

Abstract
Inappropriate activation of mammalian/mechanistic target of rapamycin complex 1 (mTORC1) is common in cancer and has many cellular consequences including elevated endoplasmic reticulum (ER) stress. Cells employ autophagy as a critical compensatory survival mechanism during ER stress. This study utilised drug‐induced ER stress through nelfinavir in order to examine ER stress tolerance in cell lines with hyper‐active mTORC1 signalling. Our initial findings in wild type cells showed nelfinavir inhibited mTORC1 signalling and upregulated autophagy, as determined by decreased rpS6 and S6K1 phosphorylation, and SQTSM1 protein expression, respectively. Contrastingly, cells with hyper‐active mTORC1 displayed basally elevated levels of ER stress which was greatly exaggerated following nelfinavir treatment, seen through increased CHOP mRNA and XBP1 splicing. To further enhance the effects of nelfinavir, we introduced chloroquine as an autophagy inhibitor. Combination of nelfinavir and chloroquine significantly increased ER stress and caused selective cell death in multiple cell line models with hyper‐active mTORC1, whilst control cells with normalised mTORC1 signalling tolerated treatment. By comparing chloroquine to other autophagy inhibitors, we uncovered that selective toxicity invoked by chloroquine was independent of autophagy inhibition yet entrapment of chloroquine to acidified lysosomal/endosomal compartments was necessary for cytotoxicity. Our research demonstrates that combination of nelfinavir and chloroquine has therapeutic potential for treatment of mTORC1‐driven tumours.
Keywords: Nelfinavir, Chloroquine, Cancer, TSC, mTOR, ER stress, Autophagy
Highlights
Therapeutic potential for nelfinavir–chloroquine in mTORC1‐driven tumours.
Nelfinavir impairs mTORC1 signalling in wild‐type cells and activates autophagy.
mTORC1‐overactive cells have increased sensitivity to nelfinavir‐induced ER stress.
Chloroquine and mefloquine sensitise mTORC1‐overactive cells to nelfinavir‐induced death.
Abbreviations
- 3‐MA
3‐methyladenine
- ACC
acetyl‐CoA carboxylase
- AMPK
AMP‐dependent protein kinase
- ANOVA
analysis of variance
- ATF4
activating transcription factor 4
- BiP
binding immunoglobulin protein
- CHOP
C/EBP homologous protein
- CQ
chloroquine
- ddCT
delta–delta comparison test
- DTT
dithiothreitol
- ETL3
Eker rat leiomyoma‐derived cells
- ER
endoplasmic reticulum
- ELISA
enzyme‐linked immunosorbent assay
- FBS
foetal bovine serum
- GADD34
growth arrest and DNA damage‐inducible protein 34
- IRE1α
inositol‐requiring and ER‐to‐nucleus signalling protein 1α
- HEK293
human embryonic kidney 293
- LAM
lymphangioleiomyomatosis
- LC3
light chain 3
- MAPK
mitogen‐activated protein kinase
- mTORC1
mammalian/mechanistic target of rapamycin complex 1
- MEF
mouse embryonic fibroblast
- PBS
phosphate buffered saline
- PI3K
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase
- PP1
protein phosphatase 1
- PTEN
phosphatase and tensin homologue
- RAS
rat sarcoma
- Rheb
Ras homologue enriched in brain
- RIPA
radio immunoprecipitation assay
- rpS6
ribosomal protein S6
- S6K1
ribosomal protein S6 kinase beta‐1
- SESN2
sestrin 2
- SD
standard deviation
- SQSTM1
sequestosome‐1
- TBS
tris buffered saline
- TSC
tuberous sclerosis complex
- XBP1
X‐box binding protein 1
1. Introduction
The mammalian/mechanistic target of rapamycin complex 1 (mTORC1) pathway is frequently hyper‐activated in cancer. mTORC1 regulates many processes linked to cancer, such as cell growth, proliferation, metastasis, autophagy, metabolism and angiogenesis. mTORC1 hyper‐activation through genetic mutations of upstream components also underlies several tumour predisposition syndromes, such as tuberous sclerosis complex (TSC) and Cowden disease/PTEN hamartoma syndrome (Krymskaya and Goncharova, 2009). TSC is an autosomal dominant condition caused through mutations in either TSC1 or TSC2 and is characterised by tumour growth in multiple organs, neurocognitive problems and epilepsy (for review see Kohrman, 2012). The TSC1 and TSC2 tumour suppressor proteins together with TBC1D7 form a functional complex which has GTPase‐activating protein activity towards Ras homologue enriched in brain (Rheb). Rheb‐GTP potently activates mTORC1, while conversion of Rheb‐GTP to an inactive GDP‐bound state by TSC1/TSC2/TBC1D7 turns off mTORC1 (Tee et al., 2003; Dibble et al., 2012). Consequently, loss‐of‐function mutations in either TSC1 or TSC2 cause aberrant signal transduction through mTORC1. Mutations of TSC1, TSC2 and mTOR occur in some sporadic cancers, but more common components within mitogenic signalling upstream of mTORC1 are altered, such as PTEN or RAS within PI3K and MAPK pathways, respectively. TSC1 is mutated in approximately 15% of bladder cancers and 3% of clear cell renal carcinomas; TSC2 is mutated in 3% of bladder cancers and in 8% of well‐differentiated pancreatic neuroendocrine tumours and activating mTOR kinase domain mutations have been identified in intestinal adenocarcinomas and clear cell renal carcinomas (Platt et al., 2009; Sjodahl et al., 2011; Jiao et al., 2011). Frequent mutations affecting the wider PI3K/PTEN‐Akt‐mTOR signalling network have also been reported in clear cell renal cancers and head and neck cancer (Sato et al., 2013; Liao et al., 2011).
Aberrant signalling through mTORC1 is known to enhance the basal levels of ER stress, which is done in part by heightened levels of de novo protein synthesis, leading to an accumulation of unfolded proteins within the ER (Kang et al., 2011; Ozcan et al., 2008). mTORC1 further enhances the burden of ER stress through autophagy repression, as autophagy is utilised by the cell to remove unfolded protein aggregates to restore the protein folding environment within the ER (Høyer‐Hansen and Jäättelä, 2007). Related work highlighting the crosstalk between autophagy and ER homeostasis showed that induction of ER stress by thapsigargin was via impairment of autophagosome–lysosome fusion (Ganley et al., 2011). Elevated cell stress is common in cancer and could potentially be exploited therapeutically (Hanahan and Weinberg, 2011). For instance, it is recognised that compromised stress recovery pathways in cancer cells may confer sensitivity to stress‐inducing drugs as many cancer cell lines are sensitive to endoplasmic reticulum (ER) stress‐inducers (Liu et al., 2012; Li et al., 2013; Zang et al., 2009), where excessive or prolonged ER stress leads to cell death (for review see Appenzeller‐Herzog and Hall, 2012).
The mTOR inhibitor and rapamycin analogue, everolimus, is licensed for the treatment of renal angiomyolipomas and subependymal giant cell astrocytomas in patients with TSC (Kohrman, 2012). However, rapalogues only induce partial regression of tumour volume and the tumours often regrow following treatment cessation (Franz and Weiss, 2012). Evidently, alternative therapeutic strategies must be explored for mTORC1‐driven tumours and targeting existing stress pathways is one possibility. Ozcan et al. (2008) and Di Nardo et al. (2009) demonstrated that Tsc2−/− cells elicited a higher level of ER stress that was dependent on aberrant signalling through mTORC1. In addition, tumours derived from TSC patients were also observed to have increased levels of ER stress. Supporting the notion that targeting this already elevated ER stress pathway in Tsc2−/− cells is a therapeutic option, both cellular and murine models of TSC have proved to be sensitive to ER stress‐inducers such as thapsigargin (Siroky et al., 2012; Kang et al., 2011; Ozcan et al., 2008). ER stress upregulates autophagy as part of the unfolded protein response; a cytoprotective process purposed to alleviate ER stress through removal of misfolded protein products (He and Klionsky, 2009). Cells lacking Tsc2 show an attenuated autophagic response due to mTORC1‐mediated repression despite elevated levels of ER stress (Siroky et al., 2012; Parkhitko et al., 2011).
In this study we utilised nelfinavir, an ER stress‐inducer that was originally clinically approved for treatment of human immunodeficiency virus infection. Nelfinavir was more recently observed to have anti‐cancer activity in pre‐clinical cell and xenograft models and is currently being investigated in multiple clinical trials (Buijsen et al., 2013; Pan et al., 2012; Rengan et al., 2012; Alonso‐Basanta et al., 2014). Nelfinavir has been shown to affect many cellular processes linked to ER homeostasis including proteasome inhibition, impairment of signal transduction through the PI3K/Akt pathway and induction of autophagy, although some actions appear cell type dependent (Gills et al., 2007). We also employed chloroquine, a widely‐used anti‐malarial drug which has multiple effects on cells including alteration of mitochondrial and lysosomal membrane potential and arrest of autophagy flux in the later stages through alteration of lysosomal pH (Poole and Ohkuma, 1981). There is current interest in the clinical use of chloroquine in combination with conventional and novel anti‐cancer agents (Goldberg et al., 2012; Solomon and Lee, 2009; Sotelo et al., 2006). The rational for the use of chloroquine in these combinations is often inhibition of a presumed protective autophagic response. However, there is emerging evidence in some contexts that the cytotoxic action of chloroquine may be largely independent of autophagy inhibition (Maycotte et al., 2012; Harhaji‐Trajkovic et al., 2012; Seitz et al., 2013). Herein, we examined the effects of nelfinavir and chloroquine in multiple cell line models with hyper‐activated mTORC1 signalling and elevated basal levels of ER stress. Unlike previous studies in cancer cell lines, we were able to test the hypothesis that loss of Tsc2 and consequent mTORC1 activation specifically sensitises cells to drug‐induced ER stress and cell death. Importantly, we directly compared the effectiveness of nelfinavir and chloroquine in Tsc2−/− cell lines with matched controls expressing wild‐type Tsc2 before validating our findings in a human non‐small cell lung cancer cell line known to have aberrant mTORC1 signalling due to an oncogenic KRAS mutation. We found chloroquine promotes nelfinavir‐induced ER stress and cell death in mTORC1‐hyperactive cells in an autophagy‐independent manner. Our data suggests a novel therapeutic approach for killing tumour cells which have aberrant levels of mTORC1 activity and higher basal levels of ER stress.
2. Materials and methods
2.1. Cell culture and reagents
Tsc2+/+ p53−/− and Tsc2−/− p53−/− mouse embryonic fibroblasts (MEFs) were kindly provided by David J. Kwiatkowski (Harvard University, Boston, USA). Tsc2−/− ETL3 (Eker rat leiomyoma‐derived cells) and control ELT3‐Tsc2 cells in which Tsc2 is re‐expressed were kindly provided by Cheryl Walker (M.D. Anderson Cancer Center, Houston, USA). Human embryonic kidney 293 (HEK293) cells and human lung carcinoma (NCI‐H460) cells were purchased from ATCC. All cell lines were cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% (v/v) foetal bovine serum (FBS), 100 U/ml penicillin and 100 μ/ml streptomycin (Life Technologies Ltd., Paisley, UK) in a humidified incubator at 37 °C, 5% (v/v) CO2. Nelfinavir mesylate hydrate, chloroquine di‐phosphate salt, mefloquine hydrochloride, bafilomycin‐A1, 3‐methyladenine (3‐MA) and thapsigargin were purchased from Sigma–Aldrich Company Ltd. (Dorset, UK). Nelfinavir, mefloquine, bafilomycin‐A1 and thapsigargin were dissolved in dimethyl sulfoxide (DMSO) at 30 mM, 50 mM, 2.5 mM and 10 mM stock solutions, respectively. Chloroquine was dissolved in fresh culture medium to a 100 mM stock and further diluted in culture medium to the required concentrations for use. 3‐MA was dissolved in dH2O to a concentration of 100 mM. Drug(s) or DMSO vehicle control was added to cell culture medium in a manner that kept the final DMSO vehicle concentration below 0.5% (v/v).
2.2. mRNA extraction and reverse transcription
Cells were plated onto 6 cm2 plates and allowed to adhere overnight. Following the appropriate treatment, cells were first washed in phosphate buffered saline (PBS) then lysed using 1 ml RNAprotect Cell Reagent (Qiagen, West Sussex, United Kingdom). RNA was extracted using the RNeasy Plus mini kit (Qiagen). Cell lysates were homogenized using Qiashredders (Qiagen) during the RNA extraction procedure. The concentration and purity of RNA was determined by measuring the absorbance at 260 nm and 280 nm in a Nanodrop spectrophotometer (Thermo Scientific, Hemel Hempsted, UK). Total RNA from each sample (1 μg) was transcribed into cDNA using Quantitect reverse transcription kit (Qiagen) in a thermal cycler (Applied Biosystems, California, USA). RNA, gDNA wipeout buffer and RNase‐free water at a final volume of 14 μl were incubated at 42 °C for 2 min. Quantiscript Reverse Transcriptase (Qiagen), Quantiscript RT buffer (Qiagen) and primers were added to a final volume of 20 μl and samples incubated at 42 °C for 15 min followed by 95 °C for 3 min.
2.3. Detection of endoplasmic reticulum stress
Quantitative PCR (Q‐PCR) reactions were conducted in 96 well plates (Thermo Fisher, Loughborough, UK) using 25 ng DNA per reaction, appropriate primer sets and SYBR Green PCR Master mix (Qiagen). Quantitect β‐actin and C/EBP Homologous Protein (CHOP) primers were purchased from Qiagen. Q‐PCR was performed using an Applied Biosystems 7500 real‐time cycler as follows: initial denaturation step (95 °C, 15 min); 40 cycles of denaturation (94 °C, 15 s); annealing step (55 °C, 30 s); extension step (72 °C, 30 s). The amplification products were quantified during the extension step in the 40th cycle. Relative quantification was performed using the comparative CT method (ddCT) with β‐actin as reference gene and DMSO vehicle‐treated Tsc2+/+ MEFs as the calibrator. Melting curve analysis was performed to verify their specificity of the RT‐PCR products. XBP1 primers [Forward: 5′‐AAA CAG AGT AGC AGC TCA GAC TGC‐3′, Reverse: 5′‐TCC TTC TGG GTA GAC CTC TGG GA‐3′] were synthesised through MWG Operon‐Eurofin (Ebersberg, Germany). PCR was performed using an Applied Biosystems GeneAmp 9700 PCR system as follows: initial denaturation step (98 °C, 3 min); 31 cycles of denaturation (9 °C, 30 s); annealing step (60 °C, 30 s); extension step (72 °C, 1 min). XBP1 products were run on 3% (w/v) agarose gels, 1× Tris–Acetate–EDTA (pH 8.0) using GelRed nucleic acid stain (Cambridge Bioscience, Cambridge, UK). DNA samples were prepared for loading by the addition of 5× loading buffer. PCR products of X‐box binding protein 1 (XBP1) were 480 bp, unspliced, and 454 bp, spliced.
2.4. Antibodies and western blotting
Antibodies towards ribosomal protein S6 (rpS6), phospho‐rpS6 (Ser235/236), p70‐S6 kinase 1 (S6K1), phospho‐S6K1 (Th389), Acetyl‐CoA carboxylase (ACC), phospho‐ACC (Ser79), AMP‐dependent protein kinase (AMPK), phospho‐AMPK (Thr172), binding immunoglobulin protein (BiP), inositol‐requiring and ER‐to‐nucleus signalling protein 1α (IRE1α), TSC2 and β‐actin were purchased from Cell Signaling Technology (Danvers, USA). LC3 antibody was purchased from Novus Ltd. (Cambridge, UK), while the antibody for Sequestesome 1 (SQSTM1) was purchased from Progen Biotecknik (Heidelberg, Germany). Cells were washed in PBS and then lysed in radio immunoprecipitation assay (RIPA) buffer (Sigma–Aldrich) supplemented with Complete Mini protease inhibitor cocktail, PhosSTOP phosphatase inhibitor cocktail (Roche Diagnostics Ltd., Burgess Hill, UK) and 1 mM dithiothreitol (DTT) at 4 °C. Following centrifugation at 13,000 rpm for 8 min at 4 °C, protein concentrations were optimised by Bradford assay (Sigma–Aldrich). Samples were diluted in 4× NuPAGE loading sample buffer (Life Technologies) with 25 mM DTT and boiled at 70 °C for 10 min. Western blot was performed as previously described (Dunlop et al., 2011). β‐actin was used to as a control for equal protein loading.
2.5. Cell viability assay
Cells were plated at 100% confluency (5 × 103/well) in a 96‐well plate. Drug or DMSO vehicle was added and the plates incubated at 37 °C for 48 h. Media was removed from the cells and the plates frozen at −80 °C. Following thawing, 200 μl CyQuant mix, prepared as described in the CyQUANT Cell Proliferation Assay (Life Technologies) manufacturer's handbook, was added to each well. Fluorescence was measured using a FLUOstar OPTIMA fluorometer (BMG LABTECH, Buckinghamshire, UK) at 485 nm and 520 nm. To determine cell number, counted cells were co‐analysed as a standard curve. Results are expressed relative to untreated Tsc2+/+ cells which are assigned a value of 100%.
2.6. DNA fragmentation ELISA
DNA fragmentation was measured with the Cell Death Detection ELISA kit (Roche) according to the manufacturer's protocol. This one‐step colorimetric sandwich ELISA allows relative quantification of histone‐complexed DNA fragments which are indicative of apoptosis. 5 × 103 cells were plated in 96‐well plates and incubated overnight. Drugs were added to the required concentration and cells were incubated for 48 h. The relative quantity of immobilized antibody–histone complex was determined photometrically with absorbance at 405 nm using 2,2′‐azino‐bis‐3‐ethylbenzothiazoline‐6‐sulfonic acid as a peroxidase substrate. Results are expressed relative to untreated Tsc2+/+ cells which were assigned a value of 1.
2.7. Late cell death assay
For assessment of cell viability, treated cells were incubated with 3 μM DRAQ7 for 10 min at 37 °C prior to flow cytometry using excitation at 488 nm and detection of fluorescence in log mode at wavelengths greater than 695 nm (far red). Flow cytometry was performed using a FACS Calibur flow cytometer (Becton Dickinson, Cowley, UK) and Cell Quest Pro software (Beckton Dickinson Immunocytometry Systems) was used for signal acquisition. To control for autofluorescence, unstained cells were also analysed. Correlated signals were collected for a minimum of 10,000 events.
2.8. Statistical analysis
Experiments were carried out at least 3 times. Where applicable, results are expressed as mean ± standard deviation (SD). Student's t‐test and ANOVA were used and significance reported at p ≤ 0.05.
3. Results
3.1. Nelfinavir induces ER stress in Tsc2−/− MEFs
We initially assessed ER stress after 3 h treatment with 20 μM nelfinavir in Tsc2+/+ and Tsc2−/− MEF cell lines by measurement of CHOP mRNA (Figure 1A). In DMSO vehicle treatment, Tsc2−/− MEFs had over 3‐fold increase in relative expression of CHOP mRNA compared to wild‐type MEFs, which indicates an elevated basal level of ER stress in the absence of Tsc2. Although nelfinavir increased CHOP mRNA levels in both cell lines, relative expression was more pronounced in the Tsc2−/− MEFs (4 times more when compared to Tsc2+/+, Figure 1A). These data suggest that the Tsc2−/− MEFs are more sensitive to nelfinavir‐induced ER stress than the wild‐type cells, which is concurrent with previous findings (Ozcan et al., 2008). Thapsigargin, a potent inducer of ER stress, served as a positive control which enhanced CHOP mRNA expression in both MEF cell lines. To confirm ER stress‐induction, we also analysed XBP1 mRNA splicing as a reliable readout of nelfinavir‐induced ER stress (Brüning, 2011). We observed a higher basal level of XBP1 mRNA splicing in Tsc2−/− cells (Figure 1B) and nelfinavir also selectively induced ER stress in the Tsc2−/− MEFs as observed by increased splicing (compare lane 5 versus lane 2). In both MEF cell lines, thapsigargin led to complete XBP1 mRNA splicing. We next measured protein expression of BiP and IRE1α which are considered common markers of ER stress (Figure 1C). Both BiP and IRE1α protein levels were elevated in the Tsc2−/− MEFs, which further confirms that cells lacking Tsc2 have a higher basal level of ER stress. However, 3 h treatment of either nelfinavir or thapsigargin failed to further enhance BiP or IRE1α protein in the Tsc2−/− MEFs, presumably as ER stress levels were already basally high (Siroky et al., 2012; Kang et al., 2011; Ozcan et al., 2008).
Figure 1.
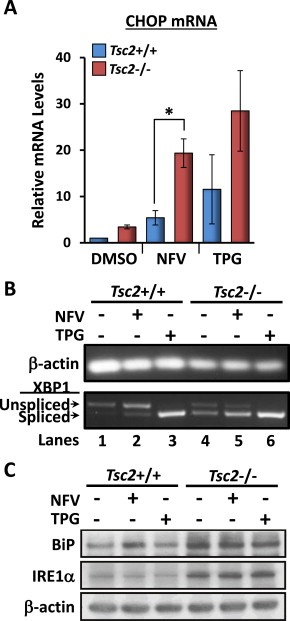
Nelfinavir‐induced ER stress is more pronounced in Tsc2−/− MEFs. Tsc2−/− and Tsc2+/+ MEFs were treated with either DMSO vehicle alone, 20 μM nelfinavir (NFV), or 1 μM thapsigargin (TPG) for 3 h. Cells were then appropriately harvested to analyse A) CHOP mRNA, which was standardised against β‐actin mRNA (n = 3, *p < 0.05), B) PCR products for XBP1 mRNA were resolved on agarose gels (unspliced = 480 bp upper band, spliced = 454 bp lower band). β‐actin is shown as a control. C) Protein extracts were analysed for BiP, IRE1α and β‐actin levels. n = 3, *p = <0.05.
3.2. Nelfinavir inhibits mTORC1 signal transduction and induces autophagy
We next analysed mTORC1 signal transduction in the Tsc2+/+ and Tsc2−/− MEFs by examining rpS6 phosphorylation (Figure 2A). In the Tsc2+/+ MEFs, we observed complete inhibition of rpS6 phosphorylation after nelfinavir treatment but inhibition of mTORC1 signalling was incomplete in the Tsc2−/− MEFs, which reveals nelfinavir requires Tsc2 for full repression of mTORC1 signalling. Cell quantification assays revealed that nelfinavir reduced the number of Tsc2−/− cells by 16%, whereas Tsc2+/+ MEFs were unaffected (Figure 2B). Furthermore, there was a 3‐fold increase in DNA fragmentation of Tsc2−/− MEFs compared to Tsc2+/+ after 48 h exposure to nelfinavir (Figure 2C), which indicates that loss of Tsc2 sensitises cells to nelfinavir‐induced cytotoxicity. Given that autophagy is modulated by both mTORC1 and ER stress, we next examined markers of autophagy after treatment with nelfinavir. Untreated Tsc2−/− MEFs had a lower level of autophagy when compared to the Tsc2+/+ MEFs, as observed by increased protein accumulation of SQSTM1 (Figure 2D). Both nelfinavir and thapsigargin increased autophagy as observed by decreased SQSTM1 protein. Increased LC3 lipidation to the lower resolving LC3‐II isoform suggest either increased autophagy or a block in late stage autophagy (or both). Furthermore, we observed marked activation of AMPK upon treatment with nelfinavir, via increased phosphorylation of AMPK and its substrate, ACC (Figure 2D).
Figure 2.
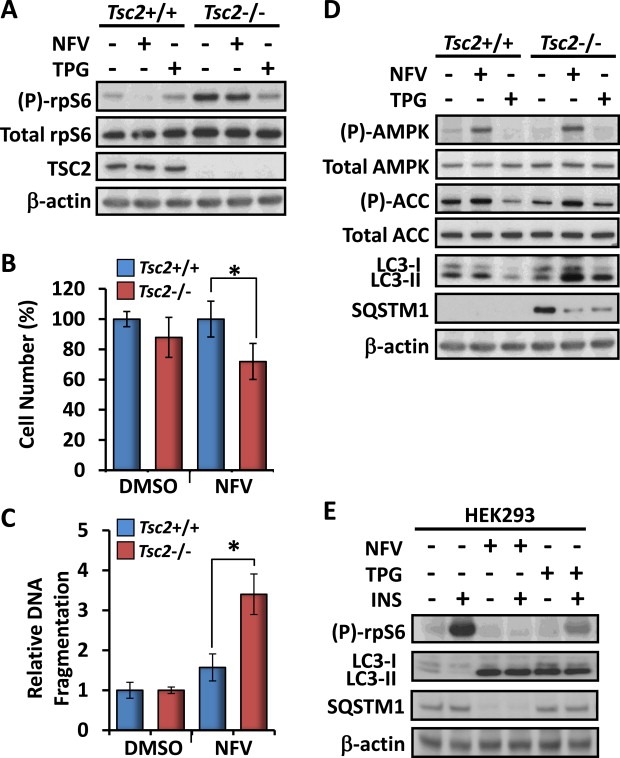
Nelfinavir inhibits mTORC1, induces autophagy and selectively kills Tsc2−/− MEFs. Tsc2−/− and Tsc2+/+ MEFs were treated with either DMSO vehicle alone, 20 μM nelfinavir (NFV), or 1 μM thapsigargin (TPG) for 3 h. A) Total and phosphorylated ‘(P)‐rpS6’, TSC2 and β‐actin were determined by western blot analysis from total protein lysates. 48 h DMSO and 20 μM nelfinavir‐treated Tsc2−/− and Tsc2+/+ MEFs were subjected to B) cell viability and C) cell death assays. D) Samples (prepared as for A) were subjected to western blot analysis to determine total and phosphorylated ‘(P)‐AMPK’, total and phosphorylated ‘(P)‐ACC’, and LC3 (‘LC3‐I’ and ‘LC3‐II’). E) HEK293 cells were pre‐treated with either 20 μM nelfinavir or 1 μM thapsigargin as indicated for 30 min prior to 100 nM insulin stimulation for 30 min. Total and phosphorylated ‘(P)‐rpS6’, LC3 (‘LC3‐I’ and ‘LC3‐II’) and β‐actin were then determined by western blot analysis from prepared cell lysates. n = 3, *p = <0.05.
To validate some of our initial findings, we examined the effects of nelfinavir on mTORC1 signalling and autophagy after acute insulin stimulation in HEK293 cells (Figure 2E). The high level of insulin‐induced rpS6 phosphorylation was completely abolished upon treatment with nelfinavir. SQSTM1 protein levels were also markedly reduced with nelfinavir, which again suggests enhanced autophagy fluxing. Similar to the effects we observed in the MEF cell lines, both nelfinavir and thapsigargin strongly induced LC3‐II accumulation.
3.3. Chloroquine enhances nelfinavir‐induced cell death in Tsc2−/− cells
Given that autophagy is a well‐established survival mechanism employed by cells to alleviate ER stress, we wanted to determine whether inhibition of autophagy would enhance the potency of nelfinavir to selectively kill Tsc2−/− cells. To do this we used chloroquine which blocks autophagy during the final stages. Analysis of S6K1 phosphorylation revealed that chloroquine alone did not affect mTORC1 signal transduction but combination with nelfinavir was particularly effective at reducing S6K1 phosphorylation in Tsc2+/+ MEFs and modestly in Tsc2−/− MEFs (Figure 3A). Chloroquine treatment blocked autophagy, more effectively in the presence of nelfinavir, as observed by an accumulation of LC3‐II (Figure 3A). In the Tsc2−/− MEFs, a combination of nelfinavir and chloroquine was much more effective at induction of CHOP mRNA levels (by 27%, Figure 3B) and XBP1 splicing (Figure 3C), than either agent alone.
Figure 3.
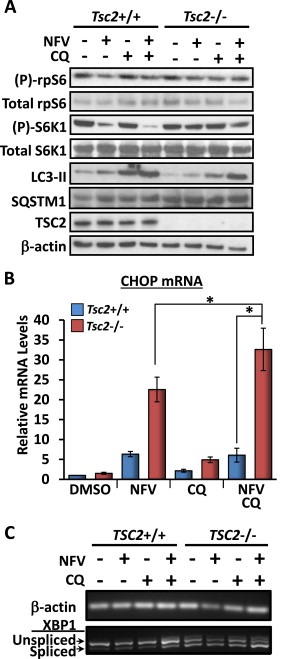
Chloroquine further sensitises Tsc2−/− MEFs to nelfinavir‐induced ER stress. Tsc2−/− and Tsc2+/+ MEFs were treated with DMSO vehicle alone, 20 μM nelfinavir (NFV), and/or chloroquine (CQ, 10 μM and 20 μM) as indicated. A) Following 3 h drug treatment, protein lysates were subjected to western blot analysis to determine levels of total and phosphorylated ‘(P)‐rpS6’, total and phosphorylated ‘(P)‐S6K1’, LC3‐II, TSC2 and β‐actin. B) CHOP mRNA was analysed and standardised against β‐actin (n = 3, *p < 0.05). C) PCR products for XBP1 mRNA were resolved on agarose gels (unspliced = 480 bp upper band, spliced = 454 bp lower band). β‐actin mRNA is shown as a control. n = 3, *p = <0.05.
We next measured cell death by flow cytometry after staining with DRAQ7. DRAQ7 is a membrane impermeable far‐red fluorescence DNA‐binding stain that is used to measure cell death as a consequence of increased membrane permeability. Events were gated to exclude fragmented cells and debris before gating viable and non‐viable populations according to DRAQ7 staining. Combined 24 h treatment with nelfinavir and chloroquine selectively killed Tsc2−/− MEFs by an average of 43% compared to 6% cell death in Tsc2+/+ MEFs (Figure 4A and B). Using DNA fragmentation ELISA we found 48 h combined nelfinavir and chloroquine led to over 5‐fold increase in relative DNA fragmentation in Tsc2−/− MEFs when compared to nelfinavir treatment alone (Figure 4C).
Figure 4.
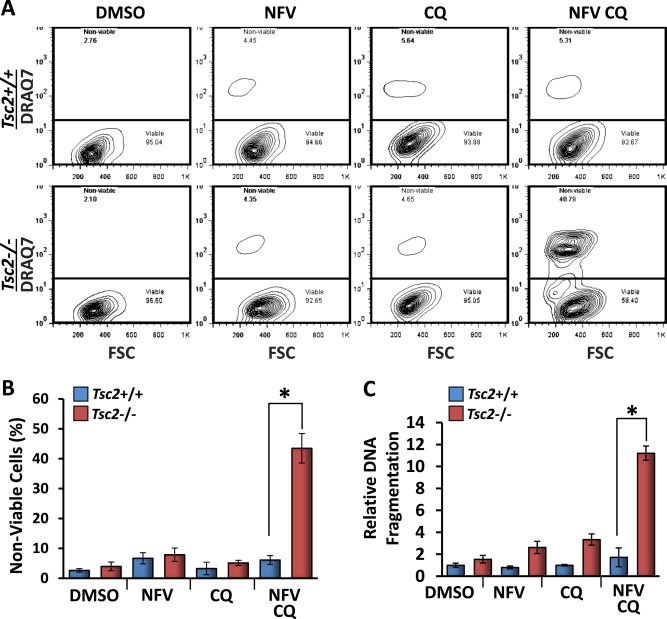
Combined treatment of nelfinavir and chloroquine selectively kills Tsc2−/− MEFs. Tsc2−/− and Tsc2+/+ MEFs were treated with either DMSO vehicle alone, 20 μM nelfinavir (NFV), 20 μM chloroquine (CQ), or both NFV and CQ over 24 h and subjected to cell death assays. A) For quantification of cell death, Tsc2+/+ and Tsc2−/− MEFs were subjected to flow cytometry analysis following DRAQ7 staining. DRAQ7 exclusion (below line) represents the viable cell population, whilst positive DRAQ7 staining (above line) indicates cell death. The number of DRAQ7‐stained cells are graphed in B. C) Relative DNA fragmentation as determined by cell death ELISA. n = 3, *p = <0.05.
3.4. Enhancement of nelfinavir‐induced cell death is not due to autophagy inhibition by chloroquine
To confirm that enhancement of nelfinavir‐induced cell death was due to autophagy inhibition by chloroquine, Tsc2+/+ and Tsc2−/− MEFs were treated with two other known inhibitors of autophagy, bafilmycin‐A1 and 3‐methyladenine (3‐MA), in combination with nelfinavir. Analysis by western blot following 3 h of treatment confirmed bafilomycin‐A1 inhibited autophagy comparable to chloroquine, as shown by increased levels of SQSTM1 and LC3‐II (Figure 5A). 3‐MA is known to have differing effects on autophagy depending on nutrient status (Wu et al., 2010). Therefore, the lack of observed autophagy inhibition by 3‐MA was unsurprising. Interestingly, 24 h treatment with combined nelfinavir and bafilomycin‐A1 or nelfinavir and 3‐MA failed to induce Tsc2−/− cell death to a similar extent as combined nelfinavir and chloroquine (on average 3% and 7% vs 25%, respectively, Figure 5B–C). To determine whether cell death was exclusive to chloroquine, we analysed the chloroquine analogue, mefloquine, and found it to be highly effective at killing Tsc2−/− MEFs in combination with nelfinavir (on average 63%). Further supporting autophagy inhibition is not the underlying mechanism by how these chloroquine compounds sensitise Tsc2−/− cells to nelfinavir, mefloquine did not repress autophagy (as observed by low levels of SQSTM1 and no accumulation of lipidated LC3‐II, Figure 5A). Collectively, our data implies that chloroquine sensitises nelfinavir‐induced cell death via a mechanism that is not autophagy inhibition. To explore other possible mechanisms of drug action, we combined nelfinavir and chloroquine treatment with bafilomycin‐A1, which blocks the v‐ATPase and prevents entrapment of chloroquine to lysosomes and late endosomes. We observed that bafilomycin‐A1 robustly rescued cell death by nelfinavir and chloroquine in the Tsc2−/− MEFs (Figure 5B). This indicates that entrapment of chloroquine to acidified lysosomes and late endosomal compartments is required for chloroquine‐mediated enhancement of nelfinavir‐induced cell death.
Figure 5.
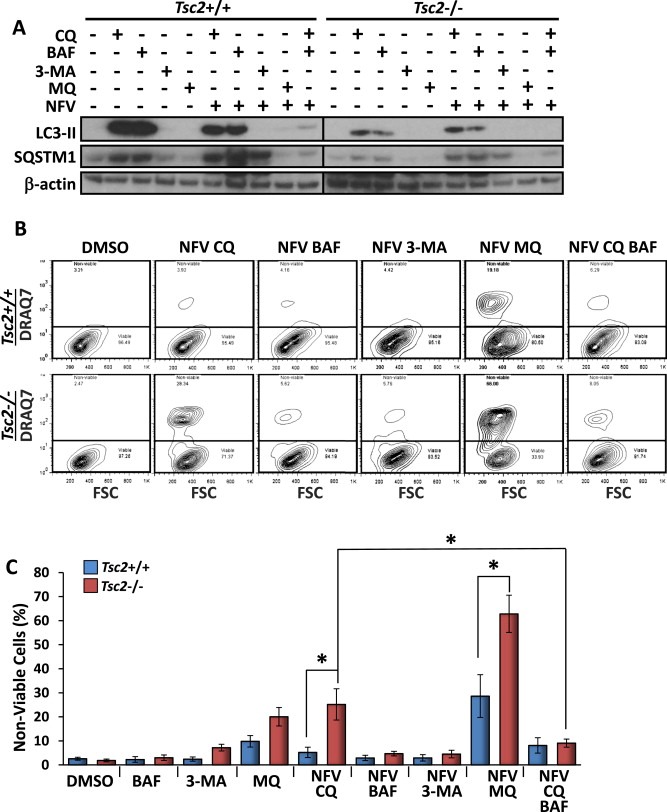
Combination treatments of nelfinavir and bafilomycin‐A1 or nelfinavir and 3‐methyladenine fail to kill Tsc2−/− MEFs. Tsc2−/− and Tsc2+/+ MEFs were treated with either DMSO vehicle alone, 2.5 μM baflilomycin‐A1 (BAF), 10 mM 3‐methyladenine (3‐MA), 10 μM mefloquine (MQ) or combined nelfinavir (NFV) BAF/3‐MA/MQ. A) Following 3 h drug treatment, protein lysates were subjected to western blot analysis to determine levels of LC3‐II, SQSTM1 and β‐actin. B) For quantification of cell death following 48 h of treatment, Tsc2−/− and Tsc2+/+ MEFs were subjected to flow cytometry analysis following DRAQ7 staining. DRAQ7 exclusion (below line) represents the viable cell population, whilst positive DRAQ7 staining (above line) indicates cell death. The number of DRAQ7‐stained cells are graphed in C. n = 3, *p = <0.05.
To confirm the cytotoxic effects of combined nelfinavir and chloroquine, we utilised another well‐established Tsc2−/− cell line model, the ELT3 rat smooth muscle cell line (Eker et al., 1983). Similar to the MEFs, 3 h combined treatment with nelfinavir and chloroquine reduced phosphorylation of S6K1, lowered SQSTM1 protein and enhanced levels of LC3‐II in ELT3‐V3 (Tsc2−/−) cells (Figure 6A). Cell viability was significantly reduced in ELT3‐V3 cells compared to ELT‐T3 cells following 48 h of combined nelfinavir and chloroquine treatment (Figure 6B). Cell death assays following 48 h of treatment showed selective cell death in ELT3‐V3 cells by an average of 34% compared to 9% in ELT3‐T3 (Tsc2+/+) cells (Figure 5C,D). Similarly to the Tsc2−/− MEFs, the ELT3‐V3 cells were almost 5‐fold more sensitive to 48 h combined treatment of nelfinavir and chloroquine when compared to ELT3‐T3 cells, as determined by DNA fragmentation (Figure 6E).
Figure 6.
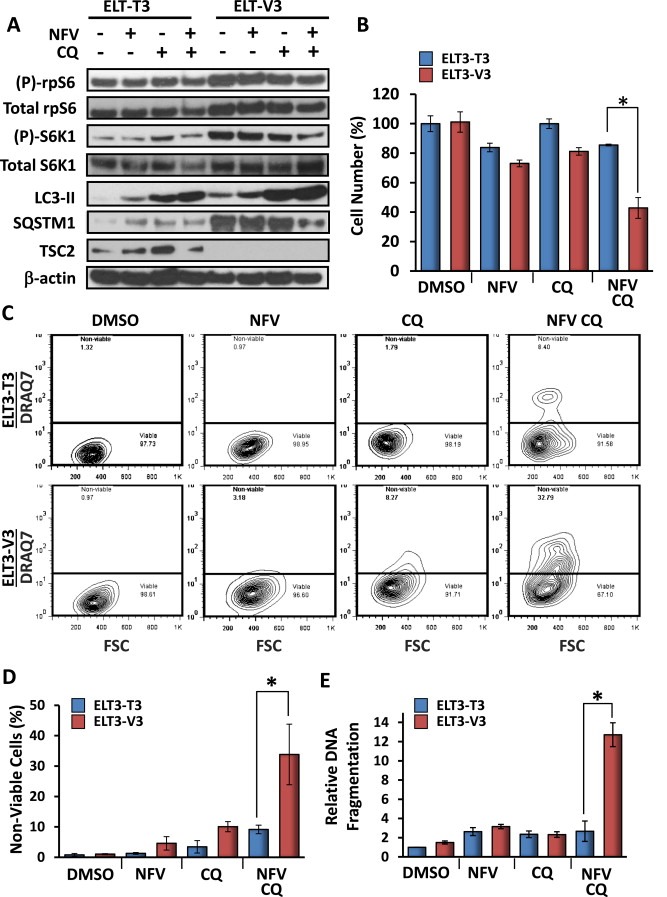
Combined treatment of nelfinavir and chloroquine selectively kills Tsc2−/− ELT3 cells. Tsc2−/− and Tsc2+/+ ELT3 cells were treated with either DMSO vehicle alone, 20 μM nelfinavir (NFV), 20 μM chloroquine (CQ), or both NFV and CQ and subjected to western blot and death assays. A) Following 3 h drug treatment, protein lysates were subjected to western blot analysis to determine levels of total and phosphorylated ‘(P)‐rpS6’, total and phosphorylated ‘(P)‐S6K1’, LC3‐II, SQSTM1, TSC2 and β‐actin. B) Quantification of cell viability. C) For quantification of cell death following 48 h of treatment, Tsc2+/+ and Tsc2−/− ELT3 cells were subjected to flow cytometry analysis following DRAQ7 staining. DRAQ7 exclusion (below line) represents the viable cell population, whilst positive DRAQ7 staining (above line) indicates cell death. The number of DRAQ7‐stained cells are graphed in D. E) Relative DNA fragmentation as determined by cell death ELISA. n = 3, *p = <0.05.
3.5. Combined nelfinavir and chloroquine effectively kills cancer cells with over‐active mTORC1
To validate our previous findings in a cancer cell line with over‐active mTORC1, NCI‐H460 human lung carcinoma cells were treated with nelfinavir and chloroquine. As a control, rapamycin was used to inhibit mTORC1 signalling in these cells. Similar to the MEF and ELT3 cell lines, 3 h combined nelfinavir and chloroquine treatment reduced both phosphorylated rpS6 and S6K1 signalling and increased levels of LC3‐II (Figure 7A). As expected, rapamycin completely inhibited both rpS6 and S6K1 phosphorylation. 48 h combination treatment with nelfinavir and chloroquine effectively killed NCI‐H460 cells by an average of 49% which appeared to be partially rescued by the addition of rapamycin to an average of 26% although this trend was not significant (P = 0.127, Figure 7B, C). This suggests mTORC1 activity contributes to induction of cell death following treatment with nelfinavir and chloroquine.
Figure 7.
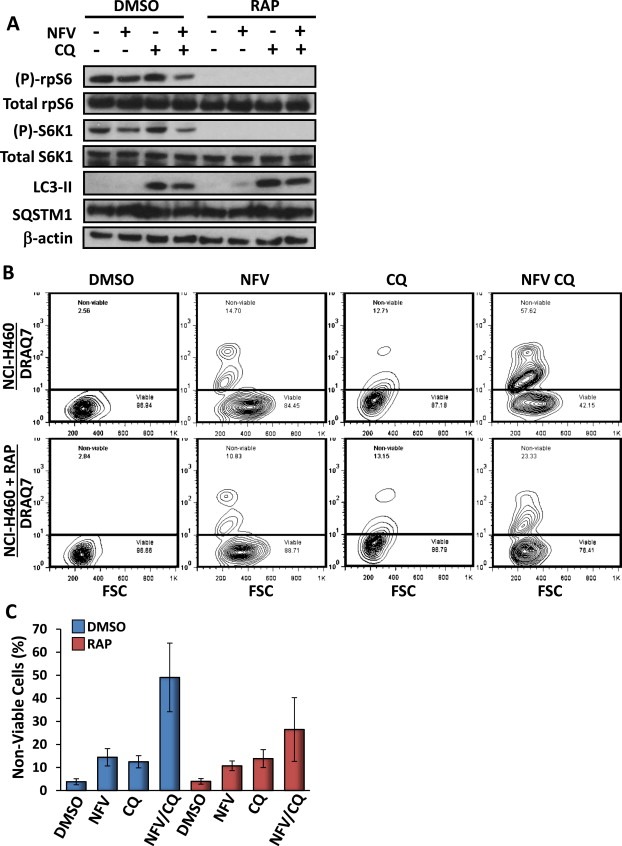
Combined treatment of nelfinavir and chloroquine kills NCI‐H460 lung cancer cells. NCI‐H460 cells were treated with either DMSO vehicle alone, 20 μM nelfinavir (NFV), 20 μM chloroquine (CQ), or both NFV and CQ and subjected to western blot and death assays. 100 nm rapamycin was used to inhibit mTORC1 signalling. A) Following 3 h drug treatment, protein lysates were subjected to western blot analysis to determine levels of total and phosphorylated rpS6, total and phosphorylated S6K1, LC3‐II, SQSTM1 and β‐actin. B) For quantification of cell death following 48 h of treatment, NCI‐H460 cells were subjected to flow cytometry analysis following DRAQ7 staining. DRAQ7 exclusion (below line) represents the viable cell population, whilst positive DRAQ7 staining (above line) indicates cell death. The number of DRAQ7‐stained cells are graphed in C. n = 3.
4. Discussion
In this study we show that combination of nelfinavir and chloroquine is selectively cytotoxic to Tsc2−/− cells. Importantly, we were able to directly compare the potency of this drug combination in Tsc2−/− cell lines with matched controls expressing wild‐type Tsc2. Tsc2−/− cells exhibit constitutive mTORC1 activation, high levels of basal ER stress and impaired autophagy. We show a combination of nelfinavir and chloroquine potentiated ER stress and affected autophagy in Tsc2−/− cells.
Unfolded proteins in the ER are not exclusively removed via the autophagy pathway; protein aggregates are also removed via the proteasome (Rubinsztein, 2006). Nelfinavir has previously been shown to inhibit the proteasome (Bono et al., 2012; Gupta et al., 2007), suggesting inhibition of the proteasome allows accumulation of unfolded protein aggregates in the ER, leading to enhanced ER stress and consequent autophagy induction. Despite observing changes to the autophagy flux upon nelfinavir and chloroquine or nelfinavir and bafilomycin‐A1 treatment, induction of cell death was exclusive to the use of nelfinavir with chloroquine or its analogue, mefloquine. In contrast to chloroquine, mefloquine did not inhibit autophagy in the MEF cell lines (Figure 5A). Supporting our findings, Shin et al. (2012) showed mefloquine to induce autophagy, and combined autophagy repression with 3‐MA aggravated mefloquine‐mediated cytotoxicity. Our data indicates an autophagy‐independent role for chloroquine is the underlying mechanism of enhanced sensitivity to nelfinavir‐induced cell death: entrapment of chloroquine to acidified lysosomal/endosomal compartments was necessary for cytotoxicity, as we observed bafilomycin‐A1 inhibition of v‐ATPase (Klionsky et al., 2008) prevented nelfinavir and chloroquine‐induced cell death. Our results are in accordance with previous data showing nutrient deprivation combined with chloroquine treatment effectively killed glioma, melanoma and fibrosarcoma cells which was not replicated with bafilomycin‐A1, and that addition of bafilomycin‐A1 was able to rescue the effects of chloroquine (Harhaji‐Trajkovic et al., 2012).
Cells lacking Tsc2 have a truncated ER stress response (Kang et al., 2011), implying that TSC2 is required for a normal response to ER stress. There are several signalling mechanisms which converge upon mTORC1 during a normal ER stress response: GADD34 recruits protein phosphatase 1 (PP1) which activates TSC2 through dephosphorylation and subsequent inhibition of Rheb/mTORC1 (Uddin et al., 2011; Watanabe et al., 2007; Kojima et al., 2003). As we observed increased AMPK activation (Figure 2D), nelfinavir likely inhibits mTORC1 through AMPK‐mediated TSC2 activation. This method of mTORC1 inhibition through AMPK has been previously established in Tsc2−/− cells (Gwinn et al., 2008). Interestingly, recent research by Brüning et al. (2013) linked AMPK activation to ER stress through the sestrin 2 (SESN2) protein. Furthermore, their research indicated that nelfinavir‐mediated ER stress results in accumulation of Activating Transcription Factor 4 (ATF4), a known regulator of CHOP and SESN2 (Brüning et al., 2013). Nelfinavir also inhibits Akt (Gupta et al., 2005; Plastaras et al., 2008), which could activate TSC2 to further repress mTORC1 signalling (Tee et al., 2003). These various ER stress and nelfinavir TSC2‐dependent signalling mechanisms may explain why rpS6 and S6K1 phosphorylation levels were markedly reduced in Tsc2+/+ MEFs, but only partially reduced in Tsc2−/− MEFs following nelfinavir treatment (Figures 2A and 3A). Therefore, nelfinavir may inhibit mTORC1 signal transduction through multiple mechanisms that require TSC2, i.e., via GADD34/PP1/TSC2, Akt/TSC2, and/or SESN2/AMPK/TSC2, all of which would be compromised in Tsc2−/− cells. We are currently investigating the drug actions of nelfinavir and chloroquine to induce cell death, and propose a mechanism in Figure 8.
Figure 8.
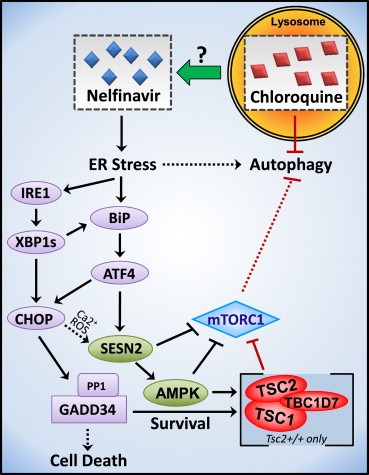
Chloroquine augments nelfinavir‐induced ER stress and cell death independently of autophagy. In normal cells, nelfinavir‐induced ER stress inhibits mTORC1 de novo protein synthesis through two survival pathways, GADD34 and AMPK, which both directly activate the TSC protein complex. In TSC‐deficient cells or when mTORC1 is oncogenically activated, ER stress is less efficient at repressing mTORC1 signalling, further enhancing ER stress through continual protein production and autophagy inhibition. Prolonged, elevated ER stress produces a lethal outcome in cells unable to alleviate mTORC1 activation. Entrapment of chloroquine to lysosomes is required for enhancement of nelfinavir‐induced cell death.
TSC2 appears vital for the survival of nelfinavir and chloroquine‐treated cells. The enhanced sensitivity of Tsc2−/− cell lines to cell death could be due in part to the inability of nelfinavir to ablate mTORC1 signalling in these cells. One important feature of the ER stress response pathway is to block mTORC1 signalling for two main purposes: firstly, mTORC1 inhibition reduces the rate of protein synthesis, lessening the burden on the ER, and secondly, mTORC1 inhibition is required for a robust autophagy response, allowing efficient removal of unfolded protein aggregates in the ER. Therefore, activation of TSC2 and inhibition of mTORC1 appears to be critically important for cell survival during acute or prolonged ER stress. Frequent constitutive activation of mTORC1 signalling in cancer suggests that our observations may have wider relevance. In line with our work, nelfinavir effectively induced cell death when combined with chloroquine or mefloquine in breast cancer models (Thomas et al., 2012), medullary thyroid cancer cell lines (Kushchayeva et al., 2014), and a model of chronic lymphocytic leukaemia (Mahoney et al., 2013). Chloroquine in combination with rapamycin was also effective in Tsc2−/− MEFs (Parkhitko et al., 2011).
In conclusion, our data shows that mTORC1‐hyperactive cells are sensitive to nelfinavir‐induced cell death, which is further enhanced by chloroquine through an autophagy‐independent mechanism involving its protonation and entrapment within lysosomes which is prevented by addition of bafilomycin‐A1. In addition to therapeutic potential in the context of TSC and LAM, dual treatment with nelfinavir and chloroquine promises to be minimally toxic and efficacious in a range of tumour types featuring hyper‐active mTORC1.
Conflict of interest
The authors have no conflicts of interest to declare.
Acknowledgements
This research was supported by the Association for International Cancer Research Career Development Fellowship [No. 06‐914/915] and the Tuberous Sclerosis Association awarded to A. Tee, a BBSRC Research Equipment Initiative [BB/E012574/1] awarded to R.J. Errington. Additional support was provided from the National Institute for Social Care and Health Research (NISCHR), the Wales Gene Park, the Tuberous Sclerosis Association and the Cardiff University School of Medicine.
Johnson Charlotte E., Hunt David K., Wiltshire Marie, Herbert Terry P., Sampson Julian R., Errington Rachel J., Davies D. Mark, Tee Andrew R., (2015), Endoplasmic reticulum stress and cell death in mTORC1‐overactive cells is induced by nelfinavir and enhanced by chloroquine, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.11.005.
References
- Alonso-Basanta, M. , Fang, P. , Maity, A. , Hahn, S.M. , Lustig, R.A. , Dorsey, J.F. , 2014. A phase I study of nelfinavir concurrent with temozolomide and radiotherapy in patients with glioblastoma multiforme. J. Neurooncol. 116, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenzeller-Herzog, C. , Hall, M.N. , 2012. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 22, 274–282. [DOI] [PubMed] [Google Scholar]
- Bono, C. , Karlin, L. , Harel, S. , Mouly, E. , Labaume, S. , Galicier, L. , Apcher, S. , Sauvageon, H. , Fermand, J.P. , Bories, J.C. , Arnulf, B. , 2012. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica. 97, 1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning, A. , 2011. Analysis of nelfinavir-induced endoplasmic reticulum stress. Method Enzymol. 491, 127–142. [DOI] [PubMed] [Google Scholar]
- Brüning, A. , Rahmeh, M. , Friese, K. , 2013. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 6, 1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijsen, J. , Lammering, G. , Jansen, R.L. , Beets, G.L. , Wals, J. , Sosef, M. , Den Boer, M.O. , Leijtens, J. , Riedl, R.G. , Theys, J. , Lambin, P. , 2013. Phase I trial of the combination of the Akt inhibitor nelfinavir and chemoradiation for locally advanced rectal cancer. Radiother. Oncol. 107, 184–188. [DOI] [PubMed] [Google Scholar]
- Di Nardo, A. , Kramvis, I. , Cho, N. , Sadowski, A. , Meikle, L. , Kwiatkowski, D.J. , Sahin, M. , 2009. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J. Neurosci. 29, 5926–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble, C.C. , Elis, W. , Menon, S. , Qin, W. , Klekota, J. , Asara, J.M. , Finan, P.M. , Kwiatkowski, D.J. , Murphy, L.O. , Manning, B.D. , 2012. TBC1D7 is a third subunit of the TSC1–TSC2 complex upstream of mTORC1. Mol. Cell. 47, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop, E.A. , Hunt, D.K. , Acosta-Jaquez, H.A. , Fingar, D.C. , Tee, A.R. , 2011. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 7, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eker, P. , Eker, R. , Johannessen, J.V. , 1983. Establishment and characterization of a new cell line from a hereditary renal rat tumor. In Vitro. 19, 495–503. [DOI] [PubMed] [Google Scholar]
- Franz, D.N. , Weiss, B.D. , 2012 Jun. Molecular therapies for tuberous sclerosis and neurofibromatosis. Curr. Neurol. Neurosci. Rep. 12, (3) 294–301. 10.1007/s11910-012-0269-4 [Review] [DOI] [PubMed] [Google Scholar]
- Ganley, I.G. , Wong, P.M. , Gammoh, N. , Jiang, X. , 2011. Distinct autophagosomal–lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell. 42, 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gills, J.J. , Lopiccolo, J. , Tsurutani, J. , Shoemaker, R.H. , Best, C.J. , Abu-Asab, M.S. , Borojerdi, J. , Warfel, N.A. , Gardner, E.R. , Danish, M. , Hollander, M.C. , Kawabata, S. , Tsokos, M. , Figg, W.D. , Steeg, P.S. , Dennis, P.A. , 2007. Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin. Cancer Res. 13, 5183–5194. [DOI] [PubMed] [Google Scholar]
- Goldberg, S.B. , Supko, J.G. , Neal, J.W. , Muzikansky, A. , Digumarthy, S. , Fidias, P. , Temel, J.S. , Heist, R.S. , Shaw, A.T. , McCarthy, P.O. , Lynch, T.J. , Sharma, S. , Settleman, J.E. , Sequist, L.V. , 2012. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J. Thorac. Oncol. 7, 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A.K. , Cerniglia, G.J. , Mick, R. , McKenna, W.G. , Muschel, R.J. , 2005. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 65, 8256–8265. [DOI] [PubMed] [Google Scholar]
- Gupta, A.K. , Li, B. , Cerniglia, G.J. , Ahmed, M.S. , Hahn, S.M. , Maity, A. , 2007. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia. 9, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn, D.M. , Shackelford, D.B. , Egan, D.F. , Mihaylova, M.M. , Mery, A. , Vasquez, D.S. , Turk, B.E. , Shaw, R.J. , 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harhaji-Trajkovic, L. , Arsikin, K. , Kravic-Stevovic, T. , Petricevic, S. , Tovilovic, G. , Pantovic, A. , Zogovic, N. , Ristic, B. , Janjetovic, K. , Bumbasirevic, V. , Trajkovic, V. , 2012. Chloroquine-mediated lysosomal dysfunction enhances the anticancer effect of nutrient deprivation. Pharm. Res. 29, 2249–2263. [DOI] [PubMed] [Google Scholar]
- He, C. , Klionsky, D.J. , 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høyer-Hansen, M. , Jäättelä, M. , 2007. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 14, 1576–1582. [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Shi, C. , Edil, B.H. , de Wilde, R.F. , Klimstra, D.S. , Maitra, A. , Schulick, R.D. , Tang, L.H. , Wolfgang, C.L. , Choti, M.A. , Velculescu, V.E. , Diaz, L.A. , Vogelstein, B. , Kinzler, K.W. , Hruban, R.H. , Papadopoulos, N. , 2011. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 331, 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, Y.J. , Lu, M.K. , Guan, K.L. , 2011. The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ. 18, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J. , Elazar, Z. , Seglen, P.O. , Rubinsztein, D.C. , 2008. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes?. Autophagy. 4, 849–850. [DOI] [PubMed] [Google Scholar]
- Kohrman, M.H. , 2012. Emerging treatments in the management of tuberous sclerosis complex. Pediatr. Neurol. 46, 267–275. [DOI] [PubMed] [Google Scholar]
- Kojima, E. , Takeuchi, A. , Haneda, M. , Yagi, A. , Hasegawa, T. , Yamaki, K. , Takeda, K. , Akira, S. , Shimokata, K. , Isobe, K. , 2003. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J. 17, 1573–1575. [DOI] [PubMed] [Google Scholar]
- Krymskaya, V.P. , Goncharova, E.A. , 2009. PI3K/mTORC1 activation in hamartoma syndromes: therapeutic prospects. Cell Cycle. 8, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushchayeva, Y. , Jensen, K. , Recupero, A. , Costello, J. , Patel, A. , Klubo-Gwiezdzinska, J. , Boyle, L. , Burman, K. , Vasko, V. , 2014. The HIV protease inhibitor nelfinavir downregulates RET signaling and induces apoptosis in medullary thyroid Cancer cells. J. Clin. Endocrinol. Metab. 99, E734–E745. [DOI] [PubMed] [Google Scholar]
- Li, T. , Su, L. , Zhong, N. , Hao, X. , Zhong, D. , Singhal, S. , Liu, X. , 2013. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy. 9, 1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y.M. , Kim, C. , Yen, Y. , 2011. Mammalian target of rapamycin and head and neck squamous cell carcinoma. Head Neck Oncol. 3, 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G. , Su, L. , Hao, X. , Zhong, N. , Zhong, D. , Singhal, S. , Liu, X. , 2012. Salermide up-regulates death receptor 5 expression through the ATF4–ATF3–CHOP axis and leads to apoptosis in human cancer cells. J. Cell. Mol. Med. 16, 1618–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney, E. , Maddocks, K. , Flynn, J. , Jones, J. , Cole, S.L. , Zhang, X. , Byrd, J.C. , Johnson, A.J. , 2013. Identification of endoplasmic reticulum stress-inducing agents by antagonizing autophagy: a new potential strategy for identification of anti-cancer therapeutics in B-cell malignancies. Leuk. Lymphoma. 54, 2685–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maycotte, P. , Aryal, S. , Cummings, C.T. , Thorburn, J. , Morgan, M.J. , Thornburn, A. , 2012. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 8, 200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan, U. , Ozcan, L. , Yilmaz, E. , Duvel, K. , Sahin, M. , Manning, B.D. , Hotamisligil, G.S. , 2008. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell. 29, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, J. , Mott, M. , Xi, B. , Hepner, E. , Guan, M. , Fousek, K. , Magnusson, R. , Tinsley, R. , Valdes, F. , Frankel, P. , Synold, T. , Chow, W.A. , 2012. Phase I study of nelfinavir in liposarcoma. Cancer Chemother. Pharmacol. 70, 791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhitko, A. , Myachina, F. , Morrison, T.A. , Hindi, K.M. , Auricchio, N. , Karbowniczek, M. , Wu, J.J. , Finkel, T. , Kwiatkowski, D.J. , Yu, J.J. , Henske, E.P. , 2011. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc. Natl. Acad. Sci. U. S. A. 108, 12455–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plastaras, J.P. , Vapiwala, N. , Ahmed, M.S. , Gudonis, D. , Cerniglia, G.J. , Feldman, M.D. , Frank, I. , Gupta, A.K. , 2008. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol. Ther. 7, 628–635. [DOI] [PubMed] [Google Scholar]
- Platt, F.M. , Hurst, C.D. , Taylor, C.F. , Gregory, W.M. , Harnden, P. , Knowles, M.A. , 2009. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin. Cancer Res. 15, 6008–6017. [DOI] [PubMed] [Google Scholar]
- Poole, B. , Ohkuma, S. , 1981. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J. Cell Biol. 90, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengan, R. , Mick, R. , Pryma, D. , Rosen, M.A. , Lin, L.L. , Maity, A.M. , Evans, T.L. , Stevenson, J.P. , Langer, C.J. , Kucharczuk, J. , Friedberg, J. , Prendergast, S. , Sharkoski, T. , Hahn, S.M. , 2012. A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a report of toxicities and clinical response. J. Thorac. Oncol. 7, 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein, D.C. , 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 443, 780–786. [DOI] [PubMed] [Google Scholar]
- Sato, Y. , Yoshizato, T. , Shiraishi, Y. , Maekawa, S. , Okuno, Y. , Kamura, T. , Shimamura, T. , Sato-Otsubo, A. , Nagae, G. , Suzuki, H. , Nagata, Y. , Yoshida, K. , Kon, A. , Suzuki, Y. , Chiba, K. , Tanaka, H. , Niida, A. , Fujimoto, A. , Tsunoda, T. , Morikawa, T. , Maeda, D. , Kume, H. , Sugano, S. , Fukayama, M. , Aburatani, H. , Sanada, M. , Miyano, S. , Homma, Y. , Ogawa, S. , 2013. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 45, 860–867. [DOI] [PubMed] [Google Scholar]
- Seitz, C. , Hugle, M. , Cristofanon, S. , Tchoghandjian, A. , Fulda, S. , 2013. The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial–lysosomal cross-talk. Int. J. Cancer. 132, 2682–2693. [DOI] [PubMed] [Google Scholar]
- Shin, J.H. , Park, S.J. , Jo, Y.K. , Kim, E.S. , Kang, H. , Park, J.H. , Lee, E.H. , Cho, D.H. , 2012. Suppression of autophagy exacerbates Mefloquine-mediated cell death. Neurosci. Lett. 515, 162–167. [DOI] [PubMed] [Google Scholar]
- Siroky, B.J. , Yin, H. , Babcock, J.T. , Lu, L. , Hellmann, A.R. , Dixon, B.P. , Quilliam, L.A. , Bissler, J.J. , 2012. Human TSC-associated renal angiomyolipoma cells are hypersensitive to ER stress. Am. J. Physiol. Renal Physiol. 303, 831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjodahl, G. , Lauss, M. , Gudjonsson, S. , Liedberg, F. , Hallden, C. , Chebil, G. , Mansson, W. , Hoglund, M. , Lindgren, D. , 2011. A systematic study of gene mutations in urothelial carcinoma; inactivating mutations in TSC2 and PIK3R1. PLoS ONE. 6, e18583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon, V.R. , Lee, H. , 2009. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 625, 220–233. [DOI] [PubMed] [Google Scholar]
- Sotelo, J. , Briceno, E. , Lopez-Gonzalez, M.A. , 2006. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343. [DOI] [PubMed] [Google Scholar]
- Tee, A.R. , Manning, B.D. , Roux, P.P. , Cantley, L.C. , Blenis, J. , 2003. Tuberous sclerosis complex gene products, tuberin and hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268. [DOI] [PubMed] [Google Scholar]
- Thomas, S. , Sharma, N. , Golden, E.B. , Cho, H. , Agarwal, P. , Gaffney, K.J. , Petasis, N.A. , Chen, T.C. , Hofman, F.M. , Louie, S.G. , Schonthal, A.H. , 2012. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 325, 63–71. [DOI] [PubMed] [Google Scholar]
- Uddin, M.N. , Ito, S. , Nishio, N. , Suganya, T. , Isobe, K. , 2011. Gadd34 induces autophagy through the suppression of the mTOR pathway during starvation. Biochem. Biophys. Res. Commun. 407, 692–698. [DOI] [PubMed] [Google Scholar]
- Watanabe, R. , Tambe, Y. , Inoue, H. , Isono, T. , Haneda, M. , Isobe, K. , Kobayashi, T. , Hino, O. , Okabe, H. , Chano, T. , 2007. GADD34 inhibits mammalian target of rapamycin signaling via tuberous sclerosis complex and controls cell survival under bioenergetic stress. Int. J. Mol. Med. 19, 475–483. [PubMed] [Google Scholar]
- Wu, Y.T. , Tan, H.L. , Shui, G. , Bauvy, C. , Huang, Q. , Wenk, M.R. , Ong, C.N. , Codogno, P. , Shen, H.M. , 2010. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 285, 10850–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang, C. , Liu, H. , Bertz, J. , Possinger, K. , Koeffler, H.P. , Elstner, E. , Eucker, J. , 2009. Induction of endoplasmic reticulum stress response by TZD18, a novel dual ligand for peroxisome proliferator-activated receptor alpha/gamma, in human breast cancer cells. Mol. Cancer Ther. 8, 2296–2307. [DOI] [PubMed] [Google Scholar]