

Abstract
An awesome number of experimental and clinical evidences indicate that constitutive activation of the Met oncogenic receptor plays a critical role in the progression of cancer toward metastasis and/or resistance to targeted therapies. While mutations are rare, the common mechanism of Met activation is overexpression, either by gene amplification (‘addiction’) or transcriptional activation (‘expedience’). In the first instance ligand‐independent kinase activation plays a major role in sustaining the transformed phenotype. Anti‐Met antibodies directed against the receptor binding site behave essentially as ligand (Hepatocyte Growth Factor, HGF) antagonists and are ineffective to counteract ligand‐independent activation. The monovalent chimeric MvDN30 antibody fragment, PEGylated to extend its half‐life, binds the fourth IPT domain and induces ‘shedding’ of the Met extracellular domain, dramatically reducing both the number of receptors on the surface and their phosphorylation. Downstream signaling is thus inhibited, both in the absence or in the presence of the ligand. In vitro, MvDN30 is a strong inhibitor not only of ligand‐dependent invasive growth, sustained by both paracrine and autocrine HGF, but notably, also of ligand‐independent growth of ‘Met‐addicted’ cells. In immunocompromised mice, lacking expression of Hepatocyte Growth Factor cross‐reacting with the human receptor – thus providing, by definition, a model of ‘ligand‐independent’ Met activation – PEGylated MvDN30 impairs growth of Met ‘addicted’ human gastric carcinoma cells. In a Met‐amplified patient‐derived colo‐rectal tumor (xenopatient) MvDN30‐PEG overcomes the resistance to EGFR targeted therapy (Cetuximab). The PEGylated MvDN30 is thus a strong candidate for targeting tumors sustained by ligand‐independent Met oncogenic activation.
Keywords: Cancer targeted therapy, Met, MvDN30, Anti-Met antibody, HGF
Highlights
Monovalency, chimerization and Pegylation improve potential clinical application of the anti‐Met MvDN30 molecule.
MvDN30‐PEG is a potent inhibitor of both HGF‐dependent and –independent Met activation.
MvDN30‐PEG overcomes the resistance to EGFR targeted therapy (Cetuximab).
MvDN30‐PEG stands as an highly promising anti‐Met therapeutic tool.
Abbreviations
- HGF
Hepatocyte Growth Factor
- mDN30 mAb
mouse DN30 monoclonal antibody
- mDN30 Fab
mouse DN30 Fab fragment
- MvDN30
monovalent chimerized DN30 Fab
- PEG
polyethylene glycol
1. Introduction
The Met oncogene encodes for the Hepatocyte Growth Factor (HGF) receptor, a transmembrane protein with tyrosine kinase activity. HGF stimulation induces a complex cellular response resulting in activation of the ‘invasive growth’ program, which is crucial during embryogenesis and tissue regeneration (Trusolino et al., 2010). The program, when aberrantly activated, sustains transformation and metastasis dissemination (Birchmeier et al., 2003; Boccaccio and Comoglio, 2006). The Met kinase was originally identified as a transforming oncoprotein activated by gene rearrangement induced by a carcinogen (Park et al., 1986). Activating point mutations of the Met gene have been initially described as a trait of hereditary and sporadic papillary renal cell carcinomas (Schmidt et al., 1997). Subsequently, Met mutations have been found in other tumor types and notably have been detected in highly metastatic diseases e.g. Head & Neck and Cancer of Unknown Primary Origin (Lorenzato et al., 2002; Stella et al., 2011). However, the screening of a large number of tumor cell lines and of patient‐derived cancer samples revealed that the Met receptor is more often activated by overexpression (Danilkovitch‐Miagkova and Zbar, 2002). Moreover, a plethora of carcinomas displays increased levels of the Met protein that are associated with poor prognosis (Blumenschein et al., 2012). Finally, it has been shown that the Met oncogene is under control of an inducible promoter (Gambarotta et al., 1994) and that over‐expression of the oncogene can result from transcriptional up‐regulation (De Bacco et al., 2011; Pennacchietti et al., 2003). Some wild‐type oncogenes, including Met, are in fact activated in cancer cells as an adaptive response to adverse micro environmental conditions (e.g. hypoxia, nutrient starvation, or ionizing radiation), favoring tumor progression and confering therapeutic resistance. This phenomenon is known as ‘expedience’ (Comoglio et al., 2008). In a number of cases (1–3%), Met overexpression is sustained by gene amplification: this has been reported among gastric‐esophageal cancers, medulloblastomas and CRC derived‐metastatic lesions (Di Renzo et al., 1995; Houldsworth et al., 1990; Tong et al., 2004). Met amplification sustains secondary resistance to Epithelial Growth Factor Receptor targeted therapies in Non‐Small Cell Lung (Bean et al., 2007; Engelman et al., 2007) and Colo‐Rectal cancers (Bardelli et al., 2013). Met amplification is responsible for the ‘Met‐addicted’ phenotype, a condition in which the transformed cells completely rely on activation of the oncogene for growth and survival (Comoglio et al., 2008). In a number of cases, it has been reported that patients with glioblastoma, esophageal or lung carcinoma carrying an amplified Met gene received substantial benefit from a specific small molecule kinase inhibitor (Chi et al., 2012; Lennerz et al., 2011; Ou et al., 2011). Met‐addiction thus represents the ideal – and possibly the unique – status for successful application of therapies targeting the oncogene.
Different strategies to inhibit Met signaling have been explored. These include low molecular weight kinase inhibitors, ligand (HGF) antagonists, receptor decoys, Short‐Harpin RNAs and antibodies against HGF or Met. Some of these molecules are either in pre‐clinical characterization or already in clinical trials (for a comprehensive list see Cui, 2014). Among those, mDN30 is a promising monoclonal anti‐Met antibody characterized by its peculiar ability to induce ‘shedding’ (i.e. release from the cell surface) of the Met receptor resulting in a dramatic inhibition of Met‐driven intracellular responses, such as anchorage independent growth and invasion in vitro and tumor growth and metastasis dissemination in vivo (Petrelli et al., 2006). Due to its bivalent nature, the native mDN30 is a partial agonist, promoting some, but not all, of the Met‐mediated biological responses. Transformation into the monovalent Fab fragment converts the molecule into a pure and potent Met antagonist (Pacchiana et al., 2010). However, the murine nature and the short half‐life of the Fab prevented further development for human therapy. To circumvent these limitations, we pursued chimerization and PEGylation to generate an inhibitor of both HGF‐dependent and ‐independent Met activation endowed with therapeutic properties.
2. Material and methods
2.1. Cell culture
A549 human lung carcinoma cells, U87‐MG human glioblastoma cells and Hs746T human gastric carcinoma cells were obtained from ATCC/LGC Standards S.r.l. (Sesto San Giovanni, Italy). GTL‐16 human gastric carcinoma cells were derived from MKN‐45 cells as described (Giordano et al., 1988). EBC‐1 human lung carcinoma cells were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan). Cells were maintained in recommended media (RPMI or DMEM, Sigma Life Science, St Louis, Missouri) as described (Pacchiana et al., 2010). M162 colon cancer cells were derived from tumor material of a patient resistant to EGFR targeted therapy (Bardelli et al., 2013) propagated (one step) in mice. Tumor extracted from the animal was chopped and then incubated in Leibovitz's L‐15 medium (Gibco® Life technologies Italia, Monza, Italy) plus 4 mg/ml Collagenase (Sigma Life Sciences) for 1 h at 37 °C. Cells were centrifuged, washed twice with Leibovitz's L‐15 medium plus 10% Fetal Bovine Serum (FBS‐Gibco® Life technologies) and incubated in the same medium without serum plus 100 mg/ml DNAasi (Sigma Life Science) for 10 min at 37 °C. After centrifugation and two washes as above, cells were plated and maintained in culture in 10% FBS F‐12 Nut Mix (Ham) (Gibco® Life technologies) medium plus Rock1 inhibitor Y‐27632‐2HCl (Selleckchem.com, Munich, Germany). Twice a week cells were treated with Trypsin (Sigma Life Science) for 2 min, to separate fibroblasts from tumor cells, and fresh medium was added. An almost pure tumor cell culture was obtained after two weeks.
2.2. MvDN30 generation, production and purification
We applied standard PCR technique to insert: i) at the 3′ of the DN30 light chain variable region (VL) and at the 5′ of the human Igk light chain constant region (huCL) the restriction site KpnI; ii) at the 3′ of the DN30 heavy chain variable region (VH) and at the 5′ of the human IgG1 heavy chain constant region 1 (CH1) the restriction site HindIII (truncation of the IgG1 heavy chain at the hinge region). By conventional molecular biology techniques, VL was joined to huCL and VH to huCH1. MvDN30 was produced by Wacker Biotech GmbH using its Escherichia coli proprietary secretory expression system ESETEC® and purified by Protein G affinity chromatography.
2.3. MvDN30 PEGylation
For PEGylation we produced a Fab2 molecule inserting a Cys residue at the 3′ of the huCH1 region. Fab2 was reduced using 1:1 molar ratio of Tris[2‐carboxyethyl] phosphine Hydrochloride (TCEP) at neutral pH for 30 min at RT. Polyethylene glycol (20 or 40 kDa) conjugated with Maleimide (PEG‐Mal) was added to the Fab solution to achieve a 1:1 Fab:PEG‐Mal molar ratio and the solutions were maintained by top‐down mixing at room temperature for 1 h. The conjugation efficiency was 70–80% with both PEG‐Mal. The bioconjugates were purified using Protein G sepharose and ion exchange chromatography and then dialyzed against PBS pH 7.0.
2.4. ELISA assays
For comparison of mDN30 Fab (Pacchiana et al., 2010) and MvDN30 binding to Met, antibodies (100 ng/well) were in solid phase and the Met‐Fc chimera (R&D Systems, Minneapolis, Minnesota) (range of dilutions 0–100 nM) was in liquid phase; revealing was done with HRP conjugated anti‐Histidine antibody (AbD Serotec, Kidlington, UK), exploiting a TAG sequence present in the chimera.
For identification of the Met region recognized by MvDN30, the antibody (100 ng/well) was in solid phase and full size or truncated Met extracellular domains (range of dilutions 0–100 nM) were in liquid phase. Revealing was done with HRP conjugated anti‐Histidine antibody, a TAG sequence expressed by the different Met domains. According to the sequence GenBank™ accession number X54559, the extracellular domain of Met (25‐932) includes: i) SEMA domain (25‐516); ii) PSI domain (517‐562); iii) four IPT domains: IPT1 (563‐656), IPT2 (657‐740), IPT3 (741‐837) and IPT4 (839‐932).
For comparison of MvDN30, MvDN30‐PEG20 and MvDN30‐PEG40 binding to Met, Met‐Fc chimera (100 ng/well) was in solid phase and antibody species (range of dilutions 0–400 nM) were in liquid phase; revealing was done with HPR‐conjugated anti‐human k light chain antibody (Sigma Life Sciences). Colorimetric assay was quantified by the multi‐label reader VictorTM X4 (Perkin Elmer Inc., Waltham, Massachusetts).
2.5. Pharmacokinetic analysis
All animal procedures were performed according to protocols approved by Italian Ministry of Health. Mice (female nu/nu on Swiss CD‐1 background mice) were purchased from Harlan (Udine, Italy). Some days before radioactive compound administration, the drinking water was supplemented with 2–3 drops of Lugol solution to block iodine thyroid uptake. One week prior to antibody administration, mice were subcutaneously transplanted with 5 × 106 GTL‐16 cells. At time 0, groups of 30 mice were intravenously injected with 2 μg/mouse of 125I‐DN30 mAb or 125I‐MvDN30 or 125I‐MvDN30‐PEG20 or 125I‐MvDN30‐PEG40. Antibody 125I radiolabeling was carried out by Iodogen method (Fraker and Speck, 1978). Five mice/group were sacrificed at each 0.25, 1, 4, 24, 48 and 72 h time point. At the sacrifice, blood, spleen, kidney, liver, lung and tumor mass samples were collected, weighed and counted in a γ‐counter. Percentage of antibody in each sample/gram of tissue was calculated referring to the injected activity. Data were also expressed as ngeq/gram of tissue. Pharmacokinetic parameters were evaluated according to a non‐compartmental approach for sparse data sampling and calculated by WinNonlin software (Pharsight Corporation, Sunnyvale, CA). Evaluated pharmacokinetic parameters: Tlast: The last sampling time at which a quantifiable concentration is found; Clast: The concentration value obtained at sampling time Tlast; AUClast: The area under the plasma concentration vs time curve up to sampling time Tlast, calculated by trapezoidal rule; AUCINF: The area under the plasma concentrations vs time curve up to infinity, calculated by the following equation: AUCINF = AUClast + Clast/λz; λz: The terminal elimination rate constant, calculated by the slope of the linear regression curve obtained by fitting the natural logarithms of terminal concentration values vs time; T1/2: The apparent terminal elimination half‐life, calculated by the equation: T1/2 = ln2/λz; Clearance (CL): The total clearance determined by the ratio Dose/AUCINF; Vss: the steady state volume of distribution calculated by the following equation: Vss = MRTINF * CL (MRTINF: mean residence time, extrapolated to infinity using the last observed concentration).
2.6. Antibody shedding activity
Sub‐confluent A549 were incubated in serum‐free medium for 48 h with the indicated concentrations of purified antibodies. Then conditioned medium was collected and cells were lysed with Laemmli Buffer. Cell extracts (same amount of total proteins for each sample, 20 μg) and cell culture supernatants (same volume, 15 μl) were resolved by SDS‐PAGE and analyzed by Western blotting.
2.7. Antibody agonistic properties
Sub‐confluent A549 were incubated in serum‐free medium for 48 h and then stimulated for 15 min at 37 °C with the indicated concentrations of recombinant human HGF (R&D Systems) or purified antibodies. Following stimulation, cells were immediately lysed with EB Buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 10% Glycerol, 1% Triton plus a cocktail of protease inhibitors and the phosphatase inhibitor Na3VO4 1 mM). Cell extracts (same amount of total proteins for each sample, 15 μg) were resolved by SDS‐PAGE and analyzed by Western blotting.
2.8. Antibody antagonistic properties
Sub‐confluent cell monolayers were washed twice with PBS and then incubated in serum‐free medium with the indicated concentrations of purified MvDN30 or MvDN30‐PEG20 for 24 h. A549 and U87‐MG were then stimulated with 100 ng/ml of recombinant human HGF for 15 min at 37 °C and then immediately lysed with EB buffer while Hs746T, GTL‐16 and EBC‐1 were directly lysed. Cell extracts (same amount of total proteins for each sample: A549, 15 μg; U87‐MG, 17 μg; Hs746T, 10 μg; GTL‐16, 5 μg; EBC‐1, 5 μg) were resolved by SDS‐PAGE and analyzed by Western blotting.
2.9. Western blot analysis
Primary antibodies for Western blot detection were: anti‐human Met mAb (3D4 Invitrogen Corporation, Camarillo, California), anti‐pMet Tyr1234/1235 (D26), anti‐pAkt Ser473, anti‐Akt, anti‐pERK1/2 Thr202/Tyr204 and anti‐ERK1/2 polyclonal Abs (Cell Signaling Technology, Beverly, Massachusetts); anti‐Vinculin clone hVIN‐1 (Sigma Life Sciences). Secondary anti‐mouse IgG or anti rabbit IgG were from GE Healthcare (Freiburg, Germany). Western blot bands were quantified using Image J software.
2.10. Biological assays in vitro
Growth assay: cells were seeded in 96‐well dishes as described (Pacchiana et al., 2010). After 16 h, medium was replaced with fresh one containing MvDN30, MvDN30‐PEG20 or DN30 mAb at increasing concentrations. Cell number was evaluated after 72 h using the CellTiter‐Glo luminescent cell viability assay (Promega Corp., Madison, Wisconsin). Chemiluminescence was detected with VictorTM X4.
Proliferation assay: cells were prepared, treated and analyzed as described for growth assay. Cellular DNA synthesis was determined measuring BrdU incorporation by a chemiluminescent‐based ELISA assay (Roche Diagnostics, Mannheim, Germany) according to manufacturer's instructions. BrdU incorporation index was calculated by dividing the data obtained in the BrdU incorporation assay with the data obtained in the cell growth assay.
Anchorage‐independent growth assays: cells were seeded in 48‐well dishes (500 cells/well) in semisolid medium as described (Pacchiana et al., 2010), except U87‐MG that were in 10% FCS. Treatments with MvDN30 or MvDN30‐PEG20 and analysis were done as described (Pacchiana et al., 2010).
Branching morphogenesis/invasion assay: cells (1000/well) were seeded in non‐adherent 96‐well plates with U‐bottom (Greiner Bio‐One GmbH, Frickenhausen, Germany) in medium containing 10% FBS plus 0.4% of Methylcellulose (Sigma Life Science) and incubated at 37 °C. After 24 h preformed spheroids were collected and plated in 96‐well costar plates (8–10 spheroids/well) in medium containing 10% FBS, 1.5 mg/ml Collagen type I (Corning Incorporated, NY USA), 0.8% Methylcellulose and HEPES 0.05 M pH 7.4. For A549, cells were incubated 15 min at 37 °C and then medium containing the indicated concentrations of MvDN30 or MvDN30‐PEG20 was added on the top of the semisolid culture. After 24 h, human recombinant HGF (50 ng/ml) was added and branches were scored after further 5 days of culture. For Hs746T, cells were incubated 24 h at 37 °C and then medium containing MvDN30 or MvDN30‐PEG20, at the indicated concentration, was added on the top of the semisolid culture. Colony areas were scored after further 48 h of culture. Analysis of branch length (A549) or colony areas (Hs746T) was done on images (at least 8 spheroids per experimental group) acquired with microscope and analyzed with Image J software.
2.11. In vivo experiments
All animal procedures were performed according to protocols approved by Ethical Committee for animal experimentation of the Fondazione Piemontese per la Ricerca sul Cancro and by Italian Ministry of Health. Mice (female nu/nu on Swiss CD‐1 background mice or female non‐obese diabetic/severe combined immunodeficient – NOD/SCID – mice) were purchased from Charles River Laboratories (Calco, Italy). Animals were maintained in hyper‐ventilated cages.
Prevention trial on Hs746T cells: 3 × 106 cells/mouse in 200 μl Iscove Modified Dulbecco Medium (IMDM, Sigma Life Science) enriched with 50% Matrigel (BD Biosciences, Franklin Lakes, New Jersey) were injected subcutaneously into the right posterior flank of 7‐week‐old nu/nu mice. Mice were immediately randomized into four groups and treated by intraperitoneal injection as follows: mDN30 mAb 375 μg/mouse (n = 4); MvDN30 250 μg/mouse (n = 7); MvDN30‐PEG20 250 μg/mouse (n = 7); Vehicle (PBS, n = 7). Considering that mDN‐30 mAb is bivalent and MvDN30 is monovalent, the amount of antibody administered was calculated so that each mouse received equal number of antigen‐binding domains. Molecules were delivered every 2 days, starting from the day of cell injection until the end of the experiment (35th day after cell injection). Tumor size was evaluated periodically with a caliper. Tumor volume was calculated as described (Pacchiana et al., 2010).
Regression trial on Hs746T cells: tumors grown in nu/nu mice were obtained injecting cells prepared as above. After 31 days (average tumor volume 106 ± 1.3 mm3), mice were randomized into three groups (n = 8) and treated twice a week by intravenous injection with mDN30 mAb (900 μg/mouse), MvDN30‐PEG20 (600 μg/mouse) or PBS until the end of the experiment (49th day after cell injection). Tumor growth was evaluated as above.
Regression trial on M162 tumor: tumor material (25–30 mm3) from a patient resistant to EGFR targeted therapy (Bardelli et al., 2013) already propagated in mice (one step) was implanted subcutaneously in NOD‐SCID mice as described (Migliardi et al., 2012). After 24 days mice carrying established tumors (average tumor volume 135 ± 46 mm3) were randomized into four groups and treated by intraperitoneal injection as follows: MvDN30‐PEG 600 μg/mouse (n = 6); MvDN30‐PEG20 600 μg/mouse + Cetuximab (Merck) 400 μg/mouse (n = 5); Cetuximab 400 μg/mouse (n = 4); Vehicle (PBS, n = 7). Mice were treated twice a week until the end of the experiment (52nd day after tumor implant). Tumor growth was evaluated as above.
2.12. Statistical analysis
Averages, standard deviations and P values obtained by Student's t Test were calculated using Microsoft Office Excel 2010 software (Microsoft Corporation, Redmond, Washington). To calculate Kd and Bmax, data from ELISA assay were analyzed and fitted according to nonlinear regression, one site binding hyperbola curve, using GraphPad Prism software (GraphPad Software, San Diego, California). To calculate IC50, data from growth and proliferation assays were analyzed and fitted according to a nonlinear regression, sigmoidal dose response curve, using GraphPad Prism software.
3. Results
3.1. Chimerization of MvDN30 does not affect the properties of the native murine antibody
To chimerize the murine DN30 Fab fragment (mDN30 Fab), the light chain constant domain was replaced by the human kappa type region and the heavy chain constant region 1 was substituted with the homologous domain derived from the human IgG1. The new chimeric molecule, called MvDN30, was produced in E. coli by expression and secretion into culture broth, then purified by affinity chromatography. In different assays, the properties of the original murine Fab and of MvDN30 were superimposable (summarized in Supplementary Information Table 1). Affinity for Met receptor was measured by ELISA assay. Antibodies were in solid phase and Met‐Fc (a recombinant protein including the entire Met extracellular domain in frame with the human IgG Fc region) was in liquid phase. Figure 1A shows that the two molecules interacted with Met with high affinity. Induction of Met down‐regulation and shedding was tested on A549 human non‐small lung carcinoma cells treated with increasing concentrations of the two different DN30‐derived Fabs. The analysis by immunoblotting of Met protein levels, in total cell lysates, and of released Met ectodomains, in the cell supernatants, showed that MvDN30 efficiently induced Met down‐regulation and shedding (Figure 1B and C). The inhibitory potency of MvDN30 and mDN30 Fab on the growth of Met‐addicted cells of different origins (Hs746T gastric carcinoma cells and EBC‐1 lung carcinoma cells) was comparable (Figure 1D and Supplementary Information Figure 1).
Figure 1.
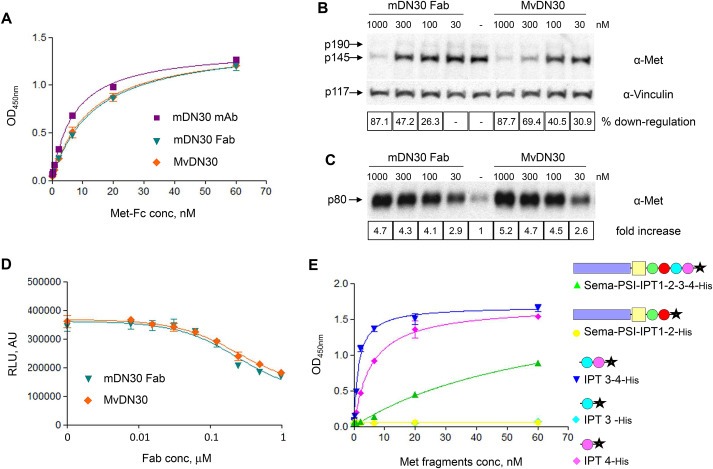
MvDN30 maintains the properties of the original murine molecule. (A) ELISA binding analysis of mDN30 mAb, mDN30 Fab and MvDN30 (solid phase) to a Met‐Fc chimera (liquid phase). O.D., optical density at 450 nm. Each point is the mean of triplicate values; bars represent standard deviation. (B) Met down‐regulation in cells treated with mDN30 Fab or MvDN30. A549 cells were incubated with the indicated concentrations of antibodies for 48 h. Total Met levels were determined by Western blot analysis of cell extracts using anti‐Met antibodies. The two Met bands correspond to the unprocessed (p190) and mature (p145) form of the receptor. To normalize protein loading, the same filter was re‐probed with anti‐Vinculin antibodies. Bands were quantified and normalized against vinculin (p117); values reported in the boxes represent the percentage reduction of Met signal in the treated samples with respect to the control, for the shown experiment. (C) Met shedding in A549 treated as above. The amount of Met extracellular domains released in the extracellular environment was determined by Western blot analysis of conditioned medium using anti‐Met antibodies. Values reported in the boxes represent fold increase of the Met signal in treated samples with respect to the control, for the shown experiment. (D) Anchorage‐dependent cell growth assay. Met‐addicted Hs746T cells were grown for 3 days in the presence of increasing concentrations of mDN30 Fab or MvDN30; cell number was determined using a luminescence‐based ATP assay. Samples are in quadruplicates, bars represent standard deviation. RLU: Relative Light Unit; AU: arbitrary unit. (E) ELISA binding analysis of MvDN30 (solid phase) to different truncated forms of the Met extracellular domain (liquid phase). Sema‐PSI‐IPT1‐2‐3‐4: full size Met extracellular region; Sema‐PSI‐IPT1‐2: N‐terminal portion of Met extracellular region. IPT 3–4: C‐terminal portion of Met extracellular region; IPT 3 and IPT 4: isolated Met IPT domains. O.D., optical density at 450 nm. Each point is the mean of triplicate values; bars represent standard deviation. Data reported in the figure are representative of at least two experiments done.
The Met domain recognized by MvDN30 was identified by an ELISA assay using the chimeric Fab in solid phase challenged by different Met extracellular domains truncated at different sites. MvDN30 binds with high affinity the IPT 4 domain (Figure 1E and Supplementary Information Table 2).
3.2. PEGylation of MvDN30 improves pharmacokinetics properties
To address the issue of Fab fragment short half‐life, polyethylene glycol (PEG) chains of different length (20 kDa or 40 kDa) were conjugated to the thiol group of a Cys artificially inserted at the c‐terminal of the MvDN30 heavy chain. MvDN30, its PEGylated derivatives and mDN30 mAb (as reference) were radiolabelled with 125I and a single dose of each test item was administered to nude mice bearing the Met‐addicted GTL‐16 tumors. The pharmacokinetic parameters are reported in Table 1 and in Supplementary Information Table 3. As expected, MvDN30 showed the shortest plasma terminal half‐life and mDN30 mAb the longest. Intermediate T½ values were observed for the PEGylated MvDN30s, with a proportional increase related to the molecular weight of the conjugated PEG moiety. Moreover, MvDN30 showed the lowest blood systemic exposure (AUC) coupled with the highest volume of distribution at steady state (Vss) and accumulation in the kidney. The AUC and clearance shown by MvDN30‐PEG20 were very similar to those of mDN30 mAb while Vss was reduced, indicating that mDN30 mAb is largely up taken by the organs, in particular liver and spleen, while MvDN30‐PEG20 is not. MvDN30‐PEG40 showed higher AUC, lower clearance and reduced Vss, indicating slow elimination but also a low propensity to distribute outside the systemic circulation.
Table 1.
Blood pharmacokinetic parameters of mDN30 mAb and its derivatives.
| mDN30 mAb | MvDN30 | MvDN30‐PEG20 | MvDN30‐PEG40 | |
|---|---|---|---|---|
| T1/2 (h) | 23.1 | 2.7 | 10.9 | 15.1 |
| CL (ml/h) | 0.18 | 2.81 | 0.2 | 0.13 |
| AUCINF (h*ngeq/ml) | 11267 | 711 | 9934 | 15839 |
| AUClast (h*ngeq/ml) | 9699 | 447 | 9850 | 15197 |
| Vss (ml) | 5.21 | 10.79 | 2.28 | 2.29 |
| λ (1/h) | 0.03 | 0.258 | 0.064 | 0.046 |
T1/2: apparent terminal elimination half‐life; CL: clearance; AUCINF: area under the plasma concentrations vs time curve up to infinity; AUClast: area under the plasma concentration vs time curve up to sampling time; Vss: steady state volume of distribution; λ: terminal elimination rate constant.
3.3. PEGylated MvDN30 species maintain Met binding and shedding properties
PEGylated MvDN30 species (20 or 40 KDa) were tested for Met binding properties, by ELISA assay, with Met‐Fc chimera in solid phase and the antibody species in liquid phase. Data reported in Figure 2A and Supplementary Information Table 4 show that PEGylation was followed by a modest (3.5–10 folds) length‐dependent reduction in affinity. When assayed for the ability to induce Met down‐regulation and shedding, MvDN30‐PEG20 proved to be similar to MvDN30 while MvDN30‐PEG40 was considerably less efficient (Figure 2B and Supplementary Information Table 4). On the basis of these data, we selected MvDN30‐PEG20 for further studies.
Figure 2.
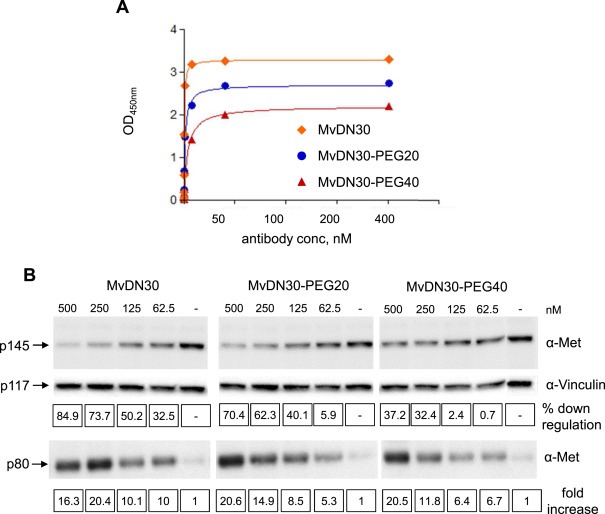
PEGylation does not result in major modifications of MvDN30 properties. (A) ELISA binding analysis of MvDN30, MvDN30‐PEG20 and MvDN30‐PEG40 (liquid phase) to a Met‐Fc chimera (solid phase). O.D.: optical density at 450 nm. Each point is the mean of triplicate values; bars represent standard deviation. (B) Met down‐regulation and shedding in A549 cells treated with increasing concentration of MvDN30, MvDN30‐PEG20 or MvDN30‐PEG40 for 48 h. Total Met levels (p145) and Met extracellular domains (p80) released in the extracellular environment were determined by Western blot analysis of cell extracts and of cell culture supernatants, using anti‐Met antibodies. The lysate filter was re‐probed with anti‐Vinculin antibodies. Bands were quantified and normalized against vinculin (p117); values in the top line boxes represent the percentage reduction of Met signal in the lysates extracted from treated samples with respect to the control and values in the bottom line boxes represent the fold increase of the Met signal in the treated samples with respect to the control, for the shown experiment. Data reported in the figure are representative of at least two experiments done.
3.4. The PEGylated MvDN30 inhibits HGF‐dependent or ‐independent Met phosphorylation
MvDN30 and MvDN30‐PEG20 agonist or antagonist properties were evaluated by analyzing phosphorylation of Met and of its downstream signal transducers. Agonist activity was ruled‐out in A549 cells, a standard system for determining Met activation in response to acute stimulation by ligands (Figure 3A). Antagonist activity was assessed in two cell lines (A549 and U87‐MG) stimulated by HGF and in three cell lines (Hs746T, GTL‐16 and EBC‐1) expressing constitutively active Met due to gene amplification and overexpression. MvDN30 and MvDN30‐PEG20 exerted a severe inhibition of both HGF‐dependent and HGF‐independent Met phosphorylation (Figure 3B and C and Supplementary Information Figure 2). This was reflected by inhibition of the Met downstream signaling pathway, as assessed by AKT and ERK phosphorylation (Figure 3B and C). Thus MvDN30 and MvDN30‐PEG20 behave as pure Met antagonists.
Figure 3.
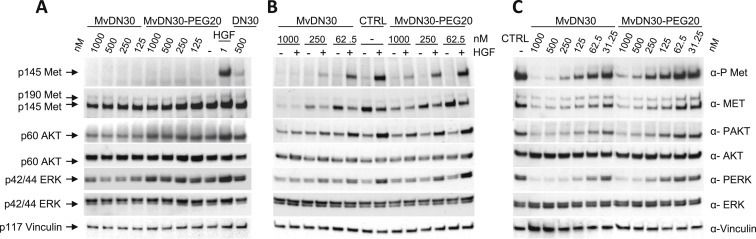
MvDN30 and MvDN30‐PEG20 inhibit both HGF‐dependent and HGF‐independent Met activation. (A) A549 cells stimulated (15 min) with increasing concentrations of MvDN30 or MvDN30‐PEG20 and analyzed for Met, AKT and ERK activation. As positive control cells were also stimulated with the indicated concentrations of purified mDN30 mAb or HGF. (B) A549 cells treated for 24 h with increasing concentrations of MvDN30 or MvDN30‐PEG20 and then stimulated with HGF for 15 min. Analysis of Met, AKT and ERK activation. (C) Hs746T cells treated for 24 h with increasing concentrations of MvDN30 or MvDN30‐PEG20. Analysis of Met, AKT and ERK activation. Met activation was determined in total cell lysates by Western blotting with anti‐phospho‐Met antibodies. The same blot was re‐probed with anti‐Met antibodies and with anti‐Vinculin antibodies. ERK and AKT activation was determined by Western blotting with anti‐phospho‐ERK and anti‐phospho‐AKT antibodies. Total ERK and AKT levels were determined using respectively anti‐ERK and anti‐AKT antibodies. Data reported in the figure are representative of at least two experiments done.
3.5. The PEGylated MvDN30 inhibits HGF‐dependent or –independent Met biological responses in vitro
MvDN30 and MvDN30‐PEG20 inhibition of HGF‐dependent biological responses was assessed in A549 cells, where the ligand sustains anchorage‐independent growth and branching morphogenesis. In HGF‐stimulated cells, anchorage‐independent growth inhibition – according to the dose treatment – ranged from 55 to 82.2% and from 20.2 to 53.2%, for MvDN30 and MvDN30‐PEG20 respectively. The PEGylated molecule is thus less potent than the parental moiety (Figure 4A and Supplementary Information Figure 3A and Table 5). The small reduction in activity following PEGylation of MvDN30‐PEG20 was confirmed in branching morphogenesis assays, with a branch length reduction, at the maximal tested concentration, of 11.4 versus 4.6 folds for MvDN30 and MvDN30‐PEG20 respectively (Figure 4B and Supplementary Information Figure 3B and Table 5). Inhibition of anchorage‐independent growth was detected also in U87‐MG, which features autocrine HGF stimulation (Figure 4C and Supplementary Information Figure 3C).
Figure 4.
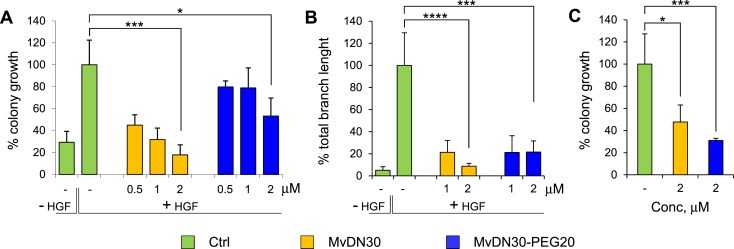
MvDN30 and MvDN30‐PEG20 inhibit HGF‐dependent Met biological responses in vitro. (A) Anchorage‐independent growth of HGF‐stimulated A549 cells treated with increasing concentrations of MvDN30 or MvDN30‐PEG20. Graph represents the percentage of the average colony growth for each treatment with respect to HGF‐treated cells. (B) Branching morphogenesis of HGF‐stimulated A549 cells treated with two different concentrations of MvDN30 or MvDN30‐PEG20. Graph represents the percentage of the average branch length for each treatment with respect to HGF‐treated cells. (C) Anchorage‐independent growth of U87‐MG cells (characterized by the presence of the HGF/Met autocrine loop) treated with MvDN30 or MvDN30‐PEG20. Graph represents the percentage of the average colony growth for each treatment with respect to untreated cells. *, Student's t Test p < 0.05; ***, Student's t Test p < 0.001; **** Student's t Test p < 0.0001. Each point is the mean of at least triplicate values, bars: standard deviation. Data reported in the figure are representative of at least two experiments done.
HGF‐independent inhibition was then tested in Met‐addicted cells. i) While the native mAb was not or poorly effective, MvDN30 and its PEGylated derivative strongly inhibited cell growth, with IC50 values ranging – in the different cell lines tested – from 98 to 351 nM for MvDN30 and from 308 to 1153 nM for MvDN30‐PEG20, indicating again a slight decrease in potency after PEGylation (Figure 5A–C and Supplementary Information Table 5). These results were further confirmed by measuring BrdU incorporation, where the IC50 difference between the two molecules was about two folds (Figure 5D and E and Supplementary Information Table 5). ii) mDN30 mAb‐derived molecules were also challenged to inhibit anchorage‐independent cell growth. Both MvDN30 and MvDN30‐PEG20 reduced GTL‐16 colony formation in a dose‐dependent manner; at the minimal tested dose, inhibition was of 88.5% for MvDN30 and of 58.9% for MvDN30‐PEG20 and, at the maximal dose, it was almost complete for both molecules (Figure 5F and Supplementary Information Figure 4A and Table 5). iii) The effect on Met‐addicted cell invasion was evaluated by seeding pre‐formed Hs746T cell spheroids in collagen. Along time, cells invaded the surrounding area and spheroid border‐lines crumbled, generating large cell islands without structural organization. When treated with the mDN30 mAb‐derived molecules, cells maintained a more compact structure compared to controls, indicating a diminished propensity to invade the extracellular matrix. The area occupied by the islands was reduced by 73.7% in the presence of MvDN30 and by 65.1% in the presence of MvDN30‐PEG20 (Figure 5G and Supplementary Information Figure 4B and Table 5). iv) Finally, the two mDN30 mAb derivatives were tested for growth inhibition of M162, Met‐amplified colon cancer cells derived from a patient resistant to EGFR targeted therapy. Again in this HGF‐independent model both MvDN30 and MvDN30‐PEG20 were effective, reducing cell growth by about 34% and 27% respectively. As previously reported (Bardelli et al., 2013), M162 cells did not respond to Cetuximab treatment (Figure 5H).
Figure 5.
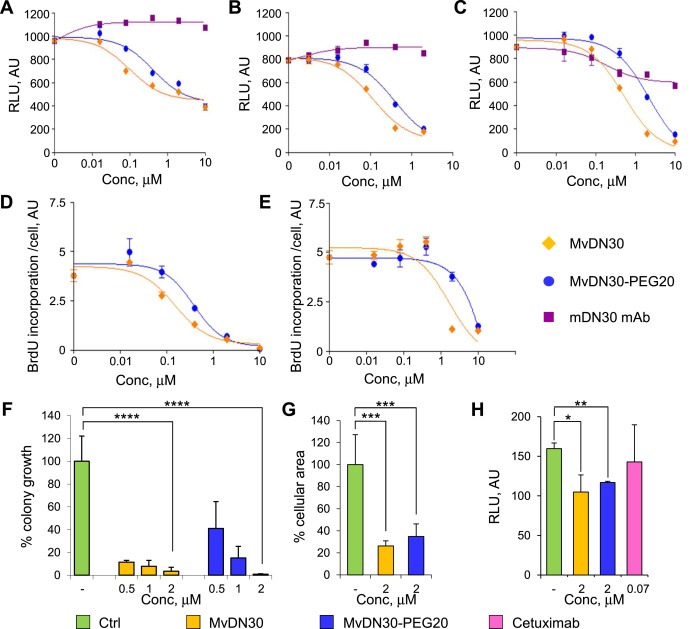
MvDN30 and MvDN30‐PEG20 inhibit HGF‐independent Met biological responses in vitro. Growth of Hs746T (A), EBC‐1 (B), GTL‐16 (C) Met‐addicted cells treated with increasing concentrations of MvDN30, MvDN30‐PEG20 or mDN30 mAb for 3 days. Samples are in quadruplicates, bars represent standard deviation. RLU: Relative Light Unit; AU: arbitrary unit. DNA synthesis determined by BrdU incorporation of Hs746T (D) and EBC‐1 (E) cells treated with increasing concentrations of MvDN30 or MvDN30‐PEG20 for 3 days; graphs report values normalized by cell number. Samples are in triplicates, bars represent standard deviation. AU: arbitrary unit. (F) Anchorage‐independent growth of GTL‐16 cells treated with increasing concentrations of MvDN30 or MvDN30‐PEG20. Graph represents percentage of average colony growth for each treatment with respect to untreated cells. (G) Three‐dimensional growth of Hs746T cells. Graph represents the percentage of average area covered by cell islands with respect to untreated cells. (H) Growth of M162 patient‐derived Met‐amplified cells treated with MvDN30, MvDN30‐PEG20 or Cetuximab for 5 days. *, Student's t Test p < 0.05; **, Student's t Test p < 0.01; ***, Student's t Test p < 0.001; **** Student's t Test p < 0.0001. Each point is the mean of at least triplicate values, bars: standard deviations. Data reported in the figure are representative of at least two experiments done.
3.6. The PEGylated MvDN30 inhibits HGF‐independent tumor growth in vivo
MvDN30‐PEG20 inhibition of tumor growth in vivo was tested on the HGF‐independent, Met‐addicted Hs746T model. First, mDN30 mAb, MvDN30 and MvDN30‐PEG20 were delivered in the peritoneal cavity starting from the day of cell injection until the end of the experiment (prevention trial). MvDN30 was not effective – as expected from its short plasma half‐life – while MvDN30‐PEG20 inhibited tumor growth: the treated masses were 74.6% smaller than controls (P = 0.021) (Figure 6A). Reduction of tumor volumes induced by mDN30 mAb was not statistically significant (P = 0.11). Then the same experimental model was tested in a regression trial, starting treatments on established tumors. Also in this setting MvDN30‐PEG20 exerted a pronounced inhibitory activity, as the tumor masses were reduced by 68% (P = 0.005), and mDN30 mAb was not significantly active (P = 0.09) (Figure 6B). MvDN30‐PEG20 inhibitory activity was also evaluated in the Met‐amplified M162 tumor by subcutaneously transplanting the patient‐derived specimen (xenopatient). Twenty‐four days after transplantation, mice were randomized into four groups and treated by intraperitoneal administration as follows: Vehicle, Cetuximab, MvDN30‐PEG20 and MvDN30‐PEG20 plus Cetuximab. MvDN30‐PEG20 was effective in inhibiting tumor growth: the average mass of the treated tumors was 35% smaller than the average of the control group (P = 0.050). Cetuximab alone, as already described (Bardelli et al., 2013), was not effective on this tumor, but in combination with MvDN30‐PEG20 induced the best response: tumor masses were 62% smaller than controls (P = 0.003 Combo versus control; P = 0.045 Combo versus MvDN30‐PEG20 alone) (Figure 6C).
Figure 6.
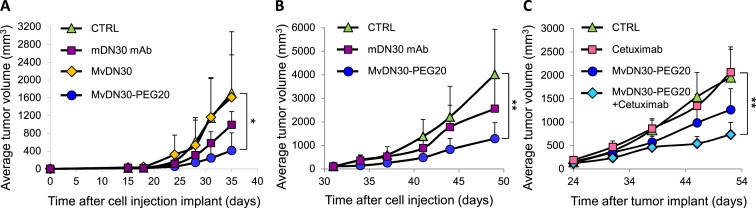
MvDN30‐PEG20 inhibits HGF‐independent tumor growth in vivo. (A) HGF‐independent Hs746T tumors, prevention trial. Cells were inoculated subcutaneously into the flank of CD1 nu/nu mice; animals were immediately randomized into four groups and treated as follows: Vehicle (CTRL), mDN30 mAb, MvDN30 and MvDN30‐PEG20. (B) HGF‐independent Hs746T tumors, regression trial. Cells were inoculated subcutaneously into the flank of CD1 nu/nu mice and after 31 days animals were randomized into three arms (n = 8) and treated as follows: Vehicle (CTRL), mDN30 mAb and MvDN30‐PEG20. (C) HGF‐independent M162 ‘xenopatient’, regression trial. Tumor specimens were implanted subcutaneously into the flank of NOD/SCID mice; after 24 days animals were randomized into four groups and treated as follows: Vehicle (CTRL), Cetuximab, MvDN30‐PEG20 and MvDN30‐PEG20 + Cetuximab. Each symbol represents the average tumor volume at the indicated time point; bars: standard deviations. *, Student's t Test P < 0.05 **, Student's t Test P < 0.01.
4. Discussion
Chimerization and PEGylation were introduced into the mDN30 Fab to fit clinical applications. Like other monoclonal antibodies with therapeutic potential, DN30 was raised in mice. The expected human anti‐murine antibody reaction (Shawler et al., 1985) was minimized by substitution of the murine constant regions of the antibody with homologous sequences derived from human immunoglobulins (antibody chimerization) (Hosono et al., 1992). ‘Chimeric’ mAbs and Fabs are successfully used in the clinic (Hildebrandt et al., 2007; Plosker and Figgitt, 2003; Tamhane and Gurm, 2008). The MvDN30 manufacturing system – expression in prokaryotes – was safe, robust, economical and suitable for large scale production (Baneyx, 1999).
Due to their small molecular weight, Fab fragments are rapidly cleared by the kidney, with the consequence of a short half‐life in the circulation. Covalent attachment of polyethylene glycol (PEG) chains to antibody fragments represents a valuable strategy to address the above shortcoming, as the polymer increases the molecular weight of the Fab above the critical glomerular filtration threshold. MvDN30 PEGylation was site‐specific, assuring the homogeneity of the molecules generated from the chemical modification. Moreover PEGylation improves pharmacokinetic properties by shielding the therapeutic molecule from the immune system recognition (Harris and Chess, 2003).
Binding data show that PEGylation slightly affects MvDN30 affinity (4 fold decrease). As a consequence, it is not surprising to measure some modest impairment in the biological responses in vitro, such as inhibition of anchorage‐dependent and ‐independent growth and invasion. On the other hand, the pharmacokinetic profile of MvDN30 was strongly improved. The PEGylated molecule showed not only blood systemic exposure (AUC) and clearance similar to the native intact monoclonal antibody but also reduced volume of distribution at steady state (Vss), indicative of a MvDN30‐PEG20 minor uptake in the tissues. Upon systemic delivery the therapeutic performance of MvDN30 in vivo was enhanced by PEGylation. Thus, while native MvDN30 is appropriate for loco‐regional applications, MvDN30‐PEG20 seems to be suitable also upon systemic administration.
To develop antibodies inhibiting Met, different immunization strategies, a variety of hosts and/or recombinant technologies have been employed. Despite these wide approaches, antibody selection was generally based on the same strategy: the antibody‐mediated inhibition of ligand–receptor interaction. This implies that the mechanism of action of most antibodies is essential the same: they behave as ligand competitors. Thus, most Met antibodies are functionally superimposable to ligand (HGF) antibodies and their use is restricted to treatment of cancers where HGF is crucial. Onartuzumab (anti‐Met) and Rilotumumab (anti‐HGF) are under investigation in glioblastomas characterized by the presence of an HGF/Met autocrine loop (Wen et al., 2011; www.clinicaltrial.com). The autocrine loop is also observed in a number of myelomas, osteosarcomas, rabdhomiosarcomas, mesotheliomas (Börset et al., 1996, 1995, 1996, 2005) but is not a common feature. In carcinomas, Met is activated mostly by receptor overexpression and in fewer instances by activating point mutations. In both cases, Met kinase constitutive activity is sustained in the absence of the ligand (Miller et al., 2001; Nakamura et al., 2008). Thus, although HGF can exacerbate the status of Met activation (Michieli et al., 1999; Naldini et al., 1991), the ligand is not the Met kinase driver. As a word of cautions it should be said, however, that the relevance of possible ligand‐dependent activation of MET in human carcinomas remains to be elucidated in‐full. Thus, in clinical trials, it should still be important to stratify patients according to HGF levels.
Three different Met antibodies do not work as ligand competitors: SAIT301, LY2875358 and MvDN30. SAIT301 promotes Met degradation by the LRIG1‐mediated lysosomal pathway (Lee et al., 2014). LY2875358 promotes Met down‐regulation, without inducing kinase activation. This antibody has been demonstrated to be functional in both HGF‐dependent and ‐independent systems (Liu et al., 2014). MvDN30 has the unique property of inducing Met shedding. This leads concomitantly to: i) reduction in the number of Met receptors exposed at the cell surface and ii) release of the soluble Met extracellular domains in the microenvironment. The latter is responsible for the so called ‘Decoy’ effect, as it sequesters free HGF and forms inactive heterodimers after interaction with the transmembrane full length Met receptors surviving cleavage (Michieli et al., 2004; Pacchiana et al., 2010; Petrelli et al., 2006). This mechanism of action explains the inhibitory activity of MvDN30 on both HGF‐dependent and –independent instances. DN30‐induced shedding most likely relies on an antibody‐induced Met conformational change exposing the motif targeted by ADAM‐10, the proteolytic enzyme responsible for cleavage of the receptor extracellular domain (Foveau et al., 2009; Schelter et al., 2010). The Met epitope recognized by MvDN30 is within the IPT4 domain, a region included in one of the identified HGF binding sites (domains IPT3‐IPT4, Basilico et al., 2008). However DN30 binding does not compete with HGF (Prat et al., 1998), concluding that the epitope and the site are separate. Recently we found that a single point mutation included in the Met IPT4 domain profoundly impairs DN30 binding without perturbing HGF/Met interaction (C. Basilico, unpublished data). The formal proof underlying the therapeutic activity of MvDN30‐PEG20 in HGF‐independent cancers was provided by the ‘xenopatient’ experiment, where a human colon carcinoma grows in an HGF‐free environment. It is known in fact that the mouse HGF does not activate the human receptor (Ikebuchi et al., 2013).
Targeted therapy, by definition and by experience, is effective only when applied to selected patients, whose tumor expresses the target playing the role of ‘driver’. The Met oncogene behaves as a driver mostly after amplification or point mutations, where constitutive activation of the kinase is ligand‐independent. In this scenario, MvDN30‐PEG20 stands as a promising therapeutic tool.
Conflict of interest
PMC and EV are authors of the international patent WO2007090807 (“Anti‐met monoclonal antibody, fragments and vectors thereof…”) owned by Metheresis Translational Research SA (Switzerland); the University of Torino received financial support from Metheresis and PMC was a consultant. The company did not interfere at all in the design of the study, collection/analysis of data and decision to publish. The other authors declare no potential conflict of interest.
Supporting information
Supplementary data
Acknowledgements
We thank Rita De Santis for helpful scientific discussion, Livio Trusolino for critical reading the manuscript, Livio Trusolino and Andrea Bertotti for providing M162 xenopatient and Simona Corso for support and suggestions in animal experiments.
This work was supported by AIRC grants (IG Project n° 11852, IG Project n° 15572 and 2010 Special Program Molecular Clinical Oncology 5xMille, Project n° 9970) to PMC and by ‘Metheresis Translational Research SA’ grant to the University of Torino.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.05.007.
Vigna Elisa, Chiriaco Cristina, Cignetto Simona, Fontani Lara, Basilico Cristina, Petronzelli Fiorella, Comoglio Paolo M., (2015), Inhibition of ligand‐independent constitutive activation of the Met oncogenic receptor by the engineered chemically‐modified antibody DN30, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.05.007.
Contributor Information
Elisa Vigna, Email: elisa.vigna@ircc.it.
Paolo M. Comoglio, Email: pcomoglio@gmail.com
References
- Baneyx, F. , 1999. Recombinant protein expression in Escherichia coli . Cur. Opin. Biotech. 10, 411–421. [DOI] [PubMed] [Google Scholar]
- Bardelli, A. , Corso, S. , Bertotti, A. , Hobor, S. , Valtorta, E. , Siravegna, G. , Sartore-Bianchi, A. , Scala, E. , Cassingena, A. , Zecchin, D. , Apicella, M. , Migliardi, G. , Galimi, F. , Lauricella, C. , Zanon, C. , Perera, T. , Veronese, S. , Corti, G. , Amatu, A. , Gambacorta, M. , Diaz, L.A. , Sausen, M. , Velculescu, V.E. , Comoglio, P.M. , Trusolino, L. , Di Nicolantonio, F. , Giordano, S. , Siena, S. , 2013. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 3, 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilico, C. , Arnesano, A. , Galluzzo, M. , Comoglio, P.M. , Michieli, P. , 2008. A high affinity hepatocyte growth factor-binding site in the immunoglobulin-like region of Met. J. Biol. Chem. 283, 21267–21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, J. , Brennan, C. , Shih, J.Y. , Riely, G. , Viale, A. , Wang, L. , Chitale, D. , Motoi, N. , Szoke, J. , Broderick, S. , Balak, M. , Chang, W.C. , Yu, C.J. , Gazdar, A. , Pass, H. , Rusch, V. , Gerald, W. , Huang, S.F. , Yang, P.C. , Miller, V. , Ladanyi, M. , Yang, C.H. , Pao, W. , 2007. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA. 104, 20932–20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier, C. , Birchmeier, W. , Gherardi, E. , Vande Woude, G.F. , 2003. Met, metastasis, motility and more. Nat. Rev. Mol. Cell. Biol. 4, 915–925. [DOI] [PubMed] [Google Scholar]
- Blumenschein, G.R. , Mills, G.B. , Gonzalez-Angulo, A.M. , 2012. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol. 30, 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaccio, C. , Comoglio, P.M. , 2006. Invasive growth: a MET-driven genetic program for cancer and stem cells. Nat. Rev. Cancer. 6, 637–645. [DOI] [PubMed] [Google Scholar]
- Börset, M. , Hjorth-Hansen, H. , Seidel, C. , Sundan, A. , Waage, A. , 1996. Hepatocyte growth factor and its receptor c-met in multiple myeloma. Blood. 88, 3998–4004. [PubMed] [Google Scholar]
- Chi, A.S. , Batchelor, T.T. , Kwak, E.L. , Clark, J.W. , Wang, D.L. , Wilner, K.D. , Louis, D.N. , Iafrate, A.J. , 2012. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 30, e30–33. [DOI] [PubMed] [Google Scholar]
- Comoglio, P.M. , Giordano, S. , Trusolino, L. , 2008. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 7, 504–516. [DOI] [PubMed] [Google Scholar]
- Cui, J.J. , 2014. Targeting receptor tyrosine kinase MET in cancer: small molecule inhibitors and clinical progress. J. Med. Chem. 57, 4427–4453. [DOI] [PubMed] [Google Scholar]
- Danilkovitch-Miagkova, A. , Zbar, B. , 2002. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J. Clin. Invest. 109, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bacco, F. , Luraghi, P. , Medico, E. , Reato, G. , Girolami, F. , Perera, T. , Gabriele, P. , Comoglio, P.M. , Boccaccio, C. , 2011. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 103, 645–661. [DOI] [PubMed] [Google Scholar]
- Di Renzo, M.F. , Olivero, M. , Giacomini, A. , Porte, H. , Chastre, E. , Mirossay, L. , Nordlinger, B. , Bretti, S. , Bottardi, S. , Giordano, S. , Plebani, M. , Gespach, C. , Comoglio, P.M. , 1995. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin. Cancer Res. 1, 147–154. [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , Kosaka, T. , Holmes, A.J. , Rogers, A.M. , Cappuzzo, F. , Mok, T. , Lee, C. , Johnson, B.E. , Cantley, L.C. , Jänne, P.A. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Ferracini, R. , Di Renzo, M.F. , Scotlandi, K. , Baldini, N. , Olivero, M. , Lollini, P. , Cremona, O. , Campanacci, M. , Comoglio, P.M. , 1995. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene. 10, 739–749. [PubMed] [Google Scholar]
- Ferracini, R. , Olivero, M. , Di Renzo, M.F. , Martano, M. , De Giovanni, C. , Nanni, P. , Basso, G. , Scotlandi, K. , Lollini, P. , Comoglio, P.M. , 1996. Retrogenic expression of the MET proto-oncogene correlates with the invasive phenotype of human rhabdomyosarcomas. Oncogene. 12, 1697–1705. [PubMed] [Google Scholar]
- Foveau, B. , Ancot, F. , Leroy, C. , Petrelli, A. , Reiss, K. , Vingtdeux, V. , Giordano, S. , Fafeur, V. , Tulasne, D. , 2009. Down-regulation of the met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Mol. Biol. Cell. 20, 2495–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraker, P.J. , Speck, J.C. , 1978. Protein and cell membrane iodinations with a sparingly soluble chloroamide,1,3,4,6-tetrachloro-3α,6α-diphenylglycoluril. Biochem. Biophys. Res. Commun. 80, 849–857. [DOI] [PubMed] [Google Scholar]
- Gambarotta, G. , Pistoi, S. , Giordano, S. , Comoglio, P.M. , Santoro, C. , 1994. Structure and inducible regulation of the human MET promoter. J. Biol. Chem. 269, 12852–12857. [PubMed] [Google Scholar]
- Giordano, S. , Di Renzo, M.F. , Ferracini, R. , Chiadò-Piat, L. , Comoglio, P.M. , 1988. p145, a protein with associated tyrosine kinase activity in a human gastric carcinoma cell line. Mol. Cell. Biol. 8, 3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, J.M. , Chess, R.B. , 2003. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2, 214–221. [DOI] [PubMed] [Google Scholar]
- Hildebrandt, B. , le Coutre, P. , Nicolaou, A. , Köble, K. , Riess, H. , Dörken, B. , 2007. Cetuximab: appraisal of a novel drug against colorectal cancer. Recent Results Cancer Res. 176, 135–143. [DOI] [PubMed] [Google Scholar]
- Hosono, M. , Endo, K. , Sakahara, H. , Watanabe, Y. , Saga, T. , Nakai, T. , Kawai, C. , Matsumori, A. , Yamada, T. , Watanabe, T. , Konishil, J. , 1992. Human/mouse chimeric antibodies show low reactivity with human anti-murine antibodies (HAMA). Br. J. Cancer. 65, 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houldsworth, J. , Cordon-Cardo, C. , Ladanyi, M. , Kelsen, D.P. , Chaganti, R.S. , 1990. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res. 50, 6417–6422. [PubMed] [Google Scholar]
- Ikebuchi, F. , Oka, K. , Mizuno, S. , Fukuta, K. , Hayata, D. , Ohnishi, H. , Nakamura, T. , 2013. Dissociation of c-Met phosphotyrosine sites in human cells in response to mouse hepatocyte growth factor but not human hepatocyte growth factor: the possible roles of different amino acids in different species. Cell Biochem. Funct. 31, 298–304. [DOI] [PubMed] [Google Scholar]
- Lee, J.M. , Kim, B. , Lee, S.B. , Jeong, Y. , Oh, Y.M. , Song, Y.J. , Jung, S. , Choi, J. , Lee, S. , Cheong, K.H. , Kim, D.U. , Park, H.W. , Han, Y.K. , Kim, G.W. , Choi, H. , Song, P.H. , Kim, K.A. , 2014. Cbl-independent degradation of Met: ways to avoid agonism of bivalent Met-targeting antibody. Oncogene. 33, 34–43. [DOI] [PubMed] [Google Scholar]
- Lennerz, J.K. , Kwak, E.L. , Ackerman, A. , Michael, M. , Fox, S.B. , Bergethon, K. , Lauwers, G.Y. , Christensen, J.G. , Wilner, K.D. , Haber, D.A. , Salgia, R. , Bang, Y.J. , Clark, J.W. , Solomon, B.J. , Iafrate, A.J. , 2011. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 29, 4803–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Zeng, W. , Wortinger, M.A. , Yan, S.B. , Cornwell, P. , Peek, V.L. , Stephens, J.R. , Tetreault, J.W. , Xia, J. , Manro, J.R. , Credille, K.M. , Ballard, D.W. , Brown-Augsburger, P. , Wacheck, V. , Chow, C.K. , Huang, L. , Wang, Y. , Denning, I. , Davies, J. , Tang, Y. , Vaillancourt, P. , Lu, J. , 2014. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin. Cancer Res. 20, 6059–6070. [DOI] [PubMed] [Google Scholar]
- Lorenzato, A. , Olivero, M. , Patanè, S. , Rosso, E. , Oliaro, A. , Comoglio, P.M. , Di Renzo, M.F. , 2002. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 62, 7025–7030. [PubMed] [Google Scholar]
- Michieli, P. , Basilico, C. , Pennacchietti, S. , Maffè, A. , Tamagnone, L. , Giordano, S. , Bardelli, A. , Comoglio, P.M. , 1999. Mutant Met-mediated transformation is ligand-dependent and can be inhibited by HGF antagonists. Oncogene. 18, 5221–5231. [DOI] [PubMed] [Google Scholar]
- Michieli, P. , Mazzone, M. , Basilico, C. , Cavassa, S. , Sottile, A. , Naldini, L. , Comoglio, P.M. , 2004. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 6, 61–73. [DOI] [PubMed] [Google Scholar]
- Migliardi, G. , Sassi, F. , Torti, D. , Galimi, F. , Zanella, E.R. , Buscarino, M. , Ribero, D. , Muratore, A. , Massucco, P. , Pisacane, A. , Risio, M. , Capussotti, L. , Marsoni, S. , Di Nicolantonio, F. , Bardelli, A. , Comoglio, P.M. , Trusolino, L. , Bertotti, A. , 2012. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin. Cancer Res. 18, 2515–2525. [DOI] [PubMed] [Google Scholar]
- Miller, M. , Ginalski, K. , Lesyng, B. , Nakaigawa, N. , Schmidt, L. , Zbar, B. , 2001. Structural basis of oncogenic activation caused by point mutations in the kinase domain of the MET proto-oncogene: modeling studies. Proteins. 44, 32–43. [DOI] [PubMed] [Google Scholar]
- Mukohara, T. , Civiello, G. , Davis, I.J. , Taffaro, M.L. , Christensen, J. , Fisher, D.E. , Johnson, B.E. , Jänne, P.A. , 2005. Inhibition of the met receptor in mesothelioma. Clin. Cancer Res. 11, 8122–8130. [DOI] [PubMed] [Google Scholar]
- Nakamura, Y. , Matsubara, D. , Goto, A. , Ota, S. , Sachiko, O. , Ishikawa, S. , Aburatani, H. , Miyazawa, K. , Fukayama, M. , Niki, T. , 2008. Constitutive activation of c-Met is correlated with c-Met overexpression and dependent on cell-matrix adhesion in lung adenocarcinoma cell lines. Cancer Sci. 99, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini, L. , Vigna, E. , Narsimhan, R.P. , Gaudino, G. , Zarnegar, R. , Michalopoulos, G.K. , Comoglio, P.M. , 1991. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene. 6, 501–504. [PubMed] [Google Scholar]
- Ou, S.H. , Kwak, E.L. , Siwak-Tapp, C. , Dy, J. , Bergethon, K. , Clark, J.W. , Camidge, D.R. , Solomon, B.J. , Maki, R.G. , Bang, Y.J. , Kim, D.W. , Christensen, J. , Tan, W. , Wilner, K.D. , Salgia, R. , Iafrate, A.J. , 2011. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J. Thorac. Oncol. 6, 942–946. [DOI] [PubMed] [Google Scholar]
- Pacchiana, G. , Chiriaco, C. , Stella, M.C. , Petronzelli, F. , De Santis, R. , Galluzzo, M. , Carminati, P. , Comoglio, P.M. , Michieli, P. , Vigna, E. , 2010. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J. Biol. Chem. 285, 36149–36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, M. , Dean, M. , Cooper, C.S. , Schmidt, M. , O'Brien, S.J. , Blair, D.G. , Vande Woude, G.F. , 1986. Mechanism of met oncogene activation. Cell. 45, 895–904. [DOI] [PubMed] [Google Scholar]
- Pennacchietti, S. , Michieli, P. , Galluzzo, M. , Mazzone, M. , Giordano, S. , Comoglio, P.M. , 2003. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 3, 347–361. [DOI] [PubMed] [Google Scholar]
- Petrelli, A. , Circosta, P. , Granziero, L. , Mazzone, M. , Pisacane, A. , Fenoglio, S. , Comoglio, P.M. , Giordano, S. , 2006. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc. Natl. Acad. Sci. USA. 103, 5090–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plosker, G.L. , Figgitt, D.P. , 2003. Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia. Drugs. 63, 803–843. [DOI] [PubMed] [Google Scholar]
- Prat, M. , Crepaldi, T. , Pennacchietti, S. , Bussolino, F. , Comoglio, P.M. , 1998. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J. Cell Sci. 111, 237–247. [DOI] [PubMed] [Google Scholar]
- Schmidt, L. , Duh, F.M. , Chen, F. , Kishida, T. , Glenn, G. , Choyke, P. , Scherer, S.W. , Zhuang, Z. , Lubensky, I. , Dean, M. , Allikmets, R. , Chidambaram, A. , Bergerheim, U.R. , Feltis, J.T. , Casadevall, C. , Zamarron, A. , Bernues, M. , Richard, S. , Lips, C.J. , Walther, M.M. , Tsui, L.C. , Geil, L. , Orcutt, M.L. , Stackhouse, T. , Lipan, J. , Slife, L. , Brauch, H. , Decker, J. , Niehans, G. , Hughson, M.D. , Moch, H. , Storkel, S. , Lerman, M.I. , Linehan, W.M. , Zbar, B. , 1997. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 16, 68–73. [DOI] [PubMed] [Google Scholar]
- Schelter, F. , Kobuch, J. , Moss, M.L. , Becherer, J.D. , Comoglio, P.M. , Boccaccio, C. , Krüger, A. , 2010. A disintegrin and metalloproteinase-10 (ADAM-10) mediates DN30 antibody-induced shedding of the met surface receptor. J. Biol. Chem. 285, 26335–26340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawler, D.L. , Bartholomew, R.M. , Smith, L.M. , Dillman, R.O. , 1985. Human immune response to multiple injections of murine monoclonal IgG. J. Immunol. 135, 1530–1535. [PubMed] [Google Scholar]
- Stella, G.M. , Benvenuti, S. , Gramaglia, D. , Scarpa, A. , Tomezzoli, A. , Cassoni, P. , Senetta, R. , Venesio, T. , Pozzi, E. , Bardelli, A. , Comoglio, P.M. , 2011. MET mutations in cancers of unknown primary origin (CUPs). Hum. Mutat. 32, 44–50. [DOI] [PubMed] [Google Scholar]
- Tamhane, U.U. , Gurm, H.S. , 2008. The chimeric monoclonal antibody abciximab: a systematic review of its safety in contemporary practice. Expert Opin. Drug Saf. 7, 809–819. [DOI] [PubMed] [Google Scholar]
- Tong, C.Y. , Hui, A.B. , Yin, X.L. , Pang, J.C. , Zhu, X.L. , Poon, W.S. , Ng, H.K. , 2004. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J. Neurosurg. 100, 87–93. [DOI] [PubMed] [Google Scholar]
- Trusolino, L. , Bertotti, A. , Comoglio, P.M. , 2010. MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell. Biol. 11, 834–848. [DOI] [PubMed] [Google Scholar]
- Wen, P.Y. , Schiff, D. , Cloughesy, T.F. , Raizer, J.J. , Laterra, J. , Smitt, M. , Oliner, K.S. , Anderson, A. , Zhu, M. , Loh, E. , Reardon, D.A. , 2011. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 13, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data