

Abstract
Obesity, owing to adiposity, is associated with increased risk and development of various cancers, and linked to their rapid growth as well as progression. Although a few studies have attempted to understand the relationship between obesity and melanoma, the consequences of controlling body weight by reducing adiposity on cancer progression is not well understood. By employing animal models of obesity, we report that controlling obesity either by orlistat treatment or by restricting caloric intake significantly slows down melanoma progression. The diminished tumor progression was correlated with decreased fat mass (adiposity) in obese mice. Obesity associated factors contributing to tumor progression were decreased in the experimental groups compared to respective controls. In tumors, protein levels of fatty acid synthase (FASN), caveolin (Cav)‐1 and pAkt, which are tumor promoting molecules implicated in melanoma growth under obese state, were decreased. In addition, increased necrosis and reduction in angiogenesis as well as proliferative markers PCNA and cyclin D1 were observed in tumors of the orlistat treated and/or calorically restricted obese mice. We observed that growth of melanoma cells cultured in conditioned medium (CM) from orlistat‐treated adipocytes was reduced. Adipokines (leptin and resistin), via activating Akt and modulation of FASN as well as Cav‐1 respectively, enhanced melanoma cell growth and proliferation. Together, we demonstrate that controlling body weight reduces adipose mass thereby diminishing melanoma progression. Therefore, strategic means of controlling obesity by reduced caloric diet or with antiobesity drugs treatment may render obesity‐promoted tumor progression in check and prolong survival of patients.
Keywords: Obesity, Melanoma, Orlistat, Diet, Adipokines, Adiposity
Abbreviations
- FASN
fatty acid synthase
- Cav‐1
caveolin‐1
- CM
conditioned medium
- HFD
high fat diet
- ND
normal diet
1. Introduction
Obesity and overweight, resulting from excessive adiposity is a serious public health problem worldwide with imminent clinical complications and economic burden (Fontaine and Barofsky, 2001; Kopelman, 2000). Epidemiological studies and meta analyses support a possible link between obesity and risk of breast, colon, pancreatic and cervical cancers as well as melanomas (Calle et al., 2003; Calle and Kaaks, 2004). Approximately 20% of all cancers are attributed to obesity and overweight causing late‐stage disease, poor prognosis, cancer aggravation and impairment in chemotherapy by imposing chemoresistance (Wolin et al., 2010). Adiposity deleteriously alters the production of proliferative, inflammatory, anti‐inflammatory factors which influence the development and growth of cancer by releasing several factors/hormones collectively termed as adipokines (Park et al., 2011; Roberts et al., 2010). The precise mechanisms of tumor progression under obesity are still not clear, and studies deciphering precise effects of altered serum profile on cancer progression are scarce. In order to counteract the tumor promoting effect of obesity and adipokines, there is growing interest in exploring the possibility of whether weight loss therapies could reduce cancer‐related deaths (McTiernan, 2008; Sirin and Kolonin, 2013).
Adipose tissue plays an important role in tumorigenesis, invasion and metastasis. In obesity, secretory profile of adipokines from adipose tissue is altered leading to development of oxidative stress, pro‐inflammatory and proliferative microenvironment (Khandekar et al., 2011). Adipokines exert their effects through receptors or membrane‐associated molecules and activate various cellular signaling pathways (van Kruijsdijk et al., 2009). Cancer cells express receptors for most of the adipokines (Balistreri et al., 2010; Paz‐Filho et al., 2011). These adipokines activate multiple signaling pathways including PI3K/Akt, MAPK and JAK/STAT. Activated status of these pathways eventually supports cancer cell growth and proliferation by modulating genes or proteins involved in tumor progression. Leptin and resistin are the major adipokines associated with obesity (Steppan et al., 2001; Vendrell et al., 2004), and their role in growth and proliferation has been extensively explored in breast and prostate cancers (Ando and Catalano, 2011; Kim et al., 2011). However, the involvement of these adipokines in melanoma is not well understood.
Melanoma is one of the most aggressive and obesity‐promoted human malignancies. It is a fatal form of skin cancer that occurs in the proximity of subcutaneous adipose tissue. Being resistant to many anticancer drugs, it accounts for about 75% of skin cancer‐related deaths worldwide (Jerant et al., 2000). Therefore, consideration of life style factors or metabolic diseases becomes integral to the management of various aspects of tumorigenesis and tumor progression. In pharmacological front, orlistat, an FDA approved antiobesity drug, is a relatively tolerable and safe agent used to induce weight loss in obese individuals (Richelsen et al., 2007). It primarily acts by preventing absorption of dietary lipids through reversible inhibition of gastrointestinal lipases. At cellular level, orlistat has also been shown to irreversibly inhibit fatty acid synthase (FASN), a key enzyme in de novo synthesis of fatty acids (Kridel et al., 2004). Orlistat, at higher dosage, has been reported to exhibit antitumor properties as cancer cells rely on availability of fatty acids and related molecules for their survival (Menendez and Lupu, 2007; Seguin et al., 2012). However, the equivalent anticancer dose of orlistat in humans, due to its severe adverse side effects, could be clinically unfeasible.
Although a number of antiobesity drugs are available, diet‐control interventions still remain to be the preferred line of therapy for effective management of obesity. Also, the role of dietary and nutritional factors towards cancer risk has been recently reported by many research groups (Kampman et al., 2012; Prieto‐Hontoria et al., 2011; Rock et al., 2012). However, the comprehensive investigations on the impact of effective management of obesity on tumor progression are lacking. Therefore, we hypothesized that controlling obesity would be an appropriate approach in minimizing the risk of obesity‐promoted cancer progression. In this study, we investigated the implications of therapeutic and dietary interventions for controlling obesity on the progression of melanoma. The underlying molecular events and role of specific adipokines were explored using appropriate in vitro and in vivo models. We demonstrate that controlling obesity is associated with normalization in levels of obesity‐associated factors which parallels with reduction in melanoma progression and it may possibly be true for other cancer types too.
2. Materials and methods
2.1. Experimental animals and diets
Mice were procured from Experimental Animal Facility (EAF) at National Centre for Cell Science (NCCS), Pune, India. High fat diet (24% fat) was purchased from Provimi Animal Nutrition Pvt. Ltd., Bangalore, India, and normal diet (5% fat) was obtained from Amrut Laboratory, Pune, India. Diet‐induced obesity was developed in the mice by feeding with high fat diet as described previously (Pandey et al., 2012). The composition of the diets used is shown in Supplementary Table 1. Briefly, male C57BL/6J or female NOD/SCID mice (6–8 weeks old) were divided into normal diet (ND) and high fat diet (HFD) group. ND group (N = 12 NOD/SCID, N = 40 C57BL/6J) was fed with normal diet and HFD group (N = 12 NOD/SCID, N = 40 C57BL/6J) was fed with high fat diet supplemented with ground nut and dried coconut for 6 months. NOD/SCID mice were fed with sterilized high fat diet, ground nut and coconut. Body weight and serum chemistry profile were measured monthly to verify obesity‐associated changes. Water and food were provided ad libitum to all the mice. All animal experiments were carried out as per the requirement and guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India, and after obtaining permission of the Institutional Animal Ethics Committee (IAEC).
2.2. Cells and culture conditions
Murine melanoma cells B16F10, human melanoma cells A375 and murine preadipocyte cells 3T3‐L1 were procured from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained at our in‐house cell repository at National Centre for Cell Science, Pune, India. Cells were routinely cultured in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum (Hyclone, UT, USA or Gibco, NY, USA), penicillin (100 U/ml) and streptomycin (100 μg/ml) (Invitrogen Life Technologies, CA, USA) and maintained at 37 °C in a 5% CO2 humidified incubator (Thermo Fisher Scientific, OH, USA).
2.3. Serum biochemical analysis
Blood glucose level was measured using rapid glucose analyzer (Accu‐Chek Sensor Comfort, Roche Diagnostics, Mannheim, Germany) by collecting through an approved tail cap method. For serum collection, blood was collected by orbital sinus puncture and centrifuged at 6000 RPM at room temperature. Triglycerides (TG), cholesterol, LDLc and free fatty acids levels in fresh serum were estimated using colorimetric kits (Spinreact, Girona, Germany) as per the manufacturer's instructions. Insulin, leptin and adiponectin levels in the serum was estimated by mouse specific respective ELISA kits as described (Pandey et al., 2012). Resistin, IL‐6 and TNF‐α levels in the serum were detected by indirect ELISA. Briefly, standard curve was prepared with different concentrations of respective recombinant proteins. ELISA plates (Becton Dickenson, NJ, USA) were coated with serum samples collected from mice. Blocking was done using 2% BSA in phosphate buffered saline (PBS, pH = 7.4). After washing with PBS, samples were incubated with primary antibodies for resistin (1:100), IL‐6 (1:100) and TNF‐α (1:100) (Santa Cruz Biotechnology, CA, USA) specified for ELISA. Following washing, samples were incubated with HRP‐conjugated secondary antibodies (1:200) (Santa Cruz Biotechnology, CA, USA). ABTS [2,2′‐azinobis‐(3‐ethylbenzothiazoline‐6‐sulfonic acid)] (Sigma–Aldrich, MO, USA) was used as a chromogenic substrate for HRP. After developing the color, absorbance was recorded at 405 nm.
2.4. Orlistat treatment and/or diet shifting in HFD mice, tumor challenge and follow‐up
To control obesity in HFD mice, we employed two strategies: (i) treatment with orlistat (ii) shifting of mice from high fat diet to normal diet. The detailed experimental plan is illustrated in Figure 1A. Obese mice were administered orally with orlistat (10 mg/kg on every alternate day) purchased from Enzo Life Sciences, NY, USA and/or shifted from high fat diet to normal diet. HFD C57BL/6J or NOD/SCID mice treated with vehicle or orlistat were termed as HFD‐HFD Ctrl and HFD‐HFD Orli (N = 6–10 per each group) respectively whereas HFD C57BL/6J mice shifted to normal diet and treated with vehicle or orlistat were grouped as HFD‐ND Ctrl and HFD‐ND Orli (N = 10 per each group) respectively. After fifteen days, these mice were injected subcutaneously (sc) with B16F10 (2 × 105) or A375 (5 × 105) cells in 100 μl of PBS and monitored daily for the presence of palpable tumors and dimensions were recorded on alternate days. Tumor volume was calculated using formula: 0.52 × length × width2, and was followed up throughout the study. At the end of the experiment, mice were sacrificed by CO2 euthanasia. Excised tumors volume and weight were recorded and the samples were immediately preserved at −80 °C until further use. For histological studies, organ and tissue samples were preserved in 10% paraformaldehyde.
Figure 1.
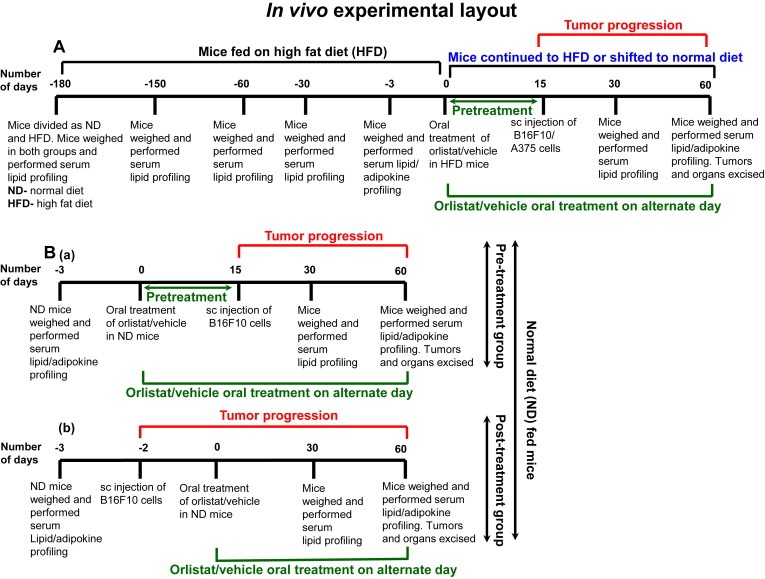
Experimental strategy to study the impact of controlling obesity on melanoma progression in mice. (A) Impact of controlling diet‐induced obesity on melanoma progression in HFD C57BL/6J or NOD/SCID mice. (B) Effect of (a) pre‐treatment, or (b) post treatment with orlistat, at antiobesity dose (10 mg/kg, oral), on melanoma progression in ND C57BL/6J mice.
2.5. Orlistat treatment in ND mice, tumor challenge and follow‐up
To evaluate whether oral treatment with orlistat directly influences melanoma progression in C57BL/6J mice fed with normal diet, these mice were divided in to two major groups (N = 10 per each group): (i) ND mice treated with vehicle or orlistat 15 days prior to injecting melanoma cells were termed as ND‐Pre Ctrl and ND‐Pre Orli respectively; (ii) ND mice treated with vehicle or orlistat after 2 days of injecting melanoma cells were grouped as ND‐Post Ctrl and ND‐Post Orli respectively. Orlistat treatment was continued and tumor progression was followed up in these mice throughout the study as mentioned above.
2.6. Immunoblotting
Melanoma tumor samples or cells were washed 3–5 times with ice‐cold PBS and lysed in ice‐cold RIPA lysis buffer as described previously (Pandey et al., 2012). Briefly, the samples were centrifuged at 12000 RPM for 40 min and clear supernatants were stored at −80 °C. Protein concentrations were determined by Coomassie Plus Protein Assay Reagent (Thermo Scientific, IL, USA). Equal amounts of protein samples (50–100 μg) were resolved on 8–10% SDS‐polyacrylamide gel and then transferred onto PVDF membrane (Millipore, MA, USA). The membranes were blocked and further probed with antibodies against Cav‐1 (1:1000), FASN (1:1000), pAkt (Ser‐473) (1:1000), total Akt (1:1000), PCNA (1:1000), cyclin D1 (1:1000), active caspase‐3 (1:1000), active caspase‐7 (1:1000) and β‐Tubulin (1:1000) (Santa Cruz Biotechnology, CA, USA). After washing, the membranes were incubated with HRP‐conjugated secondary antibodies (1:2000) and blots were developed using luminescence detection reagents (Santa Cruz Biotechnology, CA, USA).
2.7. Histological analysis
Fine sections (4 μm) from tissue/organ samples (fixed in 10% paraformaldehyde) were prepared and stained with hematoxylin and eosin (H&E) (Safeline Histology Centre, Pune, India). Stained sections of tissue/organ samples were analyzed in a blinded manner by pathologists at KEM Hospital, Pune, India. The images were taken using Olympus camera (DP‐71) attached to microscope (Olympus, Tokyo, Japan).
2.8. Immunohistochemistry
Paraffin‐embedded sections (4 μm) from tumor samples were deparaffinized using xylene. After processing in ascending and descending series with ethanol, antigen retrieval was performed by boiling the sections in citrate buffer (pH = 4.0) for 2 min. Next, BSA (2% in PBS) was used for blocking the non‐specific molecules at 37 °C for 2 h. These sections were incubated with primary antibodies for CD31 (Santa Cruz Biotechnology, CA, USA) specified for immunohistochemistry at a dilution of 1:100 at 4 °C for overnight under humidified condition. After washing with PBS, the sections were incubated with HRP‐conjugated secondary antibodies (1:200) at 37 °C for 1 h under humidified condition and again washed. Diaminobenzidine (DAB) (Sigma–Aldrich, MO, USA) was used as a substrate for HRP to detect the signals in the form of brown color corresponding to specific binding of the primary antibodies. These sections were further counterstained with hematoxylin and mounted. Normal immunoglobulin‐G was used as negative control for the primary antibodies.
2.9. Treatment with adipokines in vitro
To study the effect of adipokines, we used recombinant human leptin (Sigma–Aldrich, MO, USA) and recombinant human resistin (Calbiochem, CA, USA) to treat melanoma cells in vitro. A375 cells were plated in culture dishes or 6‐well plates in DMEM containing 10% FBS. After 24 h, medium was removed and cells were treated with varying concentrations (range 0.01–100 ng/ml) of these adipokines in DMEM containing 1% FBS for 24 or 48 h as per the experimental requirements. Treated cells were then subjected to desired analysis by MTT assay or processed for immunoblotting or RT‐PCR or confocal staining.
2.10. Reverse transcriptase (RT) PCR
A375 cells were treated with leptin or resistin for 48 h in DMEM containing 1% FBS. Thereafter, the cells were stored at −80 °C in TRIzol reagent (Invitrogen Life Technologies, CA, USA) until processing for RT‐PCR. Total RNA was extracted as per the manufacturer's instructions. Synthesis of cDNA and RT‐PCR were performed using thermal cycler (Eppendorf, Hamburg, Germany). The primer pairs were used as follows: Cav‐1: 5′‐AGACTCGGAGGGACATCTCTACAC‐3′ (F), 5′‐ACTGTGTGTCCCTTCTGGTTCTG‐3′ (R); FASN: 5′‐GGCCTGGACTCGCTCATGGG‐3′ (F), 5′‐TGGGCCTGCAGCTGGGAGCA‐3′ (R) and β‐Actin: 5′‐ATCTGGCACCACACCTTCTACAATGAGCTGCG‐3′ (F), 5′‐CGTCATACTCCTGCTTGCTGATCCACATCTGC‐3′ (R). The annealing temperature used for both Cav‐1 and β‐Actin was 58 °C, whereas it was 62 °C for FASN.
2.11. MTT assay
Melanoma cells or 3T3‐L1 cells were plated at a density of 6 × 103 cells/well in 96‐well plates and allowed to adhere. After 24 h, cells were treated with vehicle (PBS or ethanol), drugs or adipokines as per the experimental requirements. After treatment duration, medium was removed and 50 μl of MTT (methylthiazole tetrazolium, 1 mg/ml in DMEM without phenol red) (Sigma–Aldrich, MO, USA) was added in each well and further incubated for 4 h at 37 °C. Formazan crystals were solubilized in 100 μl of isopropanol and absorbance was measured at 570 nm.
2.12. Long‐term survival assay
B16F10, A375 and 3T3‐L1 were plated cells at a density of 1 × 103 cells/well in 6‐well plates. Next day, these were treated with vehicle or orlistat as per the experimental requirements. After 48 h, medium was removed and fresh medium was added. Cells were allowed to grow for 10 days with medium change on every 2–3 days. Thereafter, cells were fixed with 3% paraformaldehyde for 10 min and stained with 0.05% crystal violet for 2 h at room temperature. Images were taken using digital camera (Olympus, Tokyo, Japan).
2.13. Immunodepletion of leptin and/or resistin from serum collected from mice
Serum from HFD C57BL/6J mice was collected (as described above). Leptin or resistin or both together were immunodepleted from the serum by using respective specific antibodies (Santa Cruz Biotechnology, CA, USA) by incubating at 4 °C for overnight. Antigen–antibody complexes were precipitated using protein A/G‐plus agarose beads (Santa Cruz Biotechnology, CA, USA) by incubating at 4 °C for 4 h. Next, supernatant containing immunodepleted serum was collected by centrifuging the tubes at 10000 RPM at 4 °C. B16F10 cells (3 × 105) were seeded in 35‐mm dishes and cultured in DMEM containing 5% immunodepleted serum. After 48 h, the cells were harvested and lysates were prepared for immunoblotting.
2.14. Culture of melanoma cells in serum and conditioned medium
Approximately 1.5 × 102 B16F10 cells were plated in 24‐well plates and allowed to adhere. After 24 h, DMEM containing 5% serum collected from experimental C57BL/6J mice (as illustrated in Figure 1) was added and cells were cultured chronically for 10 days. Medium was changed on every 2–3 days. Thereafter, cells were fixed with paraformaldehyde, stained with crystal violet and images were taken (as described above).
3T3‐L1 cells were plated in 35‐mm dishes and differentiated as described (Vijayakumar et al., 2010), along with vehicle or orlistat treatment (10–50 μM). Cerulenin (Calbiochem, CA, USA) (10 μM) was used as positive control for inhibition of differentiation. The medium was changed every alternate day and fresh medium containing orlistat or cerulenin was added to the cells. After 11 days, medium was removed and cells were washed twice with DMEM. Fresh DMEM was added and cells were incubated for further 18 h. The levels of adipokines in conditioned medium (CM) collected from these cells were measured using ELISA as mentioned in the previous section. For culturing melanoma cells, CM was mixed with fresh DMEM in 1:1. Approximately 3 × 102 B16F10 cells were plated in 12‐well plates and cultured chronically for 10 days in this CM. Thereafter, crystal violet staining was performed to verify long term survival and the plates were photographed.
2.15. Statistical analysis
Statistical analysis was performed using Sigma Plot 12.0 (Systat Software Inc., CA, USA). All data were represented as the mean ± standard deviation (S.D.). In most cases, bars represent variations within the wells of an in vitro experiment and the experiments were repeated multiple times. For animal experiments, two‐tailed unpaired Student's t‐test was used to compare the different groups of mice. The values of p < 0.05, p < 0.01 and p < 0.001 were considered as statistically significant (*), very significant (**) and highly significant difference (***) respectively unless otherwise mentioned.
3. Results
3.1. Controlling obesity with orlistat or dietary intervention hampers melanoma progression in mice
We have earlier reported that diet induced obesity promotes melanoma progression (Pandey et al., 2012). Further, to study the impact of controlling obesity on melanoma progression, diet‐induced obese mouse models were used. Chronic feeding with high fat diet for six months significantly increased body weight and altered serum chemistry profile, hallmarks of obesity, in both HFD C57BL/6J and HFD NOD/SCID mice (Table shown in Supplementary Figure 1) (p < 0.05) as compared to mice exposed to normal diet (ND). Following oral administration of orlistat (10 mg/kg on every alternate day), on fifteenth day ectopic isograft and xenograft were induced in obese mice by injecting B16F10 and A375 melanoma cells respectively. Subsequently, tumor progression and obesity‐associated parameters were regularly monitored.
In both C57BL/6J and NOD/SCID mice, body weight and fat mass were significantly reduced in HFD‐HFD orlistat group in comparison to HFD‐HFD control group (Table 1; Supplementary Figure 2Aa and B) (p < 0.05). Parallel to the changes in body weight, blood glucose, serum TG, cholesterol, LDLc and free fatty acids, and the level of serum leptin, insulin, resistin, IL‐6 and TNF‐α was decreased significantly in orlistat administered obese C57BL/6J and NOD/SCID mice (p < 0.05 each), whereas adiponectin levels were increased (Table 1). No noticeable changes were observed in serum chemistry profile of ND C57BL/6J mice treated with orlistat under identical experimental set‐up (Table 1).
Table 1.
Obesity‐associated parameters in the experimental mice recorded at the end of the experiment. HFD C57BL/6J mice were divided into two major groups. One group was continuously fed with HFD, whereas the other group was shifted from HFD to normal diet. Mice of both the groups were orally treated with orlistat (10 mg/kg) or vehicle control on every alternate day for 8 weeks. Their body weight was monitored weekly throughout the study and serum was collected at the end of the experiment. Blood glucose, serum TG, serum cholesterol, serum LDLc and serum free fatty acids were measured. Serum factors including leptin, adiponectin, insulin, resistin, IL‐6 and TNF‐α were estimated by ELISA. Similarly, HFD NOD/SCID mice were treated orally with orlistat or vehicle control on every alternate day for 8 weeks and serum parameters were measured. Parallely, ND C57BL/6J mice were divided into two major groups. One group was orally treated with orlistat or vehicle control on every alternate day before inoculating B16F10 cells. The second group was orally treated with orlistat (10 mg/kg) or vehicle control on every alternate day after 2 days of inoculating B16F10 cells. The treatment was continued for 8 weeks. Their body weight was monitored weekly throughout the study and serum was collected at the end of the experiment for further biochemical analysis. The results are given as means ± standard deviation; *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant; #, comparison with C57BL/6J HFD‐HFD Ctrl; $, comparison with NOD/SCID HFD‐HFD Ctrl; §, comparison with C57BL/6J ND‐Pre Ctrl; ψ, comparison with C57BL/6J ND‐Post Ctrl.
| Parameters | C57BL/6J HFD | NOD/SCID HFD | C57BL/6J ND | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HFD‐HFD Ctrl | HFD‐HFD Orli | HFD‐ND Ctrl | HFD‐ND Orli | HFD‐HFD Ctrl | HFD‐HFD Orli | ND‐Pre Ctrl | ND‐Pre Orli | ND‐Post Ctrl | ND‐Post Orli | |
| Body weight (g) | 32.35 ± 2.61 | 25.22#∗ ± 2.36 | 26.73#∗ ± 2.6 | 22.69#∗ ± 2.13 | 33.10 ± 2.18 | 27.22$∗ ± 2.19 | 20.61 ± 1.39 | 20.67§NS ± 1.71 | 20.81 ± 1.77 | 20.78 ψNS ± 1.67 |
| Blood glucose (mg/dl) | 192.34 ± 12.31 | 146.47#∗ ± 10.21 | 168.94#∗ ± 14.51 | 129.38#∗ ± 8.96 | 171.23 ± 22.33 | 143.45$∗ ± 15.52 | 162.24 ± 11.31 | 156.4§NS ± 10.21 | 161.63 ± 14.51 | 159.24ψNS ± 11.96 |
| Serum TG (mg/dl) | 84.54 ± 4.31 | 65.32#∗ ± 7.32 | 68.56#∗ ± 7.32 | 63.45#∗ ± 5.17 | 78.36 ± 5.62 | 66.35$∗ ± 5.37 | 56.54 ± 4.31 | 58.32§NS ± 5.61 | 55.36 ± 7.32 | 57.12ψNS ± 5.17 |
| Serum cholesterol (mg/dl) | 119.36 ± 8.96 | 84.36#∗ ± 6.98 | 97.54#∗ ± 7.39 | 83.5#∗ ± 5.43 | 107.56 ± 8.96 | 89.21$∗ ± 6.94 | 85.36 ± 8.96 | 84.43§NS ± 6.98 | 87.54 ± 7.39 | 86.50ψNS ± 5.43 |
| Serum LDLc (mg/dl) | 87.36 ± 6.46 | 69.23#∗ ± 5.37 | 72.94#∗ ± 6.38 | 67.96#∗ ± 4.31 | 89.37 ± 5.68 | 72.40$∗ ± 4.71 | 53.36 ± 6.46 | 54.23§NS ± 5.37 | 52.94 ± 6.38 | 52.96ψNS ± 4.31 |
| Serum free fatty acids (mM/L) | 2.56 ± 0.20 | 2.06#∗ ± 0.08 | 1.81#∗ ± 0.09 | 1.57#∗∗ ± 0.05 | 1.08 ± 0.05 | 0.66$∗ ± 0.06 | 0.89 ± 0.07 | 0.91§NS ± 0.06 | 0.90 ± 0.05 | 0.92ψNS ± 0.07 |
| Serum leptin (ng/ml) | 42.92 ± 4.93 | 29.55#∗ ± 2.45 | 35.5#∗ ± 1.69 | 24.1#∗∗ ± 1.37 | 36.70 ± 1.99 | 23.83$∗ ± 1.50 | 14.32 ± 1.93 | 13.98§NS ± 1.46 | 13.61 ± 1.70 | 14.38ψNS ± 1.38 |
| Serum adiponectin (ng/ml) | 3820 ± 432.97 | 8650#∗ ± 174.73 | 5805#∗ ± 238.42 | 9171#∗∗ ± 505.96 | 5426 ± 360.74 | 9726$∗∗ ± 310.05 | 12982 ± 832.97 | 12246§NS ± 674.74 | 13147 ± 738.23 | 13213ψNS ± 805.97 |
| Serum insulin (μg/L) | 0.3935 ± 0.03 | 0.2527#∗ ± 0.02 | 0.309#∗ ± 0.011 | 0.2401#∗ ± 0.014 | 0.3496 ± 0.027 | 0.225$∗∗ ± 0.011 | 0.1225 ± 0.03 | 0.114§NS ± 0.02 | 0.1162 ± 0.014 | 0.121ψNS ± 0.012 |
| Serum resistin (ng/ml) | 2.13 ± 0.13 | 1.51#∗ ± 0.16 | 1.66#∗ ± 0.14 | 1.1#∗∗∗ ± 0.09 | 1.13 ± 0.06 | 0.82$∗ ± 0.04 | 0.99 ± 0.09 | 1.01§NS ± 0.09 | 0.98 ± 0.10 | 0.99ψNS ± 0.08 |
| Serum IL‐6 (pg/ml) | 40.62 ± 4.13 | 29.62#∗ ± 2.90 | 33.45#∗ ± 2.86 | 26.8#∗∗∗ ± 3.72 | 33.17 ± 1.81 | 21.42$∗ ± 0.73 | 19.23 ± 2.13 | 20.32§NS ± 2.90 | 19.86 ± 2.87 | 19.45ψNS ± 2.73 |
| Serum TNF‐α (pg/ml) | 28.35 ± 3.01 | 14.02#∗ ± 1.42 | 17.6#∗ ± 1.51 | 12.07#∗ ± 1.73 | 24.06 ± 1.40 | 10.46$∗ ± 0.23 | 10.88 ± 2.01 | 11.05§NS ± 1.82 | 10.23 ± 1.52 | 11.12ψNS ± 1.73 |
Orlistat treatment decreased not only body weight (Figure 2Aa and Ba) but also tumor progression, in terms of volume and weight, in both the HFD models as compared to untreated counter parts (Figure 2Ab–c and Bb–c). Further to assess the impact of controlling diet on melanoma progression in obese mice, HFD mice were shifted from high fat to normal chow. Consistent with findings in orlistat treated HFD mice, switch from high fat diet to normal diet led to normalization of body weight and serum chemistry profile in par with that of ND mice which was accompanied by decreased tumor volume and weight (Table 1; Figure 2Aa–c). Also, fat mass of these mice was reduced significantly as compared to the control group (Supplementary Figure 2Ab). Interestingly, the normalization in obese parameters and reduction in tumor parameters were much more effective and pronounced when dietary intervention was combined with orlistat treatment (Table 1; Figure 2Aa–c). Further, no noticeable changes were observed in H&E stained sections of liver, heart, kidney, spleen and lung tissues from orlistat treated HFD mice (Supplementary Figure 3A). In addition, we did not find any change in the levels of serum enzymes associated with organ functions (Supplementary Figure 3Ba‐e) which is suggestive of non‐toxicity of orlistat at this dose.
Figure 2.
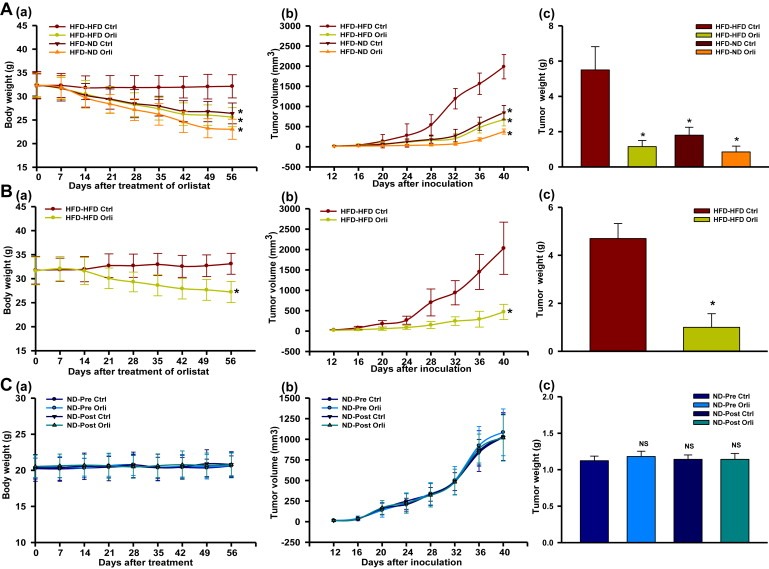
Orlistat treatment and/or diet shifting reduces tumor progression and decreases tumor size in HFD but not in ND mice. (A) HFD male C57BL/6J mice were divided into two major groups. One group was continuously fed with HFD (HFD‐HFD), whereas the other group was shifted from HFD to normal diet (HFD‐ND). Mice of both the groups were orally treated with orlistat (10 mg/kg) or vehicle control on every alternate day for 8 weeks (N = 10 mice per group). Body weight was monitored weekly throughout the study. At the end of the experiment, mice were sacrificed, and tumors as well as adipose tissues were collected. (a) Changes in body weight of the mice, (b) Tumor progression, and (c) Tumor weight. (B) HFD female NOD/SCID mice were orally treated with orlistat or vehicle control on every alternate day for 8 weeks (N = 6 mice per group). Body weight was monitored weekly throughout the study. At the end of the experiment, mice were sacrificed and tumors as wells as adipose tissues were collected. (a) Changes in body weight, (b) Tumor progression, and (c) Tumor weight. (C) ND male C57BL/6J mice (N = 10 mice per group) were orally treated with orlistat or vehicle control on every alternate day before or after inoculating B16F10 cells for 8 weeks. Body weight of the mice was monitored weekly throughout the study. At the end of the experiment, mice were sacrificed and tumors were collected. (a) Changes in body weight, (b) Tumor progression, and (c) Tumor weight. The results are given as means ± standard deviation; *, p < 0.05; NS, not significant.
3.2. Orlistat at antiobesity dose does not influence melanoma progression in mice fed with normal diet
We investigated whether orlistat affects melanoma progression by directly acting on tumor cells or indirectly through controlling obesity and adiposity by monitoring periodically body weight and melanoma progression in mice fed with normal diet as per the experimental layout shown in Figure 1B. Orlistat treatment on every alternate day was initiated in mice 15 days before injection of B16F10 cells or after two days following injection of these cells and continued till the termination of the experiment. Irrespective of whether mice were administered with orlistat prior to or after injecting melanoma cells, no significant changes in body weight, serum lipids and adipokines levels were observed in ND mice (Table 1) as compared to the respective control groups (p > 0.05 each). Also, with unaltered body weight (Figure 2Ca), no noticeable difference in tumor progression was detected in pre‐ or post‐orlistat treated mice compared to the control group as evident by unchanged tumor volume and weight (Figure 2Cb and Cc) (p > 0.05 each). No adverse effects were observed in tissue sections of vital organs excised from the mice administered with orlistat as evident by H&E stained sections of organs and by serum analysis for normal functionality of organs (Supplementary Figure 4A and Ba‐e).
3.3. Orlistat at higher concentration affects growth and proliferation of melanoma cells
To check the direct effect of orlistat, if any, on B16F10 and A375 melanoma cells, and 3T3‐L1 preadipocytes, cells were treated with varying concentrations of orlistat in vitro. Survival of these cells was unaffected by orlistat up to 100 μM. However, at 1000 μM concentration of orlistat, melanoma cell survival was reduced significantly up to ∼40%, whereas 3T3‐L1 cells remained unaffected (Supplementary Figure 5A). Long term colony formation assay demonstrates that orlistat at higher concentration (≥250 μM) significantly reduced survival of melanoma cells (Supplementary Figure 5B and C) but not in 3T3‐L1 cells (Supplementary Figure 5D). Further, to ascertain that cell growth inhibition by orlistat is not because of its direct action, but rather it is a consequence of altered adipokines levels, B16F10 cells were cultured in medium containing 5% serum collected from mice fed with normal diet or high fat diet. These cells were then treated chronically with 50 μM orlistat (concentration at which differentiation of adipocytes is inhibited in 3T3‐L1 cells) with changing of medium on every alternate day for 10 days. No difference in the survival was observed in orlistat treated cells as compared to the respective control, although increased survival was found in HFD cultured cells as compared to those cultured in ND serum. These results suggest that orlistat treatment did not affect the growth and proliferation of melanoma cells cultured in serum from ND or HFD C57BL/6J mice at this concentration (Supplementary Figure 6A and B). Indeed, in vivo effect of orlistat is associated with normalized levels of obesity‐associated factors through reduction in adiposity.
3.4. Reduction in adipocyte size influences tumor cell proliferation and angiogenesis
Fat accumulation influences the adipocyte expansion by mass and volume, and eventually adipokine secretion profile. Therefore, to investigate the changes in tumor progression after controlling obesity, fat accumulation was assessed. Both orlistat treatment and/or shifting of diet caused reduction in adipose tissue mass (Supplementary Figure 2) in HFD mice. Analysis of H&E stained adipose tissue sections indicates that size of adipocytes were reduced in the experimental groups compared to the control (Figure 3Aa). Adipocytes size determines the serum levels of adipokines which are known to modulate different aspects of tumorigenesis such as survival, proliferation, migration and metastasis (Khandekar et al., 2011). To unravel the cellular and molecular events associated with reduced tumor progression in HFD mice following orlistat treatment or diet shifting (Figure 2), H&E stained tumor sections were analyzed for occurrence of necrosis and cell density. Increased necrotic areas were clearly visible in tumors excised from mice administered with orlistat and/or shifted to normal diet as compared to their respective controls (Figure 3Ab). We also checked the expression level of CD31, a marker for angiogenesis. Immunohistochemical analysis shows that CD31 levels were significantly reduced (Figure 3Ac), which could lead to increase in necrosis in these tumors because of decreased angiogenesis (Vaupel et al., 1989).
Figure 3.
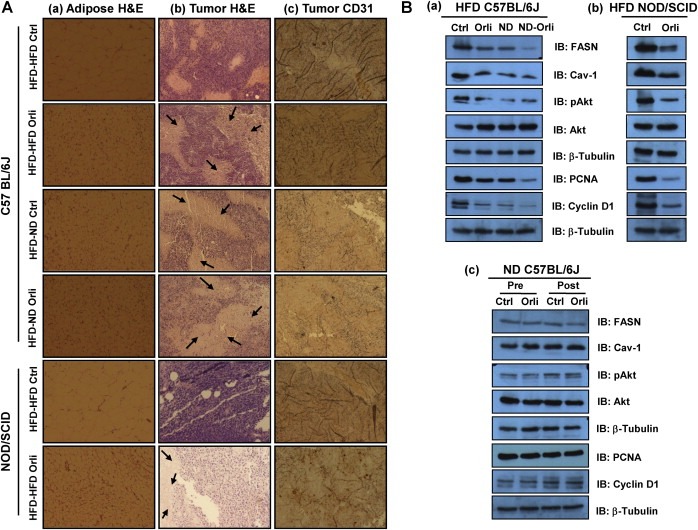
Controlling obesity in HFD mice reduces size of adipocytes in adipose tissue, diminishes angiogenesis and proliferation in tumors. (A) Histological analysis of sections of adipose tissue and tumor samples. (a) Adipose tissues from treated and untreated mice were excised and washed with PBS and then kept in 10% paraformaldehyde at room temperature and sections were stained with H&E. Images were taken in Olympus (DP‐71) camera (Magnification: 40×). (b) The tumor samples were washed with PBS and then kept in 10% paraformaldehyde at room temperature and their sections were stained with H&E. Histopathological examination was done, and images were taken. The arrow marks indicate necrotic area in the tumors (Magnification: 40×). (c) Tumor samples were subjected to immunohistochemistry of CD31. Diaminobenzidine (DAB) was used as a substrate for HRP. Images were taken in Olympus (DP‐71) camera (Magnification: 40×). (B) Western blot analysis of lysates from tumors subjected to SDS‐PAGE and probed for levels of FASN, Cav‐1, activated Akt, PCNA and cyclin D1 in (a) HFD C57BL/6J mice, (b) HFD NOD/SCID mice, and (c) ND C57BL/6J mice.
FASN plays a crucial role in growth, survival and proliferation of cancer cells as it provides fatty acids for maintenance of membrane architecture and energy generation under stress (Kuhajda, 2000). Also, Cav‐1 is a membrane associated protein which performs dynamic functions in cells including signal transduction (Liu et al., 2002). We have previously reported the role of FASN and Cav‐1 in melanoma progression in obese mice (Pandey et al., 2012). Here, we sought to explore whether these molecules are altered in our experimental system. As anticipated, significant reduction in protein level of FASN and Cav‐1 was detected in tumors from both HFD C57BL/6J and NOD/SCID experimental mice when compared to the control. These changes were associated with reduced levels of activated Akt (Figure 3Ba and Bb). To evaluate whether decrease in the tumor size is a consequence of apoptosis or because of decrease in growth and proliferation, we checked the status of apoptotic as well as proliferative markers. The level of apoptotic markers caspase‐3 and caspase‐7 were unchanged in all the tumors (Supplementary Figure 7A and B) while a significant decrease in the level of proliferating cell nuclear antigen (PCNA) and cyclin D1 was detected (Figure 3Ba and Bb). However, the levels of these molecules remained unaltered in tumors from orlistat treated ND C57BL/6J mice (Figure 3Bc; Supplementary Figure 7C).
3.5. Increased tumor progression in obese mice is attributed to the involvement of adipocyte secretory factors
Controlling obesity by orlistat treatment or diet‐shifting was associated with reduction in obese phenotype and normalization in serum chemistry profile in HFD mice (Figure 2; Table 1). To explore if the decline in obesity‐associated factors have any role in tumor progression, B16F10 cells were chronically cultured in DMEM containing 5% serum collected from experimental C57BL/6J HFD mice treated with or without orlistat and/or shifted to normal diet. Survival was reduced significantly in cells cultured in serum taken from HFD mice administered with orlistat and/or shifted to normal diet when compared to those cultured in the serum from control HFD mice (Figure 4Aa). Interestingly, no such changes in growth property of B16F10 cells were noticed upon culturing them in serum collected from ND mice administered with or without orlistat (Figure 4Ab).
Figure 4.
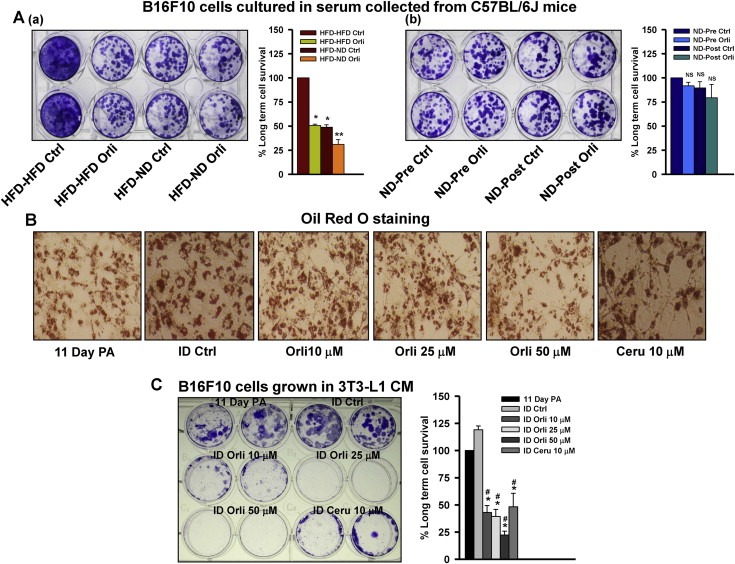
Effect of adipocyte secreted factors on the survival of melanoma cells. (A) Long term culture of B16F10 cells in serum collected from (a) HFD C57BL/6J and (b) ND C57BL/6J experimental mice. B16F10 cells (1.5 × 102 cells/well) were plated in 24‐well plate. After 24 h, medium was removed and fresh medium containing 5% serum collected from experimental mice described in Materials and methods. The medium was changed on every 2–3 days. After 10 days, the cells were stained with 0.05% crystal violet and images were taken. The cell survival was quantified using Image J software. (B) Oil Red O staining of 3T3‐L1 cells treated with orlistat. 3T3‐L1 cells were induced to differentiate and treated with indicated concentrations of orlistat. Cerulenin served as positive control for inhibition of adipocyte differentiation. Medium was changed and orlistat treatment was given on every alternate day. After 10 days, cells were stained with 0.3% Oil Red O and images were taken on Olympus (DP‐71) camera (Magnification: 40×). (C) Long term culture of B16F10 cells in conditioned medium (CM) collected from 3T3‐L1 cells. B16F10 cells (3 × 102 cells/well) were plated in 12‐well plate. After 24 h, medium was removed and conditioned medium collected from 3T3‐L1 cells was added. The medium was changed on every 2–3 days. After 10 days, the cells were stained with 0.05% crystal violet and images were taken. The cell survival was quantified using Image J software. The results are given as means ± standard deviation of the experiments repeated multiple times; *, p < 0.05; **, p < 0.01; #, comparison with ID Ctrl; NS, not significant; PA, preadipocytes; ID, differentiated 3T3‐L1 cells induced by IBMX and DEX; Orli, orlistat; Ceru, cerulenin.
Further, to consolidate these observations, we used 3T3‐L1, an in vitro model system, to study the expression of lipogenic factors and secretory adipokines. To check whether orlistat induced tumor regression is associated with its effect on adipocytes, differentiation of 3T3‐L1 adipocytes was induced in the presence and absence of orlistat (10–50 μM). Oil Red O analysis of accumulated lipid droplets in differentiated cells suggests that orlistat significantly inhibited differentiation of 3T3‐L1 preadipocytes (Figure 4B). Estimation of secretory factors in conditioned medium (CM) collected from differentiated adipocytes was found to be comparable to the levels in serum collected from mice administered with orlistat and/or shifted to normal diet (Supplementary Table 2). It indicates that orlistat, in addition to its inhibitory effect on gastrointestinal lipases, has a direct inhibitory effect on adipocyte differentiation, thereby reducing the adipose mass.
Next, to verify the involvement of adipocyte‐secreted factors in melanoma cell growth, B16F10 cells were chronically cultured for 10 days in conditioned medium collected from day 11 of differentiated 3T3‐L1 cells treated with or without orlistat. Number of colonies was drastically reduced in B16F10 cells cultured in CM from orlistat treated 3T3‐L1 cells as compared to those cultured in CM from the untreated differentiated cells (Figure 4C). This observation further confirms that orlistat by inhibiting adipocyte differentiation reduces the levels of adipocyte‐secreted factors, which in turn affect melanoma cell proliferation.
3.6. Leptin and resistin are involved in modulation of growth promoting molecules in melanoma cells
Adipocyte‐secreted factors favor growth, survival, proliferation and invasiveness of cancer cells (Khandekar et al., 2011). We therefore speculated that altered levels of these factors might be involved directly or indirectly in melanoma progression. Leptin and resistin are major adipokines reported to promote proliferation of many cancer cells (Kim et al., 2011; Somasundar et al., 2004). However, the role of these adipokines in melanoma is not very clear. As we noted decreased serum levels of these adipokines in HFD mice treated with orlistat and/or shifted from high fat to normal diet, the involvement of these adipokines on melanoma growth was explored. A375 cells were treated with varying concentrations of recombinant human leptin and resistin individually, and it was found that both these adipokines enhanced proliferation of cells (Figure 5A and B respectively).
Figure 5.
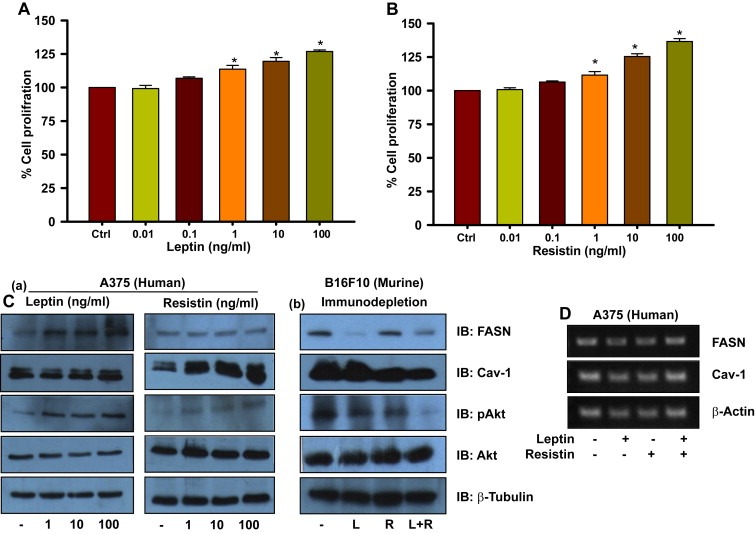
Leptin and resistin promote proliferation, and modulate levels of FASN and Cav‐1 respectively, in melanoma cells. (A) MTT assay in leptin treated melanoma cells. A375 cells were plated in 96‐well plates. After 24 h, treatment with recombinant leptin was given at indicated concentrations in DMEM containing 1% FBS and cells were incubated for 48 h. (B) MTT assay for resistin treated melanoma cells. A375 cells were plated in 96‐well plates. After 24 h, treatment with recombinant resistin was given at indicated concentrations in DMEM containing 1% FBS and cells were incubated for 48 h. (C) Western blotting analysis of FASN, Cav‐1 and activated Akt. (a) A375 cells were plated in 35‐mm culture dishes. After 24 h, treatment with recombinant leptin or resistin was given at indicated concentrations in DMEM containing 1% FBS and cells were incubated for 48 h. These cell lysates were then subjected to SDS‐PAGE and Western blotting, (b) B16F10 cells were cultured in serum (collected from HFD C57BL/6J mice) which was immuno‐depleted of leptin and/or resistin for 48 h. Lysates of these cells were prepared and were subjected to SDS‐PAGE and Western blotting. (D) RT‐PCR analysis of 3T3‐L1 cells treated with leptin and/or resistin. A375 cells were treated with leptin and/or resistin (100 ng/ml) for 48 h as described in Materials and methods. The results are given as means ± standard deviation; *, p < 0.05; L, leptin; R, resistin.
Akt pathway is reported to be hyper activated in melanoma in obese mice (Pandey et al., 2012). Interestingly, we found that leptin and resistin treatment not only caused activation of Akt but also enhanced the protein levels of FASN and Cav‐1 respectively in A375 cells (Figure 5Ca). To confirm this finding in murine melanoma cells, we collected serum from HFD C57BL/6J mice and immunodepleted it of leptin and/or resistin. As expected, in B16F10 cells cultured in serum depleted of leptin and resistin, protein levels of FASN and Cav‐1 were reduced respectively, with concomitant reduction in the levels of activated Akt (Figure 5Cb). When both the adipokines were depleted simultaneously, FASN as well as Cav‐1 protein levels were reduced (Figure 5Cb). At transcription levels, however, no differences in both FASN and Cav‐1 were detected (Figure 5D) which was also consistent with the unaltered protein levels and nuclear localization of transcription factors which regulate FASN and Cav‐1 (Data not shown).
4. Discussion
Different mechanisms have been suggested to explain the link between obesity and many cancer types. Increased amount of white adipose tissue, elevated serum levels of adipokines and lipids, and accompanying insulin resistance are considered to be contributing factors for increased cancer risk (Khandekar et al., 2011; van Kruijsdijk et al., 2009). We, therefore, in this line, speculated that controlling obesity either with antiobesity drug or dietary intervention may restrict or impede melanoma progression. Considering the close vicinity of melanoma to subcutaneous adipose tissue, present study was aimed at investigating whether reducing adiposity has any impact on rampaging melanoma malignancy.
Obesity‐associated chronic metabolic malfunctioning can be controlled by long term pharmacological or dietary interventions or through physical activity. We employed both pharmacological as well as diet‐control approaches to address the role of obesity‐associated factors in tumor progression. Although, either of the approaches is individually effective in reducing obesity, we employed combination of both these strategies in diet‐induced obese mice so as to have pronounced effect on adiposity in influencing obesity‐promoted melanoma progression. This study clearly demonstrates that controlling body weight either by pharmacological intervention or through dietary restriction halts rapid progression of melanoma. By either means, normalization in body weight as a consequence of reduction in fat accumulation (adiposity) correlated with reduced serum levels of lipids and pro‐inflammatory or proliferative factors. Employing both the strategies simultaneously had a profound impact on tumor progression than the either of the strategies exploited independently. Unaffected tumor progression under orlistat administration in ND mice substantiates the fact that orlistat exerts its effect on melanoma progression indirectly by reducing adiposity.
Reduction in adiposity and subsequent retardation of tumor progression was associated with increased necrosis in tumors of these experimental mice. Adipokines like TNF‐α and IL‐6, proangiogenic factors (Cohen et al., 1996; Fajardo et al., 1992; Park et al., 2010), were decreased in these HFD mice. It is important here to note that orlistat treatment and/or diet‐shifting not only reduces circulating levels of lipids and pro‐inflammatory cytokines, but also lowered blood glucose and serum insulin levels, which are considered to be important for cell growth and proliferation (Han et al., 2011; Okumura et al., 2002). It has also been reported that adipocytes promote tumor growth by aiding tumor favorable microenvironment and fueling tumor cells by providing free fatty acids (Nieman et al., 2011). Concommitantly, we noted that orlistat treatment or dietary intervention decreases serum free fatty acids levels. Similarly, results obtained from in vitro studies on proliferation of B16F10 cells cultured in serum collected from experimental mice or CM of differentiated adipocytes indicate that serum chemistry profile contributes to tumor progression. Together, diminished tumor growth could also be a consequence of reduced supply of nutrients like free fatty acids and glucose owing to retarded angiogensis. Decreased proliferative molecules while unaltered levels of apoptotic markers in tumors suggest that orilstat treatment and/or reduced caloric intake in obese mice exert a growth inhibitory effect rather than inducing apoptosis thereby reduction in tumor progression.
Dysregulated lipid metabolism is a characteristic feature of cancer cells (Fritz and Fajas, 2010; Santos and Schulze, 2012). Lipid metabolism‐related genes like FASN, SCD1, OLR1 and ACC are usually upregulated in cancer cells (Hirsch et al., 2010) and these lipogenic genes are known to favor cancer cell survival and growth (Lu and Archer, 2010; Yamashita et al., 2009). For rapidly growing or proliferating cells like cancer cells, enhanced membrane synthesis is required. Being an important component of plasma membrane, lipids play an integral role in cellular proliferation. At cellular level, fatty acid synthesis is regulated by FASN. Therefore, its elevated expression as well as activity is required to meet the high proliferation rate of cancer cells (Zeng et al., 2010). In this context, we have reported that FASN protein level increased in melanoma from obese mice (Pandey et al., 2012). As an inhibitor of FASN, orlistat has been reported to have anticancer activity and effectively targets tumor at an intraperitoneal dose of 240 mg/kg body weight in mice (human equivalent dose would be 20 mg/kg body weight) (Carvalho et al., 2008). However, such a high dose of orlistat could be intolerable to humans and would lead to severe side effects. Intake of relatively tolerable dose of 120 mg three times daily is reported to reduce body weight in human by 5–10% with few side effects (Sjostrom et al., 1998; Tiikkainen et al., 2004). In this study, we have shown that at a tolerable dose of orlistat (10 mg/kg, oral) in mice, significant reduction in body weight was observed and accompanied by normalization of obesity‐associated factors in HFD mice without exerting any generalized or organ‐specific toxicity. At identical orlistat dosage without any toxicity, such effect was not seen on body weight or tumor progression in ND mice suggesting the important role of adiposity in melanoma growth. Thus, controlling obesity either by pharmacological or dietary intervention could be helpful in reducing obesity‐enhanced tumorigenesis. However, dietary intervention could be more helpful in reducing tumor progression as it is clinically relevant and does not involve any side effects. In this study, we have shown that FASN protein level was reduced in tumors of obese mice subjected to orlistat treatment and/or switched to normal diet indicating the role of obesity‐associated factors in FASN modulation.
A membrane‐associated protein involved in signal transduction and maintenance of membrane architecture is Cav‐1. Depending upon the cancer type, it can be either tumor suppressor (Quann et al., 2013) or tumor promoter (Felicetti et al., 2009). Due to its involvement in drug resistance (Lavie et al., 2001; Meena et al., 2013), Cav‐1 which is stabilized by FASN through palmitoylation (Di Vizio et al., 2008), has been reported to be a tumor promoting factor in prostate cancer and in melanoma (Lavie et al., 1998). We earlier reported that mutual interdependence of Cav‐1 and FASN is integral to melanoma progression (Pandey et al., 2012). Thus, our study shows that while FASN and Cav‐1 levels are unaffected by orlistat under normal serum chemistry in ND mice, controlling adiposity reduces the protein levels of Cav‐1 which correlates with diminished FASN in HFD mice suggesting the crucial role of adipokines and nutritional factors in tumor progression.
Obesity causes elevation in the levels of many pro‐inflammatory and proliferative factors which are implicated in increased growth and proliferation of cancer cells (Balkwill, 2004; Honma et al., 2002; Lin and Karin, 2007). Also, in vitro studies have revealed the growth promoting phenomenon in melanoma cells cultured in serum of ob/ob mice (Kushiro and Nunez, 2011) suggesting the influence of adipokines on tumor progression. As observed in our study, by controlling obesity tumor growth is restricted partly through normalization in the serum levels of leptin and resistin. Also, as reported in our previous study, hyper activated Akt pathway was associated with increased protein levels of FASN and Cav‐1 (Pandey et al., 2012). Therefore, we studied the influence of leptin and resistin which are Akt‐activating adipokines (Pang et al., 2013; Yan et al., 2012) on proliferation of melanoma cells. Treatment of melanoma cells with adipokines (leptin and resistin) in vitro leads to increased levels or activation of proteins that in turn augment cellular growth and metabolism. Although both the adipokines activated Akt, leptin specifically enhanced FASN protein level and resistin influenced Cav‐1 protein level. Intriguingly, there was no change in the transcript levels of these two molecules in cells treated with leptin and resistin. The modulation of FASN and Cav‐1 by these adipokines could be due to increased stability or through reduced degradation which is yet to be verified and further investigated. Normalized serum levels of these adipokines, via reduced adiposity, could be associated with reduction in FASN, Cav‐1 and activated Akt levels leading to reduced progression of melanoma in the experimental HFD mice.
Conclusively, our study establishes a link between controlling obesity and melanoma progression through involvement of obesity‐associated factors. The study suggests that controlling obesity could be crucial to prevention of obesity‐promoted cancer progression by virtue of normalizing the serum levels of adipokines that affect tumor promoting molecules and signaling pathways (see the schematic overview in Figure 6).
Figure 6.
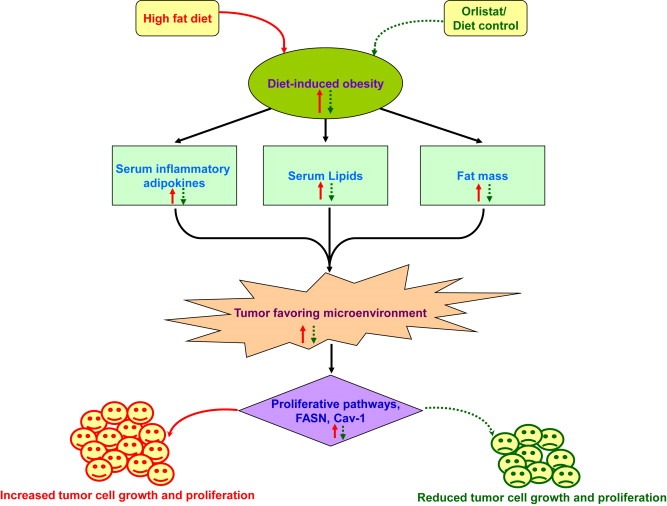
Proposed model of impact of targeting obesity on melanoma progression. Obesity‐associated factors increase melanoma progression by inducing chronic low‐grade inflammation and tumor‐favoring microenvironment (solid arrow marks). On the other hand, controlling obesity causes reduction in fat mass, serum lipids and pro‐inflammatory adipokines. This in turn reduces melanoma growth and progression through modulating molecules and pathways associated with increased tumor progression (dotted arrow marks).
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
The authors thank Dr. S.C. Mande, Director, NCCS, Pune, India and Dr. G.C. Mishra, former Director, NCCS, Pune, India for being very supportive and giving all the encouragement to carry out this work. P.M. and S.V.S. thank University Grants Commission (UGC), New Delhi, India; B.C., V.P. and N.M. thank Council for Scientific and Industrial Research (CSIR), India for research fellowship. The support from Experimental Animal Facility (EAF), Central Instrumental Facility and technical staff of NCCS is also duly acknowledged. This work was supported in part by intramural grant from National Centre for Cell Science (NCCS) funded by Department of Biotechnology (DBT), Government of India; and an extramural grant from Department of Science and Technology (DST), Government of India (Grant No. SR/SO/HS‐0136/2012). The funding agencies had no involvement in study design, data collection, interpretation and analysis, decision to publish, or writing of the manuscript.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.11.006.
Malvi Parmanand, Chaube Balkrishna, Pandey Vimal, Vijayakumar Maleppillil Vavachan, Boreddy Purushotham Reddy, Mohammad Naoshad, Singh Shivendra Vikram, Bhat Manoj Kumar, (2015), Obesity induced rapid melanoma progression is reversed by orlistat treatment and dietary intervention: Role of adipokines, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.11.006.
This work was carried out in partial fulfillment of a Ph.D. thesis (of P.M.) to be submitted to Savitribai Phule Pune University, Pune, India. The work was partly presented in the special conference on “Metabolism and Cancer” organized by American Association for Cancer Research (AACR) in Baltimore, USA in 2011, and in 1st Indian Cancer Congress (ICC), New Delhi, India in 2013.
References
- Ando, S. , Catalano, S. , 2011. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 8, 263–275. [DOI] [PubMed] [Google Scholar]
- Balistreri, C.R. , Caruso, C. , Candore, G. , 2010. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. 2010, (802078) 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill, F. , 2004. Cancer and the chemokine network. Nat. Rev. Cancer. 4, 540–550. [DOI] [PubMed] [Google Scholar]
- Calle, E.E. , Rodriguez, C. , Walker-Thurmond, K. , Thun, M.J. , 2003. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 348, 1625–1638. [DOI] [PubMed] [Google Scholar]
- Calle, E.,E. , Kaaks, R. , 2004. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer. 4, 579–591. [DOI] [PubMed] [Google Scholar]
- Carvalho, M.A. , Zecchin, K.G. , Seguin, F. , Bastos, D.C. , Agostini, M. , Rangel, A.L. , Veiga, S.S. , Raposo, H.F. , Oliveira, H.C. , Loda, M. , Coletta, R.D. , Graner, E. , 2008. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int. J. Cancer. 123, 2557–2565. [DOI] [PubMed] [Google Scholar]
- Cohen, T. , Nahari, D. , Cerem, L.W. , Neufeld, G. , Levi, B.Z. , 1996. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 271, 736–741. [DOI] [PubMed] [Google Scholar]
- Di Vizio, D. , Adam, R.M. , Kim, J. , Kim, R. , Sotgia, F. , Williams, T. , Demichelis, F. , Solomon, K.R. , Loda, M. , Rubin, M.A. , Lisanti, M.P. , Freeman, M.R. , 2008. Caveolin-1 interacts with a lipid raft-associated population of fatty acid synthase. Cell Cycle. 7, 2257–2267. [DOI] [PubMed] [Google Scholar]
- Fajardo, L.F. , Kwan, H.H. , Kowalski, J. , Prionas, S.D. , Allison, A.C. , 1992. Dual role of tumor necrosis factor-alpha in angiogenesis. Am. J. Pathol. 140, 539–544. [PMC free article] [PubMed] [Google Scholar]
- Felicetti, F. , Parolini, I. , Bottero, L. , Fecchi, K. , Errico, M.C. , Raggi, C. , Biffoni, M. , Spadaro, F. , Lisanti, M.P. , Sargiacomo, M. , Care, A. , 2009. Caveolin-1 tumor-promoting role in human melanoma. Int. J. Cancer. 125, 1514–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine, K.R. , Barofsky, I. , 2001. Obesity and health-related quality of life. Obes. Rev. 2, 173–182. [DOI] [PubMed] [Google Scholar]
- Fritz, V. , Fajas, L. , 2010. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene. 29, 4369–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, L. , Ma, Q. , Li, J. , Liu, H. , Li, W. , Ma, G. , Xu, Q. , Zhou, S. , Wu, E. , 2011. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS One. 6, e27074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, H.A. , Iliopoulos, D. , Joshi, A. , Zhang, Y. , Jaeger, S.A. , Bulyk, M. , Tsichlis, P.N. , Shirley Liu, X. , Struhl, K. , 2010. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. 17, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma, S. , Shimodaira, K. , Shimizu, Y. , Tsuchiya, N. , Saito, H. , Yanaihara, T. , Okai, T. , 2002. The influence of inflammatory cytokines on estrogen production and cell proliferation in human breast cancer cells. Endocr. J. 49, 371–377. [DOI] [PubMed] [Google Scholar]
- Jerant, A.F. , Johnson, J.T. , Sheridan, C.D. , Caffrey, T.J. , 2000. Early detection and treatment of skin cancer. Am. Fam. Physician. 62, 357–368. 375-376, 381-382 [PubMed] [Google Scholar]
- Kampman, E. , Vrieling, A. , van Duijnhoven, F.J. , Winkels, R.M. , 2012. Impact of diet, body mass Index, and physical activity on Cancer survival. Curr. Nutr. Rep. 1, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandekar, M.J. , Cohen, P. , Spiegelman, B.M. , 2011. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer. 11, 886–895. [DOI] [PubMed] [Google Scholar]
- Kim, H.J. , Lee, Y.S. , Won, E.H. , Chang, I.H. , Kim, T.H. , Park, E.S. , Kim, M.K. , Kim, W. , Myung, S.C. , 2011. Expression of resistin in the prostate and its stimulatory effect on prostate cancer cell proliferation. BJU Int. 108, E77–E83. [DOI] [PubMed] [Google Scholar]
- Kopelman, P.G. , 2000. Obesity as a medical problem. Nature. 404, 635–643. [DOI] [PubMed] [Google Scholar]
- Kridel, S.J. , Axelrod, F. , Rozenkrantz, N. , Smith, J.W. , 2004. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 64, 2070–2075. [DOI] [PubMed] [Google Scholar]
- Kuhajda, F.P. , 2000. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 16, 202–208. [DOI] [PubMed] [Google Scholar]
- Kushiro, K. , Nunez, N.P. , 2011. Ob/ob serum promotes a mesenchymal cell phenotype in B16BL6 melanoma cells. Clin. Exp. Metastasis. 28, 877–886. [DOI] [PubMed] [Google Scholar]
- Lavie, Y. , Fiucci, G. , Liscovitch, M. , 1998. Up-regulation of caveolae and caveolar constituents in multidrug-resistant cancer cells. J. Biol. Chem. 273, 32380–32383. [DOI] [PubMed] [Google Scholar]
- Lavie, Y. , Fiucci, G. , Liscovitch, M. , 2001. Upregulation of caveolin in multidrug resistant cancer cells: functional implications. Adv. Drug Deliv. Rev. 49, 317–323. [DOI] [PubMed] [Google Scholar]
- Lin, W.W. , Karin, M. , 2007. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 117, 1175–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P. , Rudick, M. , Anderson, R.G. , 2002. Multiple functions of caveolin-1. J. Biol. Chem. 277, 41295–41298. [DOI] [PubMed] [Google Scholar]
- Lu, S. , Archer, M.C. , 2010. Sp1 coordinately regulates de novo lipogenesis and proliferation in cancer cells. Int. J. Cancer. 126, 416–425. [DOI] [PubMed] [Google Scholar]
- McTiernan, A. , 2008. Mechanisms linking physical activity with cancer. Nat. Rev. Cancer. 8, 205–211. [DOI] [PubMed] [Google Scholar]
- Meena, A.S. , Sharma, A. , Kumari, R. , Mohammad, N. , Singh, S.V. , Bhat, M.K. , 2013. Inherent and acquired resistance to paclitaxel in hepatocellular carcinoma: molecular events involved. PLoS One. 8, e61524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez, J.A. , Lupu, R. , 2007. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 7, 763–777. [DOI] [PubMed] [Google Scholar]
- Nieman, K.M. , Kenny, H.A. , Penicka, C.V. , Ladanyi, A. , Buell-Gutbrod, R. , Zillhardt, M.R. , Romero, I.L. , Carey, M.S. , Mills, G.B. , Hotamisligil, G.S. , Yamada, S.D. , Peter, M.E. , Gwin, K. , Lengyel, E. , 2011. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura, M. , Yamamoto, M. , Sakuma, H. , Kojima, T. , Maruyama, T. , Jamali, M. , Cooper, D.R. , Yasuda, K. , 2002. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: reciprocal involvement of PKC-alpha and PPAR expression. Biochim. Biophys. Acta. 1592, 107–116. [DOI] [PubMed] [Google Scholar]
- Pandey, V. , Vijayakumar, M.V. , Ajay, A.K. , Malvi, P. , Bhat, M.K. , 2012. Diet-induced obesity increases melanoma progression: involvement of Cav-1 and FASN. Int. J. Cancer. 130, 497–508. [DOI] [PubMed] [Google Scholar]
- Pang, L. , Zhang, Y. , Yu, Y. , Zhang, S. , 2013. Resistin promotes the expression of vascular endothelial growth factor in ovary carcinoma cells. Int. J. Mol. Sci. 14, 9751–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, E.J. , Lee, J.H. , Yu, G.Y. , He, G. , Ali, S.R. , Holzer, R.G. , Osterreicher, C.H. , Takahashi, H. , Karin, M. , 2010. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 140, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. , Euhus, D.M. , Scherer, P.E. , 2011. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr. Rev. 32, 550–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Filho, G. , Lim, E.L. , Wong, M.L. , Licinio, J. , 2011. Associations between adipokines and obesity-related cancer. Front. Biosci. (Landmark Ed.). 16, 1634–1650. [DOI] [PubMed] [Google Scholar]
- Prieto-Hontoria, P.L. , Perez-Matute, P. , Fernandez-Galilea, M. , Bustos, M. , Martínez, J.A. , Moreno-Aliaga, M.J. , 2011. Role of obesity-associated dysfunctional adipose tissue in cancer: a molecular nutrition approach. Biochim. Biophys. Acta. 1807, 664–678. [DOI] [PubMed] [Google Scholar]
- Quann, K. , Gonzales, D.M. , Mercier, I. , Wang, C. , Sotgia, F. , Pestell, R.G. , Lisanti, M.P. , Jasmin, J.F. , 2013. Caveolin-1 is a negative regulator of tumor growth in glioblastoma and modulates chemosensitivity to temozolomide. Cell Cycle. 12, 1510–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richelsen, B. , Tonstad, S. , Rossner, S. , Toubro, S. , Niskanen, L. , Madsbad, S. , Mustajoki, P. , Rissanen, A. , 2007. Effect of orlistat on weight regain and cardiovascular risk factors following a very-low-energy diet in abdominally obese patients: a 3-year randomized, placebo-controlled study. Diabetes Care. 30, 27–32. [DOI] [PubMed] [Google Scholar]
- Roberts, D.L. , Dive, C. , Renehan, A.G. , 2010. Biological mechanisms linking obesity and cancer risk: new perspectives. Annu. Rev. Med. 61, 301–316. [DOI] [PubMed] [Google Scholar]
- Rock, C.L. , Doyle, C. , Demark-Wahnefried, W. , Meyerhardt, J. , Courneya, K.S. , Schwartz, A.L. , Bandera, E.V. , Hamilton, K.K. , Grant, B. , McCullough, M. , Byers, T. , Gansler, T. , 2012. Nutrition and physical activity guidelines for cancer survivors. CA Cancer J. Clin. 62, 243–274. [DOI] [PubMed] [Google Scholar]
- Santos, C.R. , Schulze, A. , 2012. Lipid metabolism in cancer. FEBS J. 279, 2610–2623. [DOI] [PubMed] [Google Scholar]
- Seguin, F. , Carvalho, M.A. , Bastos, D.C. , Agostini, M. , Zecchin, K.G. , Alvarez-Flores, M.P. , Chudzinski-Tavassi, A.M. , Coletta, R.D. , Graner, E. , 2012. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br. J. Cancer. 107, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirin, O. , Kolonin, M.G. , 2013. Treatment of obesity as a potential complementary approach to cancer therapy. Drug Discov. Today. 18, 567–573. [DOI] [PubMed] [Google Scholar]
- Sjostrom, L. , Rissanen, A. , Andersen, T. , Boldrin, M. , Golay, A. , Koppeschaar, H.P. , Krempf, M. , 1998. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 352, 167–172. [DOI] [PubMed] [Google Scholar]
- Somasundar, P. , Frankenberry, K.A. , Skinner, H. , Vedula, G. , McFadden, D.W. , Riggs, D. , Jackson, B. , Vangilder, R. , Hileman, S.M. , Vona-Davis, L.C. , 2004. Prostate cancer cell proliferation is influenced by leptin. J. Surg. Res. 118, 71–82. [DOI] [PubMed] [Google Scholar]
- Steppan, C.M. , Bailey, S.T. , Bhat, S. , Brown, E.J. , Banerjee, R.R. , Wright, C.M. , Patel, H.R. , Ahima, R.S. , Lazar, M.A. , 2001. The hormone resistin links obesity to diabetes. Nature. 409, 307–312. [DOI] [PubMed] [Google Scholar]
- Tiikkainen, M. , Bergholm, R. , Rissanen, A. , Aro, A. , Salminen, I. , Tamminen, M. , Teramo, K. , Yki-Järvinen, H. , 2004. Effects of equal weight loss with orlistat and placebo on body fat and serum fatty acid composition and insulin resistance in obese women. Am. J. Clin. Nutr. 79, 22–30. [DOI] [PubMed] [Google Scholar]
- van Kruijsdijk, R.C. , van der Wall, E. , Visseren, F.L. , 2009. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol. Biomarkers Prev. 18, 2569–2578. [DOI] [PubMed] [Google Scholar]
- Vaupel, P. , Kallinowski, F. , Okunieff, P. , 1989. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 49, 6449–6465. [PubMed] [Google Scholar]
- Vendrell, J. , Broch, M. , Vilarrasa, N. , Molina, A. , Gomez, J.M. , Gutierrez, C. , Simon, I. , Soler, J. , Richart, C. , 2004. Resistin, adiponectin, ghrelin, leptin, and proinflammatory cytokines: relationships in obesity. Obes. Res. 12, 962–971. [DOI] [PubMed] [Google Scholar]
- Vijayakumar, M.V. , Pandey, V. , Mishra, G.C. , Bhat, M.K. , 2010. Hypolipidemic effect of fenugreek seeds is mediated through inhibition of fat accumulation and upregulation of LDL receptor. Obesity (Silver Spring). 18, 667–674. [DOI] [PubMed] [Google Scholar]
- Wolin, K.Y. , Carson, K. , Colditz, G.A. , 2010. Obesity and cancer. Oncologist. 15, 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita, T. , Honda, M. , Takatori, H. , Nishino, R. , Minato, H. , Takamura, H. , Ohta, T. , Kaneko, S. , 2009. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 50, 100–110. [DOI] [PubMed] [Google Scholar]
- Yan, D. , Avtanski, D. , Saxena, N.K. , Sharma, D. , 2012. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires β-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J. Biol. Chem. 287, 8598–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, L. , Biernacka, K.M. , Holly, J.M. , Jarrett, C. , Morrison, A.A. , Morgan, A. , Winters, Z.E. , Foulstone, E.J. , Shield, J.P. , Perks, C.M. , 2010. Hyperglycaemia confers resistance to chemotherapy on breast cancer cells: the role of fatty acid synthase. Endocr. Relat. Cancer. 17, 539–551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data