer constituted an exception, with 67% being classified correctly. A complete list of classifier predictions is given in Supplementary Table S1. Approximately 40% (17/43) of the misclassified samples were classified as normal liver (reactive liver or cirrhosis) and 35/40 misclassified metastases from non‐liver derived primary tumors were classified as either primary liver cancer or normal liver. This strongly indicated that contamination with normal liver in core biopsies impeded correct classification. A principal component plot (Supplementary Figure S2) illustrates how core biopsies independent of class clustered together with liver derived samples.
Table 2.
Performance of the PRIM classifier, CCM classifier and CCM+CB classifier on the 79‐core biopsy sample set. Each assay class was represented by 2–15 samples, as marked in brackets. The number of correctly classified samples according to the reference diagnosis is listed for each assay class. The sample set constituted a test set for the PRIM and CCM classifier. For the CCM + CB classifier, performance was estimated by eight‐fold cross validation. Squamous, squamous cell carcinoma (mixed population). Normal liver, 8 cirrhotic and 7 reactive liver samples. CCA, cholangiocarcinoma; CRC, colorectal carcinoma; GC, gastric or cardia carcinoma; HCC, hepatocellular carcinoma.
| Reference site | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bladder (2) | Breast (7) | CCA (4) | CRC (12) | GC (12) | HCC (3) | Lung (2) | Pancreas (10) | Squamous cell (12) | Normal liver (15) | Overall accuracy | ||
| Classifier | PRIM | 0 | 2 | 4 | 8 | 2 | 1 | 1 | 1 | 2 | 14 | 44.3% |
| CCM | 0 | 0 | 2 | 9 | 8 | 1 | 1 | 3 | 8 | 14 | 58.2% | |
| CCM + CB | 1 | 4 | 4 | 8 | 8 | 1 | 1 | 4 | 9 | 13 | 67.1% | |
3.2. Application of a contamination model for classifier training improves classification of core biopsies
To improve classification of liver core biopsies, we applied a statistical contamination model as part of classifier development. Based on the contamination model, computationally constructed samples, mimicking liver core biopsies, were developed. These samples were constructed for each assay class, only using miRNA profiles of normal liver and primary tumor resections from the 199‐sample training set. By exchanging the original primary tumors with the computationally constructed samples as a training set, a classifier consisting of 104 miRNAs (CCM classifier) was developed.
To test the performance of the CCM classifier on liver core biopsies, we applied the 79‐liver core biopsy test set. The accuracy showed an improvement across most assay classes, with a pronounced effect on non‐liver derived malignancies, resulting in an overall accuracy of 58.2% (Table 2). The improved classification accuracy was largely due to a reduction in samples being misclassified as normal liver (8/32) and fewer metastases from non‐liver derived primary tumors being misclassified as derived from the liver (11/30) (Supplementary Table S1).
3.3. Metastases may feature important information for correct classification
miRNA signatures may differ between primary tumors and metastases, not only due to normal tissue contamination but also due to underlying biological differences. Such biological differences will obviously not be present in the computationally constructed samples. Therefore, to encompass a potential molecular difference between primary tumors and metastases, we used the same computationally constructed training samples as described for the CCM classifier together with the 79 liver core biopsies. From this combined training set, we developed a classifier consisting of 116 miRNAs (CCM + CB classifier). To estimate the performance of this CCM + CB classifier, 8‐fold cross‐validation was performed, which showed 67.1% overall accuracy (Table 2 and Supplementary Figure S3). Further, an independent validation using 55 liver core biopsies was performed, demonstrating an overall accuracy of 74.5%. The lowest performance upon independent validation was obtained for cholangiocarcinomas (CCA), while an intermediate performance was achieved for bladder‐, gastric‐, lung and squamous cell cancers (SCC). Figure 2 shows the independent validation results of the CCM + CB classifier illustrated by a confusion matrix. CCA were generally misclassified as normal liver (reactive or cirrhotic), while the remaining errors occurred randomly. We observed no difference in classification accuracy due to primary tumor site, sample age or tumor percentage, as presented in Supplementary Table 1.
Figure 2.
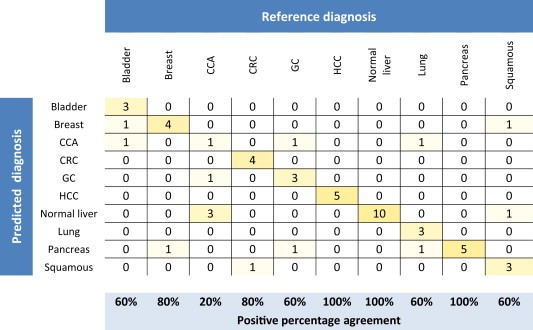
Confusion matrix showing CCM + CB classifier predictions upon the independent validation set consisting of 55 liver core biopsies. Each row and column corresponds to one of the assay classes included in the classifier. Columns indicate classes according to the reference diagnosis; rows indicate the diagnosis predicted by the CCM + CB classifier. Numbers on the diagonal indicate cases for which the predicted diagnosis matched the reference diagnosis, whereas off‐diagonal numbers were in disagreement and counted as test errors. The positive percentage agreement for each class was calculated. Squamous, Squamous cell carcinoma (mixed population); CCA, cholangiocarcinoma; CRC, colorectal carcinoma; GC, gastric or cardia carcinoma; HCC, hepatocellular carcinoma.
For comparison, we applied the independent validation set to the PRIM classifier and the CCM classifier and obtained overall accuracies of 38.2% and 67.3%, respectively. A comparison of validation results between the three classifiers is shown in Table 3.
Table 3.
Results of the independent validation of the PRIM classifier, CCM classifier and CCM+CB classifier. The performance of each of the three classifiers on the independent validation set consisting of 55 liver core biopsies is shown. Each class was represented by 5 samples, except the Normal liver class, which consisted of 5 reactive liver samples and 5 cirrhotic liver samples. The number of correctly classified samples according to the reference diagnosis is listed for each assay class. Squamous, squamous cell carcinoma (mixed population); CCA, cholangiocarcinoma; CRC, colorectal carcinoma; GC, gastric or cardia carcinoma; HCC, hepatocellular carcinoma.
| Reference site | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bladder (5) | Breast (5) | CCA (5) | CRC (5) | GC (5) | HCC (5) | Lung (5) | Pancreas (5) | Squamous cell (5) | Normal liver (10) | Overall accuracy | ||
| Classifier | PRIM | 1 | 1 | 1 | 3 | 1 | 3 | 0 | 0 | 2 | 9 | 38.2% |
| CCM | 3 | 4 | 3 | 4 | 2 | 4 | 3 | 0 | 4 | 10 | 67.3% | |
| CCM + CB | 3 | 4 | 1 | 4 | 3 | 5 | 3 | 5 | 3 | 10 | 74.5% | |
An important feature of a clinical applicable classifier is the ability to deliver a single high‐confidence prediction. By proposing two or more differential diagnoses, uncertainty and subjectivity may be imposed. Due to these considerations, the 79 training biopsies were used to establish a threshold for high‐confidence predictions. Based on this threshold, a prediction was defined as high‐confidence if the class probability was equal to or larger than 0.6. When applied to the independent validation set, 65% of the samples were high‐confidence predictions, with 92% being classified according to the reference diagnosis. The positive percentage agreement reached 100% for all classes except CCA, GC and SCC (Table 4).
Table 4.
High confidence predictions of the CCM + CB classifier. The number of high confidence predictions (estimated class probability ≥ 0.6) and the number of high confidence predictions in agreement with the reference diagnosis are listed for the 55 liver core biopsies constituting the independent validation set. The overall positive percentage agreement and the positive percentage agreement for each class was calculated. Squamous, squamous cell carcinoma (mixed population); CCA, cholangiocarcinoma; CRC, colorectal carcinoma; GC, gastric or cardia carcinoma; HCC, hepatocellular carcinoma.
| Reference site | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bladder (5) | Breast (5) | CCA (5) | CRC (5) | GC (5) | HCC (5) | Lung (5) | Pancreas (5) | Squamous cell (5) | Normal liver (10) | Total (55) | |
| High confidence predictions | 3 | 3 | 2 | 3 | 3 | 5 | 2 | 1 | 4 | 10 | 36 |
| Agreement with reference diagnosis | 3 | 3 | 1 | 3 | 2 | 5 | 2 | 1 | 3 | 10 | 33 |
| 100% | 100% | 50% | 100% | 67% | 100% | 100% | 100% | 75% | 100% | 92% | |
| Positive percentage agreement | |||||||||||
The miRNAs included in the 55‐miRNA PRIM classifier, the 104‐miRNA CCM classifier and the 116‐miRNA CCM + CB classifier are listed in Supplementary Table S2. Forty‐two miRNAs were included in all three classifiers, whereas 5 miRNAs were represented only in the PRIM classifier, including miR‐122, which is known to be specifically expressed in liver tissue. Supplementary Figure S4 illustrates how two selected miRNAs (miR 122 and miR 196‐b) were expressed in primary tumor and normal liver resections, in samples constructed using the contamination model and in liver core biopsies, respectively.
4. Discussion
We have developed and validated a microRNA (miRNA) classifier, designed as a supplementary diagnostic tool to histopathological evaluation and imaging, during the diagnostic work‐up of patients suspected of a malignant liver tumor (either metastases or primary liver cancer). The classifier is trained on primary tumors, liver metastases and normal liver tissue and consists of expression profiles from 116 miRNAs. The classifier performs on formalin‐fixed and paraffin‐embedded (FFPE) liver core biopsies with limited amounts of tumor tissue and varying amounts of normal liver tissue. It distinguishes between eight primary tumor classes, squamous cell cancer (mixed population) and normal liver tissue with an overall accuracy of 67.1% upon cross‐validation and 74.5% upon an independent validation. Sixty‐five % of the validation samples in the present study were classified with high confidence, and classification accuracy of those samples reached 92% (Table 4).
To mimic the daily diagnostic routine, we validated the classifier using small sections of liver core biopsies with as little as 10% tumor tissue and refrained from microdissection. As opposed to previously reported classifiers, we limited the application to a single biopsy site. This allowed us to study the impact of surrounding normal tissue on primary tumor site classification, avoiding classification bias caused by biopsy site and reducing the number of validation samples needed. The liver was chosen because: (i) it is a common site for metastatic disease and the most common single site of metastatic involvement in patients with carcinoma of unknown primary site (Pavlidis et al., 2012); (ii) it represents the most common metastatic site for gastrointestinal (GI) cancers (Hess et al., 2006) and (iii) the liver is easily accessible for a biopsy.
Tumor samples contain varying amounts of malignant cells, stromal cells and surrounding (contaminating) normal tissue from the biopsy/resection site. The influence of surrounding normal tissue on molecular classification is not clear, although a potential systematic classification bias, caused by normal tissue, has been reported (Elloumi et al., 2011; Staub et al., 2010). Most previously developed diagnostic classifiers require a high tumor content (≥50% tumor) (Meiri et al., 2012; Pillai et al., 2011; Ferracin et al., 2011; Kerr et al., 2012; Rosenfeld et al., 2008; Rosenwald et al., 2010) and use microdissection for tumor cell enrichment, prior to gene expression analysis. Although microdissection reduces the surrounding normal tissue, it also holds several disadvantages. Most importantly, microdissection is time consuming, costly and it may not always be possible due to a relatively small number of tumor cells located dispersedly in the core biopsy (Cheng et al., 2013). Secondly, microdissection precludes use of potentially important information hidden in the stromal cells surrounding the neoplastic cells.
To investigate the influence of normal liver tissue contamination, we constructed a miRNA classifier (PRIM classifier) trained on primary tumor and normal liver resections. Although a high cross‐validation accuracy of 90% was achieved, the PRIM classifier showed a disappointing classification accuracy of 38.2% on the independent validation set consisting of liver core biopsies. The low classification accuracy was predominantly caused by samples being misclassified as normal liver or primary liver malignancies. By adjusting for liver contamination, the accuracy improved significantly to 67.3% upon independent validation. Hence, our results indicate that a miRNA signature is sustained in metastases compared to corresponding primary tumors, but contamination with surrounding normal tissue must be considered a potential cause of error in molecular primary tumor site classification based on miRNA.
Increasing evidence support a key role for miRNAs in cancer cell invasion, migration and metastasis (Baranwal et al., 2010), and studies have reported altered miRNA signatures in metastases compared to matched primary tumors (Chen et al., 2012; Gravgaard et al., 2012). By including metastatic liver core biopsies in the training set, we observed an increase in classification accuracy from 67.3% to 74.5% upon independent validation (Table 3), with a notable effect on pancreatic cancer classification. This improvement could be due to different genetic information in metastases, compared to primary tumors.
Metastases from the GI tract, especially pancreatic and gastric cancers, are usually difficult to distinguish from one another and from cholangiocarcinomas by histopathology alone (Oien 2009; Park et al., 2007). As illustrated in Supplementary Figure S3, our classification demonstrates a similar tendency, especially upon cross‐validation. This tendency is less clear from the independent validation results (Figure 2). Still, a significant proportion of GI tract cancers were correctly classified from their miRNA signature.
Cholangiocarcinomas constituted an exception, since only 1 out of 5 validation samples was correctly classified. The poor classification accuracy was predominantly due to misclassification as normal liver.
We included a class of squamous cell cancers (SCC) defined by histology and not by site of origin. A correct classification was obtained for 9/12 SCC samples in the test set and 3/5 SCC samples in the validation set. Among the 17 SCC samples included, only one sample was misclassified according to the primary tumor site, since a test sample representing SCC of the esophagus was misclassified as GC. The two misclassified validation samples represented a metastatic cervix cancer classified as breast and a metastatic esophagus cancer classified as cirrhotic liver. Indeed, the later sample constituted a substantial amount of cirrhosis. A complete list of classifier predictions is given in Supplementary Table S1.
In recent years, several molecular tissue of origin classifiers have been developed, of which two are based on miRNAs (Meiri et al., 2012; Ferracin et al., 2011). The second generation tissue classifier developed by Rosetta Genomics uses 64 miRNAs to distinguish between 42 tumor types (Meiri et al., 2012). This classifier requires samples with a minimum of 50% tumor tissue and it uses two algorithms to predict the tissue of origin. With this classifier, the authors reached an overall accuracy of 85% upon independent validation (Meiri et al., 2012). However, this overall accuracy reflects the union of the predictions made by the two algorithms, and with metastatic samples constituting 30% of the validation set, a directly comparison to results obtained by our classifier is difficult.
Ferracin et al. (Ferracin et al., 2011) identified a 47 miRNA signature, which predicts the primary tumor site of metastases belonging to 10 different tumor classes. Independent validation performed on 45 microdissected metastases, reached an overall accuracy of 73.3%. The validation set included samples from 9 tumor classes, with a preponderance of metastases originating from the lower gastrointestinal tract. A second validation, performed on a publically available data set (Rosenfeld et al., 2008), resulted in an overall classification accuracy of 69%.
We have previously shown, how the ANOVA + PAM classifier reported in the study by Ferracin et al. (Ferracin et al., 2011), like the multinomial group lasso classifier applied in our study, was unable to generalize from primary tumor samples to non‐microdissected liver core biopsies with a heterogeneous tumor content distribution. Applying the contamination model improved classification accuracy for both classifiers (Vincent et al., 2014).
Performance comparison across different classifiers must be interpreted with caution, due to different configurations of assay classes. Obviously, differences in number and sample distribution among the included assay classes affect the performance estimate, but other important considerations need to be highlighted. First, the performance of most classifiers is often estimated on a combination of primary tumor samples and metastatic samples (Ma et al., 2006; Meiri et al., 2012; Pillai et al., 2011; Kerr et al., 2012; Rosenfeld et al., 2008; Rosenwald et al., 2010), with metastases contributing merely 1/3 of the total validation set. In the present study, we showed that primary tumor site classification from primary tumor samples reached a 90% overall accuracy but only 38.2% accuracy was achieved when the classifier was applied to an independent set of liver core biopsies, predominantly constituting metastases. Second, most classifiers are validated on a combination of resections and biopsies. Since the relative amount of normal surrounding tissue often is higher in biopsies than in resections, the influence of normal tissue contamination may be overlooked by this approach. The importance of validating a molecular classifier on representative samples on which it is intended to perform, remains essential in order to avoid potential overestimation of classifier performance.
Histopathology remains the cornerstone in primary tumor site identification, but metastatic disease constitutes a persistent diagnostic challenge (Oien et al., 2012). The efficacy of IHC analysis in determining the primary site of metastatic tumors is difficult, since few controlled, blinded studies are available. Though, a meta‐analysis based on a small number of older series, reported a mean accuracy of 65.6% (Anderson et al., 2010). Increasing evidence support superior overall accuracy of gene expression profiling compared to IHC in primary tumor site identification of metastases (Handorf et al., 2013; Weiss et al., 2013). Hence, molecular classifiers have the potential to improve primary tumor site classification in patients with metastatic disease when conventional diagnostics draw a blank, when IHC provides inadequate classification (none or multiple diagnoses) or when histopathology and imaging offers discordant diagnostic suggestions.
The perhaps most important use of molecular classifiers is to define the tumor of origin in patients with carcinoma of unknown primary site (CUP) (Hainsworth et al., 2014). The feasibility of primary tumor site classification by gene expression profiling has been shown for CUP (Greco et al., 2010, 2013 Jun 5, 2010 Jan 13, 2011 Apr 29) and recently a possible benefit of site‐specific treatment in CUP patients directed by gene expression profiling was suggested (Hainsworth et al., 2013).
The aim of this study was to develop and validate a clinical applicable diagnostic tool, which predicts the primary tumor site of liver core biopsies. The classifier was developed with CUP in mind, although not restricted to this group of patients.
With an overall accuracy of 74.5% and an accuracy of 92% on high‐confidence predictions upon independent validation, our classifier compares favorably to what is reported by histopathology alone (Anderson et al., 2010) and the overall accuracy is in line with first prediction accuracies obtained in former studies (Meiri et al., 2012; Ferracin et al., 2011; Rosenfeld et al., 2008; Rosenwald et al., 2010). Notably, we performed a validation on small sections of liver biopsies, with limited tumor content, predominantly representing metastatic tumors. A prospectively conducted study is planned to explore if the miRNA classifier improves the diagnostic accuracy, reduces the diagnostic work‐up time and the number of investigations needed.
In conclusion, we have developed a miRNA classifier, which is able to determine the primary tumor site of FFPE liver core biopsies. Based on our data set, the signal provided by the surrounding normal liver hampered correct classification significantly. We show that application of a statistical model which adjusts for the signal provided by normal liver tissue is essential for obtaining a valid classification. Notably, the statistical contamination model ensured that classification could be established on samples containing limited tumor tissue, making prior microdissection redundant.
Funding
This work was supported by The Danish National Advanced Technology Foundation, The Danish Cancer Research Foundation, The Copenhagen University Hospital, The Preben and Anna Simonsens Foundation, the Svend HA Schrøder and Ketty L Larsen Schrøder foundation and The Beckett Foundation.
Conflicts of interest
No conflicts to disclose by any of the authors.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
The authors wish to thank Ewa Futoma‐Kazmierczak, Mette Hedegaard Moldaschl and Jonas Vikeså for laboratory and technical support and Snjòlaug Nielsdottir, MD for pathology assistance.
1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.07.015.
Perell Katharina, Vincent Martin, Vainer Ben, Petersen Bodil Laub, Federspiel Birgitte, Møller Anne Kirstine, Madsen Mette, Hansen Niels Richard, Friis-Hansen Lennart, Nielsen Finn Cilius, Daugaard Gedske, (2015), Development and validation of a microRNA based diagnostic assay for primary tumor site classification of liver core biopsies, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.07.015.
References
- Anderson, G.G. , Weiss, L.M. , 2010 Jan. Determining tissue of origin for metastatic cancers: meta-analysis and literature review of immunohistochemistry performance. Appl. Immunohistochem. Mol. Morphol. 18, (1) 3–8. [DOI] [PubMed] [Google Scholar]
- Baranwal, S. , Alahari, S.K. , 2010 Mar 15. miRNA control of tumor cell invasion and metastasis. Int. J. Cancer. 126, (6) 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Tang, Z. , Sun, Y. , Zhang, Y. , Wang, X. , Shen, Z. , Liu, F. , Qin, X. , 2012 Feb. miRNA expression profile in primary gastric cancers and paired lymph node metastases indicates that miR-10a plays a role in metastasis from primary gastric cancer to lymph nodes. Exp. Ther. Med. 3, (2) 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, L. , Zhang, S. , MacLennan, G.T. , Williamson, S.R. , Davidson, D.D. , Wang, M. , Jones, T.D. , Lopez-Beltran, A. , Montironi, R. , 2013 Jan. Laser-assisted microdissection in translational research: theory, technical considerations, and future applications. Appl. Immunohistochem. Mol. Morphol. 21, (1) 31–47. [DOI] [PubMed] [Google Scholar]
- Elloumi, F. , Hu, Z. , Li, Y. , Parker, J.S. , Gulley, M.L. , Amos, K.D. , Troester, M.A. , 2011. Systematic bias in genomic classification due to contaminating non-neoplastic tissue in breast tumor samples. BMC Med. Genomics. 4, 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferracin, M. , Pedriali, M. , Veronese, A. , Zagatti, B. , Gafa, R. , Magri, E. , Lunardi, M. , Munerato, G. , Querzoli, G. , Maestri, I. , Ulazzi, L. , Nenci, I. , 2011 Apr 13. MicroRNA profiling for the identification of cancers with unknown primary tissue-of-origin. J. Pathol. 225, (1) 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan, E.F. , Pasquinelli, A.E. , 2013 Jan. MicroRNA biogenesis: regulating the regulators. Crit. Rev. Biochem. Mol. Biol. 48, (1) 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravgaard, K.H. , Lyng, M.B. , Laenkholm, A.V. , Sokilde, R. , Nielsen, B.S. , Litman, T. , Ditzel, H.J. , 2012 Jul. The miRNA-200 family and miRNA-9 exhibit differential expression in primary versus corresponding metastatic tissue in breast cancer. Breast Cancer Res. Treat. 134, (1) 207–217. [DOI] [PubMed] [Google Scholar]
- Greco, F.A. , Spigel, D.R. , Yardley, D.A. , Erlander, M.G. , Ma, X.J. , Hainsworth, J.D. , 2010. Molecular profiling in unknown primary cancer: accuracy of tissue of origin prediction. Oncologist. 15, (5) 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco, F.A. , Lennington, W.J. , Spigel, D.R. , Hainsworth, J.D. , 2013 Jun 5. Molecular profiling diagnosis in unknown primary cancer: accuracy and ability to complement standard pathology. J. Natl. Cancer Inst. 105, (11) 782–790. [DOI] [PubMed] [Google Scholar]
- Hainsworth, J.D. , Rubin, M.S. , Spigel, D.R. , Boccia, R.V. , Raby, S. , Quinn, R. , Greco, F.A. , 2013 Jan 10. Molecular gene expression profiling to predict the tissue of origin and direct site-specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah Cannon Research Institute. J. Clin. Oncol. 31, (2) 217–223. [DOI] [PubMed] [Google Scholar]
- Hainsworth, J.D. , Greco, F.A. , 2014 Apr. Gene expression profiling in patients with carcinoma of unknown primary site: from translational research to standard of care. Virchows Arch. 464, (4) 393–402. [DOI] [PubMed] [Google Scholar]
- Hall, J.S. , Taylor, J. , Valentine, H.R. , Irlam, J.J. , Eustace, A. , Hoskin, P.J. , Miller, C.J. , West, C.M. , 2012 Aug 7. Enhanced stability of microRNA expression facilitates classification of FFPE tumour samples exhibiting near total mRNA degradation. Br. J. Cancer. 107, (4) 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handorf, C.R. , Kulkarni, A. , Grenert, J.P. , Weiss, L.M. , Rogers, W.M. , Kim, O.S. , Monzon, F.A. , Halks-Miller, M. , Anderson, G.G. , Walker, M.G. , Pillai, R. , Henner, W.D. , 2013 Jul. A multicenter study directly comparing the diagnostic accuracy of gene expression profiling and immunohistochemistry for primary site identification in metastatic tumors. Am. J. Surg. Pathol. 37, (7) 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, K.R. , Varadhachary, G.R. , Taylor, S.H. , Wei, W. , Raber, M.N. , Lenzi, R. , Abbruzzese, J.L. , 2006 Apr 1. Metastatic patterns in adenocarcinoma. Cancer. 106, (7) 1624–1633. [DOI] [PubMed] [Google Scholar]
- Hosmer, D.W. , Lemeshow, S. , Sturdivant, R.X. , 2013. Applied Logistic Regression Wiley; [Google Scholar]
- Kerr, S.E. , Schnabel, C.A. , Sullivan, P.S. , Zhang, Y. , Singh, V. , Carey, B. , Erlander, M.G. , Highsmith, W.E. , Dry, S.M. , Brachtel, E.F. , 2012 Jul 15. Multisite validation study to determine performance characteristics of a 92-gene molecular cancer classifier. Clin. Cancer Res. 18, (14) 3952–3960. [DOI] [PubMed] [Google Scholar]
- Ma, X.J. , Patel, R. , Wang, X. , Salunga, R. , Murage, J. , Desai, R. , Tuggle, J.T. , Wang, W. , Chu, S. , Stecker, K. , Raja, R. , Robin, H. , 2006 Apr. Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Arch. Pathol. Lab Med. 130, (4) 465–473. [DOI] [PubMed] [Google Scholar]
- Meiri, E. , Mueller, W.C. , Rosenwald, S. , Zepeniuk, M. , Klinke, E. , Edmonston, T.B. , Werner, M. , Lass, U. , Barshack, I. , Feinmesser, M. , Huszar, M. , Fogt, F. , 2012. A second-generation microRNA-based assay for diagnosing tumor tissue origin. Oncologist. 17, (6) 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzon, F.A. , Medeiros, F. , Lyons-Weiler, M. , Henner, W.D. , 2010 Jan 13. Identification of tissue of origin in carcinoma of unknown primary with a microarray-based gene expression test. Diagn. Pathol. 5, (3) 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oien, K.A. , 2009 Feb. Pathologic evaluation of unknown primary cancer. Semin. Oncol. 36, (1) 8–37. [DOI] [PubMed] [Google Scholar]
- Oien, K.A. , Dennis, J.L. , 2012 Sep. Diagnostic work-up of carcinoma of unknown primary: from immunohistochemistry to molecular profiling. Ann. Oncol. 23, (Suppl. 10) x271–x277. [DOI] [PubMed] [Google Scholar]
- Park, S.Y. , Kim, B.H. , Kim, J.H. , Lee, S. , Kang, G.H. , 2007 Oct. Panels of immunohistochemical markers help determine primary sites of metastatic adenocarcinoma. Arch. Pathol. Lab Med. 131, (10) 1561–1567. [DOI] [PubMed] [Google Scholar]
- Pavlidis, N. , Pentheroudakis, G. , 2012 Apr 14. Cancer of unknown primary site. Lancet. 379, (9824) 1428–1435. [DOI] [PubMed] [Google Scholar]
- Pillai, R. , Deeter, R. , Rigl, C.T. , Nystrom, J.S. , Miller, M.H. , Buturovic, L. , Henner, W.D. , 2011 Jan. Validation and reproducibility of a microarray-based gene expression test for tumor identification in formalin-fixed, paraffin-embedded specimens. J. Mol. Diagn. 13, (1) 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy, S. , Tamayo, P. , Rifkin, R. , Mukherjee, S. , Yeang, C.H. , Angelo, M. , Ladd, C. , Reich, M. , Latulippe, E. , Mesirov, J.P. , Poggio, T. , Gerald, W. , 2001 Dec 18. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. U S A. 98, (26) 15149–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld, N. , Aharonov, R. , Meiri, E. , Rosenwald, S. , Spector, Y. , Zepeniuk, M. , Benjamin, H. , Shabes, N. , Tabak, S. , Levy, A. , Lebanony, D. , Goren, Y. , 2008 Apr. MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol. 26, (4) 462–469. [DOI] [PubMed] [Google Scholar]
- Rosenwald, S. , Gilad, S. , Benjamin, S. , Lebanony, D. , Dromi, N. , Faerman, A. , Benjamin, H. , Tamir, R. , Ezagouri, M. , Goren, E. , Barshack, I. , Nass, D. , 2010 Jun. Validation of a microRNA-based qRT-PCR test for accurate identification of tumor tissue origin. Mod. Pathol. 23, (6) 814–823. [DOI] [PubMed] [Google Scholar]
- Staub, E. , Buhr, H.J. , Grone, J. , 2010 May 31. Predicting the site of origin of tumors by a gene expression signature derived from normal tissues. Oncogene. 29, (31) 4485–4492. [DOI] [PubMed] [Google Scholar]
- Su, A.I. , Welsh, J.B. , Sapinoso, L.M. , Kern, S.G. , Dimitrov, P. , Lapp, H. , Schultz, P.G. , Powell, S.M. , Moskaluk, C.A. , Frierson, H.F. , Hampton, G.M. , 2001 Oct 15. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 61, (20) 7388–7393. [PubMed] [Google Scholar]
- Talantov, D. , Baden, J. , Jatkoe, T. , Hahn, K. , Yu, J. , Rajpurohit, Y. , Jiang, Y. , Choi, C. , Ross, J.S. , Atkins, D. , Wang, Y. , Mazumder, A. , 2006 Jul. A quantitative reverse transcriptase-polymerase chain reaction assay to identify metastatic carcinoma tissue of origin. J. Mol. Diagn. 8, (3) 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadhachary, G.R. , Spector, Y. , Abbruzzese, J.L. , Rosenwald, S. , Wang, H. , Aharonov, R. , Carlson, H.R. , Cohen, D. , Karanth, S. , Macinskas, J. , Lenzi, R. , Chajut, A. , 2011 Apr 29. Prospective gene signature study using microRNA to identify the tissue of origin in patients with carcinoma of unknown primary (CUP). Clin. Cancer Res. 17, (12) 4063–4070. [DOI] [PubMed] [Google Scholar]
- Vincent, M. , Hansen, N.R. , 2014 Mar. Sparse group lasso and high dimensional multinomial classification. Comput. Stat. Data Anal. 71, (0) 771–786. [Google Scholar]
- Vincent, M. , Perell, K. , Nielsen, F.C. , Daugaard, G. , Hansen, N.R. , 2014 May. Modeling tissue contamination to improve molecular identification. Bioinformatics. 30, (10) 1417–1423. [DOI] [PubMed] [Google Scholar]
- Weiss, L.M. , Chu, P. , Schroeder, B.E. , Singh, V. , Zhang, Y. , Erlander, M.G. , Schnabel, C.A. , 2013 Mar. Blinded comparator study of immunohistochemical analysis versus a 92-gene cancer classifier in the diagnosis of the primary site in metastatic tumors. J. Mol. Diagn. 15, (2) 263–269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data