

Abstract
Promoter DNA hypermethylation is an important biomarker of hepatocellular carcinoma (HCC), supporting the potential utility of demethylating agents in this disease. Guadecitabine (SGI‐110) is a second‐generation hypomethylating agent formulated as a dinucleotide of decitabine and deoxyguanosine that yields longer half‐life and more extended decitabine exposure than decitabine IV infusion. Here we performed preclinical evaluation of SGI‐110 in HCC models to guide the design of a phase I/II clinical trial. HCC cell lines and xenograft models were used to determine the antitumor activity of SGI‐110 as a single agent and in combination with oxaliplatin. Pretreatment with low doses of SGI‐110 significantly synergized with oxaliplatin yielding enhanced cytotoxicity. The combination of SGI‐110 and oxaliplatin was well tolerated and significantly delayed tumor growth in mice compared to oxaliplatin alone. Bromouridine‐labeled RNA sequencing (Bru‐seq) was employed to elucidate the effects of SGI‐110 and/or oxaliplatin on genome‐wide transcription. SGI‐110 and the combination treatment inhibited the expression of genes involved in WNT/EGF/IGF signaling. DNMT1 and survivin were identified as novel PD markers to monitor the efficacy of the combination treatment. In conclusion, SGI‐110 priming sensitizes HCC cells to oxaliplatin by inhibiting distinct signaling pathways. We expect that this combination treatment will show low toxicity and high efficacy in patients. Our study supports the use of the combination of low doses of SGI‐110 and oxaliplatin in HCC patients.
Keywords: SGI-110, Oxaliplatin, Hepatocellular carcinoma, Combination therapy, DNMT1
Highlights
SGI‐110, a second generation HMA, significantly inhibits HCC cell proliferation.
Cytotoxicity of oxaliplatin is enhanced by SGI‐110 priming in HCC cell lines.
Combination of SGI‐110 and oxaliplatin at low doses delays HCC tumor growth.
WNT/EGF/IGF signaling are implicated in the synergistic response to SGI‐110 and oxaliplatin.
DNMT1 and survivin are potential PD markers to monitor efficacy.
Abbreviations
- HCC
hepatocellular carcinoma
- HMA
hypomethylating agent
- Bru-seq
bromouridine-labeled RNA sequencing
- GSEA
geneset enrichment analysis
1. Introduction
Liver cancer claimed 746,000 lives worldwide in 2012 (Ferlay J, 2013). With poor survival statistics and a growing rate of incidence due to an increase in HCV infections and other liver diseases in the population, liver cancer has now become the fifth leading cause of cancer‐related deaths in the United States (Siegel et al., 2014). Eighty percent of all liver cancer cases are hepatocellular carcinoma (HCC). Patients diagnosed at advanced stage of the disease are not eligible for potential curative treatment or transarterial chemoembolization, leaving systemic treatment as the major remaining therapeutic option. Sorafenib, a VEGFR, PDGFR and Raf kinase inhibitor, is the only FDA approved drug since 2007 for use as palliative treatment for these patients (Forner et al., 2012). Treatment with sorafenib has been shown to improve median survival and time to progression by 3 months in HCC patients compared to placebo (Llovet et al., 2008). Unfortunately, drug resistance and adverse events have limited its applicability in the clinic. New effective treatments for HCC are in urgent need.
Overexpression of DNA methyltransferases DNMT1 and DNMT3A is a characteristic of HCC (Figure S1) (Wurmbach et al., 2007; Fan et al., 2009; Roessler et al., 2010). Knocking down DNMT1 significantly inhibits HCC cell proliferation (Fan et al., 2009), further implicating an oncogenic role of DNMT1. In line with overexpression of DNMTs, DNA hypermethylation in the promoter regions of tumor suppressor genes like CDKN2A (p16) and CDH1 (E‐cadherin) has been associated with HCC. A series of DNA methylation‐regulated biomarkers specific for HCC have been identified by microarray analyses and next generation sequencing (Nishida et al., 2012; Shitani et al., 2012). Treatment with decitabine restores transcription of many tumor suppressor genes silenced by promoter hypermethylation and inhibits cell proliferation (Suh et al., 2000; Neumann et al., 2012; Zhang et al., 2012). Taken together, these results provide the impetus for the therapeutic targeting of DNMTs in HCC.
Guadecitabine (SGI‐110) is a dinucleotide comprising of deoxyguanosine and the DNA demethylating agent decitabine (2‐deoxy‐5′‐aza‐cytidine), an FDA approved agent for myelodisplastic syndrome (MDS). When activated, decitabine is incorporated into DNA and the presence of nitrogen at the 5 position of the pyrimidine leads to formation of covalent DNA‐protein adducts with DNMTs (Jones and Taylor, 1980; Song et al., 2012). DNMT proteins bound to decitabine are degraded, resulting in a down‐regulation of total DNMT protein levels and the reduction in the hypermethylation phenotype. Unfortunately, decitabine is rather chemically unstable in vivo. Catalyzed by cytidine deaminase (CDA), 2‐deoxy‐5′‐aza‐cytidine is rapidly converted into the inactive metabolite 2‐deoxy‐5′‐aza‐uridine. Importantly, SGI‐110 is a dinucleotide of deoxyguanosine and decitabine to protect the latter from CDA inactivation. SGI‐110 is formulated as a pharmaceutically stable subcutaneous injection formulation that yields longer half‐life and more extended decitabine exposure than decitabine IV infusion (Yoo et al., 2007; Chuang et al., 2010; Tellez et al., 2014). Such desirable features make SGI‐110 a clinically appealing demethylating drug.
Combination treatments have advantages over single agent applications in that they attack multiple targets making it less likely for the tumor to develop resistance, and allow anti‐cancer agents to be used at lower doses reducing adverse events. Oxaliplatin has been evaluated in combination with gemcitabine (GEMOX) (Louafi et al., 2007) or with leucovorin and fluorouracil (FOLFOX4) (Qin et al., 2013) in HCC. Although no significant survival benefit was observed in the FOLFOX4 phase III trial, the efficacy reported in the GEMOX phase II trial and the favorable safety profile shared by both studies suggest the potential of oxaliplatin‐based treatments for HCC patients. The combination of SGI‐110 and cisplatin (Fang et al., 2014) and carboplatin (Wang et al., 2014) showed encouraging anti‐tumor activity in ovarian cancer, implying potential therapeutic utility of such combinations. However, the combination of SGI‐110 and oxaliplatin has never been tested in HCC. In this study we evaluated the in vivo efficacy of SGI‐110 as a single agent and in combination with oxaliplatin at low doses as a novel therapy for HCC. Using Bru‐seq, a recently developed next generation sequencing technique measuring the newly synthesized RNA (Paulsen et al., 2014, 2013), we elucidated the effects these agents have on the transcriptome in HCC either alone or in combination. We discovered the WNT/EGF/IGF signaling pathways as potential targets of the combination treatment and identified DNMT1 and survivin as novel PD markers. The findings from this study will be used to guide the design of clinical studies of the use of SGI‐110 in combination with oxaliplatin for the treatment of HCC.
2. Materials and methods
2.1. Cell culture
SNU‐398, SNU‐449, SNU‐387, SNU‐475, Hep‐3B, Hep‐G2 hepatocellular carcinoma cell lines were obtained from ATCC (Manassas, VA) in June 2012. Isoenzymology and STR analyses were performed by ATCC to confirm species and cell line identity. No further authentication was performed in‐house. Cells were expanded into 10 tubes (1 × 106/tube) and frozen immediately. All cell lines were cultured as monolayers and maintained in RPMI1640 supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere with 5% CO2 at 37 °C. Cells were kept in culture for 20 passages and discarded, then a new batch of cells was used in subsequent experiments. PlasmoTest™ (InvivoGen, San Diego, CA) were performed every three weeks to confirm all cell lines were mycoplasma‐free.
2.2. Compound preparation
For in vitro experiments, 10 mM stock solution was prepared by dissolving SGI‐110 (Astex Pharmaceuticals, Dublin, CA) in PBS. Solution was kept at −80 °C for storage. For in vivo experiments, SGI‐110 was diluted in reconstitution solvent (65% propylene glycol, 10% ethanol and 25% glycerin). Solution was stored at 4 °C. Oxaliplatin was purchased from BIOTANG Inc. (Lexington, MA) and freshly dissolved in DMSO to prepare a 10 mM stock solution. Z‐VAD‐fmk (Tocris, Minneapolis, MN) and Necrostatin‐1 (Cayman, Ann Arbor, MI) were freshly dissolved in DMSO to make 40 mM stock solutions.
2.3. MTT assay
Cytotoxicity of compounds was evaluated with 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. Cells were placed in 96‐well plate at 1000 cells/well on Day 1. After overnight attachment, SGI‐110 was added to the wells at sequential dilutions (10 nM–1 μM for most cell lines) on Day 2. Due to the hydrolysis of the compound, SGI‐110 treatment was repeated every 24 h. After 72 h treatment (on Day 5), SGI‐110 containing media was carefully removed and fresh cell culture media was added to the plate. For combination treatment, oxaliplatin was added on Day 5 after changing the media, and kept in culture for 72 h treatment. On Day 8, compound‐containing media was carefully removed and fresh cell culture media was added to the plates. On Day 12, MTT was added into the media to a final concentration of 300 μg/mL. Cells were incubated for 3 h at 37 °C, and the insoluble formazan converted by viable cells was dissolved in 150 μL of DMSO. Absorbance at 570 nm was read on a microplate reader (Molecular Devices, Sunnyvale, CA), and inhibition of cell proliferation was calculated using the following formula:
Synergistic effect of the combination treatment was evaluated by computing combination index (CI) using the Chou‐Talalay method (Chou, 2006). CI values lower than 1 indicates synergistic effect.
2.4. Colony formation assay
Cells were placed in a 96‐well plate at 200 cells/well or in a 6‐well plate at 5000 cells/well on Day 1. Treatment schedules were performed as described for the MTT assay. After treatment, cells were kept in culture until colonies were observed in control wells. Colonies were then fixed and stained with 0.05% crystal violet solution (2% formaldehyde, 40% methanol in distilled water), washed with water to remove excess stain, imaged with Odyssey Imaging Systems (LI‐COR Biosciences, Lincoln, NE), and quantified with Image J software.
2.5. Western blotting
Cells (4 × 105) were cultured in 60 mm tissue culture dishes and treated with SGI‐110 or oxaliplatin at designated concentrations. After treatment, cells were lysed with cell lysis buffer at 4 °C for 30 min and centrifuged (12,000 rpm, 10 min, 4 °C). Protein concentrations of supernatants were measured with BCA assay (Thermo Fisher Scientific, Rockford, IL). 40 μg of protein per sample was subjected to SDS‐PAGE analysis. Proteins were electro‐transferred to methanol activated immobilon‐FL PVDF membranes (EMD Millipore, Billerica, MA). Membranes were blocked with 5% skim milk in TBST buffer and incubated with primary antibodies (anti‐E‐Cadherin, anti‐survivin, anti‐PARP and anti‐cleaved caspase 3 from Cell Signaling (Beverly, MA), anti‐DNMT1 and anti‐β‐tubulin from Santa Cruz Biotechnology (Santa Cruz, CA), anti‐ephrin‐B2 and anti‐PCNA from Sigma–Aldrich (Saint Louis, MO)) 1:1000 dilutions overnight at 4 °C. Membranes were then washed with TBST (10 min × 3), incubated with Dylight 800‐conjugated secondary antibodies (Thermo Fisher Scientific) 1:5000 dilutions in 5% milk for 1 h at room temperature, and washed with TBST (10 min × 2) and TBS (10 min). Fluorescent signal was then scanned by Odyssey Imaging Systems (LI‐COR Biosciences).
2.6. Bru‐seq analysis for nascent RNA synthesis
Bru‐seq analysis was performed as previously reported (Paulsen et al., 2014). Briefly, 4 × 106 SNU‐398 cells were placed in 10 cm dishes on Day 1. On Day 2, cells were treated with PBS or SGI‐110 at 100 nM for 72 h with fresh drug addition every 24 h. Cells were changed to fresh media on Day 5 and treated with DMSO or oxaliplatin at 3 μM for 4 h. Bromouridine, at a final concentration of 2 mM, was added into the media to label newly synthesized nascent RNA in the last 30 min of treatment. Cells were then collected in TRIZOL and total RNA was isolated. Bromouridine containing RNA population was further isolated and sequenced. Sequencing reads were mapped to the HG19 reference genome. Pre‐ranked gene lists were generated for each treatment through ranking genes by fold changes in gene synthesis levels compared to control, and analyzed with GSEA (Broad Institute, MA) (Mootha et al., 2003; Subramanian et al., 2005).
2.7. Xenograft study
SNU‐398 cells (2.0 × 106) in a 100 μL suspension of RPMI1640 were injected subcutaneously into the dorsal flank of 6‐week old athymic nude mice (The Jackson Laboratory, Bar Harbor, ME). Tumor size was monitored twice a week by caliper measurement using the following equation: V = d2×D/2, where d represents width and D represents length of the tumor. Mice were randomly grouped (n = 5 per group) when average tumor size reached 100 mm3. Treatment was given in 14‐day cycles. On Day 1–5, SGI‐110 was administrated by subcutaneous injection in a 100 μL vehicle to SGI‐110 single treatment and combination groups. On Day 5 (4 h after SGI‐110 administration) and Day 12, oxaliplatin treatment was given by intraperitoneal injection in 100 μL saline to oxaliplatin single treatment and combination group. Control mice received vehicle only. Study was concluded when tumor size in the group reached 2000 mm3. Unpaired Student's t‐test was performed for data analysis and p < 0.05 was considered significant.
2.8. Histochemical analysis
On necropsy, tumors, hearts, kidneys, livers, lungs, spleens and pancreases were collected, fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned. Sections (5 μm) were stained with hematoxylin and eosin to facilitate histologic examination. For Ki67 expression level, immunohistochemistry staining was performed on sections with Ki67 antibody. Embedding, sectioning and staining of samples were performed by the ULAM pathology core for animal research at the University of Michigan. Representative images were taken on an Olympus IX83 microscope with 20X magnification.
3. Results
3.1. SGI‐110 inhibits HCC cell proliferation
To evaluate the potency of SGI‐110 in HCC, we used six cell lines with different genetic backgrounds as in vitro models. Decitabine and SGI‐110 showed similar cytotoxicity in Hep‐3B cells (Figure S2). After 72 h treatment, SGI‐110 at 10 μM inhibited cell proliferation by no more than 27% in the six cell lines (Figure 1A, C). However, significant inhibition of cell proliferation was observed when cells were treated with SGI‐110 for 72 h and placed in fresh cell culture media for 7 days (Day 12 of the experiment). Four of the HCC cell lines Hep‐3B, SNU‐398, SNU‐449 and Hep‐G2 were more sensitive to SGI‐110, with IC50 values lower than 500 nM (Figure 1A). SGI‐110 also significantly inhibited colony formation in these four cell lines at low μM (Figure 1B). The SNU‐475 and SNU‐387 cells having doubling time of over 60 h (Park et al., 1995) are more resistant to SGI‐110 treatment. In long‐term treatment, SGI‐110 showed an IC50 value of 54 μM in SNU‐475 and 63 μM in SNU‐387 in MTT and 10 μM in colony formation assays (Figure 1C, D). Since SGI‐110 acts by incorporating into DNA and modifying methylation patterns during DNA synthesis, the long doubling time of SNU‐475 and SNU‐387 might be responsible for their lower sensitivity. SNU‐475 cells are more sensitive to SGI‐110 treatment in colony formation assay than in MTT assay supporting the notion that SGI‐110's mechanism of cytotoxicity is a combination of apoptosis, necrosis, and other modes of cell death. As the direct target of SGI‐110, DNMT1 protein level is a robust marker for assessing treatment efficacy. DNMT1 protein levels were depleted within 24 h of SGI‐110 treatment (100 nM) in both SNU‐398 and Hep‐G2 cells, and started to recover after SGI‐110 removal, confirming that SGI‐110 directly targets DNMT1 for protein degradation (Figure 1E). In conclusion, SGI‐110 targets DNMT1 and inhibits HCC cell proliferation in a long‐term treatment similar to other epigenetic‐targeted drugs.
Figure 1.
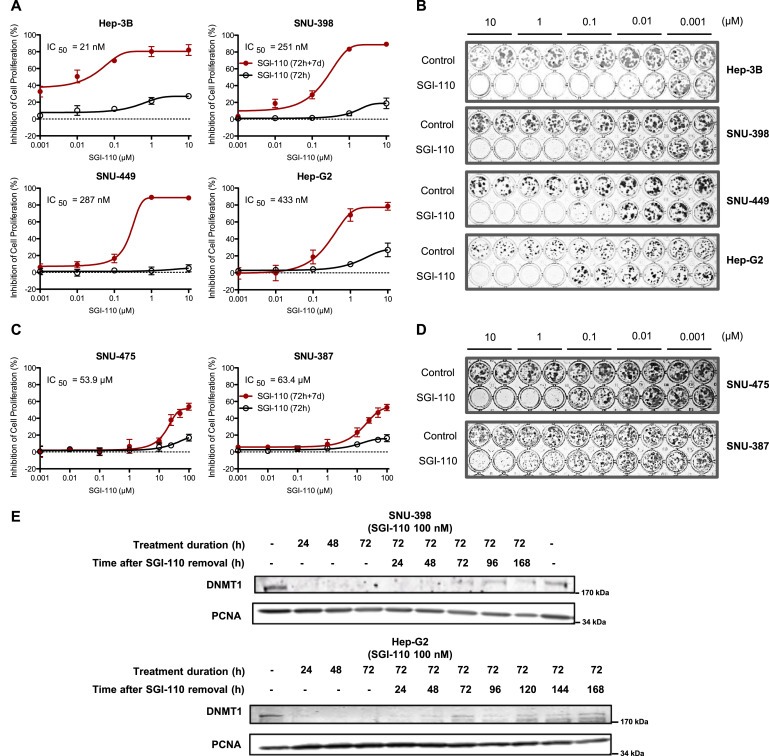
SGI‐110 is cytotoxic to hepatocellular carcinoma cell lines. A) Dose‐response curves for SGI‐110 in 4 sensitive HCC cell lines. For acute cytotoxicity (72 h), cells were treated with SGI‐110 for 72 h (fresh drug added every 24 h) and subjected to MTT assay. For long‐term cytotoxicity (72 h + 7 d), cells were treated with SGI‐110 for 72 h as above and changed to fresh cell culture media for seven days. MTT assay was then performed to evaluate the number of live cells under each treatment condition. Inhibition of cell proliferation was calculated against PBS‐treated controls. Data points are shown as Mean ± SD from three independent experiments. B) Colony formation assay for SGI‐110 in 4 sensitive HCC cell lines. Cells were treated with SGI‐110 for 72 h and left in culture in fresh media until colonies formed in PBS‐treated controls. Colonies were stained with crystal violet and imaged. C) Dose‐response curves for SGI‐110 in 2 resistant HCC cell lines from MTT assay as described in panel A. D) Colony formation assay for SGI‐110 in 2 resistant HCC cell lines as described in panel B. E) In SNU‐398 cells and Hep‐G2 cells, DNMT1 levels were reduced by SGI‐110 treatment in a time dependent manner. DNMT1 levels recovered after SGI‐110 removal. Cells were treated with SGI‐110 at 100 nM for up to 72 h. Cell culture media containing SGI‐110 were then removed and cells were changed into fresh media. Samples were collected at indicated time points from 24 h to 168 h after SGI‐110 removal.
3.2. Pretreatment with SGI‐110 sensitizes HCC cell lines to oxaliplatin
Oxaliplatin was selected as a candidate for combination with SGI‐110 because in our studies it demonstrated superior synergistic effect when compared to several other FDA approved drugs under similar conditions. Both oxaliplatin and the current standard of care, sorafenib, produced higher IC50 values in SNU‐475 and SNU‐387 cell lines than in SNU‐398 and Hep‐G2 cells (Figure 2A). Initially, we observed that pretreatment with SGI‐110 sensitized HCC cells to oxaliplatin treatment. The combination was hence further tested in three independent schedules: 72 h SGI‐110 pretreatment, where oxaliplatin was given either with SGI‐110 (schedule 1), immediately after SGI‐110 removal (schedule 2), or 72 h after SGI‐110 removal (schedule 3) (Figure 2B). In colony formation assay, best synergy was observed with combination treatment using schedule 2. Following this schedule, low‐dose oxaliplatin single treatment only inhibited colony formation by 15%, while pretreatment with SGI‐110 at 50 nM increased the inhibition to 54%, and pretreatment with SGI‐110 at 100 nM further increased the inhibition to 94% (Figure 2C).
Figure 2.
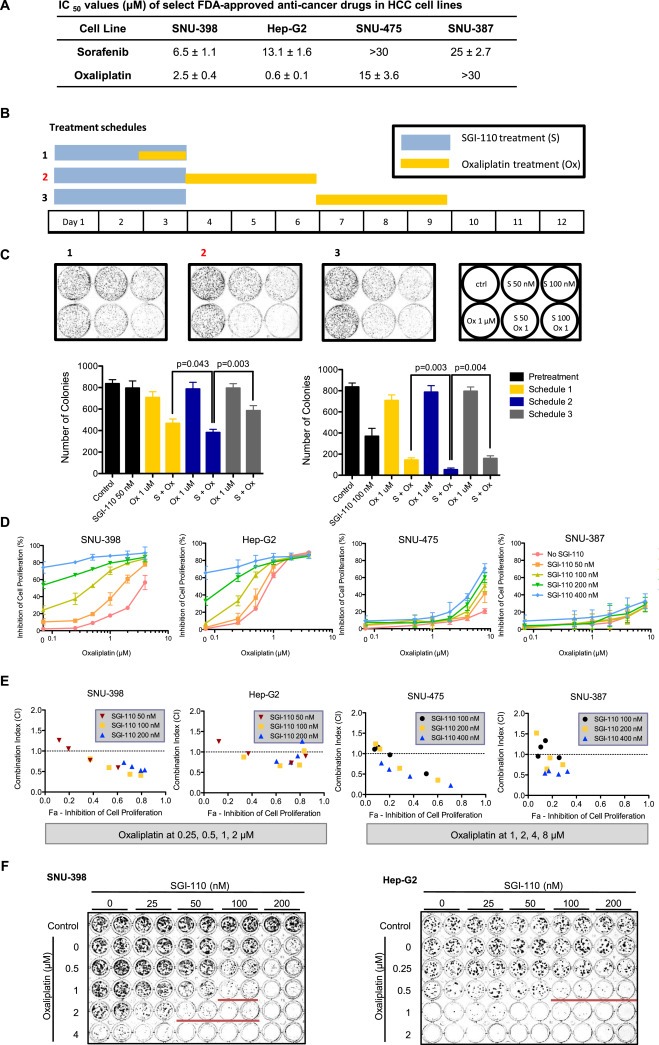
SGI‐110 pretreatment sensitizes SNU‐398 cells to oxaliplatin treatment. A) IC50 values of sorafenib and oxaliplatin in HCC cell lines. B) Schematic of three treatment schedules. SGI‐110 was given in the first 72 h (fresh drug added every 24 h), and oxaliplatin was added at different time points after SGI‐110 pretreatment. C) Representative images and quantitation of colony formation assay for the 3 treatment schedules. SNU‐398 cells were coated on Day 0 and treated according to schedules 1, 2, 3 as indicated. Cells were kept in culture until colonies were observed in controls. Colonies were stained with crystal violet and imaged. Number of colonies at each treatment condition was quantified with Image J. Results are shown as Mean ± SD (n = 3). P‐values were calculated using Student's t‐test. D) Dose‐response curves for oxaliplatin in 2 sensitive (SNU‐398, Hep‐G2) and 2 less sensitive (SNU‐475, SNU‐387) HCC cell lines pretreated with SGI‐110. Cells were treated with SGI‐110 for 72 h followed by oxaliplatin for 72 h, and placed in fresh cell culture media. Considering different sensitivity of the cell lines, SGI‐110 was given at 50, 100, 200, 400 nM in all cell lines, while oxaliplatin was given at 0.25–4 μM in SNU‐398 and Hep‐G2, and 0.5–8 μM in SNU‐475 and SNU‐387. MTT assay was performed on Day 12 to assess the number of live cells under each treatment condition. Inhibition of cell proliferation was calculated against controls. Data points were shown as Mean ± SD from three independent experiments. E) Combination Index (CI) of SGI‐110‐oxaliplatin combination treatment at different concentrations (non‐constant ratio) was calculated using Chou‐Talalay method. CI < 1 indicates synergistic effect of two compounds. F) Colony formation assay for SGI‐110 and oxaliplatin in SNU‐398 and Hep‐G2 cells. Cells were treated with SGI‐110 for 72 h followed by oxaliplatin for 72 h, and kept in culture until colonies were observed in PBS‐treated control. Colonies were stained and imaged. Red lines indicate wells with significant synergistic effect from the combination treatment.
We further tested the combination effect of SGI‐110 and oxaliplatin in two SGI‐110 sensitive cell lines (SNU‐398, Hep‐G2) and two SGI‐110 resistant cell lines (SNU‐475, SNU‐387) using MTT assay performed on Day 12 following schedule 2 (Figure 2B). Pretreatment with SGI‐110 sensitized SNU‐398, Hep‐G2 and SNU‐475 cells to oxaliplatin treatment, achieving over 50% inhibition of cancer cell proliferation with significant synergism (Figure 2D, E), but was not optimal in the other less sensitive cell line SNU‐387. The synergistic effect was further confirmed with colony formation assay (Figure 2F), suggesting the utility of combination of SGI‐110 and oxaliplatin as an effective therapy for HCC.
Pretreatment with the apoptosis inhibitor Z‐VAD‐fmk and/or with the necroptosis inhibitor necrostatin‐1 partially rescued SNU‐398 cells from oxaliplatin induced cytotoxicity in the MTT assay (Figure S3A). When cells were pre‐treated with Z‐VAD‐fmk and/or necrostatin‐1 and then treated with SGI‐110 in combination with oxaliplatin, only partial protection was observed mainly when inhibitors were given 1 h before oxaliplatin (Figure S3B, S3C). At higher doses of drugs no significant protection was observed with these inhibitors. The partial protection by these two inhibitors demonstrates that apoptosis and necroptosis are partly responsible for SGI‐110 and oxaliplatin‐induced cytotoxicity and suggests that other death pathways are also implicated. A cytostatic mechanism may also play a role on the effect of combination treatment.
3.3. SGI‐110 inhibits WNT3A and IGF/EGF signaling
To better understand the potential mechanism of SGI‐110 and oxaliplatin synergy, we performed Bru‐seq to examine the global changes in transcription in HCC cells. Pre‐ranked gene lists were analyzed with GSEA (Tables S1–S6). In SGI‐110 treatment, we identified enrichment of gene sets that were similarly upregulated by decitabine treatment in pancreatic cancer cells (Figure S4) (Missiaglia et al., 2005). This finding validates the similar transcriptional regulation by SGI‐110 and decitabine, and it also suggests overlap of methylation‐regulated genes in HCC and pancreatic cancer.
Using the false discovery rate (FDR, q‐value) of 0.25 as the cut‐off, oxaliplatin uniquely up‐regulated a cluster of hypermethylated genes characterized in AML, but no other gene sets were observed as positively associated with the treatment (Figure 3A). In cells treated with SGI‐110, the WNT3A gene set was among the most highly upregulated sets by SGI‐110 alone, and it was the only significantly enriched gene set in the combination treatment (Figure 3B). Heat map showing relative synthesis levels indicates up‐regulated expression of listed genes by SGI‐110 or its combination with oxaliplatin (Figure 3C). WNT/β‐catenin signaling is a major signature pathway of liver cancer, where nearly half of HCC patients exhibit activation of the pathway (Lachenmayer et al., 2012). The enriched gene set represents genes down‐regulated following WNT3A treatment (Labbe et al., 2007). Increased synthesis of genes negatively regulated by WNT3A suggests potential inhibition of WNT/β‐catenin signaling by SGI‐110 treatment. In support of this hypothesis, we found that the expression levels of the endogenous β‐catenin inhibitor, E‐cadherin, were gradually upregulated after SGI‐110 treatment. In contrast, the β‐catenin target gene, survivin, was down‐regulated consistent with the findings from GSEA (Figure 3D).
Figure 3.
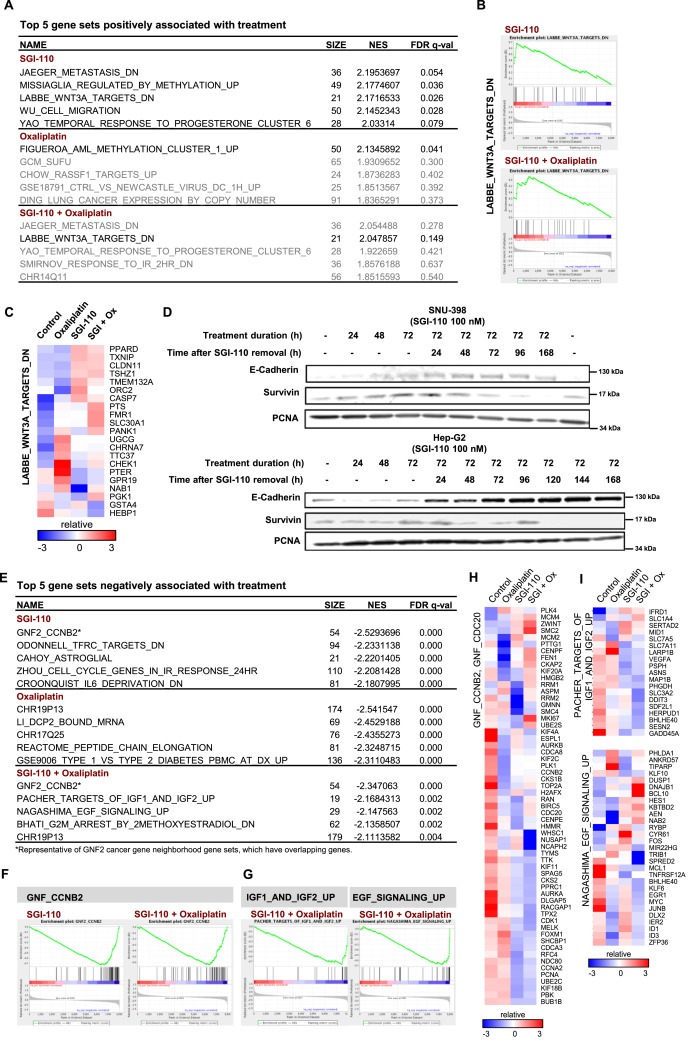
Bru‐Seq reveals inhibition of Wnt, IGF and EGF signaling by the combination of SGI‐110 and oxaliplatin. SNU‐398 cells were treated with SGI‐110 at 100 nM for 72 h, followed by oxaliplatin at 3 μM for 4 h. Nascent RNA was labeled by bromouridine 30 min before sample collection, and subjected to RNA sequencing. 22,984 genes were analyzed and filtered by gene size (>300 bp) and synthesis level (RPKM > 0.5). The gene list were then pre‐ranked by fold change of treatment over control and subjected to Gene Set Enrichment Analysis. A) Top 5 gene sets positively associated with SGI‐110, oxaliplatin or the combination treatment. Gene sets with false discovery rate (FDR) q‐value lower than 0.25 are considered true enrichment. B) Enrichment plots of LABBE_WNT3A_TARGETS_DN gene set over‐represented on the top of pre‐ranked gene lists from both SGI‐110 and combination treatment. C) Heat map for relative transcription level of genes in the LABBE_WNT3A_TARGETS_DN gene set over‐represented on the top of pre‐ranked gene lists from both drugs and combination treatment. D) In SNU‐398 and Hep‐G2 cells, E‐Cadherin and survivin were modulated by SGI‐110 in a time dependent manner. Cells were treated with SGI‐110 at 100 nM for up to 72 h. Cell culture media containing SGI‐110 were then removed and cells were left to recover in fresh cell media. Samples were collected at indicated time points from 24 h to 168 h after SGI‐110 removal. E) Top 5 gene sets negatively associated with SGI‐110, oxaliplatin or the combination treatment. F) Enrichment plots of the GNF2_CCNB2 gene set over‐represented on the bottom of pre‐ranked gene lists from both SGI‐110 and combination treatment. Several computational gene sets for cancer gene neighborhood were enriched in the same manner, and GNF2_CCNB2 was selected as a representative. G) Enrichment plots of IGF/EGF gene sets over‐represented on the bottom of pre‐ranked gene list from the combination treatment only. H) Heat map for relative transcription level of genes in the additive list of GNF2_CCNB2 and GNF2_CDC20 gene sets, which were over‐represented on the bottom of pre‐ranked gene lists from both SGI‐110 and combination treatment. I) Heat maps for relative transcription level of genes in the IGF/EGF gene sets over‐represented on the bottom of pre‐ranked gene list from the combination treatment only.
Among the gene sets negatively associated with treatments (Figure 3E), GNF2_CCNB2 was selected as representative for cancer gene neighborhood sets, where gene synthesis was downregulated by SGI‐110 alone or with the combination treatment (Figure 3F, H). Interestingly, the combination treatment induced unique enrichment of the PACHER_TARGETS_OF_IGF1_ AND_IGF2_UP and MAGASHIMA_EGF_SIGNALING _UP gene sets (Figure 3G), downregulating target genes from Insulin‐like growth factor (IGF) and epidermal growth factor (EGF) signaling (Figure 3I), whose activation or overexpression are often observed in HCC (Psyrri et al., 2012).
As revealed by GSEA, simultaneous inhibition of WNT/β‐catenin, IGF, and EGF signaling contributed to the synergistic effect of the combination treatment.
3.4. SGI‐110 treatment induces EFNB2 transcription levels
Using Bru‐seq to investigate the rates of transcription genome‐wide we observed that the transcription of the EFNB2 gene was significantly upregulated in SNU‐398 cells by SGI‐110 treatment (15.0 fold) and when combined with oxaliplatin (13.5 fold) (Figure 4A). When SNU‐398 cells were treated with SGI‐110 for 72 h, protein levels of the EFNB2 gene product ephrin‐B2 were increased dose‐dependently, where significant induction was observed at doses as low as 100 nM (Figure 4B). Upon treatment with SGI‐110 (100 nM), the expression of ephrin‐B2 protein increased from 72 h post treatment and achieved maximal induction at 96 h (Figure 4C), consistent with the EFNB2 up‐regulation after 72 h of SGI‐110 treatment revealed by Bru‐seq. Ephrin‐B2 is a ligand for tyrosine kinase receptor EPHB4 and EPHA4. In neuroblastoma, cells treated with decitabine re‐expressed ephrin‐B2 and exhibited impaired proliferation (Tang et al., 2004). In MethHC, a database of DNA methylation and gene expression in human cancers, EFNB2 is ranked 29th among the most differentially methylated genes in the promoter region between tumor and normal samples. The increased average methylation level was 0.2674 (in a 0–1 scale) in 204 hepatocellular carcinoma samples from TCGA (Huang et al., 2015) (Figure S5). This data suggests that EFNB2 can be a potential biomarker for SGI‐110 treatment in HCC.
Figure 4.
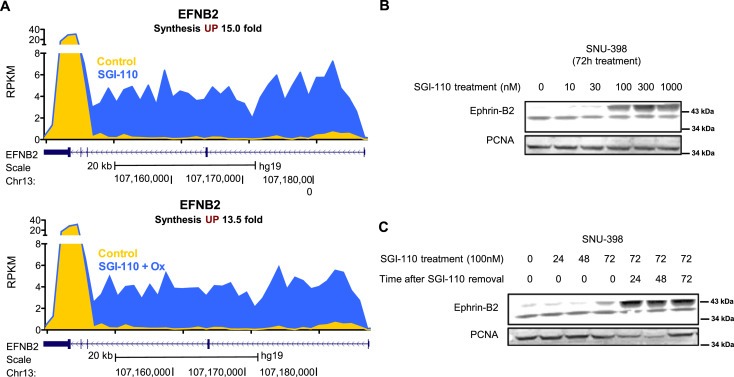
EFNB2 expression is up‐regulated by SGI‐110 and its combination treatment with oxaliplatin. A) Synthesis of EFNB2 nascent RNA is up‐regulated by SGI‐110 and its combination treatment with oxaliplatin in SNU‐398 cells as identified by Bru‐Seq. The gene map is from RefSeq Genes (UCSC genome browser, http://genome.ucsc.edu/). B) Ephrin‐B2 protein levels were up‐regulated dose‐dependently by SGI‐110 treatment. C) Ephrin‐B2 protein levels were up‐regulated time‐dependently by SGI‐110 treatment, and the induced ephrin‐B2 expression remained up to 72 h after SGI‐110 removal.
However, in our efforts to further validate the role of EFNB2 in HCC, we found that SGI‐110‐ induced ephrin‐B2 expression was only observed in SNU‐398 among a panel of 12 cancer cell lines that included 6 HCC cell lines (SNU‐398, Hep‐3B, SNU‐449, Hep‐G2, SNU‐475, SNU‐387), 3 pancreatic cancer cell lines (BxPC‐3, Panc‐1, MiaPaCa‐2) and 3 other cancer cell lines (HCT‐116, LNCaP, U87), indicating that upregulation of ephrin‐B2 is cell line specific (Figure S6). Therefore, EFNB2 is not a robust PD marker for efficacy of SGI‐110 treatment, but its importance in HCC needs to be further characterized.
3.5. Survivin is a new PD marker for SGI‐110 and oxaliplatin combination treatment
DNMT1 and survivin expression levels changed after SGI‐110 treatment in a time dependent manner as discussed above. To address their potential as PD markers, we evaluated the expression levels of these proteins over different times of SGI‐110 and/or oxaliplatin treatments (Figure 5A). While oxaliplatin treatment did not affect DNMT1 protein levels in SNU‐398 cells (Figure 5B), SGI‐110 treatment reduced DNMT1 levels in these cells. Following drug removal, DNMT1 protein levels started to recover from SGI‐110‐induced depletion within 72 h. Importantly, when oxaliplatin was given after SGI‐110 pretreatment, no recovery of DNMT1 protein levels occurred. Survivin is a member of the inhibitor of apoptosis family that directly inhibits apoptosis and promotes cell survival (Cheung et al., 2013). Survivin levels were downregulated after long‐term SGI‐110 treatment as mentioned above. Interestingly, survivin levels were not affected within 72 h of SGI‐110 or oxaliplatin treatment, but a significant decrease was detected within 48 h of the combination treatment, suggesting a robust disruption of the survival signaling in these cells. We further evaluated the levels of the apoptotic markers cleaved PARP and caspase 3 after 72 h combination treatment. While significant dose‐dependent decrease in DNMT1 and survivin levels were detected with the combination, only mild increases of the two apoptotic markers were observed (Figure S7), demonstrating that DNMT1 and survivin are more robust biomarkers in SNU398 at this early time point.
Figure 5.
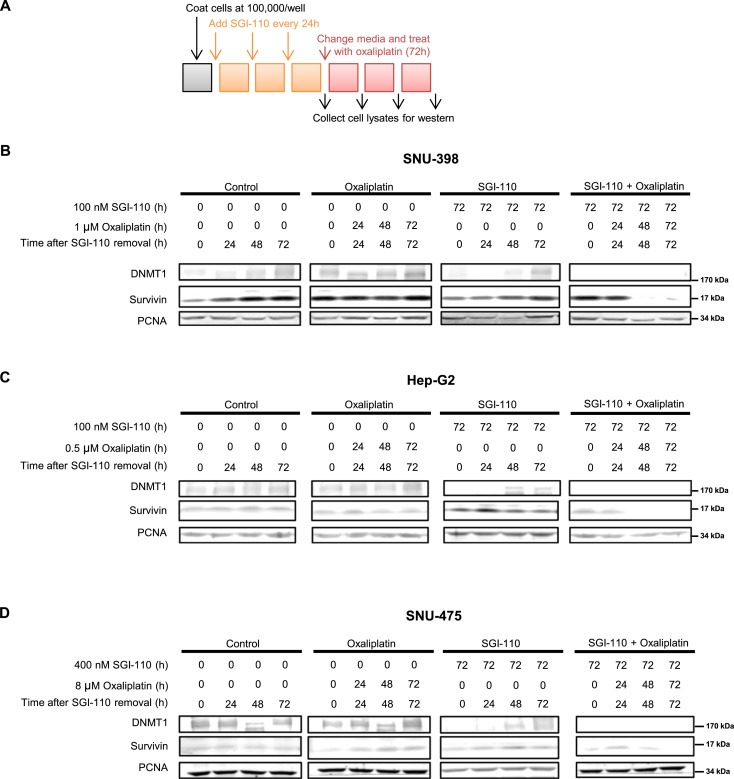
Oxaliplatin treatment down‐regulates DNMT1 and survivin levels in SGI‐110 pre‐treated cells. A) Schematic for SGI‐110 and oxaliplatin treatment. Cells were treated with SGI‐110 for 72 h, changed to fresh media and treated with oxaliplatin for up to 72 h. Samples were collected every 24 h after SGI‐110 treatment to study the time course effects of SGI‐110 and oxaliplatin treatments. Control, oxaliplatin, SGI‐110 and combination treated cell lysates were blotted for DNMT1 and survivin. Representative results are shown for B) SNU‐398, C) Hep‐G2 and D) SNU‐475 cells.
To further validate these potential markers for the combination treatment, we performed the same experiment in another sensitive cell line Hep‐G2, and the least sensitive cell lines SNU‐475 and SNU‐387. Similarly, the combination treatment decreased the levels of survivin and blocked DNMT1 protein recovery following SGI‐110 removal in Hep‐G2 cells (Figure 5C). In order to achieve similar blockade of DNMT1 recovery and decreased survivin levels in the oxaliplatin‐resistant SNU‐475 cells, higher doses were required when compared to the sensitive SNU‐398 cells (Figure 5D). In the other less sensitive cell line SNU‐387, where no significant synergism was detected there was no difference in DNMT1 and survivin levels between single and combination treatments (Figure S8).
Considering their expression profiles across the 4 HCC cell lines, the protein levels of DNMT1 and survivin reflect the molecular responses of these cells to tested drugs, suggesting that they could be used as novel PD markers for the effectiveness of the combination treatment.
3.6. SGI‐110 as single agent and in combination with oxaliplatin delays tumor growth without systemic toxicity
To investigate the in vivo antitumor efficacy of SGI‐110 alone and in combination with oxaliplatin, xenograft studies were performed in athymic nude mice. Subcutaneous human HCC xenografts from SNU‐398 cells were established on the dorsal flank of the immunodeficient mice, and treated with SGI‐110, oxaliplatin, combination or vehicle until tumor size in the group reached 2000 mm3 (Figure 6A). Treatments were given in 14‐day cycles, where SGI‐110 (2 mg/kg) was given daily on Day 1–5, and oxaliplatin (5 mg/kg) was given on Day 5 (4 h after SGI‐110 administration) and Day 12. Weekly oxaliplatin treatment was selected based on its efficacy and safety profile reported in previous studies (Selvakumaran et al., 2013; Gaur et al., 2014; Zeng et al., 2014). In our experience, oxaliplatin shows significant toxicity when used at doses higher than 10 mg/kg or using repetitive dosing. In an ongoing phase II trial (NCT01752933), SGI‐110 was given on five consecutive days followed by a 23‐day recovery, rather than a more frequent schedule. Considering the in vivo half‐life of SGI‐110 of 4 h (Yoo et al., 2007; Tellez et al., 2014), the first oxaliplatin treatment in the 14‐day cycle was given 4 h after the last daily SGI‐110 treatment on Day 5, matching the optimal oxaliplatin treatment schedule established in Figure 2. Although oxaliplatin (5 mg/kg) did not show efficacy, SGI‐110 (2 mg/kg) treatment significantly suppressed growth of tumors after 15 days of treatment. Combination with oxaliplatin further delayed tumor growth, where significant difference in tumor sizes was achieved as early as day 8. On day 19, when average tumor sizes in control and oxaliplatin treatment groups passed the experiment endpoint of 2000 mm3, the average tumor size was 1010 ± 247 mm3 (p=0.0039) for SGI‐110 treatment alone, and only 391 ± 100 mm3 (p=0.0001) for combination with oxaliplatin. SGI‐110 treatment was able to delay endpoint from day 19 to day 26, and combination treatment further delayed the endpoint to day 31 (Figure 6B), indicating substantial survival benefits from the treatments.
Figure 6.
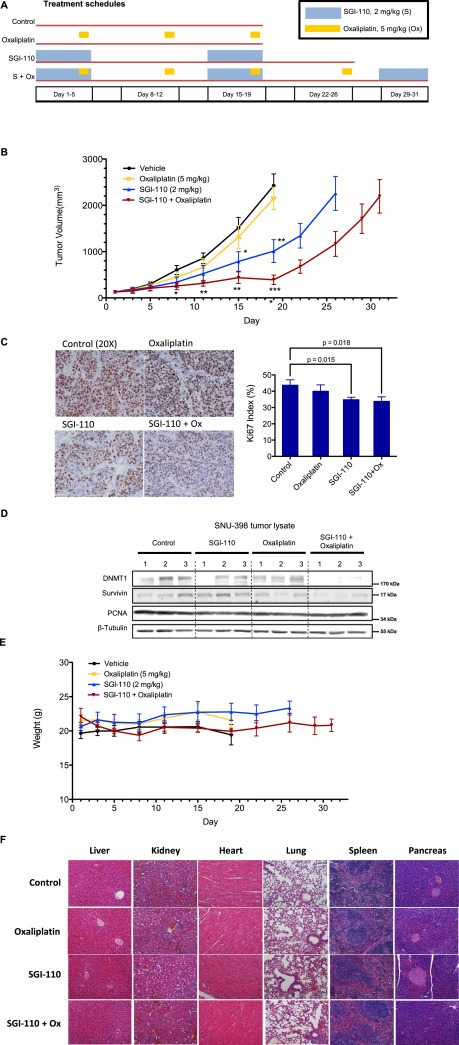
Single agent SGI‐110 and its combination treatment with oxaliplatin inhibit tumor growth in SNU‐398 liver cancer xenograft model. A) Treatment schedule. Mice with SNU‐398 tumors were randomized into 4 treatment groups (n = 5) and received vehicle, oxaliplatin (5 mg/kg weekly), SGI‐110 (2 mg/kg from day 1 to day 5 in a 2‐week cycle) or combination treatment. Mice were sacrificed when tumor size reached 2000 mm3. B) Tumor sizes were significantly reduced in mice treated with SGI‐110, and further delayed in mice with SGI‐110 and oxaliplatin combination treatment. Statistical significance was calculated using Student's t‐test. Error bars indicate Mean ± SEM. and *p < 0.05, ****p < 0.0001. C) Ki67 immunohistochemistry staining in tumor sections. Ki67 index was calculated as percentage of Ki67 positive cells in total number of cells in the field (n = 6, 3 fields of view from 2 tumors per group). Graphical data is presented as Mean ± SD. P‐values were calculated using Student's t‐test. D) Lysates from 3 tumors per treatment group were blotted for DNMT1 and survivin. E) SGI‐110 and its combination treatment with oxaliplatin did not exert systemic toxicity in vivo. Animal weights did not change significantly during the course of treatment. Error bars indicate Mean ± SEM. F) Representative micrographs of hematoxylin and eosin (H&E)‐stained organ sections. Images were taken with an Olympus IX83 inverted microscope at 20X magnification. In histopathology study, no significant morphological changes were detected in major organs after SGI‐110 or combination treatment.
Mice were sacrificed and samples were collected when the tumor sizes reached 2000 mm3 in all groups. The tumor samples from control and oxaliplatin treatment groups were collected on Day 19, while the samples from the SGI‐110 and combination groups were collected on Day 26 and Day 31, respectively. Despite reaching the same maximum tumor volume of 2000 mm3, the tumors receiving SGI‐110 treatment as a single agent or in combination with oxaliplatin showed a significant decrease in Ki67 levels, suggesting reduction in cell proliferation (Figure 6C).
Evaluation of the potential PD markers in tumor tissues unveiled that DNMT1 levels were significantly downregulated in the combination group and mildly decreased with SGI‐110 treatment, whereas no significant changes were detected with vehicle or oxaliplatin treatment. Similar to our results in in vitro experiments, survivin protein levels were markedly decreased with the combination (Figure 6D). These findings further validate these two PD markers to measure efficacy of the combination treatment, suggesting their potential use in future clinical studies.
No systemic symptoms of toxicity such as weakness, weight loss or lethargy were observed in any treatment group (Figure 6E). H&E stained organ sections of liver, kidney, heart, lung, spleen and pancreas did not reveal major histopathological changes, further confirming the safety of the treatments (Figure 6F).
4. Discussion
Accumulating pre‐clinical data has suggested epigenetic therapy as an appealing strategy to target HCC. However, when decitabine was tested in several clinical trials in solid tumors such as colon and lung cancers (Graham et al., 2009; Fan et al., 2014), no efficacy was observed as single agent. Compelling preclinical data has prompted phase II trials of combination treatment with decitabine and carboplatin in relapsed ovarian cancer patients. When decitabine was given at 90 mg/m2, combination‐induced neutropenia became a major issue that lead to closure of the study (Glasspool et al., 2014). However, low dose (10 mg/m2) decitabine treatment successfully re‐sensitized heavily pretreated ovarian cancer to carboplatin, achieving 35% objective response rate and progression‐free survival of 10.2 months among 17 patients (Matei et al., 2012), suggesting great potential for low dose hypomethylating agent combination treatment in solid tumors. The development of the second generation hypomethylating agent, SGI‐110, represents an improvement over decitabine. SGI‐110 treatment provides a longer exposure window than decitabine with lower maximal concentrations. These features may make it more amenable to combine with anticancer agents. Our preclinical studies with SGI‐110 in the six HCC cell lines and the xenograft model demonstrated significant antitumor activity of SGI‐110 and synergism when used in combination with oxaliplatin. These preclinical data provide a strong rationale for the further testing of SGI‐110 in clinical trials.
As a demethylating agent, SGI‐110 is expected to change cellular DNA methylation profiles to restore a more normal transcriptome leading to antitumor activity. To elucidate the potential mechanisms of SGI‐110 as single agent and in combination with oxaliplatin, we have employed Bru‐seq, which captures gene synthesis without RNA post‐transcription processing, allowing direct assessment on gene transcription status after epigenetic modulation. Instead of studying only the select genes or targets predicted by previous studies, Bru‐seq provides an un‐biased method to explore changes in nascent RNA synthesis for the whole genome. Gene sets enrichment analysis (GSEA) of the Bru‐seq data revealed inhibition of WNT/β‐catenin signaling with SGI‐110 treatment. It is noteworthy that synergistic effects of SGI‐110 and oxaliplatin were only observed in cell lines with mutation induced WNT/β‐catenin pathway activity (Hep‐G2 and SNU‐398 both possess mutation in CTNNB1, and SNU‐475 has deletion in AXIN1 (Satoh et al., 2000; Yuzugullu et al., 2009)), but not in SNU‐387 that has no such genetic aberrations. Suggested by these data, the WNT/β‐catenin signaling might be a major targeted pathway as well as a potential patient selection marker for SGI‐110 treatment. Another interesting finding is the down‐regulation of cancer gene neighborhood sets by SGI‐110. Defined by correlated expression with certain cancer‐associated genes in the human tissue compendia, such computational gene sets represent cancer‐oriented features of the transcriptome. It is still unclear whether it is the direct effect from SGI‐110 or secondary effect from a primary modulation of up‐stream targets; however, this significant enrichment of cancer‐associated gene sets at the bottom of our pre‐ranked gene list implies an overall suppression of cancer‐specific features by SGI‐110 treatment.
In the combination treatment, the EGF and IGF signaling pathways were uniquely inhibited as revealed by Bru‐seq, implying a potential mechanism for the synergistic effect of SGI‐110 and oxaliplatin. Considering the substantial molecular changes in liver cancer, targeting multiple signature pathways with one treatment regimen is a plausible strategy. In HCC, overexpression of EGFR proteins and amplification of the EGFR gene were confirmed with Immunohistochemistry and FISH studies by Buckley et al. (Buckley et al., 2008). However, single treatment targeting EGFR with gefitinib (O'Dwyer et al., 2006), cetuximab (Zhu et al., 2007) or lapatinib (Bekaii‐Saab et al., 2009) has not shown efficacy in Phase II studies. In a recent phase III clinical trial of erlotinib in combination with sorafenib that evaluated 720 advanced HCC patients, no significant improvement in survival was observed with erlotinib plus sorafenib compared to placebo plus sorafenib (median overall survival of 9.5 vs. 8.5 months) (Zhu et al., 2015). This study highlights the refractory nature of HCC and suggests that EGFR inhibition is not an effective treatment for HCC. For the Insulin‐like growth factor (IGF) axis, overexpression or aberrant activity of the pathway has been reported in HCC (Yang et al., 2003; Hopfner et al., 2006; Desbois‐Mouthon et al., 2009). Although preclinical studies suggested growth inhibitory effects of the IGF‐1R monoclonal antibody cixutumumab (Tovar et al., 2010), a Phase II study with cixutumumab monotherapy did not show clinically meaningful efficacy in unselected HCC patient populations (Abou‐Alfa et al., 2014). Interestingly, treatment with the IGF‐1R antibody AVE1642 caused activation of HER3 in an EGFR‐dependent manner counteracting its growth inhibitory effects. When AVE1642 was combined with the EGFR inhibitor gefitinib, significant reduction of HCC cell viability was achieved (Desbois‐Mouthon et al., 2009). On the other hand, activation and nuclear translocation of IGF‐1R was observed in an induced gefitinib‐resistant HCC cell line (Bodzin et al., 2012). These studies highlight the compensatory nature of the two signature pathways, suggesting that simultaneous inhibition of EGFR and IGF‐1R could be effective in HCC treatments. In our studies, the combination of SGI‐110 and oxaliplatin suppressed activity of both pathways in addition to the inhibition of the expression of WNT/β‐catenin signaling genes. Simultaneous suppression of these three major signature pathways supports the great potential for the combination of SGI‐110 and oxaliplatin in treating HCC.
Another important goal of this preclinical study was to establish potential PD markers for future clinical evaluations. EFNB2 was identified by Bru‐seq to be increased by SGI‐110 treatment in the HCC cell line SNU‐398. Ephrin‐B2/EPHB4 signaling is known to suppress tumor growth in neuroblastoma (Tang et al., 2004), breast (Noren et al., 2006) and colon cancers (Liu et al., 2002). This is the first study to report upregulation of ephrin‐B2 levels by hypomethylating agents in liver cancer. We observed significant induction of ephrin‐B2 expression in SNU‐398 cells by SGI‐110 as single agent and in combination treatment with oxaliplatin. Also hypermethylation in EFNB2 promoter regions in HCC patient samples was observed in TCGA studies (Figure S5), suggesting the potential for SGI‐110 mediated regulation of EFNB2 gene transcription in HCC. However, studies with additional HCC cell lines did not support ephrin‐B2 induction as a PD marker for efficacy of the SGI‐110 treatment.
In addition to the decitabine targets DNMT1, survivin was also identified as a PD marker for the efficacy of the SGI‐110 and oxaliplatin combination treatment. As a member of the inhibitor of apoptosis protein (IAP), survivin is overexpressed in most cancers. Inhibition of survivin promotes cell death in cancer cells (Kelly et al., 2011). In liver cancer, survivin was identified as a target gene of Wnt/β‐catenin pathway (Gedaly et al., 2014). In our studies, combination treatment in SNU‐398, Hep‐G2 and SNU‐475 cells decreased survivin levels rapidly where neither SGI‐110 nor oxaliplatin single treatment affected survivin levels. Downregulation of survivin levels was also observed following the combination treatment in SNU‐398 tumors. Interestingly, no change in survivin levels was observed following the combination treatment in SNU‐387 cells, in which the combination did not show synergistic effect. We conclude from our data that the expression levels of survivin correlated well with the cytotoxicity of the combination treatments, suggesting that survivin may be an important effector of the synergism. These results imply that survivin can be used as a PD marker in response to SGI‐110 and oxaliplatin treatment.
Since toxicity is an important factor for combination treatments in vivo as compared with in vitro studies, we have chosen to give weekly oxaliplatin treatment and precede the first oxaliplatin treatment with SGI‐110 in a 14‐day cycle. Oxaliplatin weekly treatment has been routinely used for in vivo studies, while it is mostly given once every two weeks in the clinic due to its toxicity. To best evaluate clinical relevant schedules and doses, as well as to avoid potential toxicity introduced by the combination, we chose to give oxaliplatin at 5 mg/kg once a week. Decitabine or SGI‐110 can be toxic as long‐term repeated treatment. Based on previous studies, QD5 treatment or bi‐weekly treatment is used to achieve therapeutic effect without systemic toxicity. In SCID mice model, SGI‐110 given as daily treatment for 5 consecutive days at 3 mg/kg showed consistent and robust hypomethylation and induction of Cancer Testis Antigen (CTA) genes in leukemia as well as ovarian cancer xenografts without significant toxicity (Srivastava et al., 2014, 2015), suggesting good balance between efficacy and toxicity at this treatment schedule. In the ongoing phase II clinical trial of SGI‐110 for the treatment of advanced HCC (NCT01752933), SGI‐110 is given daily on Days 1–5 every 28 days. In our studies, SGI‐110 at 2 mg/kg was administered on Days 1–5 in a 14‐day cycle.
SGI‐110 treatment delayed tumor growth endpoint from 19 to 26 days supporting a benefit in survival. More importantly, the combination with oxaliplatin further delayed tumor growth endpoint to 31 days, demonstrating synergism. The combination treatment was well tolerated with no apparent weight loss or gross toxicity in major organs. Based on our in vitro data and the safety of combination treatment, we believe that higher doses of oxaliplatin or SGI‐110 will improve the therapeutic effect for the combination treatment. This can be further investigated in future preclinical or clinical studies.
In this preclinical study, we have shown significant antitumor effect of SGI‐110 alone or in combination with oxaliplatin in HCC models. Application of Bru‐seq led to the identification of Wnt/β‐catenin, EGFR and IGFR signaling as key pathways inhibited by the combination treatment. Such simultaneous inhibition of three liver cancer signature pathways by the combination treatment supports the use of a DNA demethylating agent in combination with a cytotoxic agent as an effective therapy for HCC. We expect that the combination of SGI‐110 and oxaliplatin at low doses will delay disease progression and prolong overall survival without significant toxicity. Our findings provide strong rationale for a Phase I/II clinical trial with SGI‐110 and oxaliplatin in HCC patients.
Disclosure of potential conflicts of interest
Y. Kuang is a predoctoral fellow at the University of Southern California and a visiting researcher at the University of Michigan. P. Taverna is an employee of Astex Pharmaceuticals, Inc. No potential conflicts of interest are disclosed by the other authors.
Authors' contributions
Conception and design: Y. Kuang, N. Neamati, A. El‐Khoueiry, P. Taverna.
Data acquisition and analysis: Y. Kuang, N. Neamati, M. Ljungman.
Writing or revision of the manuscript: Y. Kuang, N. Neamati, M. Ljungman, P. Taverna, A. El‐Khoueiry
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
This work was supported in part by a grant from Astex Pharmaceuticals, Inc.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.06.002.
Kuang Yuting, El-Khoueiry Anthony, Taverna Pietro, Ljungman Mats, Neamati Nouri, (2015), Guadecitabine (SGI‐110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.06.002.
References
- Abou-Alfa, G.K. , Capanu, M. , O'Reilly, E.M. , Ma, J. , Chou, J.F. , Gansukh, B. , 2014. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J. Hepatol. 60, (2) 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekaii-Saab, T. , Markowitz, J. , Prescott, N. , Sadee, W. , Heerema, N. , Wei, L. , 2009. A multi-institutional phase II study of the efficacy and tolerability of lapatinib in patients with advanced hepatocellular carcinomas. Clin. Cancer Res. 15, (18) 5895–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodzin, A.S. , Wei, Z. , Hurtt, R. , Gu, T. , Doria, C. , 2012. Gefitinib resistance in HCC mahlavu cells: upregulation of CD133 expression, activation of IGF-1R signaling pathway, and enhancement of IGF-1R nuclear translocation. J. Cell. Physiol. 227, (7) 2947–2952. [DOI] [PubMed] [Google Scholar]
- Buckley, A.F. , Burgart, L.J. , Sahai, V. , Kakar, S. , 2008. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am. J. Clin. Pathol. 129, (2) 245–251. [DOI] [PubMed] [Google Scholar]
- Cheung, C.H. , Huang, C.C. , Tsai, F.Y. , Lee, J.Y. , Cheng, S.M. , Chang, Y.C. , 2013. Survivin – biology and potential as a therapeutic target in oncology. Onco. Targets Ther. 6, 1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T.C. , 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58, (3) 621–681. [DOI] [PubMed] [Google Scholar]
- Chuang, J.C. , Warner, S.L. , Vollmer, D. , Vankayalapati, H. , Redkar, S. , Bearss, D.J. , 2010. S110, a 5-Aza-2'-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol. Cancer Ther. 9, (5) 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbois-Mouthon, C. , Baron, A. , Blivet-Van Eggelpoel, M.J. , Fartoux, L. , Venot, C. , Bladt, F. , 2009. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. 15, (17) 5445–5456. [DOI] [PubMed] [Google Scholar]
- Fan, H. , Lu, X. , Wang, X. , Liu, Y. , Guo, B. , Zhang, Y. , 2014. Low-dose decitabine-based chemoimmunotherapy for patients with refractory advanced solid tumors: a phase I/II report. J. Immunol. Res. 2014, 10.1155/2014/371087 Article ID 371087, 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H. , Zhao, Z.J. , Cheng, J. , Su, X.W. , Wu, Q.X. , Shan, Y.F. , 2009. Overexpression of DNA methyltransferase 1 and its biological significance in primary hepatocellular carcinoma. World J. Gastroenterol. 15, (16) 2020–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, F. , Munck, J. , Tang, J. , Taverna, P. , Wang, Y. , Miller, D.F. , 2014. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin. Cancer Res. 20, (24) 6504–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay, J.S.I. , Ervik, M. , Dikshit, R. , Eser, S. , Mathers, C. , Rebelo, M. , Parkin, D.M. , Forman, D. , Bray, F. , 2013. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No.11 International Agency for Research on Cancer; Lyon, France: Available from: http://globocan.iarc.fr [Google Scholar]
- Forner, A. , Llovet, J.M. , Bruix, J. , 2012. Hepatocellular carcinoma. Lancet. 379, (9822) 1245–1255. [DOI] [PubMed] [Google Scholar]
- Gaur, S. , Chen, L. , Ann, V. , Lin, W.C. , Wang, Y. , Chang, V.H. , 2014. Dovitinib synergizes with oxaliplatin in suppressing cell proliferation and inducing apoptosis in colorectal cancer cells regardless of RAS-RAF mutation status. Mol. Cancer. 13, 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedaly, R. , Galuppo, R. , Daily, M.F. , Shah, M. , Maynard, E. , Chen, C. , 2014. Targeting the Wnt/beta-catenin signaling pathway in liver Cancer stem cells and hepatocellular carcinoma cell lines with FH535. PLoS One. 9, (6) e99272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasspool, R.M. , Brown, R. , Gore, M.E. , Rustin, G.J. , McNeish, I.A. , Wilson, R.H. , 2014. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br. J. Cancer. 110, (8) 1923–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, J.S. , Kaye, S.B. , Brown, R. , 2009. The promises and pitfalls of epigenetic therapies in solid tumours. Eur. J. Cancer. 45, (7) 1129–1136. [DOI] [PubMed] [Google Scholar]
- Hopfner, M. , Huether, A. , Sutter, A.P. , Baradari, V. , Schuppan, D. , Scherubl, H. , 2006. Blockade of IGF-1 receptor tyrosine kinase has antineoplastic effects in hepatocellular carcinoma cells. Biochem. Pharmacol. 71, (10) 1435–1448. [DOI] [PubMed] [Google Scholar]
- Huang, W.Y. , Hsu, S.D. , Huang, H.Y. , Sun, Y.M. , Chou, C.H. , Weng, S.L. , 2015. MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic. Acids Res. 43, (Database issue) D856–D861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P.A. , Taylor, S.M. , 1980. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 20, (1) 85–93. [DOI] [PubMed] [Google Scholar]
- Kelly, R.J. , Lopez-Chavez, A. , Citrin, D. , Janik, J.E. , Morris, J.C. , 2011. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol. Cancer. 10, 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe, E. , Lock, L. , Letamendia, A. , Gorska, A.E. , Gryfe, R. , Gallinger, S. , 2007. Transcriptional cooperation between the transforming growth factor-beta and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res. 67, (1) 75–84. [DOI] [PubMed] [Google Scholar]
- Lachenmayer, A. , Alsinet, C. , Savic, R. , Cabellos, L. , Toffanin, S. , Hoshida, Y. , 2012. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 18, (18) 4997–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , Ahmad, S.A. , Jung, Y.D. , Reinmuth, N. , Fan, F. , Bucana, C.D. , 2002. Coexpression of ephrin-Bs and their receptors in colon carcinoma. Cancer. 94, (4) 934–939. [DOI] [PubMed] [Google Scholar]
- Llovet, J.M. , Ricci, S. , Mazzaferro, V. , Hilgard, P. , Gane, E. , Blanc, J.F. , 2008. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, (4) 378–390. [DOI] [PubMed] [Google Scholar]
- Louafi, S. , Boige, V. , Ducreux, M. , Bonyhay, L. , Mansourbakht, T. , de Baere, T. , 2007. Gemcitabine plus oxaliplatin (GEMOX) in patients with advanced hepatocellular carcinoma (HCC): results of a phase II study. Cancer. 109, (7) 1384–1390. [DOI] [PubMed] [Google Scholar]
- Matei, D. , Fang, F. , Shen, C. , Schilder, J. , Arnold, A. , Zeng, Y. , 2012. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 72, (9) 2197–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaglia, E. , Donadelli, M. , Palmieri, M. , Crnogorac-Jurcevic, T. , Scarpa, A. , Lemoine, N.R. , 2005. Growth delay of human pancreatic cancer cells by methylase inhibitor 5-aza-2'-deoxycytidine treatment is associated with activation of the interferon signalling pathway. Oncogene. 24, (1) 199–211. [DOI] [PubMed] [Google Scholar]
- Mootha, V.K. , Lindgren, C.M. , Eriksson, K.F. , Subramanian, A. , Sihag, S. , Lehar, J. , 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, (3) 267–273. [DOI] [PubMed] [Google Scholar]
- Neumann, O. , Kesselmeier, M. , Geffers, R. , Pellegrino, R. , Radlwimmer, B. , Hoffmann, K. , 2012. Methylome analysis and integrative profiling of human HCCs identify novel protumorigenic factors. Hepatology. 56, (5) 1817–1827. [DOI] [PubMed] [Google Scholar]
- Nishida, N. , Kudo, M. , Nagasaka, T. , Ikai, I. , Goel, A. , 2012. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology. 56, (3) 994–1003. [DOI] [PubMed] [Google Scholar]
- Noren, N.K. , Foos, G. , Hauser, C.A. , Pasquale, E.B. , 2006. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat. Cell Biol. 8, (8) 815–825. [DOI] [PubMed] [Google Scholar]
- O'Dwyer, P.J.L.D. , Kauh, J.S. , Fitzgerald, D.B. , Benson, A.B. , 2006. Gefitinib in advanced unresectable hepatocellular carcinoma: results from the Eastern cooperative oncology group's study E1203. J. Clin. Oncol. 24, Abstract 4143 [Google Scholar]
- Park, J.G. , Lee, J.H. , Kang, M.S. , Park, K.J. , Jeon, Y.M. , Lee, H.J. , 1995. Characterization of cell lines established from human hepatocellular carcinoma. Int. J. Cancer. 62, (3) 276–282. [DOI] [PubMed] [Google Scholar]
- Paulsen, M.T. , Veloso, A. , Prasad, J. , Bedi, K. , Ljungman, E.A. , Magnuson, B. , 2014. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 67, (1) 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen, M.T. , Veloso, A. , Prasad, J. , Bedi, K. , Ljungman, E.A. , Tsan, Y.C. , 2013. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc. Natl. Acad. Sci. U. S. A. 110, (6) 2240–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psyrri, A. , Arkadopoulos, N. , Vassilakopoulou, M. , Smyrniotis, V. , Dimitriadis, G. , 2012. Pathways and targets in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 12, (10) 1347–1357. [DOI] [PubMed] [Google Scholar]
- Qin, S. , Bai, Y. , Lim, H.Y. , Thongprasert, S. , Chao, Y. , Fan, J. , 2013. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J. Clin. Oncol. 31, (28) 3501–3508. [DOI] [PubMed] [Google Scholar]
- Roessler, S. , Jia, H.L. , Budhu, A. , Forgues, M. , Ye, Q.H. , Lee, J.S. , 2010. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 70, (24) 10202–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh, S. , Daigo, Y. , Furukawa, Y. , Kato, T. , Miwa, N. , Nishiwaki, T. , 2000. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 24, (3) 245–250. [DOI] [PubMed] [Google Scholar]
- Selvakumaran, M. , Amaravadi, R.K. , Vasilevskaya, I.A. , O'Dwyer, P.J. , 2013. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 19, (11) 2995–3007. [DOI] [PubMed] [Google Scholar]
- Shitani, M. , Sasaki, S. , Akutsu, N. , Takagi, H. , Suzuki, H. , Nojima, M. , 2012. Genome-wide analysis of DNA methylation identifies novel cancer-related genes in hepatocellular carcinoma. Tumour Biol. 33, (5) 1307–1317. [DOI] [PubMed] [Google Scholar]
- Siegel, R. , Ma, J. , Zou, Z. , Jemal, A. , 2014. Cancer statistics, 2014. CA Cancer J. Clin. 64, (1) 9–29. [DOI] [PubMed] [Google Scholar]
- Song, J. , Teplova, M. , Ishibe-Murakami, S. , Patel, D.J. , 2012. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 335, (6069) 709–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, P. , Paluch, B.E. , Matsuzaki, J. , James, S.R. , Collamat-Lai, G. , Karbach, J. , 2014. Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leuk. Res. 38, (11) 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, P. , Paluch, B.E. , Matsuzaki, J. , James, S.R. , Collamat-Lai, G. , Taverna, P. , 2015. Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics. 10, (3) 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V.K. , Mukherjee, S. , Ebert, B.L. , Gillette, M.A. , 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, (43) 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh, S.I. , Pyun, H.Y. , Cho, J.W. , Baek, W.K. , Park, J.B. , Kwon, T. , 2000. 5-Aza-2'-deoxycytidine leads to down-regulation of aberrant p16INK4A RNA transcripts and restores the functional retinoblastoma protein pathway in hepatocellular carcinoma cell lines. Cancer Lett. 160, (1) 81–88. [DOI] [PubMed] [Google Scholar]
- Tang, X.X. , Robinson, M.E. , Riceberg, J.S. , Kim, D.Y. , Kung, B. , Titus, T.B. , 2004. Favorable neuroblastoma genes and molecular therapeutics of neuroblastoma. Clin. Cancer Res. 10, (17) 5837–5844. [DOI] [PubMed] [Google Scholar]
- Tellez, C.S. , Grimes, M.J. , Picchi, M.A. , Liu, Y. , March, T.H. , Reed, M.D. , 2014. SGI-110 and entinostat therapy reduces lung tumor burden and reprograms the epigenome. Int. J. Cancer. 135, (9) 2223–2231. [DOI] [PubMed] [Google Scholar]
- Tovar, V. , Alsinet, C. , Villanueva, A. , Hoshida, Y. , Chiang, D.Y. , Sole, M. , 2010. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J. Hepatol. 52, (4) 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Cardenas, H. , Fang, F. , Condello, S. , Taverna, P. , Segar, M. , 2014. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 74, (17) 4922–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmbach, E. , Chen, Y.B. , Khitrov, G. , Zhang, W. , Roayaie, S. , Schwartz, M. , 2007. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 45, (4) 938–947. [DOI] [PubMed] [Google Scholar]
- Yang, J.M. , Chen, W.S. , Liu, Z.P. , Luo, Y.H. , Liu, W.W. , 2003. Effects of insulin-like growth factors-IR and -IIR antisense gene transfection on the biological behaviors of SMMC-7721 human hepatoma cells. J. Gastroenterol. Hepatol. 18, (3) 296–301. [DOI] [PubMed] [Google Scholar]
- Yoo, C.B. , Jeong, S. , Egger, G. , Liang, G. , Phiasivongsa, P. , Tang, C. , 2007. Delivery of 5-aza-2'-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 67, (13) 6400–6408. [DOI] [PubMed] [Google Scholar]
- Yuzugullu, H. , Benhaj, K. , Ozturk, N. , Senturk, S. , Celik, E. , Toylu, A. , 2009. Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol. Cancer. 8, 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.L. , Luo, H.Y. , Yang, J. , Wu, W.J. , Chen, D.L. , Huang, P. , 2014. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin. Cancer Res. 20, (4) 1042–1052. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Yang, B. , Du, Z. , Bai, T. , Gao, Y.T. , Wang, Y.J. , 2012. Aberrant methylation of SPARC in human hepatocellular carcinoma and its clinical implication. World J. Gastroenterol. 18, (17) 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, A.X. , Rosmorduc, O. , Evans, T.R. , Ross, P.J. , Santoro, A. , Carrilho, F.J. , 2015. SEARCH: a phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 33, (6) 559–566. [DOI] [PubMed] [Google Scholar]
- Zhu, A.X. , Stuart, K. , Blaszkowsky, L.S. , Muzikansky, A. , Reitberg, D.P. , Clark, J.W. , 2007. Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer. 110, (3) 581–589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data