

Abstract
As cancer immunotherapy continues to benefit from novel approaches which cut immune ‘brake pedals’ (e.g. anti‐PD1 and anti‐CTLA4 antibodies) and push immune cell gas pedals (e.g. IL2, and IFNα) there will be increasing need to develop immune ‘steering wheels’ such as vaccines to guide the immune system specifically toward tumor associated antigens. Two primary hurdles in cancer vaccines have been: identification of universal antigens to be used in ‘off‐the‐shelf’ vaccines for common cancers, and 2) logistical hurdles of ex vivo production of individualized whole tumor cell vaccines. Here we summarize approaches using ‘in situ vaccination’ in which intratumoral administration of off‐the‐shelf immunomodulators have been developed to specifically induce (or amplify) T cell responses to each patient's individual tumor. Clinical studies have confirmed the induction of systemic immune and clinical responses to such approaches and preclinical models have suggested ways to further potentiate the translation of in situ vaccine trials for our patients.
Keywords: <i>In situ</i> vaccination, Cancer immunotherapy, Oncolytic viruses, Toll like receptors, Checkpoint blockade, Dendritic cells
1. Introduction
Prophylactic vaccinations have been one of the greatest advances in modern medicine, both eradicating disease and reducing mortality. The translation of this advance into cancer therapy has been challenging and dates back to the turn of the 20th century (Currie, 1972). Cancer cells, derived from an aberrant clone, bear predominantly self‐antigens and thus avoid alerting the immune system. In addition, the tumor microenvironment can be severely immunosuppressive; adding an extra layer of protection against the host immune response. Tumor cells can actively suppress immune responses through the downregulation of antigen presentation and the production of membrane‐bound and secreted immune‐regulatory molecules (Upadhyay et al., 2015). To overcome such obstacles, a successful cancer vaccine must be able to induce a powerful immune response against tumor‐associated antigens (TAAs) while avoiding normal host cells. This strategy has proven difficult because TAAs are highly variable in their immunogenicity and undergo immune‐editing to escape recognition. In addition, they can differ between tumor types and more importantly between individuals (Escors, 2014). The presence of antigen‐presenting cells (APC) is generally low in the tumor microenvironment. Some efficacy in the treatment of cancer has been demonstrated by the use of autologous dendritic cells (DC) pulsed with tumor cell lysates containing a whole array of antigens as well as single TAAs (Reichardt et al., 2004). DC can be differentiated and expanded from peripheral blood ex vivo, and a resected tumor mass can be used to subsequently load the DC with TAAs. These strategies, while successful in developing a patient specific vaccine, are labor and time intensive limiting the ability to experiment with numerous iterations to optimize the approach.
“In situ vaccination” represents an alternative approach in which the cancer vaccine is generated in vivo without the need to previously identify and isolate the TAA. Herein, in situ vaccination refers to any approach which exploits TAA available at a tumor site to induce a TAA‐specific adaptive immune response. TAAs are commonly released upon tumor cell death and may be subsequently processed and presented by APCs. This can be augmented by stimulating tumor cell death leading to release of TAAs and subsequent presentation by APCs while at the same time administering immunomodulators to enhance particular steps of the process. Such an approach allows for the development of vaccines in patients themselves, thereby minimizing the resource allocation required in ex vivo processing. Furthermore, this strategy can take advantage of the complete antigenic repertoire of a tumor and not be limited to a single TAA. In order to elicit a strong memory anti‐tumor immune response, an in situ vaccine should ideally be able to induce immunogenic cancer cell death, facilitate the release of TAAs, as well as enhance antigen uptake by, and activation of, antigen‐presenting cells to induce anti‐tumor T cell responses that will result in systemic anti‐tumor immunity. The generation of potent anti‐tumor T cells at one tumor site should allow them to also attack distant tumor lesions since fully activated effector T cells do not need a costimulatory signal to kill their target cells and are less susceptible to inhibitory signals (Suresh et al., 2001; London et al., 2000; Gudmundsdottir et al., 1999). Therefore, effective development of an in situ vaccination will result in a systemic response in the setting of localized treatment similar to the abscopal effect that is described in radiation therapy and felt to be driven by a T cell response (Formenti and Demaria, 2009).
One tool which can be part of in situ vaccination, oncolytic viruses – viruses that preferentially infect and kill cancer cells – are being explored for the treatment of various malignancies (Nemunaitis, 1999). They display either a natural or engineered tumor tropism and are able to kill tumor cells via direct and indirect mechanisms (Elsedawy and Russell, 2013; Bartlett et al., 2013). The host immune system directly kills viral protein‐expressing tumor cells, leading to regression in infected tumors. Uninfected cells are indirectly killed through cross‐priming of cytotoxic T‐lymphocytes (CTL) and disruption of the tumor vasculature. However, the antitumor efficacy of naturally occurring viruses has been limited, suggesting that the degree of the induced immune response depends on several factors, including the particular virus used, the tumor burden, and the immunogenicity (Elsedawy and Russell, 2013). An advantage of killing tumor cells with virus is their abundance of innately immunostimulatory components e.g. viral proteins and nucleic acid which have been shown to activate Toll‐like receptors (TLRs) expressed on APC (Zhu et al., 2008, 2014, 2007, 2007). Another advantage of oncolytic viruses is that they can be engineered to express transgenes which can influence the anti‐tumoral immune responses. This includes (i) enhancing the cross‐presentation of tumor antigens, (ii) increasing the maturation of antigen‐presenting cells, especially DC, and (iii) reducing immune suppression in the tumor microenvironment.
Herein, we examine pre‐clinical and clinical data of in situ vaccination strategies and their emerging role in the treatment of cancer as well as new developments in tumor immunology that will lead to ongoing translational research.
1.1. Manipulation of intratumoral myeloid cells
1.1.1. Increasing the number of APC at the tumor site
1.1.1.1. Autologous DC
Increasing the number of effective APC within the tumor microenvironment yields greater capacity for cross presentation of TAA to CD8+ T cells, potentially augmenting the immune response against malignant cells. As a result, several studies have recently focused on strategies to increase the number of DC at the tumor site through administration of DC growth/differentiation factors or local administration of DC themselves. In mice with subcutaneous colon cancer or lymphoma, systemic chemotherapy followed by intratumoral injection of immature DC resulted in complete regression of treated and distant tumors and protected mice from rechallenge with the same tumor cells (Tong et al., 2001; Song and Levy, 2005). This effect was also observed when mice with the same colon cancer were treated with photodynamic therapy and injection of naïve DC (Saji et al., 2006). In patients with advanced melanoma, treatment with local hyperthermia and injection of autologous DC reduced tumor growth and increased infiltration of CD8+ T cells, but overall survival was not improved (Guo et al., 2007). However, in patients with stage III/IV treatment naïve or relapsed follicular lymphoma local radiotherapy followed by intranodal injection of autologous DC, low‐dose rituximab and GM‐CSF caused durable remission in a subset of patients. Clinical response correlated closely with evidence of an immune response to autologous tumor (Kolstad et al., 2015). A phase I/II clinical trial is currently investigating the intratumoral injection of autologous DC for the treatment of solid tumors (NCT01882946). Similar studies are combining intratumoral administration of autologous DC combined with chemotherapy for patients with breast cancer (NCT02018458) and combined with local cryotherapy for patients with prostate cancer (NCT02423928).
1.1.1.2. Allogeneic DC
Pre‐clinically, it has been shown that allogeneic leukocyte co‐cultures generate immature DC‐recruiting chemokines and pro‐inflammatory cytokines which induce CD40 upregulation, increase TLRa induced IL‐12 production, and deviated T‐cell responses towards Th1 {16179010}. Additionally, allogeneic DC‐based vaccines demonstrated induction of melanoma‐protective immunity in multiple animal models {20443871}.
In an early phase clinical trial, allogeneic DC activated with a formulation including TLRa and IFNγ (COMBIG) were administered intratumorally (INTUVAK) in 12 patients with metastatic renal cell cancer (RCC) prior to nephrectomy. At the time of nephrectomy, a marked infiltration of CD8+ T cells was demonstrated and a majority of patients exhibited an increase in circulating tumor‐specific T cells after vaccination. Median overall survival in patients with poor prognosis compared favorably with historical controls. {http://meetinglibrary.asco.org/content/126079‐144} The approach has progressed to an ongoing randomized trial of post‐nephrectomy sunitinib with or without pre‐nephrectomy INTUVAK (NCT02432846) as well as an ongoing phase I study in patients with hepatoma (NCT01974661)
1.1.1.3. GM‐CSF
Granulocyte‐macrophage colony‐stimulation factor (GM‐CSF), a hematopoietic growth factor that increases DC differentiation, maturation and function has been administered intratumorally to promote the number of DC and stimulating DC activation at the tumor site. A seminal study compared anti‐tumor immunity of melanoma cells transduced with a panel of immunostimulatory genes including IL‐2, IL‐4, IL‐5, IL‐6, GM‐CSF, IFNγ, IL1‐RA, ICAM, CD2, and TNFα. GM‐CSF‐expressing tumors showed the greatest effects and induced systemic tumor rejection. Treatment also protected mice from rechallenge with non‐transduced melanoma cells (Dranoff et al., 1993). Using a related approach, mice with colon cancer received macrophages engineered to express GM‐CSF intratumorally, and developed antitumor immunity (Yizhi et al., 1998). Vaccination of melanoma patients with irradiated autologous tumor cells engineered to express GM‐CSF also resulted in tumor destruction. Those with tumor response were found to have profound infiltration of activated T cells. The number of CD11c+ in GM‐CSF‐expressing tumor lesions was significantly increased in both mice and patients (Mach et al., 2000). In patients with melanoma, intratumoral or peritumoral injection of recombinant GM‐CSF has been investigated with mixed results. While some demonstrated tumor regression in a majority of patients, including some complete responses, others reported only partial responses in a minority of patients (Si et al., 1996; Nasi et al., 1999; Hoeller et al., 2001). Regardless of response rate or degree of response, all three studies observed an increase of DC in tumor lesions. As described earlier, GM‐CSF also has promising results when combined with autologous DC and a monoclonal antibody in the treatment of low‐grade follicular lymphoma (Kolstad et al., 2015). As described GM‐CSF has therapeutic potential in both monotherapy and combination with other immune modulating agents stimulating DC activation, immune response and systemic tumor regressions.
1.1.1.4. Flt3L
Fms‐like tyrosine kinase 3 ligand (Flt3L) is a crucial growth factor in DC development (Shortman and Naik, 2007) and particularly important in the development of the DC2 subset –remarkably capable CD8 T cell stimulators whose intratumoral proportion correlates with patient clinical outcomes (Broz et al., 2014) {Salmon et al., in press}. Subcutaneous injection of Flt3L stimulates mobilization of DC to the peripheral blood of patients with melanoma or colon cancer (Marroquin et al., 2002; Morse et al., 2000). Expansion of DC is observed in mice treated with adenovirally expressed or recombinant Flt3L in colon cancer or leukemia, respectively (Pawlowska et al., 2001; Riediger et al., 2013). However, while vaccination with Flt3L prior to tumor challenge is able to prevent tumor development in both models, therapeutic application of Flt3L could not eliminate established tumors. In contrast, the combination of intratumoral adeno‐Flt3L with systemic chemotherapy inhibits the growth and induces complete remissions of murine hepatoma and colon cancer (Hou et al., 2007). Similarly, systemic administration of Flt3L followed by intranodal injection of antigen‐encoding naked RNA – thereby delivering an antigen as well as TLR stimulation – resulted in significantly enhanced tumor clearance and survival in mice with melanoma (Kreiter et al., 2011). When combined with cytotoxic therapies or DC activation, Flt3L‐induced DC expansion leads to clinical response as opposed to the administration of Flt3L alone. Several clinical trials combining Flt3L with cytotoxic and/or other concomitant treatments are currently underway (NCT02129075, NCT01811992, NCT01976585). The latter study combining intratumoral injection of recombinant Flt3L and poly‐ICLC in combination with low‐dose radiotherapy has reported increased DC numbers, partial and complete remissions of treated and untreated tumor sites in patients with advanced‐stage follicular lymphoma (Bhardwaj et al., 2014). In this study, cell killing appeared to be tumor‐specific since concomitant with malignant B cell clearing from peripheral blood, non‐malignant B cells actually increased with time. This is in contrast to standard lymphoma therapies such anti‐CD20 antibodies which induce B cell aplasia (Maloney et al., 1994) and prompts inquiry into what TAA might be targeted by T cells which spare non‐malignant B cells. Preliminary results report only mild adverse effects from such therapy suggesting that Flt3L‐primed in situ vaccination is safe as well as immunologically and clinically active.
1.1.1.5. Oncolytic viruses that increase the number of APC at the tumor site
The most extensively studied cytokine for this purpose is GM‐CSF. A vaccinia virus engineered to express GM‐CSF, JX‐594, selectively replicates in tumor cells as compared to normal cells (Parato et al., 2012) and has demonstrated anti‐tumor efficacy in pre‐clinical models and several early phase clinical trials. Rabbits and rats with liver cancer have improved survival and decreased metastatic burden when treated with JX‐594 systemically or intratumorally. These models reveal increased infiltration of T cells into the tumor microenvironment (Kim et al., 2006) and disruption of tumor‐associated vasculature in mice and humans, leading to reduced blood flow, ischemia, and rapid necrosis of tumor cells (Breitbach et al., 2011; Breitbach et al., 2013). Again, this effect is tumor‐specific as endothelial cells of normal blood vessels are not affected. Clinical trials in liver cancer and melanoma demonstrate that intratumoral treatment with JX‐594 is well tolerated and results in encouraging survival times and overall response of treated and untreated tumors (Heo et al., 2013; Park et al., 2008; Hwang et al., 2011; Breitbach et al., 2015; Cripe et al., 2015).
Similarly promising results have been observed with an oncolytic herpes simplex virus (HSV) deleted for genes that block antigen presentation on MHC molecules or support viral replication in normal cells. This HSV strain specifically replicates in tumor cells with high oncolytic potential enhancing anti‐tumor efficacy. Immunogenicity was enhanced by inserting GM‐CSF into the viral genome (Liu et al., 2003). Liu et al. have demonstrated that intratumoral administration in murine lymphoma tumors results in tumor shrinkage and anti‐tumor immunity in both treated tumors and distant, non‐injected tumors. In addition, it protects mice against rechallenge with tumor cells after the primary tumor has been cured. Clinical studies using the same HSV strain referred to as talimogene laherparepvec or T‐VEC‐ (previously OncoVEX‐GM‐CSF) have shown durable response rates and increases survival time in patients with melanoma, head and neck cancer, and other metastatic cancers (Harrington et al., 2010; Hu et al., 2006; Senzer et al., 2009; Kaufman et al., 2010). A recent phase III study in patients with advanced stage melanoma, T‐VEC demonstrates superior durable response rate compared to GM‐CSF and a trend towards improvement in overall survival (p = 0.051) (Andtbacka et al., 2015a). In concordance with the pre‐clinical studies, treated patients appear to develop systemic anti‐tumor immunity associated with an increase in tumor‐specific T cells resulting responses at both injected tumors and distant sites (Senzer et al., 2009; Kaufman et al., 2010). Clinical investigation of combining intratumoral virotherapy with systemic CTLA‐4 blockade for melanoma is currently ongoing (NCT01740297) with preliminary report of overall response rate of 41%, higher than historical results with ipilimumab alone (Puzanov et al., 2014) and studies combining T‐VEC with systemic PD1 blockade are currently accruing (NCT02263508).
1.1.2. Activation of APC
1.1.2.1. TLR9
TLRs are a class of pattern recognition receptors expressed primarily by leukocytes, and especially by antigen‐presenting cells, including DC and macrophages. They bind to pathogen‐associated molecular patterns (e.g. lipopolysaccharide, CpG‐enriched DNA, double stranded RNA), and their ligation results in APC activation, enhanced presentation to T cells and initiation of immune responses. In addition, they can have a direct influence on tumor cells themselves when expressing TLRs. B‐cell lymphomas are derived from mature B cells ‐a potential APC population‐ and therefore can express multiple TLR, including TLR9 (Bourke et al., 2003). As a result, hypomethylated CG‐enriched oligonucleotides (CpG) resembling prokaryotic DNA can activate TLR9 and increase expression of costimulatory molecules on lymphoma cells in vitro (Jahrsdörfer et al., 2001; Li et al., 2007), though differentially amongst different lymphoma subtypes (Jahrsdorfer et al., 2005). At the same time, CpG inhibited proliferation of these cells (Li et al., 2007). Furthermore, systemic chemotherapy combined with intratumoral vaccination with CpG induced tumor‐reactive, memory CD8 T cells and tumor regression at both local and distant sites in a murine lymphoma model. Both local and systemic anti‐tumor response depended on CD8+ T cells (Li et al., 2007). Interestingly, this effect was seen in mice with TLR9 deficiency, which had TLR9‐expressing tumors. This finding suggests that antigen presentation by tumor cells, not host APC, may be sufficient to induce potent immune responses. This therapy could be further enhanced by transferring splenocytes from vaccinated mice into lymphodepleted mice (‘immunotransplantation’) bearing the same tumor cell line, demonstrating that transferred CD8 T cells were both necessary and sufficient (Brody et al., 2009). Treg cells could also be depleted locally by intratumoral injection of anti‐CTLA‐4 and anti‐OX‐40 during CpG vaccination, generating a systemic antitumor immune response able to cure disseminated disease (Marabelle et al., 2013). Mice were cured in the absence of systemic chemotherapy, validating the potency of combination treatment (Houot and Levy, 2009).
In a murine glioma model, intratumoral injection of CpG also resulted in tumor regression. Again increased T cell infiltration was seen, and cured mice were protected from rechallenge with tumor cells (Grauer et al., 2008). In contrast to the lymphoma model, TLR9 expression is required in non‐tumor host cells since GL261 glioma cells are negative for TLR9. Furthermore, beneficial effects of intratumoral CpG are also observed in mice with mesothelioma, breast cancer, or melanoma tumors (Stone et al., 2009a; Sharma et al., 2008; Furumoto et al., 2004). In breast cancer models, CpG injection even leads to tumor regression in old mice – an effect not observed with other TLR agonists used in the study (Sharma et al., 2008). In the case of B16 melanoma, CpG injection alone is not sufficient to induce tumor regression but results in antitumor immunity when combined with CCL20‐mediated accumulation of DC within the tumor (Furumoto et al., 2004). Of note, a different synthetic TLR9 agonist (with modified CpG motifs) reduced tumor growth and increased survival in a murine lung cancer model when combined with local tumor irradiation (Zhang et al., 2012). Since these modified CpG motifs use a 3′‐3′‐attached structure and a dCp7‐deaza‐dG have shown greater potency in murine and human studies, they may be incorporated into future in situ approaches.
Type B TLR9 agonists preferentially activate B cells (versus plasmacytoid DC). The type B TLR9 agonist Agatolimod (PF‐ 3512676) has been used in the greatest number of clinical trials, including three trials using a combination of low‐dose irradiation and intratumoral CpG administration showing promising results in 60 patients with low‐grade B‐cell lymphoma and mycosis fungoides including partial and complete remissions, lasting up to >4 years (Brody et al., 2010; Kim et al., 2012; Kohrt et al., 2014). A study of intracerebral (intratumoral) infusion of another type B TLR9 agonist Litenimod (CpG‐28) in 34 patients with recurrent glioblastoma multiforme also showed efficacy in controlling disease burden yielding 1 and 2 year survival rates higher than historical controls (Carpentier et al., 2010). Type C TLR9 agonists –by contrast– comparably activate B cells and plasmacytoid DC, thus inducing higher amounts of both IFNα and IFNλ. One such molecule ‐SD‐101‐ is currently being studied for the treatment of relapsed or refractory, low‐grade lymphoma in combination with radiotherapy and/or intratumoral CTLA‐4 blockade (NCT02266147, NCT01745354, NCT02254772). In the completed clinical trials treatment was generally well‐tolerated, with a dose‐related incidence of injection site reactions and the approaches warrant further studies on the use of this in situ vaccination for the treatment of human cancer.
1.1.2.2. TLR7/8
TLR7 agonists such as imiquimod (an imidazoquinolinamine derivative) were first investigated in the context of cutaneous viral infections, due to their induction of type I interferon (IFNα/β). Imiquimod can be applied topically and is approved for the treatment of genital warts, actinic keratoses, and basal cell carcinoma (Miller et al., 1999). Beginning in the early 1990s, systemic treatment with imiquimod had been assessed in the treatment of various murine cancers, with oral treatment leading to significant inhibition of growth in colon carcinoma and more modest effects in lung carcinoma and sarcoma. This effect was mediated by IFN induction and could be further enhanced when combined with cyclophosphamide (Sidky et al., 1992). More recently, imiquimod has been investigated as a potential adjuvant for in situ cancer vaccination for metastatic disease. In case reports and prospective series of melanoma and superficial breast cancer metastases, topical application of imiquimod induces a pro‐immunogenic tumor microenvironment with histologic tumor regression (Adams et al., 2012; Henriques et al., 2014; Smyth et al., 2011). Similarly, numerous case reports and series have demonstrated anti‐tumor effects of imiquimod in cutaneous T‐cell (Calista et al., 2015; Didona et al., 2004; Ehst et al., 2008; Ariffin and Khorshid, 2006; Dummer et al., 2003; Suchin et al., 2002; Chong et al., 2004; Deeths et al., 2005; Ardigo et al., 2006) and B‐cell (Richmond et al., 2008; Stavrakoglou et al., 2007; Coors et al., 2006; Spaner et al., 2005) lymphomas. In a murine model of cutaneous breast cancer, topical application of imiquimod combined with local radiotherapy resulted in complete regression of locally treated tumors and inhibited growth at untreated sites. This response is associated with increase in T cell infiltration into tumor lesions, and the effect depended on CD8+ T cells. Pre‐treatment with low‐dose cyclophosphamide augments the anti‐tumor effect and protects the mice from tumor rechallenge, suggesting that a long‐term memory response against the tumor was induced (Dewan et al., 2012). Similar results have been observed with R848/resiquimod, another imidazoquinolinamine derivative, in mouse models of lymphoma. Systemic delivery of R848 combined with local tumor irradiation induces durable anti‐tumor immune responses leading to clearance of T‐ and B‐cell lymphomas and improved survival (Dovedi et al., 2013). Combined therapy also increased tumor antigen‐specific CD8+ T cells and protected mice from tumor rechallenge after clearance of the primary tumor. A novel injectable imidazoquinoline, 3M‐052, formulated in a lipid‐based vehicle to allow tissue retention has been shown to suppress melanoma growth in mice, both of injected and distant untreated sites. This anti‐tumor effect was enhanced by CTLA‐4 or PD‐L1 blockade, even in tumor models in which systemic checkpoint blockade was ineffective as monotherapy (Singh et al., 2014).
1.1.2.3. TLR3
TLR3 has an important role in host defense against viruses by recognizing dsRNA, activating IRF‐3 and ultimately increasing the production of type I interferons. Polyinosinic:polycytidylic acid (poly‐IC) is a synthetic TLR3 and MDA‐5 agonist due to its structural similarity to double‐stranded RNA (Gitlin et al., 2006) and can be stabilized with poly‐lysine and carboxymethylcellulose (poly‐ICLC) for in vivo use. Poly‐ICLC has been shown to be a promising vaccine adjuvant because it promotes type 1 immune responses (Salem et al., 2005; Salem et al., 2006). There is a significant set of clinical and pre‐clinical studies of in situ vaccination with TLR3 agonists. Several peptide‐based vaccine studies on brain cancer (Zhu et al., 2010, 2007, 2010, 2007) suggest that poly‐ICLC might also have positive effects in the context of in situ vaccination. When used as an adjuvant for an intratumoral HPV peptide‐based vaccine, poly‐IC is able to significantly improve therapeutic anti‐tumor effects and increase the proportion of tumor‐specific T cells (Wu et al., 2010). Additionally, in murine melanoma, intratumoral injection of poly‐IC markedly reduced tumor growth and prolonged survival, leading to complete eradication of tumors when combined with transfer of tumor‐peptide‐specific T cells, CD40L‐expressing plasmids, or systemic Flt3L (Amos et al., 2011; Fujimura et al., 2006; Stone et al., 2009b){Salmon et al., in press}. There is also pre‐clinical data to suggest that it may augment anti‐EGFR antibody therapy of head and neck cancer indicating potential for near‐term clinical translation (Ming Lim et al., 2013). In a case report of a patient with an advanced facial rhabdomyosarcoma, treatment with intratumoral injections of poly‐ICLC resulted in tumor inflammation and necrosis followed by marked tumor regression (Salazar et al., 2014); such results have prompted an ongoing phase II study investigating this approach in patients with advanced, unresectable solid tumors (NCT01984892). Additionally, the above described study combining intratumoral FLt3L and poly‐ICLC with radiotherapy for low‐grade lymphoma demonstrated that the poly‐ICLC induced order‐of‐magnitude increase in intratumoral, activated APCs while having minimal effect on systemic APC activation status (Marron et al., 2014).
1.1.2.4. TLR4
Bacterial lipopolysaccharide (LPS) is the primary ligand for TLR4 and ligation of TLR4 leads to activation of DC with improved processing and presentation of antigens and consequently the production of various pro‐inflammatory cytokines (Lu et al., 2008; Blander and Medzhitov, 2006). While systemically administered LPS has antitumor effects in mice and humans (Berendt et al., 1978; Goto et al., 1996; Otto et al., 1996), it also leads to cytokine storm with clinical symptoms of fevers, chills, hypotension, hepatic and hematologic effects (Engelhardt et al., 1990). Intratumoral administration of LPS maintains antitumor efficacy while avoiding significant toxicity. Mice with subcutaneous glioblastoma or rats with subcutaneous glioma tumors treated with intratumoral LPS experience partial and complete tumor regression and these responses are lessened in T cell deficient animals (Chicoine et al., 2001; Won et al., 2003; Mariani et al., 2007). Similarly, intratumoral injection of LPS in B16 melanoma mice results in tumor regression in those mice with increased activation of DC and T cells (Maito et al., 2012). By contrast, another study on the same model reported that LPS only reduced tumor growth when given in the context of a GM–CSF–expressing whole‐cell vaccine (Davis et al., 2011). This treatment regimen is, however, sufficient to induce regression of CT26 colon carcinomas and protected mice from tumor rechallenge. Another approach using Glucopyranosyl Lipid A (GLA‐SE), a small synthetic TLR4 agonist, is currently being studied by intratumoral injection in patients with Merkel cell carcinoma (NCT02035657) and sarcoma used in combination with radiotherapy (NCT02180698). A different TLR4 agonist (OK‐432/Picibanil) is currently being investigated in clinical trials for treatment of pancreatic adenocarcinoma in combination with intratumoral DC injection (NCT00795977) and for treatment of head and neck cancer in combination with chemotherapy and DC transfer (NCT01149902).
1.1.2.5. Live bacteria
Clostridia –as gram positive bacteria‐ are able to activate DC via TLR2 (and likely other mechanisms) (Kashiwagi et al., 2015) and –as an anaerobe‐ are well suited to proliferate and disseminate in the hypoxic environment within tumors. Clostridium novyi are endospore‐ forming, obligate anaerobic gram‐positive bacteria closely related to C. botulinum and a genetically engineered, lethal‐toxin deficient strain, has demonstrated CD8 T cell mediated anti‐tumor effects in pre‐clinical tumor renal, colon, and anaplastic squamous cell carcinoma models (Agrawal et al., 2004). Further studies demonstrated that intratumoral C. novyi induced tumor regressions in rat glioma tumors, spontaneously occurring canine sarcomas, and –in early, preliminary report from an ongoing clinical trial (NCT01924689)– marked tumor regression associated with fevers and local tissue necrosis, requiring intravenous narcotic analgesics and antibiotics (Roberts et al., 2014).
1.1.2.6. Anti‐CD40 monoclonal antibody
DC signaling pathways downstream of CD40 include recruitment of TNF Receptor Associated Factor family of proteins (TRAFs) –particularly TRAF6 (Kobayashi et al., 2003)– followed by p38 MAPK, JNK (Pullen et al., 1999), and NFκB and culminate in cytokine production, and co‐stimulatory molecule (e.g. CD80/CD86) upregulation (Ma and Clark, 2009). Overall, DC signaling nodes downstream of CD40 are similar to TLRs and RANK‐RANKL pathways, but are sufficiently distinct that activation of multiple pathways can lead to synergistic DC activation (Kerkmann et al., 2003).
In numerous small trials of systemic agonistic anti‐CD40 mAb, there have been data demonstrating immune activation but with some concerns of toxicity or pro‐tumorigenic effects (Vonderheide and Glennie, 2013) though a pre‐clinical model of local anti‐CD40 mAb showed decrease in liver inflammation compared to systemic therapy (Fransen et al., 2011) A recently developed agonistic mAb with high affinity for CD40 –ADC‐1013– has been tested intratumorally in human and murine in vitro models and in vivo bladder cancer models, inducing DC activation and IL12 secretion as well as antigen‐specific T‐cell proliferation and long‐term tumor‐specific immunity (Mangsbo et al., 2015) and have led to an ongoing study of intratumoral ADC‐1013 for patients with solid tumors (NCT02379741).
1.1.3. Oncolytic viruses that enhance the cross‐presentation of tumor antigens
Under conditions of cellular stress, heat shock proteins (HSPs) are produced and function as molecular chaperones mediating the folding and refolding of proteins. When HSP‐peptide complexes are released by dying tumor cells, they are taken up by APC via receptor‐mediated endocytosis, leading to cross‐presentation of tumor peptides on MHC class I molecules (Singh‐Jasuja et al., 2000; Noessner et al., 2002). Though HSP – autologous tumor derived peptide complexes have shown limited success (Wood et al., 2008) recent pre‐clinical and clinical studies of oncolytic adenoviruses, expressing HSP70 or heat shock transcription factor 1 (HSF1), have shown encouraging results. Intratumoral injection of these viruses eradicated primary tumors in mice, induced tumor‐specific immune responses that inhibited metastatic tumor growth, and protected mice from tumor rechallenge (Huang et al., 2003; Fan et al., 2012; Wang et al., 2010). A phase I clinical study demonstrated that intratumoral administration of recombinant oncolytic adenovirus expressing HSP70 is safe and exhibited some promising clinical anti‐tumor activity, though tumor regressions observed were predominantly at the treated sites (Li et al., 2009).
One potential obstacle with these approaches is that oncolytic viruses can drive an anti‐viral immune response which may divert the immune system from tumor‐specific antigens; a process called “immunodominance” (Alemany, 2014). Immunodominance occurs because the large numbers of viral peptides introduced into the patient overwhelm the APC system, thereby diverting APCs from TAAs. Immunodominant viral epitopes also have the ability to induce neutralizing antibodies which preclude subsequent booster vaccinations with the same vector. A ‘prime‐boost’ approach is an elegant method to address both of these issues; this refers to a vaccination using two different virus strains expressing the same tumor antigen in sequence. Priming with VSV expressing a melanoma‐associated antigen followed by a booster immunization with adenovirus expressing the same antigen (or vice versa) significantly increased tumor‐specific T cell responses in B16 murine melanoma (Bridle et al., 2009). Furthermore, this combination approach shifted the immune response from viral antigens to tumor antigens and reduced viral replication in normal tissues, thereby increasing efficacy as well as safety (Bridle et al., 2010). A clinical trial using intratumoral fowlpox administration has been completed (Kaufman et al., 2014) and randomized trials of prime‐boost strategies are ongoing (NCT02285816).
Even intratumorally administered oncolytic viruses may also be cytotoxic to healthy cells which might drive an immune response to self‐antigens. Increasing viral specificity for tumor cells may be an important way to increase the proportion of TAA presented on local APC. Several approaches in viral engineering have worked towards tumor‐specific infection or lysis.
An adenovirus engineered to replicate in Rb‐deficient cells and with increased binding for αVβ3 and αVβ5 integrins demonstrated induction of anti‐tumor immunity in pre‐clinical glioma model. In a phase 1 study, high grade glioma patients received a single intratumoral injection of DNX‐2401 followed by tumor resection 2 weeks later (and repeat DNX‐2401 injection into the surgical site). Complete responses were observed in 3 or 25 treated patients, all of whom are experienced prolonged (>1.5 years) remission. Resected tumor demonstrated abundant macrophages and CD8 T cells and ‐amongst the clinical responder‐ serum concentrations of IL‐12p70 were increased 10 to 10,000‐fold {Lang, 2014 #2700}. Ongoing trials are investigating combination of DNX‐2401 with temozolomide (NCT01956734) and IFNγ (NCT02197169).
A modified herpesvirus, HSV‐1716 –because of genetic deletion of ICP34.5– is unable to maintain protein translation in terminally differentiated cells and thus replicates selectively in tumor tissue. Infection by HSV‐1716 induces high levels of viral glycoprotein B and D expression and HSP70 in ovarian cancer cells which are efficiently phagocytosed by DC, upregulating DC costimulatory molecules and migration (Benencia et al., 2008). Intratumoral HSV‐1716 injection induces IFNγ expression and increase in intratumoral activated NK and CD8+ T cells (Benencia et al., 2005). HSV‐1716 administered intratumorally in five melanoma patients yielded microscopic evidence of tumor necrosis (MacKie et al., 2001) and in 12 glioma patients confirmed that viral replication can occur at the tumor site and possibly within distant tumors (Papanastassiou et al., 2002). Ongoing studies in high grade glioma (NCT02031965) and solid tumors (NCT00931931) will further assess the efficacy of the approach. More recently, additional genetic modifications of HSV‐1716 have incorporated the enzyme nitroreductase (which converts the prodrug CB1954 into an active alkylating agent) (Braidwood et al., 2009) or selected a clone which expresses inhibitor of growth 4 (Ing4) (a tumor suppressor protein) (Conner and Braidwood, 2012) significantly augmenting its oncolytic potency.
1.1.4. Oncolytic viruses that increase the maturation of antigen‐presenting cells
As previously described, TLR agonists activate APC functions and through this mechanism can initiate anti‐tumor immune responses hence their study as potential vaccine adjuvants and cancer immunotherapies. Oncolytic viruses enriched for CpG motifs, a TLR9 agonist, have shown higher anti‐tumor activity than their parental viral strains. Raykov et al. demonstrated that the oncolytic parvovirus H‐1PV enriched for CpG motifs is a potent anti‐cancer vaccine when used in a metastatic lung cancer rat model (Raykov et al., 2008). Compared to the original virus, subcutaneous injection of tumor cells with CpG enriched virus leads to increased immune responses and decrease in metastatic disease burden. This clinical and immune response correlates with high levels of IFNγ and activated DC in the tumor‐draining lymph nodes. Similar effects were observed by Cerrullo et al. when utilizing a CpG‐enriched adenovirus in murine lung cancer and melanoma (Cerullo et al., 2012). Intratumoral administration of the enriched virus led to a reduction in tumor growth in both models, with increased numbers of tumor‐specific T cells in tumors and spleen. In a related approach using the same melanoma model, Rommelfanger et al. combined intratumoral injection of VSV with subsequent injection of a TLR4 agonist. As compared to treatment with the virus alone, this approach led to improvement in survival and increased numbers of tumor‐specific T cells in the tumors and draining lymph nodes (Rommelfanger et al., 2013).
Engaging costimulatory molecules such as CD40 on the surface of the APC, which provide a secondary stimulus for activation is a different approach to increasing their capacity to present TAA. Oncolytic adenovirus expressing CD40 ligand (CD40L, CD154) has shown significant anti‐tumor effect in mice as well as patients with solid tumors (Diaconu et al., 2012; Pesonen et al., 2012). Intratumoral injection of the virus led to increases in tumor cell apoptosis and delayed tumor growth in murine melanoma and bladder cancer. Treatment was also associated with enhanced recruitment of APC and CD8+ T cells to tumor lesions locally and systemically (Diaconu et al., 2012). An early phase study of nine solid tumor patients received intratumoral injections of CD40L‐encoding adenovirus and demonstrated the induction of IFN‐γ‐producing survivin‐specific (as well as adenovirus‐specific) T in the peripheral blood. Some patients experienced prolonged stable disease in addition to regression of untreated tumors and clearance of serum tumor markers (e.g. the breast cancer marker Ca15‐3) (Pesonen et al., 2012). Similar results have been observed with oncolytic herpes simplex or adenovirus encoding CD80, a costimulatory ligand for CD28 on T cells, resulting in enhanced T cell activation and anti‐tumor immune responses (Fukuhara et al., 2005; Ino et al., 2006; Todo et al., 2001; Lee et al., 2006; Choi et al., 2006).
Another approach has been the intratumoral administration of coxsackie virus A21 which binds intercellular adhesion molecule (ICAM‐1) and decay‐accelerating factor (DAF) – molecules that are frequently upregulated on malignant melanoma cells. This genetically unmodified, wild‐type, common cold‐producing human enterovirus (CAVATAK™) has demonstrated oncolytic activity in pre‐clinical and early phase clinical studies and induces immune cell infiltration in melanoma (Andtbacka et al., 2015b). A recently reported phase II study (NCT01227551) demonstrated responses in 16 of 57 evaluable and median duration of response not reached after 17 months (Andtbacka et al., 2015c). Biomarker analyses have shown an association of serum IL8 and IFNγ with systemic responses and pre‐clinical melanoma models have demonstrated marked synergy between intratumoral CAVATAK and PD‐1 blockade (Shafren et al., 2014).
1.2. Manipulation of intratumoral lymphocytes
1.2.1. Activation of T cells
1.2.1.1. Interleukin‐12
Interleukin‐12 (IL‐12) is a cytokine with an important role in the regulation of adaptive T cell responses (Colombo and Trinchieri, 2002). It is released by various immune cells ‐particularly myeloid APC such as monocytes (D'Andrea et al., 1992) upon stimulation by infection or inflammation to induce differentiation of type 1 helper T cells; thus increasing activity of cytotoxic T lymphocytes as well as IFNγ production. For more than 2 decades, IL‐12 has been known to have potent anti‐tumor activity (Brunda et al., 1993) and has been extensively investigated for use in cancer treatments. In murine transplantable tumors IL‐12 anti‐tumor activity is dependent on a variety of factors, including tumor type, dose, and route of injection (Colombo and Trinchieri, 2002). Clinical studies have used systemic administration of IL‐12 in patients with melanoma, RCC and colon cancer, unfortunately this approach was associated with considerable hepatic and hematologic toxicity with only modest efficacy at doses tested (Atkins et al., 1997; Leonard et al., 1997). Intratumoral administration of IL‐12 is associated with less toxicity and has shown to cause partial or complete tumor regression at the locally injected tumor site, but little to no response at distant sites (Rook et al., 1999; Sangro et al., 2004; Mahvi et al., 2007; Zapała et al., 2013). By contrast, when IL‐12 is encapsulated into polymer microspheres, which release their contents over an extended period of time, regression of primary and secondary murine tumors as well as regression of metastasic lesions has been achieved (Egilmez et al., 2000). In glioblastoma models, complete regression can also be achieved by combining intratumoral IL‐12 administration with systemic immune checkpoint blockade using an antibody against CTLA‐4 (Vom Berg et al., 2013).
IL‐12 is also under investigation with oncolytic viruses for the development of cancer vaccines. In pre‐clinical murine cancer models including prostate, colon, liver, and brain cancer, viruses modified to express IL‐12 induce strong anti‐tumor immune responses with reduced tumor growth and increased survival (Toda et al., 1998; Bennett et al., 2001; Varghese et al., 2006; Zhang et al., 2013; Cheema et al., 2013).
Early phase clinical studies are currently investigating the use of intratumoral IL‐12 plasmid electroporation for the treatment of cutaneous lymphoma (NCT01579318) and advanced melanoma (NCT01502293) and have demonstrated no grade 3/4 drug‐related AEs, a doubling of intratumoral NK, activation of circulating NK cells, clinical response in 9 of 28 patients and regression of non‐injected lesions in 13 of 21 evaluable patients (Daud et al., 2014). A related approach is the use of lipopolymer to increase plasmid delivery into cells which has been administered intra‐ or peri‐tumorally by intraperitoneal delivery in patients with ovarian cancer in an early study of 13 patients, demonstrating increase IL‐12 and IFNγ levels in peritoneal fluid (but not serum) and a minority of patients with treatment‐related decrease in serum CA‐125 levels (Anwer et al., 2010). More recently this approach has been combined with systemic liposomal doxorubicin resulting in partial remissions and stable disease in 2 and 4 of the 7 patients in the highest‐dose cohort, warranting further study of the combination (Thaker et al., 2015).
Adenovirally delivered IL‐12 is being investigated for the treatment of glioblastoma (NCT02026271), breast cancer (NCT01703754, NCT02423902), and melanoma (NCT01397708). Because of the variable persistence of adenoviral vectors and potential toxicity of IL‐12, an elegant approach has been the development of vectors incorporating an ecdysone‐inducible expression system with a goal of titrating IL‐12 levels after administration. Preliminary report on intratumoral administration of Ad‐RTS‐hIL‐12 in 38 patients with breast cancer or melanoma followed by increasing doses of veledimex (an orally administered ecdysone analog) resulted in a veledimex dose‐dependent increases in intratumoral and serum IL‐12 transcript and protein as well as serum IFNγ levels. A minority of patients experienced partial remission or sub‐clinical tumor regressions and a small number of patients had grade ≥3 adverse events (e.g. hepatic or hematologic), but –perhaps the most significant proof of principle– was the rapid reversibility of adverse events with the discontinuation of veledimex (Nemunaitis et al., 2014).
1.2.1.2. Interleukin‐2
Interleukin‐2 (IL‐2) is important in the development of adaptive immune response as it mediates expansion of T cells and differentiation into effector lymphocytes and has been amongst the most thoroughly investigated cancer immunotherapies. Systemic administration of IL‐2 is FDA approved for the treatment of RCC and melanoma (Fyfe et al., 1995; Atkins et al., 1999), though systemic treatment is associated with significant toxicity, prompting the investigation of intratumoral IL‐2 therapies. Intratumoral injections of soluble IL‐2 result in increased infiltration of CD8+ T cells, reduced tumor growth and increased survival in mice bearing transplantable tumors (Fiszer‐Maliszewska et al., 1998; Jackaman et al., 2003). The clinical and immune response can be further accentuated by using expression vectors encoding IL‐2 to allow prolonged production of the cytokine (Horton et al., 1999; Slos et al., 2001). Intratumoral injection of an adenovirally encoded IL‐2 also inhibits growth of distant, untreated tumors and protects mice from rechallenge with the same tumor type (Slos et al., 2001). This effect is also observed with a slow‐release, liposomal formulation of IL‐2 in mice with a non‐immunogenic B16 melanoma tumor, inducing systemic immune responses not observed with injections of soluble IL‐2 (Neville et al., 2001). In mice with lung or hepatocellular carcinoma, using a combined treatment with microparticles encapsulating IL‐2 and microwave coagulation to induce tumor cell death results in a systemic tumor‐specific immune response (Kuang et al., 2005). Clinical trials reveal promising results in patients with RCC or melanoma who received intratumoral treatment with IL‐2 encoding plasmids or recombinant IL‐2, respectively. Compared to systemic treatment, this treatment modality generally resulted in low toxicity. While plasmid treatment of RCC led to a low number of responses (Hoffman and Figlin, 2000; Galanis et al., 1999), injection of recombinant IL‐2 into melanoma metastases induced high response rates as measured by tumor regression (Radny et al., 2003; Weide et al., 2010; Gutwald et al., 1994). However, although most injected lesions regressed and did not recur, this treatment failed to cause complete regression of untreated melanoma lesions or prevent subsequent development of metastases (Radny et al., 2003; Weide et al., 2010). Clinical studies investigating the combination of intratumoral IL‐2 and systemic or intratumoral treatment with anti‐CTLA‐4 are currently ongoing (NCT02076633, NCT01672450, NCT01480323).
1.2.1.3. T‐cell activating oncolytic viruses
Intratumorally administered modified herpesvirus has been shown to induce CD8‐dependent tumor lysis and regression of untreated tumors in pre‐clinical models (Meshii et al., 2013) and early phase clinical trials in patients with pancreatic (Nakao et al., 2011) and breast cancer (Kimata et al., 2006; Sahin et al., 2012) have yielded local tumor regressions. Recent pre‐clinical studies have demonstrated incremental benefit of HF10 herpesvirus combination with agonistic monoclonal antibody for murine glucocorticoid‐induced tumor necrosis factor receptor (Ishihara et al., 2014), the anti‐VEGF mAb bevacizumab (Tan et al., 2015), and GM‐CSF (Goshima et al., 2014).
1.2.2. Intratumoral checkpoint blockade
Immune checkpoint molecules, such as CTLA‐4 or PD‐1, are receptors expressed on T cells and down regulate their activity. In normal homeostatic settings, these molecules protect the host from auto‐immunopathology due to unchecked immune responses. However, in the tumor microenvironment, they contribute to TAA tolerance. Toxicity associated with systemic CTLA‐4 blockade (Hodi et al., 2010; Robert et al., 2011; Postow et al., 2015) has been avoided in pre‐clinical models of intratumoral anti‐CTLA4 antibody, along an increase in the CTL:Treg ratio within tumors (Tuve et al., 2007; Simmons et al., 2008). Similarly, in a mouse model of colon cancer, peritumoral injection of anti‐CTLA‐4 in a slow‐release formulation induced regression of treated as well as untreated nodules (Fransen et al., 2013). In addition, checkpoint blockade antibodies can be delivered intratumorally by using oncolytic viruses encoding these antibodies. As described in section iii, adenovirus or measles virus encoding anti‐CTLA‐4 have shown encouraging anti‐tumor efficacy in murine models of colon cancer and melanoma (Engeland et al., 2014; Du et al., 2014).
Finally, intratumoral injection of anti‐CTLA‐4 also depletes Treg at the injected site, and combination treatment with anti‐OX40 and CpG results in eradication of disseminated CNS lymphoma (Marabelle et al., 2013). An ongoing phase I/II clinical trial is investigating the combination of local irradiation and intratumoral injection of ipilimumab for the treatment of melanoma, lymphoma, colon, and rectal cancer (NCT01769222) as well as using combined intratumoral ipilimumab and IL‐2 (NCT01672450).
1.2.3. Oncolytic viruses to reduce immune suppression in the tumor microenvironment
As a result of the immunosuppressive nature of tumors, effective therapy requires not only the induction of immune responses, but also the downregulation of immune inhibitory mechanisms. Therefore, oncolytic viruses are being used in combination with agents that inhibit the induction of immunosuppressive cells – i.e. regulatory T cells (Treg) – or counteract the inhibition of effector T cells (Teff). Low dose cyclophosphamide is a chemotherapeutic agent with immune modulatory function and has been shown to specifically inhibit Treg but not Teff cells (Motoyoshi et al., 2006). In patients with treatment refractory solid malignancies, compared to administration of the virus alone, administration of intratumoral oncolytic adenovirus and low‐dose cyclophosphamide resulted in higher disease control and overall survival (Cerullo et al., 2011). This improved response was associated with a reduction in Treg cells and an increase in cytotoxic T cells. A different chemotherapeutic drug, temozolomide, has also been shown to reduce Treg numbers at low doses (Banissi et al., 2009). The combination of temozolomide with an oncolytic adenovirus is currently under investigation for the treatment of glioblastoma (NCT01956734).
In addition, monoclonal antibodies (mAb) against immune checkpoint proteins have shown encouraging results in cancer immunotherapy and have been combined with oncolytic viruses. In a murine melanoma model, intratumoral injection of Newcastle Disease virus accompanied by systemic treatment with an mAb against CTLA‐4 reduced tumor growth, improved survival, and protected mice from tumor rechallenge (Zamarin et al., 2014). However, systemic treatment with checkpoint‐blocking antibodies can lead to severe immune‐related adverse effects (Hodi et al., 2010; Robert et al., 2011; Postow et al., 2015; Voskens et al., 2013), this driving investigators to discover more effective local therapies that will have systemic responses. Dias et al. demonstrates that intratumoral treatment with an adenovirus engineered to express a complete human mAb against CTLA‐4 leads to 43‐fold higher mAb concentration in tumors vs. plasma of mice. The average plasma concentration is below what has been reported to be tolerated in humans (Toda et al., 1998). More recently, attenuated measles virus encoding mAb against CTLA‐4 or PD‐1 was shown to delay tumor growth and prolong survival in murine melanoma (Engeland et al., 2014). Similarly, Du et al. reveals that in lung cancer and melanoma murine models treatment with adenovirus expressing anti‐CTLA‐4 significantly delays tumor growth and results in complete regression when combined with a second virus encoding GM‐CSF (Du et al., 2014).
In addition to immunosuppressive cells in the tumor microenvironment, components of the extracellular matrix (ECM) such as hyaluronan contribute to tumor immune evasion. Hyaluronan synthase 2 expressed in breast cancer stem‐like cells is critical for the interaction of tumor cells with tumor‐associated macrophages (TAM) and its deficiency or inhibition reduced metastatic disease (Okuda et al., 2012). VCN‐01, a novel adenovirus engineered to avoid tropism for liver and spleen and to express hyaluronidase to degrade ECM accumulates in murine tumors after systemic administration with only transient presence in the liver. Furthermore, anti‐tumor efficacy after systemic administration of the virus was comparable to that observed after intratumoral delivery (Rodríguez‐García et al., 2015). Two phase I clinical trials with VCN‐01 alone or in combination with gemcitabine in patients with pancreatic cancer and advanced solid tumors are currently underway (NCT02045589, NCT02045602).
2. Conclusions
The primary goal of cancer immunotherapy is to induce a durable anti‐tumor immune response while leaving normal host cells unharmed. Personalized vaccines have shown encouraging results, since they can be directed against patient‐specific TAAs (Di Nicola et al., 2008; Timmerman et al., 2002; Schuster et al., 2011). A whole tumor cell approach obviates the need to identify TAAs, but manufacturing an ex vivo product is resource intensive, and time consuming limiting opportunity to optimize the vaccine in iterative clinical trials. In contrast, “off‐the‐shelf” vaccines are easier to manufacture, but mandate the identification of shared TAAs – ideally ones in which tumors are highly dependent – to minimize risk of escape variants. An in situ vaccine combines the best aspects of each strategy. By inducing immunogenic tumor cell death and antigen release at a single tumor site, in situ vaccination is personalized, and thus patient‐specific. The approach obviates the need for patient‐specific TAA‐identification. Patients can be vaccinated against the tumor's entire TAA repertoire, as opposed to a single TAA. Furthermore, injectable immunomodulators such as cytokines, TLR agonists or viruses can be mass‐produced, is not resource intensive and is therefore practical to optimize in iterative patient cohorts.
As the field of cancer therapy continues to incorporate immunotherapy as an integral treatment strategy, the effort to identify therapies that augment the benefit of checkpoint‐blocking agents will continue to develop. Intratumoral therapies will be among the most likely to induce this desired effect as they may increase the response rate, while minimizing toxicity (Simmons et al., 2008). Intratumoral therapy effectively, converts patients from ‘low intratumoral T cell infiltration type’ to a ‘T cell inflamed phenotype’ increasing the likelihood of response to checkpoint blockade therapy (Tumeh et al., 2014; Sharma and Allison, 2015). In a field of potential candidates to be combined with checkpoint blockade, in situ vaccination represents one of the promising strategies. Ultimately, the opportunities to expand the armamentarium of tumor‐specific immunotherapies with in situ vaccination strategies are continuing to increase with ongoing scrutiny in the laboratory and continued success in our patients (see Figure 1).
Figure 1.
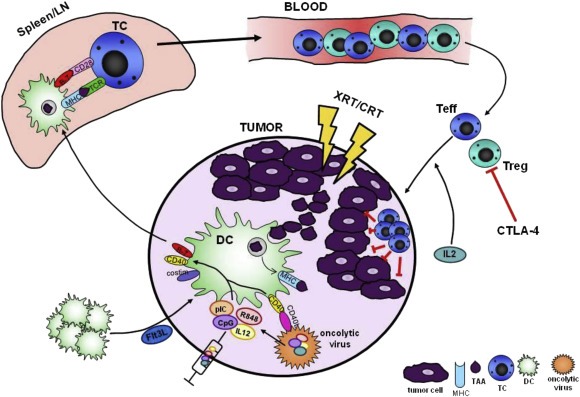
Effective in situ vaccination uses local or intratumoral immunomodulation to produce abundant, highly activated, antigen presenting cells such as dendritic cells (DC) able to present the full repertoire of tumor‐associated antigens (TAA) to tumor‐reactive T cells which, in turn, mount a systemic anti‐tumor immune response. Optimally, this approach occurs through: 1) increasing intratumoral DC populations, by recruitment or local proliferation (e.g. with intratumoral Flt3L administration); 2) inducing immunogenic tumor cell death locally or systemically to release TAA (e.g. with radiotherapy, chemotherapy, or oncolytic virus); 3) enhancing TAA uptake, processing and presentation or cross‐presentation by DC (e.g. with heat shock proteins or using prime‐boost oncolytic viral strategies); 4) promoting antigen‐loaded DC to trafficking to secondary lymphoid tissue 5) presentation or cross‐presentation to antigen‐specific T cells; 6) trafficking of T cells to tumor sites systemically; 7) inhibition of multiple levels of immunosuppression, including cells and molecules suppressing T cells both at the vaccine site as well as distant tumor sites (e.g. with intratumoral or systemic checkpoint blockade).
Hammerich Linda, Binder Adam, Brody Joshua D., (2015), In situ vaccination: Cancer immunotherapy both personalized and off‐the‐shelf, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.10.016.
This is a contribution to the special issue edited by Johanna Olweus, Cancer Immunotherapy.
References
- Adams, S. , 2012. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 18, (24) 6748–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal, N. , 2004. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc. Natl. Acad. Sci. U.S.A. 101, (42) 15172–15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemany, R. , 2014. Oncolytic adenoviruses in cancer treatment. Biomedicines. 2, (1) 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos, S.M. , 2011. Adoptive immunotherapy combined with intratumoral TLR agonist delivery eradicates established melanoma in mice. Cancer Immunol. Immunother. 60, (5) 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andtbacka, R.H. , Kaufman, H.L. , Collichio, F. , Amatruda, T. , Senzer, N. , Chesney, J. , Delman, K.A. , Spitler, L.E. , Puzanov, I. , Agarwala, S.S. , Milhem, M. , Cranmer, L. , Curti, B. , Lewis, K. , Ross, M. , Guthrie, T. , Linette, G.P. , Daniels, G.A. , Harrington, K. , Middleton, M.R. , Miller, W.H. , Zager, J.S. , Ye, Y. , Yao, B. , Li, A. , Doleman, S. , VanderWalde, A. , Gansert, J. , Coffin, R.S. , 2015. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 33, (25) 2780–2788. 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- Andtbacka, Robert , Curti, Brendan , Hallmeyer, Sigrun , Shafren, Darren R. , 2015. Phase II CALM study: changes in the tumor microenvironment induced by the immunotherapeutic agent coxsackievirus A21 delivered intratumorally in patients with advanced melanoma. [abstract] Proceedings of the 106th Annual Meeting of the American Association for Cancer Research. AACR; Philadelphia, PA: Cancer Res 2015;75(15 Suppl):Abstract nr CT214. http://dx.doi.org/10.1158/1538-7445.AM2015-CT214 [Google Scholar]
- Andtbacka, R.H.I. , 2015. Final data from CALM: a phase II study of Coxsackievirus A21 (CVA21) oncolytic virus immunotherapy in patients with advanced melanoma. J. Clin. Oncol. (Meeting Abstracts). 33, No 15_suppl (May 20 Supplement), 2015: 9030 [Google Scholar]
- Anwer, K. , 2010. Phase-I clinical trial of IL-12 plasmid/lipopolymer complexes for the treatment of recurrent ovarian cancer. Gene Ther. 17, (3) 360–369. [DOI] [PubMed] [Google Scholar]
- Appledorn, D.M. , 2008. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol. 181, (3) 2134–2144. [DOI] [PubMed] [Google Scholar]
- Ardigo, M. , Cota, C. , Berardesca, E. , 2006. Unilesional mycosis fungoides successfully treated with imiquimod. Eur. J. Dermatol. 16, (4) 446 [PubMed] [Google Scholar]
- Ariffin, N. , Khorshid, M. , 2006. Treatment of mycosis fungoides with imiquimod 5% cream. Clin. Exp. Dermatol. 31, (6) 822–823. [DOI] [PubMed] [Google Scholar]
- Atkins, M.B. , 1997. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin. Cancer Res. 3, (3) 409–417. [PubMed] [Google Scholar]
- Atkins, M.B. , 1999. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 17, (7) 2105–2116. [DOI] [PubMed] [Google Scholar]
- Banchereau, R. , 2014. Transcriptional specialization of human dendritic cell subsets in response to microbial vaccines. Nat. Commun. 5, 5283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banissi, C. , 2009. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol. Immunother. 58, (10) 1627–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett, D.L. , 2013. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer. 12, (1) 103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benencia, F. , 2005. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol. Ther. 12, (5) 789–802. [DOI] [PubMed] [Google Scholar]
- Benencia, F. , 2008. Herpes virus oncolytic therapy reverses tumor immune dysfunction and facilitates tumor antigen presentation. Cancer Biol. Ther. 7, (8) 1194–1205. [DOI] [PubMed] [Google Scholar]
- Bennett, J.J. , 2001. Interleukin 12 secretion enhances antitumor efficacy of oncolytic herpes simplex viral therapy for colorectal cancer. Ann. Surg. 233, (6) 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendt, M.J. , North, R.J. , Kirstein, D.P. , 1978. The immunological basis of endotoxin-induced tumor regression. Requirement for T-cell-mediated immunity. J. Exp. Med. 148, (6) 1550–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj, N. , 2014. Converting tumors into vaccine manufacturing factories: DC recruitment, activation and clinical responses with a flt3L-primed in situ vaccine for low-grade lymphoma [nct01976585]. J. Immunother. Cancer. 2, (Suppl. 3) 45 [Google Scholar]
- Blander, J.M. , Medzhitov, R. , 2006. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 440, (7085) 808–812. [DOI] [PubMed] [Google Scholar]
- Bourke, E. , 2003. The toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood. 102, (3) 956–963. [DOI] [PubMed] [Google Scholar]
- Braidwood, L. , 2009. Antitumor activity of a selectively replication competent herpes simplex virus (HSV) with enzyme prodrug therapy. Anticancer Res. 29, (6) 2159–2166. [PubMed] [Google Scholar]
- Breitbach, C.J. , 2011. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 477, (7362) 99–102. [DOI] [PubMed] [Google Scholar]
- Breitbach, C.J. , 2013. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 73, (4) 1265–1275. [DOI] [PubMed] [Google Scholar]
- Breitbach, C.J. , 2015. A phase 2, open-label, randomized study of Pexa-Vec (JX-594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol. Biol. 1317, 343–357. [DOI] [PubMed] [Google Scholar]
- Bridle, B.W. , 2009. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol. Ther. 17, (10) 1814–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridle, B.W. , 2010. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 18, (8) 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody, J.D. , 2009. Immunotransplantation preferentially expands T-effector cells over T-regulatory cells and cures large lymphoma tumors. Blood. 113, (1) 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody, J.D. , 2010. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J. Clin. Oncol. 28, (28) 4324–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz, M.L. , 2014. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 26, (5) 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunda, M.J. , 1993. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J. Exp. Med. 178, (4) 1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calista, D. , Riccioni, L. , Bagli, L. , Valenzano, F. , 2015. Long-term remission of primary cutaneous neutrophil-rich CD30+ anaplastic large cell lymphoma treated with topical imiquimod. A case report. J. Eur. Acad. Dermatol. Venereol. 10.1111/jdv.13070 [DOI] [PubMed] [Google Scholar]
- Carpentier, A. , 2010. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 12, (4) 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerullo, V. , 2011. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol. Ther. 19, (9) 1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerullo, V. , 2012. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 20, (11) 2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema, T.A. , 2013. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl. Acad. Sci. U S A. 110, (29) 12006–12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicoine, M.R. , Won, E.K. , Zahner, M.C. , 2001. Intratumoral injection of lipopolysaccharide causes regression of subcutaneously implanted mouse glioblastoma multiforme. Neurosurgery. 48, (3) 607–614. discussion 614-5 [DOI] [PubMed] [Google Scholar]
- Choi, K.J. , 2006. Concurrent delivery of GM-CSF and B7-1 using an oncolytic adenovirus elicits potent antitumor effect. Gene Ther. 13, (13) 1010–1020. [DOI] [PubMed] [Google Scholar]
- Chong, A. , 2004. Imiquimod 5% cream in the treatment of mycosis fungoides–a pilot study. J. Dermatolog Treat. 15, (2) 118–119. [DOI] [PubMed] [Google Scholar]
- Colombo, M.P. , Trinchieri, G. , 2002. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 13, (2) 155–168. [DOI] [PubMed] [Google Scholar]
- Conner, J. , Braidwood, L. , 2012. Expression of inhibitor of growth 4 by HSV1716 improves oncolytic potency and enhances efficacy. Cancer Gene Ther. 19, (7) 499–507. [DOI] [PubMed] [Google Scholar]
- Coors, E.A. , Schuler, G. , Von Den Driesch, P. , 2006. Topical imiquimod as treatment for different kinds of cutaneous lymphoma. Eur. J. Dermatol. 16, (4) 391–393. [PubMed] [Google Scholar]
- Cripe, T.P. , 2015. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 23, (3) 602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie, G.A. , 1972. Eighty years of immunotherapy: a review of immunological methods used for the treatment of human cancer. Br. J. Cancer. 26, (3) 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea, A. , 1992. Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J. Exp. Med. 176, (5) 1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adil Daud, Alain Patrick Algazi, Michelle T. Ashworth, Lawrence Fong, Jera Lewis, Stephen E Chan, Michael Buljan, Manuel Alberto Molina, Kathryn Toshimi Takamura, Tuan Tu Diep, Richard Heller, Robert H Pierce, Shailender Bhatia, Systemic antitumor effect and clinical response in a phase 2 trial of intratumoral electroporation of plasmid interleukin-12 in patients with advanced melanoma, J. Clin. Oncol. 32 (5s), 2014 (suppl; abstr 9025).
- Davis, M.B. , 2011. Intratumoral administration of TLR4 agonist absorbed into a cellular vector improves antitumor responses. Clin. Cancer Res. 17, (12) 3984–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeths, M.J. , 2005. Treatment of patch and plaque stage mycosis fungoides with imiquimod 5% cream. J. Am. Acad. Dermatol. 52, (2) 275–280. [DOI] [PubMed] [Google Scholar]
- Dewan, M.Z. , 2012. Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin. Cancer Res. 18, (24) 6668–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaconu, I. , 2012. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 72, (9) 2327–2338. [DOI] [PubMed] [Google Scholar]
- Didona, B. , 2004. Primary cutaneous CD30+ T-cell lymphoma responsive to topical imiquimod (Aldara). Br. J. Dermatol. 150, (6) 1198–1201. [DOI] [PubMed] [Google Scholar]
- Dovedi, S.J. , 2013. Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood. 121, (2) 251–259. [DOI] [PubMed] [Google Scholar]
- Dranoff, G. , 1993. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. U S A. 90, (8) 3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, T. , 2014. Tumor-specific oncolytic adenoviruses expressing granulocyte macrophage colony-stimulating factor or anti-CTLA4 antibody for the treatment of cancers. Cancer Gene Ther. 21, (8) 340–348. [DOI] [PubMed] [Google Scholar]
- Dummer, R. , 2003. Imiquimod induces complete clearance of a PUVA-resistant plaque in mycosis fungoides. Dermatology. 207, (1) 116–118. [DOI] [PubMed] [Google Scholar]
- Egilmez, N.K. , 2000. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 60, (14) 3832–3837. [PubMed] [Google Scholar]
- Ehst, B.D. , Dreno, B. , Vonderheid, E.C. , 2008. Primary cutaneous CD30+ anaplastic large cell lymphoma responds to imiquimod cream. Eur. J. Dermatol. 18, (4) 467–468. [DOI] [PubMed] [Google Scholar]
- Elsedawy, N.B. , Russell, S.J. , 2013. Oncolytic vaccines. Expert Rev. Vaccines. 12, (10) 1155–1172. [DOI] [PubMed] [Google Scholar]
- Engeland, C.E. , 2014. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 22, (11) 1949–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt, R. , 1990. Biological response to intravenously administered endotoxin in patients with advanced cancer. J. Biol. Response Mod. 9, (5) 480–491. [PubMed] [Google Scholar]
- Escors, D. , 2014. Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J. Sci. 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, R. , 2012. Enhanced antitumoral efficacy and immune response following conditionally replicative adenovirus containing constitutive HSF1 delivery to rodent tumors. J. Transl Med. 10, 101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszer-Maliszewska, L. , 1998. Therapeutic potential of biological response modifiers against transplantable mouse tumors of spontaneous origin. II. Local interleukin 2 treatment of tumors of different immunogenic strength. Arch. Immunol. Ther. Exp. (warsz). 46, (5) 293–300. [PubMed] [Google Scholar]
- Formenti, S.C. , Demaria, S. , 2009. Systemic effects of local radiotherapy. Lancet Oncol. 10, (7) 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen, M.F. , 2011. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin. Cancer Res. 17, (8) 2270–2280. [DOI] [PubMed] [Google Scholar]
- Fransen, M.F. , 2013. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin. Cancer Res. 19, (19) 5381–5389. [DOI] [PubMed] [Google Scholar]
- Fujimura, T. , 2006. Inhibitory effect of the polyinosinic-polycytidylic acid/cationic liposome on the progression of murine B16F10 melanoma. Eur. J. Immunol. 36, (12) 3371–3380. [DOI] [PubMed] [Google Scholar]
- Fukuhara, H. , 2005. Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res. 65, (23) 10663–10668. [DOI] [PubMed] [Google Scholar]
- Furumoto, K. , 2004. Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J. Clin. Invest. 113, (5) 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe, G. , 1995. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 13, (3) 688–696. [DOI] [PubMed] [Google Scholar]
- Galanis, E. , 1999. Immunotherapy of advanced malignancy by direct gene transfer of an interleukin-2 DNA/DMRIE/DOPE lipid complex: phase I/II experience. J. Clin. Oncol. 17, (10) 3313–3323. [DOI] [PubMed] [Google Scholar]
- Gitlin, L. , 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U S A. 103, (22) 8459–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima, F. , 2014. Oncolytic viral therapy with a combination of HF10, a herpes simplex virus type 1 variant and granulocyte-macrophage colony-stimulating factor for murine ovarian cancer. Int. J. Cancer. 134, (12) 2865–2877. [DOI] [PubMed] [Google Scholar]
- Goto, S. , 1996. Intradermal administration of lipopolysaccharide in treatment of human cancer. Cancer Immunol. Immunother. 42, (4) 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauer, O.M. , 2008. TLR ligands in the local treatment of established intracerebral murine gliomas. J. Immunol. 181, (10) 6720–6729. [DOI] [PubMed] [Google Scholar]
- Gudmundsdottir, H. , Wells, A.D. , Turka, L.A. , 1999. Dynamics and requirements of T cell clonal expansion in vivo at the single-cell level: effector function is linked to proliferative capacity. J. Immunol. 162, (9) 5212–5223. [PubMed] [Google Scholar]
- Guo, J. , 2007. Intratumoral injection of dendritic cells in combination with local hyperthermia induces systemic antitumor effect in patients with advanced melanoma. Int. J. Cancer. 120, (11) 2418–2425. [DOI] [PubMed] [Google Scholar]
- Gutwald, J.G. , Groth, W. , Mahrle, G. , 1994. Peritumoral injections of interleukin 2 induce tumour regression in metastatic malignant melanoma. Br. J. Dermatol. 130, (4) 541–542. [DOI] [PubMed] [Google Scholar]
- Harrington, K.J. , 2010. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin. Cancer Res. 16, (15) 4005–4015. [DOI] [PubMed] [Google Scholar]
- Henriques, L. , 2014. Imiquimod in the treatment of breast cancer skin metastasis. J. Clin. Oncol. 32, (8) e22–e25. [DOI] [PubMed] [Google Scholar]
- Heo, J. , 2013. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 19, (3) 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi, F.S. , 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, (8) 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller, C. , 2001. Perilesional injection of r-GM-CSF in patients with cutaneous melanoma metastases. J. Invest Dermatol. 117, (2) 371–374. [DOI] [PubMed] [Google Scholar]
- Hoffman, D.M. , Figlin, R.A. , 2000. Intratumoral interleukin 2 for renal-cell carcinoma by direct gene transfer of a plasmid DNA/DMRIE/DOPE lipid complex. World J. Urol. 18, (2) 152–156. [DOI] [PubMed] [Google Scholar]
- Horton, H.M. , 1999. IL-2 plasmid therapy of murine ovarian carcinoma inhibits the growth of tumor ascites and alters its cytokine profile. J. Immunol. 163, (12) 6378–6385. [PubMed] [Google Scholar]
- Hou, S. , 2007. Eradication of hepatoma and colon cancer in mice with Flt3L gene therapy in combination with 5-FU. Cancer Immunol. Immunother. 56, (10) 1605–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houot, R. , Levy, R. , 2009. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood. 113, (15) 3546–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J.C. , 2006. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 12, (22) 6737–6747. [DOI] [PubMed] [Google Scholar]
- Huang, X.F. , 2003. A broadly applicable, personalized heat shock protein-mediated oncolytic tumor vaccine. Cancer Res. 63, (21) 7321–7329. [PubMed] [Google Scholar]
- Hwang, T.H. , 2011. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol. Ther. 19, (10) 1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino, Y. , 2006. Triple combination of oncolytic herpes simplex virus-1 vectors armed with interleukin-12, interleukin-18, or soluble B7-1 results in enhanced antitumor efficacy. Clin. Cancer Res. 12, (2) 643–652. [DOI] [PubMed] [Google Scholar]
- Ishihara, M. , 2014. Systemic CD8+ T cell-mediated tumoricidal effects by intratumoral treatment of oncolytic herpes simplex virus with the agonistic monoclonal antibody for murine glucocorticoid-induced tumor necrosis factor receptor. PLoS One. 9, (8) e104669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackaman, C. , 2003. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J. Immunol. 171, (10) 5051–5063. [DOI] [PubMed] [Google Scholar]
- Jahrsdörfer, B. , 2001. CpG DNA increases primary malignant B cell expression of costimulatory molecules and target antigens. J. Leukoc. Biol. 69, (1) 81–88. [PubMed] [Google Scholar]
- Jahrsdorfer, B. , 2005. B-cell lymphomas differ in their responsiveness to CpG oligodeoxynucleotides. Clin. Cancer Res. 11, (4) 1490–1499. [DOI] [PubMed] [Google Scholar]
- Kashiwagi, I. , Morita, R. , Schichita, T. , Komai, K. , Saeki, K. , Matsumoto, M. , Takeda, K. , Nomura, M. , Hayashi, A. , Kanai, T. , Yoshimura, A. , 2015. Smad2 and Smad3 inversely regulate TGF-β autoinduction in Clostridium butyricum-activated dendritic cells. Immunity. 43, (1) 65–79. 10.1016/j.immuni.2015.06.010 [DOI] [PubMed] [Google Scholar]
- Kaufman, H.L. , 2010. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 17, (3) 718–730. [DOI] [PubMed] [Google Scholar]
- Kaufman, H.L. , 2014. Primary overall survival (OS) from OPTiM, a randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J. Clin. Oncol. 32, 5s, 2014 (suppl; abstr 9008a) [Google Scholar]
- Kerkmann, M. , 2003. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J. Immunol. 170, (9) 4465–4474. [DOI] [PubMed] [Google Scholar]
- Kim, J.H. , 2006. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 14, (3) 361–370. [DOI] [PubMed] [Google Scholar]
- Kim, Y.H. , 2012. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: a phase 1/2 study. Blood. 119, (2) 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata, H. , 2006. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann. Surg. Oncol. 13, (8) 1078–1084. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T. , 2003. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 19, (3) 353–363. [DOI] [PubMed] [Google Scholar]
- Kohrt, H.E. , 2014. Dose-escalated, intratumoral TLR9 agonist and low-dose radiation induce abscopal effects in follicular lymphoma. 124, 3092 [Google Scholar]
- Kolstad, A. , 2015. Sequential intranodal immunotherapy induces antitumor immunity and correlated regression of disseminated follicular lymphoma. Blood. 125, (1) 82–89. [DOI] [PubMed] [Google Scholar]
- Kreiter, S. , 2011. FLT3 ligand enhances the cancer therapeutic potency of naked RNA vaccines. Cancer Res. 71, (19) 6132–6142. [DOI] [PubMed] [Google Scholar]
- Kuang, M. , 2005. Microwave tumour coagulation plus in situ treatment with cytokine-microparticles: induction of potent anti-residual tumour immunity. Int. J. Hyperthermia. 21, (3) 247–257. [DOI] [PubMed] [Google Scholar]
- Lee, Y.S. , 2006. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 12, (19) 5859–5868. [DOI] [PubMed] [Google Scholar]
- Leonard, J.P. , 1997. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 90, (7) 2541–2548. [PubMed] [Google Scholar]
- Liu, B.L. , 2003. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 10, (4) 292–303. [DOI] [PubMed] [Google Scholar]
- London, C.A. , Lodge, M.P. , Abbas, A.K. , 2000. Functional responses and costimulator dependence of memory CD4+ T cells. J. Immunol. 164, (1) 265–272. [DOI] [PubMed] [Google Scholar]
- Lu, Y.C. , Yeh, W.C. , Ohashi, P.S. , 2008. LPS/TLR4 signal transduction pathway. Cytokine. 42, (2) 145–151. [DOI] [PubMed] [Google Scholar]
- Ma, D.Y. , Clark, E.A. , 2009. The role of CD40 and CD154/CD40L in dendritic cells. Semin. Immunol. 21, (5) 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach, N. , 2000. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 60, (12) 3239–3246. [PubMed] [Google Scholar]
- MacKie, R.M. , Stewart, B. , Brown, S.M. , 2001. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet. 357, (9255) 525–526. [DOI] [PubMed] [Google Scholar]
- Mahvi, D.M. , 2007. Intratumoral injection of IL-12 plasmid DNA–results of a phase I/IB clinical trial. Cancer Gene Ther. 14, (8) 717–723. [DOI] [PubMed] [Google Scholar]
- Maito, F.L.D.M. , 2012. Intratumoral TLR-4 agonist injection is critical for modulation of tumor microenvironment and tumor rejection. ISRN Immunol. 2012 [Google Scholar]
- Maloney, D.G. , 1994. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood. 84, (8) 2457–2466. [PubMed] [Google Scholar]
- Mangsbo, S.M. , 2015. The human agonistic CD40 antibody ADC-1013 eradicates bladder tumors and generates T-cell-dependent tumor immunity. Clin. Cancer Res. 21, (5) 1115–1126. [DOI] [PubMed] [Google Scholar]
- Marabelle, A. , 2013. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J. Clin. Invest. 123, (6) 2447–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani, C.L. , 2007. Nonspecific immunotherapy with intratumoral lipopolysaccharide and zymosan A but not GM-CSF leads to an effective anti-tumor response in subcutaneous RG-2 gliomas. J. Neurooncol. 85, (3) 231–240. [DOI] [PubMed] [Google Scholar]
- Marron, T. , 2014. Turning a tumor into a vaccine factory: in situ vaccination for low-grade lymphoma. Am. Soc. Hematol. 124, 5473 [Google Scholar]
- Marroquin, C.E. , 2002. Mobilization of dendritic cell precursors in patients with cancer by flt3 ligand allows the generation of higher yields of cultured dendritic cells. J. Immunother. 25, (3) 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshii, N. , 2013. Enhancement of systemic tumor immunity for squamous cell carcinoma cells by an oncolytic herpes simplex virus. Cancer Gene Ther. 20, (9) 493–498. [DOI] [PubMed] [Google Scholar]
- Miller, R.L. , 1999. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int. J. Immunopharmacol. 21, (1) 1–14. [DOI] [PubMed] [Google Scholar]
- Ming Lim, C. , 2013. TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR head and neck cancer cells. Oncoimmunology. 2, (6) e24677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse, M.A. , 2000. Preoperative mobilization of circulating dendritic cells by Flt3 ligand administration to patients with metastatic colon cancer. J. Clin. Oncol. 18, (23) 3883–3893. [DOI] [PubMed] [Google Scholar]
- Motoyoshi, Y. , 2006. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol. Rep. 16, (1) 141–146. [PubMed] [Google Scholar]
- Nakao, A. , 2011. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 18, (3) 167–175. [DOI] [PubMed] [Google Scholar]
- Nasi, M.L. , 1999. Intradermal injection of granulocyte-macrophage colony-stimulating factor (GM-CSF) in patients with metastatic melanoma recruits dendritic cells. Cytokines Cell Mol Ther. 5, (3) 139–144. [PubMed] [Google Scholar]
- Nemunaitis, J. , 1999. Oncolytic viruses. Invest New Drugs. 17, (4) 375–386. [DOI] [PubMed] [Google Scholar]
- Nemunaitis, J.J. , 2014. Ad-RTS-hIL-12 + veledimex regulation of IL-12 expression in advanced breast cancer (BC) and melanoma patients. AACR - Tumor Immunol. Immunother. A New Chapter Poster Session B (meeting abstract # B11). [Google Scholar]
- Neville, M.E. , Robb, R.J. , Popescu, M.C. , 2001. In situ vaccination against a non-immunogenic tumour using intratumoural injections of liposomal interleukin 2. Cytokine. 16, (6) 239–250. [DOI] [PubMed] [Google Scholar]
- Di Nicola M., Zappasodi R., Carlo-Stella C., Mortarini R., Pupa S.M., Magni M., Devizzi L., Matteucci P., Baldassari P., Ravagnani F., Cabras A., Anichini A., Gianni A.M. Vaccination with autologous tumor-loaded dendritic cells induces clinical and immunologic responses in indolent B-cell lymphoma patients with relapsed and measurable disease: a pilot study, Blood 113(1), 2009, 18–27. http://dx.doi.org/10.1182/blood-2008-06-165654. [DOI] [PubMed]
- Noessner, E. , 2002. Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J. Immunol. 169, (10) 5424–5432. [DOI] [PubMed] [Google Scholar]
- Li, J. , 2007. Lymphoma immunotherapy with CpG oligodeoxynucleotides requires TLR9 either in the host or in the tumor itself. J. Immunol. 179, (4) 2493–2500. [DOI] [PubMed] [Google Scholar]
- Li, J.L. , 2009. A phase I trial of intratumoral administration of recombinant oncolytic adenovirus overexpressing HSP70 in advanced solid tumor patients. Gene Ther. 16, (3) 376–382. [DOI] [PubMed] [Google Scholar]
- Okuda, H. , 2012. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 72, (2) 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, F. , 1996. Phase II trial of intravenous endotoxin in patients with colorectal and non-small cell lung cancer. Eur. J. Cancer. 32A, (10) 1712–1718. [DOI] [PubMed] [Google Scholar]
- Papanastassiou, V. , 2002. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 9, (6) 398–406. [DOI] [PubMed] [Google Scholar]
- Parato, K.A. , 2012. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 20, (4) 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, B.H. , 2008. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 9, (6) 533–542. [DOI] [PubMed] [Google Scholar]
- Pawlowska, A.B. , 2001. In vitro tumor-pulsed or in vivo Flt3 ligand-generated dendritic cells provide protection against acute myelogenous leukemia in nontransplanted or syngeneic bone marrow-transplanted mice. Blood. 97, (5) 1474–1482. [DOI] [PubMed] [Google Scholar]
- Pesonen, S. , 2012. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res. 72, (7) 1621–1631. [DOI] [PubMed] [Google Scholar]
- Postow, M.A. , 2015. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 372, (21) 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen, S.S. , 1999. CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J. Biol. Chem. 274, (20) 14246–14254. [DOI] [PubMed] [Google Scholar]
- Puzanov, I. , 2014. Primary analysis of a phase 1b multicenter trial to evaluate safety and efficacy of talimogene laherparepvec (T-VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB-IV melanoma. J. Clin. Oncol. 32, 5s, 2014 (suppl; abstr 9029) [Google Scholar]
- Radny, P. , 2003. Phase II trial of intralesional therapy with interleukin-2 in soft-tissue melanoma metastases. Br. J. Cancer. 89, (9) 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raykov, Z. , 2008. Arming parvoviruses with CpG motifs to improve their oncosuppressive capacity. Int. J. Cancer. 122, (12) 2880–2884. [DOI] [PubMed] [Google Scholar]
- Reichardt, V.L. , Brossart, P. , Kanz, L. , 2004. Dendritic cells in vaccination therapies of human malignant disease. Blood Rev. 18, (4) 235–243. [DOI] [PubMed] [Google Scholar]
- Richmond, H.M. , 2008. Primary cutaneous follicle center lymphoma associated with alopecia areata. Clin. Lymphoma Myeloma. 8, (2) 121–124. [DOI] [PubMed] [Google Scholar]
- Riediger, C. , 2013. Fms-like tyrosine kinase 3 receptor ligand (Flt3L)-based vaccination administered with an adenoviral vector prevents tumor growth of colorectal cancer in a BALB/c mouse model. J. Cancer Res. Clin. Oncol. 139, (12) 2097–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert, C. , 2011. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, (26) 2517–2526. [DOI] [PubMed] [Google Scholar]
- Roberts, N.J. , 2014. Intratumoral injection of Clostridium novyi-NT spores induces antitumor responses. Sci. Transl Med. 6, (249) 249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-García, A. , 2015. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin. Cancer Res. 21, (6) 1406–1418. [DOI] [PubMed] [Google Scholar]
- Rommelfanger, D.M. , 2013. The efficacy versus toxicity profile of combination virotherapy and TLR immunotherapy highlights the danger of administering TLR agonists to oncolytic virus-treated mice. Mol. Ther. 21, (2) 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook, A.H. , 1999. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood. 94, (3) 902–908. [PubMed] [Google Scholar]
- Rosenfeld, M.R. , 2010. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol. 12, (10) 1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin, T.T. , 2012. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. 19, (4) 229–237. [DOI] [PubMed] [Google Scholar]
- Saji, H. , 2006. Systemic antitumor effect of intratumoral injection of dendritic cells in combination with local photodynamic therapy. Clin. Cancer Res. 12, (8) 2568–2574. [DOI] [PubMed] [Google Scholar]
- Salazar, A.M. , 2014. Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: case report, hypothesis, and clinical trial. Cancer Immunol. Res. 2, (8) 720–724. [DOI] [PubMed] [Google Scholar]
- Salem, M.L. , 2005. Defining the antigen-specific T-cell response to vaccination and poly(I: C)/TLR3 signaling: evidence of enhanced primary and memory CD8 T-cell responses and antitumor immunity. J. Immunother. 28, (3) 220–228. [DOI] [PubMed] [Google Scholar]
- Salem, M.L. , 2006. The adjuvant effects of the toll-like receptor 3 ligand polyinosinic-cytidylic acid poly (I: C) on antigen-specific CD8+ T cell responses are partially dependent on NK cells with the induction of a beneficial cytokine milieu. Vaccine. 24, (24) 5119–5132. [DOI] [PubMed] [Google Scholar]
- Sangro, B. , 2004. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J. Clin. Oncol. 22, (8) 1389–1397. [DOI] [PubMed] [Google Scholar]
- Schuster, S.J. , 2011. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 29, (20) 2787–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senzer, N.N. , 2009. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 27, (34) 5763–5771. [DOI] [PubMed] [Google Scholar]
- Shafren, D. , 2014. Combination of a novel oncolytic immunotherapeutic agent, CAVATAK (coxsackievirus A21) and immune-checkpoint blockade significantly reduces tumor growth and improves survival in an immune competent mouse melanoma model. J. Immunother. Cancer. 2, (Suppl. 3) P125 [Google Scholar]
- Sharma, P. , Allison, J.P. , 2015. The future of immune checkpoint therapy. Science. 348, (6230) 56–61. [DOI] [PubMed] [Google Scholar]
- Sharma, S. , 2008. CpG-ODN but not other TLR-ligands restore the antitumor responses in old mice: the implications for vaccinations in the aged. Cancer Immunol. Immunother. 57, (4) 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman, K. , Naik, S.H. , 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7, (1) 19–30. [DOI] [PubMed] [Google Scholar]
- Si, Z. , Hersey, P. , Coates, A.S. , 1996. Clinical responses and lymphoid infiltrates in metastatic melanoma following treatment with intralesional GM-CSF. Melanoma Res. 6, (3) 247–255. [DOI] [PubMed] [Google Scholar]
- Sidky, Y.A. , 1992. Inhibition of murine tumor growth by an interferon-inducing imidazoquinolinamine. Cancer Res. 52, (13) 3528–3533. [PubMed] [Google Scholar]
- Simmons, A.D. , 2008. Local secretion of anti-CTLA-4 enhances the therapeutic efficacy of a cancer immunotherapy with reduced evidence of systemic autoimmunity. Cancer Immunol. Immunother. 57, (8) 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, M. , 2014. Effective innate and adaptive antimelanoma immunity through localized TLR7/8 activation. J. Immunol. 193, (9) 4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Jasuja, H. , 2000. Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J. Exp. Med. 191, (11) 1965–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slos, P. , 2001. Immunotherapy of established tumors in mice by intratumoral injection of an adenovirus vector harboring the human IL-2 cDNA: induction of CD8(+) T-cell immunity and NK activity. Cancer Gene Ther. 8, (5) 321–332. [DOI] [PubMed] [Google Scholar]
- Smyth, E.C. , 2011. Treatment of locally recurrent mucosal melanoma with topical imiquimod. J. Clin. Oncol. 29, (33) e809–e811. [DOI] [PubMed] [Google Scholar]
- Song, W. , Levy, R. , 2005. Therapeutic vaccination against murine lymphoma by intratumoral injection of naive dendritic cells. Cancer Res. 65, (13) 5958–5964. [DOI] [PubMed] [Google Scholar]
- Spaner, D.E. , 2005. Regression of lymphomatous skin deposits in a chronic lymphocytic leukemia patient treated with the Toll-like receptor-7/8 agonist, imiquimod. Leuk. Lymphoma. 46, (6) 935–939. [DOI] [PubMed] [Google Scholar]
- Stavrakoglou, A. , Brown, V.L. , Coutts, I. , 2007. Successful treatment of primary cutaneous follicle centre lymphoma with topical 5% imiquimod. Br. J. Dermatol. 157, (3) 620–622. [DOI] [PubMed] [Google Scholar]
- Stone, G.W. , 2009. Regression of established AB1 murine mesothelioma induced by peritumoral injections of CpG oligodeoxynucleotide either alone or in combination with poly(I: C) and CD40 ligand plasmid DNA. J. Thorac. Oncol. 4, (7) 802–808. [DOI] [PubMed] [Google Scholar]
- Stone, G.W. , 2009. Nanoparticle-delivered multimeric soluble CD40L DNA combined with Toll-Like Receptor agonists as a treatment for melanoma. PLoS One. 4, (10) e7334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchin, K.R. , Junkins-Hopkins, J.M. , Rook, A.H. , 2002. Treatment of stage IA cutaneous T-Cell lymphoma with topical application of the immune response modifier imiquimod. Arch. Dermatol. 138, (9) 1137–1139. [DOI] [PubMed] [Google Scholar]
- Suresh, M. , 2001. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J. Immunol. 167, (10) 5565–5573. [DOI] [PubMed] [Google Scholar]
- Tan, G. , 2015. Combination therapy of oncolytic herpes simplex virus HF10 and bevacizumab against experimental model of human breast carcinoma xenograft. Int. J. Cancer. 136, (7) 1718–1730. [DOI] [PubMed] [Google Scholar]
- Thaker, P.H. , 2015. Phase I study of intraperitoneal IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer administered in combination with pegylated liposomal doxorubicin in recurrent or persistent epithelial ovarian, Fallopian tube, or primary peritoneal cancer patients: an NRG/GOG study. J Clin Oncol (Meeting Abstracts). J. Clin. Oncol. 33, 2015 (suppl; abstr 5541) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman, J.M. , 2002. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 99, (5) 1517–1526. [DOI] [PubMed] [Google Scholar]
- Toda, M. , 1998. In situ cancer vaccination: an IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activity. J. Immunol. 160, (9) 4457–4464. [PubMed] [Google Scholar]
- Todo, T. , 2001. In situ expression of soluble B7-1 in the context of oncolytic herpes simplex virus induces potent antitumor immunity. Cancer Res. 61, (1) 153–161. [PubMed] [Google Scholar]
- Tong, Y. , Song, W. , Crystal, R.G. , 2001. Combined intratumoral injection of bone marrow-derived dendritic cells and systemic chemotherapy to treat pre-existing murine tumors. Cancer Res. 61, (20) 7530–7535. [PubMed] [Google Scholar]
- Tumeh, P.C. , 2014. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 515, (7528) 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuve, S. , 2007. Combination of tumor site-located CTL-associated antigen-4 blockade and systemic regulatory T-cell depletion induces tumor-destructive immune responses. Cancer Res. 67, (12) 5929–5939. [DOI] [PubMed] [Google Scholar]
- Upadhyay, R. , 2015. Lymphoma: immune evasion strategies. Cancers (Basel). 7, (2) 736–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese, S. , 2006. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 13, (3) 253–265. [DOI] [PubMed] [Google Scholar]
- Vom Berg, J. , 2013. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 210, (13) 2803–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide, R.H. , Glennie, M.J. , 2013. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 19, (5) 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskens, C.J. , 2013. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 8, (1) e53745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , 2010. HSF1 overexpression enhances oncolytic effect of replicative adenovirus. J. Transl Med. 8, 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weide, B. , 2010. High response rate after intratumoral treatment with interleukin-2: results from a phase 2 study in 51 patients with metastasized melanoma. Cancer. 116, (17) 4139–4146. [DOI] [PubMed] [Google Scholar]
- Won, E.K. , 2003. Analysis of the antitumoral mechanisms of lipopolysaccharide against glioblastoma multiforme. Anticancer Drugs. 14, (6) 457–466. [DOI] [PubMed] [Google Scholar]
- Wood, C. , 2008. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet. 372, (9633) 145–154. [DOI] [PubMed] [Google Scholar]
- Wu, C.Y. , 2010. Improving therapeutic HPV peptide-based vaccine potency by enhancing CD4+ T help and dendritic cell activation. J. Biomed. Sci. 17, 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhi, Y. , 1998. The therapeutic effect of intratumoral injection of GM-CSF gene-modified allogenic macrophages on tumor-bearing mice. Chines J. Cancer Res. 10, (1) 1–5. [Google Scholar]
- Zamarin, D. , 2014. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl Med. 6, (226) 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapała, Ł. , 2013. Synergistic antitumor effect of JAWSII dendritic cells and interleukin 12 in a melanoma mouse model. Oncol. Rep. 29, (3) 1208–1214. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , 2012. An in situ autologous tumor vaccination with combined radiation therapy and TLR9 agonist therapy. PLoS One. 7, (5) e38111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , 2013. Combination of oncolytic herpes simplex viruses armed with angiostatin and IL-12 enhances antitumor efficacy in human glioblastoma models. Neoplasia. 15, (6) 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , 2007. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J. Transl Med. 5, 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , 2010. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol. Immunother. 59, (9) 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, J. , Huang, X. , Yang, Y. , 2007. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 81, (7) 3170–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]