

Abstract
Chimeric antigen receptors are genetically encoded artificial fusion molecules that can re‐program the specificity of peripheral blood polyclonal T‐cells against a selected cell surface target. Unparallelled clinical efficacy has recently been demonstrated using this approach to treat patients with refractory B‐cell malignancy. However, the approach is technically challenging and can elicit severe toxicity in patients. Moreover, solid tumours have largely proven refractory to this approach. In this review, we describe the important structural features of CARs and how this may influence function. Emerging clinical experience is summarized in both solid tumours and haematological malignancies. Finally, we consider the particular challenges imposed by solid tumours to the successful development of CAR T‐cell immunotherapy, together with a number of innovative strategies that have been developed in an effort to reverse the balance in favour of therapeutic benefit.
Keywords: Immunotherapy, Cancer, Chimeric antigen receptor, T-cells, Solid tumours
Highlights
CAR T‐cells are genetically engineered to recognize tumour cells.
CD19‐targeted CAR T‐cells have demonstrated unprecedented success in clinical trials.
Solid tumours are more challenging to treat.
We review CAR design and clinical experience with CAR T‐cells.
We also discuss the challenges facing the future of CAR T‐cell immunotherapy.
1. Introduction
Overwhelming evidence supports the existence of sophisticated immune surveillance mechanisms for cancer (Swann and Smyth, 2007). By definition however, these natural defense systems have been overwhelmed once cancer is clinically detectable, an event that will afflict 50% of individuals born after the year 1960 (Ahmad et al., 2015). One fundamental difficulty stems from the poorly immunogenic nature of transformed cells, in which most tumour‐associated antigens are derived from self, or subtle modifications thereof. In addition to this, malignant cell populations deploy several mechanisms to evade immune detection (Vinay et al., 2015). These include the impairment of antigen‐processing machinery and down‐regulated expression of tumour antigens themselves and the HLA molecules that are required for their cell surface presentation. As tumours advance and acquire stromal support, they establish a local and a systemic environment that is very poorly conducive to effective sterilizing immune responses. Indeed, tumours have been compared to “wounds that do not heal” (Dvorak, 1986), emphasizing their ability to corrupt a natural process during which cell growth, stroma formation, matrix deposition, angiogenesis and the down‐regulation of both innate and adaptive immune mechanisms all occur in a physiologically controlled manner (Ayala et al., 2003).
The demonstration that transformed cells are subject to immune surveillance has important practical significance since it opens a window of therapeutic opportunity. After many false dawns, cancer immunotherapy is now entering the mainstream of clinical medicine. The advent of immune checkpoint blockade (reviewed elsewhere in this Special Issue) represents a paradigm shift in the clinical management of cancer. For the first time, this emerging modality places emphasis on pharmacological manipulation of the host, rather than of the disease. Recently, the bi‐specific T‐cell engager (biTE) antibody blinatumomab, which is specific for CD19 and CD3, was approved for the treatment of acute lymphoblastic leukaemia. Within the broad theme of cancer immunotherapy, an equally exciting development entails the genetic engineering of peripheral blood T‐cells using chimeric antigen receptor technology. Chimeric antigen receptors (CARs) are HLA‐independent fusion molecules that couple the binding of a native target(s), displayed on the surface of tumour cells, to the delivery of a precisely defined T‐cell activating signal (Figure 1). The ability to engineer large numbers of T‐cells to acquire a direct antibody‐like recognition system for antigen circumvents or mitigates many of the immune evasion mechanisms mentioned above.
Figure 1.
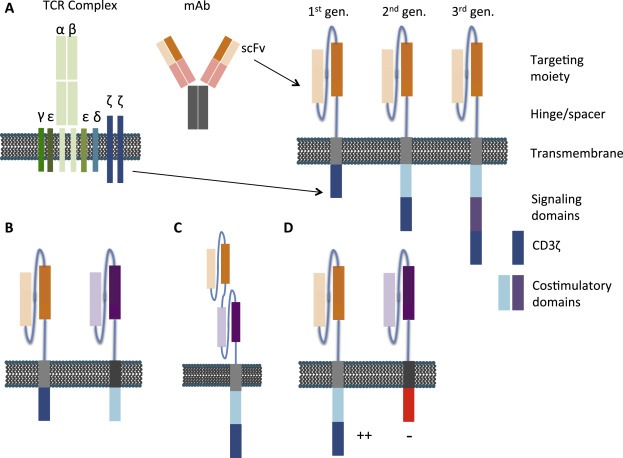
Structural features of chimeric antigen receptors. A) An antigen‐binding domain is typically comprised of antibody variable heavy (VH) and light (VL) chains from a monoclonal antibody that are able to self‐assemble through inclusion of a linker sequence to form a single chain variable fragment (scFv). In a first generation CAR, the targeting moiety is coupled via a hinge and transmembrane domain to an intracellular T‐cell signalling domain, typically the CD3ζ chain of the TCR complex. Second and third generation CARs contain an additional one or two costimulatory endodomains, respectively. B) A representative dual‐targeting CAR with two CARs expressed by the same T‐cell, each targeting an independent antigen. T‐cell signalling domains are split between the two CARs to enable optimal T‐cell activation only upon simultaneous engagement with both antigens. C) A bispecific CAR where two scFvs are expressed from the same CAR in tandem. The CAR is able to bind either antigen alone, or both antigens simultaneously. D) An inhibitory CAR that serves to attenuate positive signals delivered by activating CARs may be co‐expressed. By this means, T‐cell activation is restricted to target cells that only express the ligand for the activating but not the inhibitory CAR.
Immunotherapy using CAR T‐cells has been under development for almost three decades, but has witnessed a substantial increase in interest over the past 5 years or so. This development has been largely driven by the unprecedented success of CAR T‐cells in Phase I trials targeted against a variety of B‐cell malignancies (reviewed in Maher, 2014 and discussed further in section 4 below). Against this backdrop, it is timely to review progress and hurdles that hinder the further clinical development of this emerging therapeutic modality for cancer. Here, we focus on CAR design, recent clinical experience in both solid and haematological malignancies and will highlight some emerging approaches that aim to improve the efficacy of this approach.
2. Chimeric antigen receptor design
The overall structure of a CAR consists of four domains joined in series, namely: (i) an antigen recognition domain (targeting moiety), (ii) a hinge/spacer, (iii) a transmembrane element and (iv) a signalling endodomain (Figure 1). These will be considered in turn in the sections that follow.
2.1. The targeting moiety
The CAR ectodomain determines target specificity and, most commonly, contains elements derived from a monoclonal antibody. Initial CAR designs were somewhat cumbersome owing to the need to co‐express both antibody variable heavy (VH) and light chains (VL) in two separate polypeptide chains (Gross et al., 1989; Kuwana et al., 1987). Realizing the translational potential of non‐HLA restricted antigen recognition by T‐cells, Eshhar addressed this issue using a linker that allows VH and VL chains to self‐associate, thereby creating a single chain antibody fragment (scFv) (Eshhar et al., 1993). Use of murine hybridoma‐derived scFvs is disadvantaged however by their ability to elicit production of human anti‐mouse antibodies (HAMAs) in the host. Such antibodies can block CAR T‐cell function and may contribute to the accelerated clearance of these cells in‐vivo (Kershaw et al., 2006). Furthermore, HAMAs may also lead to life‐threatening complications such as anaphylaxis, as reported recently in a patient treated with repeated doses of mesothelin‐specific CAR T‐cells (Maus et al., 2013). In order to circumvent this issue, several humanized and human scFvs have been generated, including those that recognize HER2 (Sun et al., 2014; Zhao et al., 2009), the variant III mutation of epidermal growth factor receptor (EGFRvIII) (Johnson et al., 2015) and mesothelin (Adusumilli et al., 2014). Alternatively, targeting may be achieved using peptides (Pameijer et al., 2007), single domain antibodies (Sharifzadeh et al., 2013) or natural or chimeric ligands. These include interleukin (IL)‐13 (Kahlon et al., 2004; Kong et al., 2012), heregulin (Altenschmidt et al., 1996; Muniappan et al., 2000), or the T1E peptide, which binds several ErbB hetero‐ and homodimers (Davies et al., 2012). Use of non‐antibody based targeting moieties has the advantage that neither anti‐mouse nor anti‐idiotypic antibodies will be generated although there remains a theoretical risk that antibodies may be generated against junctional elements within the CAR ectodomain.
2.2. The hinge/spacer and trans‐membrane domain
These elements play a predominantly structural role in the CAR, although they may also affect function in some cases. The spacer physically separates the targeting moiety from the T‐cell membrane. However, the optimum distance required is likely to be different for each antigen. Illustrating this, Guest et al. studied the effect of spacer inclusion in four different CARs, targeting the pan‐B‐cell antigen CD19, the onco‐foetal antigen 5T4, carcinoembryonic antigen (CEA) or neural cell adhesion molecule (NCAM). Whilst the inclusion of a spacer enhanced cytokine release and cytotoxicity of 5T4‐and NCAM‐targeted CARs, it had the opposite effect in CD19‐and CEA‐targeted CARs (Guest et al., 2005). To enable efficient target access, a spacer appears to be required if a CAR binds an epitope that lies close to the target cell membrane, or when an antigen is complex in size and glycosylation status, such as MUC1 (Wilkie et al., 2008). Furthermore, human IgG‐derived spacers are commonly used due to their stabilizing action on CAR expression but interactions between the Fc domain of the spacer and Fc gamma receptors (FcγRs) on myeloid cells can lead to activation‐induced cell death of T‐cells and limited persistence in‐vivo (Almasbak et al., 2015; Hudecek et al., 2015). This can be overcome by deleting or modifying regions of the constant heavy (CH)2 domain that are essential for FcγR binding, thereby improving CAR T‐cell persistence and anti‐tumour activity in‐vivo in pre‐clinical models (Hudecek et al., 2015; Jonnalagadda et al., 2015). Similarly, the trans‐membrane domain may also have a functional role since this portion of CD3ζ is required for dimerization and association with other elements that form the TCR/CD3 complex, potentially refining the nature of the activation signal that is delivered (Bridgeman et al., 2010). Other transmembrane domains commonly used include those derived from T‐cell molecules such as CD28 or CD8.
2.3. The CAR signalling domain
The activating signal delivered by a CAR is largely dictated by the signalling endodomain. The modular structure of the CAR endodomain has been the subject of considerable iterative modification, in an effort to improve function and therapeutic efficacy. Nonetheless, a broad consensus on the optimal configuration has not yet been reached.
First generation CAR T‐cells contain a single T‐cell activating domain, most commonly derived from the ζ chain of the TCR/CD3 complex. Evidence suggests that CD3ζ alone provides a sufficiently potent “signal 1” from its three immunoreceptor tyrosine‐based activation motifs (ITAMs) to substitute for the global signal provided by the entire CD3 complex (Haynes et al., 2001; Irving and Weiss, 1991). It is well known however that an additional co‐stimulatory signal (“signal 2”) is required to allow activated T‐cells to undergo optimal proliferation, accompanied by cytokine release. To provide this, second and third generation CARs have been developed in which one or two co‐stimulatory domains, respectively, are placed in series with CD3ζ. Typically, CD28 is included as a co‐stimulatory domain as this provides an early second signal and promotes high‐level IL‐2 secretion (Finney et al., 1998; Maher et al., 2002). Multiple other second generation CAR T‐cells have been described, incorporating co‐stimulatory elements as diverse as CD27 (Duong et al., 2013; Song et al., 2012), OX40 (Brentjens et al., 2007; Finney et al., 2004), 4‐1BB (Brentjens et al., 2007; Finney et al., 2004; Imai et al., 2004; Song et al., 2011), ICOS (Finney et al., 2004), DAP10 (Brentjens et al., 2007; Duong et al., 2013) or 2B4(CD244) (Altvater et al., 2009). Second generation CAR T‐cells are consistently more active in pre‐clinical models, compared to first generation counterparts (Brentjens et al., 2007; Milone et al., 2009; Song et al., 2012). Nonetheless, it is difficult to determine the most suitable co‐stimulatory molecule to use as head‐to‐head comparisons are not frequently performed, and evidence is sometimes conflicting in different model systems. Production of IL‐2 is potentiated most strongly by CD28 containing second generation CARs (Brentjens et al., 2007), although this may achieve a double edged effect in some systems, owing to enhanced intra‐tumoural infiltration by regulatory T‐cells (Kofler et al., 2011). Co‐stimulation by ICOS is distinguished by its ability to potentiate cytotoxicity (Finney et al., 2004) and Th17 differentiation (Guedan et al., 2012). Chimeric antigen receptors containing 4‐1BB may promote multi‐functionality of cytokine release (Carpenito et al., 2009) and constitutive signalling leading to sustained T‐cell survival (Milone et al., 2009), although these findings have been less apparent when evaluated using other CARs (Brentjens et al., 2007).
Clinical evaluation of the optimum co‐stimulatory module has been very limited. When first and CD28‐based second generation CAR T‐cells were co‐infused, the latter exhibited more sustained persistence in patients (Savoldo et al., 2011). Second generation CARs used in clinical trials most often incorporate CD28 or 4‐1BB signalling domains, both of which demonstrate prolonged persistence when expressed in CD19‐targeted CARs (Brentjens et al., 2013; Porter et al., 2011). Emerging clinical experience with these two second generation configurations suggests that CD28‐containing constructs undergo more rapid in‐vivo expansion and decline whereas 4‐1BB‐containing fusions may persist for longer periods (van der Stegen et al., 2015). Third generation CARs incorporate two co‐stimulatory modules placed in series within the CAR endodomain (Geiger et al., 2001; Pule et al., 2005). Since CD28 and TNF receptor family members each initiate different signalling pathways (PI3K for CD28 compared to tumour necrosis family receptor‐associated factor (TRAF) adaptor proteins for 4‐1BB), this approach may enhance overall T‐cell activity (Tammana et al., 2010; Zhao et al., 2009). Third generation CAR T‐cells have recently commenced clinical evaluation (Till et al., 2012), although it remains too soon to comment as to whether these represent a significant advance over second generation configurations. Another recent innovation that warrants further investigation entails the co‐expression in T‐cells of killer immunoglobulin‐like receptor‐based CARs together with DAP‐12 (Wang et al., 2015).
Provision of co‐stimulatory signals to CAR T‐cells may also be provided in‐trans. Proof of concept for the functional activity of co‐stimulatory fusion receptors in human T‐cells was demonstrated several years ago (Krause et al., 1998). Building on this, a number of groups have co‐expressed a first generation CD3ζ‐based CAR with a co‐stimulatory fusion receptor that engages a second target molecule. This “dual targeting” CAR approach has been used pre‐clinically to target the MUC‐1 mucin and HER‐2 (Wilkie et al., 2012), prostate‐specific membrane antigen (PSMA) and prostate stem cell antigen (PSCA) (Kloss et al., 2013), and the combination of folate receptor (FR)α and mesothelin (Lanitis et al., 2013). Separating the co‐stimulatory module from the CD3ζ endodomain in this manner may enhance signalling efficiency, in addition to providing a higher level of tumour specificity. Alternatively, CD80 and CD137 ligand may be co‐expressed within the same T‐cell as a first generation CAR to deliver a combination of co‐stimulation to the same (auto‐costimulation) and neighbouring T‐cells (trans‐costimulation). This approach has also proven to be strongly efficacious in pre‐clinical studies (Stephan et al., 2007).
3. Target selection
T‐cell receptors (TCRs) recognize antigens derived from all cellular locations that have been processed to generate peptide fragments, which are presented on the cell surface by HLA antigens. By contrast, CARs can only engage native molecules that are expressed on the cell surface. While this limits antigen repertoire that may be targeted, CARs have the ability to bind not only proteins but also carbohydrates, gangliosides and tumour‐associated glycosylation variants (Pule et al., 2008; Wilkie et al., 2008).
3.1. Targets under study using CAR T‐cell immunotherapy
The ideal target for CAR T‐cells is an antigen that is expressed exclusively by tumour cells, thus eliminating risk of recognition and damage of healthy tissue. One such example is the tumour‐specific variant of the epidermal growth factor receptor (EGFR), EGFRvIII, which arises as a result of mutation in glioblastoma (Heimberger et al., 2005) and is absent in normal tissue. EGFRvIII is the target of several CARs under development (Johnson et al., 2015; Miao et al., 2014; Morgan et al., 2012; Sampson et al., 2014). However, there are very few true tumour‐specific antigens and the majority of targets are also present at low levels in normal tissue, emphasizing the need to balancing tumour targeting against the risk of unacceptable toxicity. Consequently, the majority of candidate targets under investigation for CAR T‐cell immunotherapy of solid tumours are over‐expressed in tumours, but are present at lower levels in healthy tissues. Examples include HER‐2 (Ahmed et al., 2015, 2010, 2012) and other ErbB family members (Davies et al., 2012; Papa et al., 2015; Zhou et al., 2013), EphA‐2 (Chow et al., 2013), PSMA (Gong et al., 1999; Maher et al., 2002; Zhong et al., 2010), folate receptor‐α (Kandalaft et al., 2012; Song et al., 2011), PSCA (Katari et al., 2011; Morgenroth et al., 2007), the GD2 ganglioside (Krause et al., 1998; Pule et al., 2008), TAG72 (Hombach et al., 1997), carcinoembryonic antigen (Hombach et al., 1999; Sheen et al., 2003), MUC‐1 (Wilkie et al., 2008) and mesothelin (Adusumilli et al., 2014; Beatty et al., 2014; Lanitis et al., 2012).
In the context of haematological malignancy, targets include B‐lineage‐specific antigens such as CD19 (Brentjens et al., 2003), CD20 (Till et al., 2008) and CD23 (Giordano Attianese et al., 2011). Expression of CD19 is a particularly attractive target for CAR T‐cell immunotherapy since it is found in all B‐cell malignancies and at all stages of physiological B‐lineage differentiation but, crucially, is not found on haematopoietic stem cells or other tissues. CD19 specific T‐cells represent the most advanced CAR T‐cell therapy and clinical data arising from clinical trials will be discussed in section 4. More recently, the receptor for thymic stromal lymphopoietin receptor has been advanced as a potential target for some aggressive forms of B‐cell acute lymphoblastic leukaemia (ALL) (Davies and Maher, 2015; Qin et al., 2015). In the context of myeloid malignancy, CD33 (Kenderian et al., 2015; Pizzitola et al., 2014), Lewis Y antigen (Brenner, 2013; Ritchie et al., 2013) and CD123 (Gill et al., 2014; Pizzitola et al., 2014) have all attracted interest. Targets of interest for treatment of Hodgkin's lymphoma include CD30 (Di Stasi et al., 2009) and the receptor for colony‐stimulating factor‐1 (Achkova et al., 2015).
3.2. The problem of target loss/antigen escape
Targeting of a single antigen is associated with a risk of immune escape due to emergence or outgrowth of antigen null tumour cells. This phenomenon has been observed in pre‐clinical work (Anurathapan et al., 2014; Hegde et al., 2013) and, more recently, in clinical studies involving the use of CD19‐targeted CAR T‐cells (Grupp et al., 2013; Maude et al., 2014b). In keeping with this, treatment with pooled CAR T‐cell targeting separate antigens such as HER2 and the IL13Rα (Hegde et al., 2013) or MUC1 and PSCA (Anurathapan et al., 2014) demonstrate enhanced activity in mouse models. Alternatively, it is possible to target two antigens with T‐cells co‐expressing two separate CARs (Hegde et al., 2013) or by a single CAR that has specificity for two antigens owing to expression of two tandemly arranged scFvs coupled to the same signalling endodomain (TanCAR) (Grada et al., 2013). Proof of concept for the TanCAR approach was demonstrated in a model system using a fusion with specificity for both CD19 and HER2. Whilst CAR T‐cells were activated following engagement with either antigen alone, anti‐tumour activity was greater when both targets were expressed (Grada et al., 2013). Strategies such as this may provide a means to minimize the risk of immune escape through antigen loss.
3.3. Universal targeting systems
Pre‐clinical and clinical development of individual CAR T‐cell approaches is a time consuming and costly process. In each case, there is a need to optimize the targeting system and then generate clinical grade batches of gene transfer vector and master cell banks as appropriate. Owing to this complexity, there has been increasing interest in the development of universal targeting CAR T‐cell or natural killer (NK) cell systems that may be combined with clinical available targeting moieties such as monoclonal antibodies (e.g. cetuximab, trastuzumab or rituximab). Illustrating this concept, CARs may be targeted against biotin (using avidin) (Urbanska et al., 2012) or fluorescein (using an appropriate scFv) (Tamada et al., 2012). This enables their use in conjunction with antibodies that, respectively, have been coupled to biotin or fluorescein (used routinely in retinal angiography). An alternative universal targeting approach exploits the natural ability of Fcγ receptor III (CD16)‐expressing cells such as NK cells to mediate antibody‐dependent cell mediated cytotoxicity. Building on this principle, a high affinity CD16 variant has been coupled in series to 4‐1BB and CD3ζ to create a second generation CAR that may be used in combination with a similar spectrum of monoclonal antibodies (Kudo et al., 2014).
4. Clinical studies of CAR T‐cell immunotherapy
Over 75 clinical trials investigating CAR T‐cell therapies are currently listed on clinicaltrials.gov.uk (search date 03/08/15). The majority of studies involve the use of CD19‐targeted CARs targeted against B‐cell malignancy, although several trials involving solid tumours are also ongoing. Published data from trials targeting both haematological and solid malignancies will be discussed briefly in the following sections and are summarized in 1, 2, respectively.
Table 1.
Published clinical experience of CAR‐based cancer immunotherapy of haematologic malignancies.
| Institution | Identifier | Disease | Target | CAR signalling | Number patients | Cond. | Dose | Response | CRS | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| NIH | NCT01593696 | ALL or B‐NHL | CD19 | CD28‐CD3ζ | 20 ALL1 B‐NHL | CF | 1–3 × 106/kg | CR 14SD 3PD 4 | Yes | Lee et al. (2015) |
| UPenn | NCT01626495 NCT01029366 | ALL | CD19 | 41BB‐CD3ζ | 30 | None or PC | 0.76–20.6 × 106/kgsplit over 1–3 days | CR 27PD 3 | Yes | Maude et al. (2014b) |
| UPenn | NCT01626495 | ALL | CD19 | 41BB‐CD3ζ | 2 | CE (n = 1) | 1.4–12 × 106/kg | CR 2 | Yes | Grupp et al. (2013) |
| MSKCC | NCT01044069 | ALL | CD19 | CD28‐CD3ζ | 16 | C | 1.5–3 × 106/kgsplit over 2 days | CR 14PD 2 | Yes | Davila et al. (2014) |
| MSKCC | NCT01044069 | ALL | CD19 | CD28‐CD3ζ | 5 | C | 1.5–3 × 106/kgsplit over 2 days | CR 5 | Yes | Brentjens et al. (2013) |
| MSKCC | NCT00466531 (CLL)NCT01044069 (ALL) | ALL and CLL | CD19 | CD28‐CD3ζ | 8 CLL2 ALL | C | 0.4–3 × 107/kg | CR 1PR 1SD 2PD 4NE 2 | Yes | Brentjens et al. (2011) |
| UPenn | NCT01029366 | CLL | CD19 | 41BB‐CD3ζ | 3 | BR or CP | 0.146–16 × 106/kg split over 3 days | CR 2PR 1 | Yes | Porter et al. (2011) Kalos et al. (2011) |
| NIH | NCT00924326 | CLL, F‐NHL and SMZL | CD19 | CD28‐CD3ζ | 4 CLL4 F‐NHL1 SMZL | CF | 0.5–5.5 × 107/kg | CR 1PR 6SD 1NE 1 | Yes | Kochenderfer et al. (2012) |
| NIH | NCT00924326 | CLL, DLBCL and indolent cell lymphoma | CD19 | CD28‐CD3ζ | 4 CLL9 DLBCL2 indolent lymphoma | CF | 1 × 106/kg | CR 8PR 4SD 1NE 2 | Yes | Kochenderfer et al. (2015) |
| NIH | NCT01087294 | CLL, DLBCL and MCL | CD19 | CD28‐CD3ζ | 4 CLLDLBCL 2MCL 4 | None, allo HSCT | 1–10 × 106/kg | CR 1PR 1SD 6PD 2 | Yes | Kochenderfer et al. (2013) |
| NIH | NCT00840853 | ALL and CLL | CD19 | CD28‐CD3ζ | 4 ALL4 CLL | None, allo HSCT | Dose escalation from 1.5 × 107/m2, 4.5 × 107/m2 and 1.2 × 108/m2 | CR 3PR 1SD 1PD 3 | Yes | Cruz et al. (2013) |
| NIH | SLL and DLBCL | CD19 | CD28‐CD3ζ and CD3ζ | 1 SLL5 DLBCL | None | 0.2–2 × 108/m2 per dose (1–2 doses) | SD 2PD 4 | No | Savoldo et al. (2011) | |
| Peter MacCallum Cancer Centre | CTX 08‐0002 | AML | LeY | CD28‐CD3ζ | 4 | F | Up to 1.3 × 109 total | 2 PR2 SD1 NE | Yes | Ritchie et al. (2013) |
| NIH | NCT00621452 | F‐NHL and MCL | CD20 | CD28‐41BB‐CD3ζ | 3 MCL1 NHL | C | Dose escalation of 3 infusions108, 109, and 3.3 × 109 cells/m2 | 1 PR3 NE | Yes | Till et al. (2012) |
| Chinese PLA General Hospital | NCT01735604 | DLBCL | CD20 | 41BB‐CD3ζ | 7 | Various | 18–55 × 108 cells total, split over 5 days | 1 CR4 PR1 PD1 NE | Yes | Wang et al. (2014b) |
Abbreviations: ALL – acute lymphoblastic leukaemia; allo HSCT – allogeneic haematopoietic stem cell transplant; AML – acute myeloid leukaemia; B – bendamustine; B‐NHL – B‐cell non‐Hodgkin's lymphoma; BR – bendamustine + rituximab; C – cyclophosphamide; CAIX – carbonic anhydrase IX; CE – cyclophosphamide and etoposide; CEA – carcinoembryonic antigen; CF – cyclophosphamide and fludarabine‐based lymphodepletion; CLL – chronic lymphocytic leukaemia; Cond. – conditioning therapy; CP – cyclophosphamide and pentostatin; CR – complete remission; CRS – cytokine release syndrome; DLBCL – diffuse large B‐cell lymphoma; F – fludarabine; F‐NHL – follicular NHL; LeY – Lewis Y antigen; MCL – mantle cell lymphoma; MSKCC – Memorial Sloan‐Kettering Cancer Center; MPM – malignant pleural mesothelioma; NE – non evaluable; Nec. – necrosis; NIH – National Institutes of Health; PC – Physician's choice; PD – progressive disease; PDAC – pancreatic adenocarcinoma; PR – partial remission; SD – stable disease; SLL – small lymphocytic lymphoma; SMZL – splenic marginal zone lymphoma; UPenn – University of Pennsylvania.
Table 2.
Published clinical experience of CAR‐based cancer immunotherapy of solid tumours.
| Institution | Identifier | Disease | Target | CAR signalling | Number patients | Cond. | Dose | Response | CRS | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| NIH | Ovarian cancer | FRα | FcRγ | 14 | None | 3–169 × 109 in 1–3 doses | None | Yes | Kershaw et al. (2006) | |
| NIH | BB‐IND 9149 (FDA) and 0006‐402 (NIH) | Neuroblastoma | CD171 | CD3ζ | 6 | None | 0.1–11 × 109/m2 in 1–3 doses | PR 1 | No | Park et al. (2007) |
| NIH | NCT00085930 | Neuroblastoma | GD2 | CD3ζ | 19 | None | 2–20 × 107/m2 | CR 3PR 1SD 1NE 8PD 6 | No | Louis et al. (2011) and Pule et al. (2008) |
| Erasmus University Medical Center – Daniel den Hoed Cancer Center | DDHK97‐29/P00.0040C | Renal cell carcinoma | CAIX | FcRγ | 12 | None | 0.2–2.1 × 109 cells in up to 10 infusions | None | Yes | Lamers et al. (2013), Lamers et al. (2006) and Lamers et al. (2011) |
| NIH | NCI‐09‐C‐0041 | Colon carcinoma | HER2 | CD28‐41BB‐CD3ζ | 1 | CF | 7.9 × 109 (one dose) | None | Yes, fatal. | Morgan et al. (2010) |
| UPenn | NCT01355965 | MPM and PDAC | Mesothelin | 41BB‐CD3ζ | 2 | None | 1 × 108 to 1 × 109 per dose | PR 1 | Yes | Beatty et al. (2014) |
| NIH | NCT00902044 | Sarcoma | HER | CD28‐CD3ζ | 19 | None | 1 × 104 to 1 × 108/m2 | SD 4 | No | Ahmed et al. (2015) |
| NIH | NCT01373047 | Liver metastases | CEA | CD28‐CD3ζ | 6 | None | Up to 3 × 1010 over 3 doses | SD 1 | No | Katz et al. (2015) and Saied et al. (2014) |
| NIH | NCT02208362 | Glioblastoma | IL13Rα2 | 41BB‐CD3ζ | 3 | None | Up to 12 infusions of 108 T‐cells | Transient response in 2 patients | No | Brown et al. (2015) |
Abbreviations: ALL – acute lymphoblastic leukaemia; allo HSCT – allogeneic haematopoietic stem cell transplant; AML – acute myeloid leukaemia; B – bendamustine; B‐NHL – B‐cell non‐Hodgkin's lymphoma; BR – bendamustine + rituximab; C – cyclophosphamide; CAIX – carbonic anhydrase IX; CE – cyclophosphamide and etoposide; CEA – carcinoembryonic antigen; CF – cyclophosphamide and fludarabine‐based lymphodepletion; CLL – chronic lymphocytic leukaemia; Cond. – conditioning therapy; CP – cyclophosphamide and pentostatin; CR – complete remission; CRS – cytokine release syndrome; DLBCL – diffuse large B‐cell lymphoma; F – fludarabine; F‐NHL – follicular NHL; LeY – Lewis Y antigen; MCL – mantle cell lymphoma; MSKCC – Memorial Sloan‐Kettering Cancer Center; MPM – malignant pleural mesothelioma; NE – non evaluable; Nec. – necrosis; NIH – National Institutes of Health; PC – Physician's choice; PD – progressive disease; PDAC – pancreatic adenocarcinoma; PR – partial remission; SD – stable disease; SLL – small lymphocytic lymphoma; SMZL – splenic marginal zone lymphoma; UPenn – University of Pennsylvania.
4.1. CAR T‐cell immunotherapy of haematological malignancy
Most published trials of CAR T‐cell immunotherapy of haematological malignancy involve CD19‐targeted therapies. CD19 is a B‐cell surface protein expressed throughout B‐cell development and is expressed on nearly all B‐cell malignancies. This ubiquitous expression, and the fact that B‐cell aplasia as a result of toxicity to healthy B‐cells is both tolerable and treatable makes CD19 an attractive target for immunotherapy. Initial trials using CD19‐directed CAR T‐cells were conducted on small numbers of patients but were noteworthy for striking examples of complete remission (CR) induction in patients with refractory chronic lymphocytic leukaemia (CLL) and ALL (Brentjens et al., 2011; Grupp et al., 2013; Kalos et al., 2011; Porter et al., 2011). These studies all involve the use of second generation CAR constructs containing either CD28 or 4‐1BB signalling endodomains, coupled in series to CD3ζ.
4.1.1. CD19‐targeted CAR T‐cells in ALL
Larger studies have been published by groups at Memorial Sloan Kettering Cancer Center (MSKCC), University of Pennsylvania (UPenn) and the National Cancer Institute (NCI) and all highlight the feasibility of attainment of striking CR rates in patients with B‐cell ALL (Davila et al., 2014; Lee et al., 2015; Maude et al., 2014b).
The first report of a patient with ALL to be treated with CD19 CAR T‐cells was in 2011 (Brentjens et al., 2011). In 2013, the same group at MSKCC published an update, which indicated that 5 patients with ALL had achieved CR (Brentjens et al., 2013). One patient subsequently relapsed, an event that may have been linked to the use of high dose steroids to treat cytokine release syndrome (Brentjens et al., 2013). These patients are included in a recent update, indicating an 88% CR rate in a series of 16 ALL patients. To consolidate this remission, most of the patients went on to receive an allogeneic haematopoietic stem cell transplant (HSCT) (Davila et al., 2014). More recent data presented at the 2015 annual conference of the American Society of Clinical Oncology (ASCO) reports that 33 patients have been treated to date at MSKCC in a Phase I trial with 29/32 patients evaluable for response achieving complete remission, a CR rate of 91% (Park et al., 2015). CR rates appear to be associated with disease burden at the time of treatment, as 100% patients with minimal residual disease (MRD) achieved CR compared to 81% of those with >5% blasts at the time of CAR T‐cell infusion. However, such clinical success in refractory disease where a large percentage of patients had undergone ≥3 prior lines of therapy is remarkable.
In parallel, similar success in the clinic has been seen at UPenn. An early report demonstrated attainment of CR in two children with relapsed and highly refractory ALL following treatment with CD19‐CAR T‐cells. One of these patients had ongoing remission although the other patient relapsed after two months, with blast cells that no longer expressed CD19 (Grupp et al., 2013). A more comprehensive report followed, indicating the achievement of a 90% CR rate (27/30) in a patient population, some of whom had failed prior previous HSCT (Maude et al., 2014b). Nineteen of these patients remained in CR (durable to 24 months) and seven patients subsequently relapsed. Notably, three of the relapses were characterized by the emergence of CD19 null disease, demonstrating the intense selective pressure applied by the CAR T‐cells in‐vivo (Maude et al., 2014b). A further update at the 2014 annual meeting of the American Society of Hematology showed a 92% CR rate attained in 39 paediatric patients with ALL (Maude et al., 2015). In further support of these findings, Lee et al. at the NCI recently reported that their CD19 CAR T‐cell approach had achieved CR in 14/20 children and young adults with ALL (Lee et al., 2015). Such high response rates are unprecedented for a novel agent in first‐in‐human clinical trials of otherwise untreatable malignancy. These findings are all the more remarkable considering the differences in CAR targeting moiety, hinge/spacer/transmembrane domain and co‐stimulatory module used by the three groups (CD28 at MSKCC and NCI; 4‐1BB at UPenn; described further in Maher, 2014). Although differences in disease burden, prior treatment history and conditioning regimen make comparison of data between different centres challenging, there is an indication that CD19 CAR T‐cells containing the 41BB costimulatory domain persist longer than their CD28 counterparts. CD19‐28z CAR T‐cells typically persist for 1–3months post‐infusion whereas there are reports of CD19‐41BBz CAR T‐cells persisting for up to 2 years Whether these differences truly reflect differences in CAR design or are due to other reasons (more paediatric patients were treated using the 41BB CAR for instance) (Maude et al., 2015) remains to be seen. Nonetheless, durable responses have been observed using both CARs and induction of a CR even in the absence of sustained T‐cell survival provides a bridge to allogeneic stem cell transplantation.
4.1.2. CD19‐targeted CAR T‐cells in CLL
Initial clinical experience targeting CLL with CD19‐targeted CAR T‐cells was reported in 2011 in a pilot study involving 3 patients (Kalos et al., 2011), one of whom was described in detail in another report (Porter et al., 2011). Long‐term persistence of CAR T‐cells was observed in the absence of IL‐2 cytokine support and impressive anti‐tumour activity was observed, even at a low T‐cell dose (Porter et al., 2011). Patients had received conditioning therapy using lymphodepleting agents (cyclophosphamide and pentostatin, bendamustine alone, or in combination with rituximab). In the same year, Brentjens et al. reported results involving 8 patients with CLL and demonstrated the importance of conditioning therapy. Patients treated with CAR T‐cells without prior cyclophosphamide all had progressive disease, with limited evidence of T‐cell persistence in‐vivo. By contrast, enhanced persistence and efficacy of CD19 CAR T‐cells was observed in patients pre‐treated with cyclophosphamide, corresponding to stable disease (SD) in 2 patients and 1 partial remission (PR) seen in 4 patients treated (Brentjens et al., 2011). Other trials have also reported efficacy of CD19‐targeted CAR T‐cells in CLL patients (Kochenderfer et al., 2012, 2015). Response rates are generally lower in CLL compared to ALL but The UPenn group has nonetheless reported CR rates of 45% from over 45 patients treated, with some remissions extending beyond 4 years (Maude et al., 2015). In addition, two trials have investigated the use of allogeneic donor‐derived CAR T‐cells to treat patients with CLL (Cruz et al., 2013; Kochenderfer et al., 2013). These demonstrated that CAR T‐cells could be administered following allogeneic HSCT, demonstrating efficacy without exacerbation of graft versus host disease (GvHD).
4.1.3. CD19‐targeted CAR T‐cells in B‐cell non‐Hodgkin's lymphomas (NHL)
Kochenderfer et al. reported efficacy in 6/8 patients with B‐cell malignancy of whom 4 had B‐cell NHL (follicular NHL, n = 3; splenic marginal zone lymphoma (SMZL), n = 1). However, significant toxicity was also observed, accompanied by cytokine release (Kochenderfer et al., 2012). A subsequent trial used an amended protocol, including a lower dose of CAR T‐cells given without IL‐2 cytokine support, and was the first to demonstrate efficacy of CD19‐specific CAR T‐cells against diffuse large B‐cell lymphomas (n = 5), in addition to primary mediastinal B‐cell lymphoma (n = 4), CLL (n = 4), SMZL (n = 1) and one other low‐grade NHL. Overall, 12/13 evaluable patients showed a partial or complete response to therapy in this study (Kochenderfer et al., 2015). Recent data from ASCO 2015 highlights a 67% overall response rate in a Phase IIa trial against non‐Hodgkins lymphoma, with a 100% response rate in 6 patients with follicular lymphoma (FL) (Schuster et al., 2015).
4.1.4. Additional clinical studies of CAR T‐cells in haematological malignancy
Four patients with relapsed indolent B‐cell and mantle cell lymphomas were treated with third generation CD20‐specific CAR T‐cells containing a CD28/4‐1BB/CD3ζ endodomain. The target cell dose was met for 3 patients who received 3 doses of CAR T‐cells in a dose escalation manner following lymphodepletion (Till et al., 2012). Of these, one patient had a PR and two patients without evaluable disease remained disease free for 12 and 24 months (Till et al., 2012). Wang et al. also treated patients with diffuse large B‐cell lymphoma with second‐generation anti‐CD20 CAR T‐cells containing a 4‐1BB co‐stimulatory endodomain and achieved 1 CR and several PR (Wang et al., 2014b). A further study showed some partial responses against AML targeting the LeY antigen (Ritchie et al., 2013). In addition, several currently active trials of CAR T‐cell immunotherapy are targeted again22 and CD30 in lymphoma. Recently positive Phase 1 data as been reported by Cellular Biomedicine Group Incorporatest CDd (Shanghai, China) with PR in 2 and SD observed in 3 of a total of 7 patients with refractory Hodgkin's disease who were treated (NCT02259556; Clinicaltrials.gov; http://globenewswire.com/news‐release/2015/05/22/738528/10135712/en/Cellular‐Biomedicine‐Group‐Announces‐Positive‐Phase‐I‐Results‐From‐CAR‐T‐CD30‐Immuno‐Oncology‐Clinical‐Development‐Program.html, accessed 8/8/2015).
4.2. CAR T‐cell immunotherapy of solid tumours
Solid tumours were the first to be treated with CAR T‐cells. However, clinical trials using CAR T‐cell therapy against solid tumours have not achieved the same success seen in haematological cancers. Initial clinical studies used first generation CAR T‐cells targeted to folate receptor‐α in ovarian cancer (Kershaw et al., 2006) and the L1‐cell adhesion molecule CD171 in neuroblastoma (Park et al., 2007). No clinical efficacy was observed besides one PR in a patient with neuroblastoma, in whom chemotherapy was also given after adoptive transfer of T‐cells (Park et al., 2007). Furthermore, T‐cells demonstrated limited persistence in‐vivo and dropped to low or undetectable levels within days to weeks after infusion (Kershaw et al., 2006; Park et al., 2007). In order to overcome this, Epstein–Barr virus (EBV) specific cytotoxic T‐cells were transduced with a CAR specific for the ganglioside GD2 that is over‐expressed in neuroblastoma. The rationale was that engagement of native T‐cell receptor within CAR T‐cells by EBV infected B‐cells would provide co‐stimulation in‐vivo. Patients were treated with both autologous CAR T‐cells and EBV‐CAR T‐cells to directly compare their persistence after infusion. A first report of 11 patients indicated an initial survival advantage of EBV CAR T‐cells but by 6 weeks, levels of both were low or undetectable (Pule et al., 2008). Follow‐up of a larger cohort indicated long‐term persistence (up to 192 weeks), which was associated with clinical benefit including 3 complete remissions in patients with active disease at the time of treatment (Louis et al., 2011). Intriguingly, T‐cell persistence correlated with the presence of both CD4+ and memory T‐cells in the cell product. In keeping with this, there has been considerable recent interest in the expression of CARs in defined combinations of T‐cell subsets (Riddell et al., 2014), or in potentially long‐lived populations such as memory stem cells (TSCM) (Cieri et al., 2013; Gattinoni and Restifo, 2013).
Safety concerns were also raised in initial trials against targeting CAR T‐cells to antigens that are overexpressed in tumours yet also present in normal tissue at low levels. Carbonic anhydrase‐IV is frequently overexpressed in renal carcinomas of the clear cell subtype and was targeted in a trial using first generation CAR T‐cells (Lamers et al., 2013, 2006, 2011). However, significant hepatotoxicity was observed in initially treated patients, requiring discontinuation of therapy and/or corticosteroid treatment. A liver biopsy revealed CAIX expression in bile duct epithelial cells and, in an attempt to circumvent on‐target liver toxicity, the protocol was amended to include pre‐treatment with a low dose of CAIX blocking antibody in future cohorts. This abrogated liver toxicity although no clinical anti‐tumour activity has been observed in any patients (Lamers et al., 2013).
Fatal on‐target off‐tumour toxicity was observed in a patient treated with third generation HER‐2 CAR T‐cells, where recognition of low levels of HER‐2 in lung endothelium (Heslop, 2010) and/or epithelial cells by CAR T‐cells led to respiratory and multi‐organ failure, cytokine release syndrome and death (Morgan et al., 2010). This event occurred rapidly following a single high dose (7.9 × 109) of CAR T‐cells comprising an scFv derived from trastuzumab, coupled to a fused CD28/4‐1BB/CD3ζ endodomain. However, HER‐2 has been targeted safely in a subsequent trial involving patients with sarcomas that express this target. A cautious dose escalation protocol was followed, starting at a dose of 1 × 104 CAR T‐cells/m2, rising to 1 × 108/m2. Patients did not receive prior lymphodepletion or IL‐2 therapy. The CAR was distinct in that it contained scFv derived from the FRP5 antibody (binds a distinct epitope to trastuzumab), coupled to a second generation CD28/CD3ζ endodomain (Ahmed et al., 2015). In this trial, 19 patients were treated with no adverse effects other than a single febrile episode in the highest dose‐level cohort, which responded to ibuprofen. There was no evidence of expansion of CAR T‐cells following infusion. Instead CAR T‐cells declined in peripheral blood after just 3 h. Nonetheless, long‐term persistence of these cells could be demonstrated for up to 18 months at doses higher than 1 × 106 CAR T‐cells/m2. Furthermore, CAR T‐cells were identified in resected tumours from two patients despite their absence in peripheral blood at that time, suggesting homing to and/or expansion within tumour sites. Although stable disease was reported in 4 patients, no other clinical responses were seen (Ahmed et al., 2015). However, this trial was important in demonstrating safe systemic delivery of HER2‐specific CAR T‐cells, especially given the previous fatality.
In an attempt to mitigate on‐target off‐tumour toxicity, Beatty et al. recently described the use of mRNA electroporated CAR T‐cells in a pilot study of two patients with malignant pleural mesothelioma (MPM) and pancreatic ductal adenocarcinoma (PDAC) (Beatty et al., 2014). By this means, T‐cells were engineered to achieve transient expression of a mesothelin‐specific second‐generation CAR containing a 4‐1BB/CD3ζ endodomain. Patients were treated with differing dosing regimens ranging from a total of three intravenous injections over 5 weeks (MPM) to three intravenous doses per week for 3 weeks, followed by 2 intra‐tumoural injections (PDAC). A partial response was observed in the patient with mesothelioma following the third infusion before disease subsequently progressed, whereas stable disease after 3 weeks of treatment was not sustained and disease progressed in the patient with PDAC. Safety was deemed acceptable with transient cytokine release syndrome after the third T‐cell infusion in the patient with MDM (Beatty et al., 2014). However, anaphylactic shock and cardiac arrest developed in the patient with MPM upon reinitiating treatment after a 4‐week treatment break (Beatty et al., 2014; Maus et al., 2013). This was due to induction of an IgE isotype anti‐murine antibody response to the CAR. Based on this, the protocols of ongoing clinical trials have been modified to prohibit infusion breaks of more than 10 days in an attempt to bias against IgE class switching. More recently, stable expression of the same CAR has been achieved in lentivirus‐transduced T‐cells, which have been infused safely in patients with PDAC (n = 6), MPM (n = 2) and epithelial ovarian cancer (n = 2). Patients did not receive lymphodepletion. An efficacy signal is suggested by SD in limited numbers of patients (Tanyi et al., 2015; G.B et al., 2015).
Results from two further clinical trials were published in 2015. Katz et al. treated 6 patients with liver metastases by intrahepatic delivery of CEA‐targeted CAR T‐cells through percutaneous hepatic artery infusions (Katz et al., 2015; Saied et al., 2014). Liver biopsies revealed necrosis or fibrosis in 4 of 6 patients and one patient had SD for 23 months. Local delivery was also used in the treatment of three patients with glioblastoma with anti‐IL‐13Rα2 CAR T‐cells. Patients received up to 12 local infusions with a transient response observed in 2 patients. Treatment was also well tolerated with only temporary and manageable CNS inflammation (Brown et al., 2015).
Whilst solid tumours have largely proven refractory to CAR T‐cell therapy to date, there is encouraging data to support further clinical development. Evidence of T‐cell persistence, localization within tumours, and some evidence of anti‐tumour activity have all been observed, with mostly acceptable safety profiles. Nonetheless, it is clear that there are still challenges in treating solid malignancies. These include refining systems to encourage trafficking to tumour deposits, enhanced persistence, intra‐tumoural penetration and improved effector function. These particular challenges will be discussed in section 6.
5. Toxicity induced by CAR T‐cell immunotherapy
Immunotherapy using CAR T‐cells has significant toxic potential, which broadly may be considered in four major categories, namely tumour lysis syndrome, cytokine release syndrome, neurotoxicity and on‐target off tumour toxicity. Tumour lysis syndrome comprises a combination of metabolic disturbances, including elevated serum levels of uric acid, potassium and phosphate, accompanied by hypocalcaemia and sometimes progressing to renal failure. It may occur following the rapid elimination of large numbers of tumour cells, most notably in the context of haematological malignancy and can present surprisingly late in some patients following delivery of CAR T‐cells (Kochenderfer et al., 2013; Porter et al., 2011), presumably as CAR T‐cells expand in‐vivo and cause tumour destruction. The latter three categories are each considered in turn, together with strategies that may be used to mitigate such toxicity.
5.1. Cytokine release syndrome
Cytokine release syndrome (CRS) is the most frequent life‐threatening event induced by CAR T‐cell immunotherapy (Maude et al., 2014a). This term refers to the release of a cascade of cytokines from several leukocyte populations, most notably T‐cells and cells of the mononuclear phagocyte lineage. High circulating levels of many pro‐inflammatory cytokines may be detected, including tumour necrosis factor‐α (TNF‐α), interferon‐γ and IL‐6 (Grupp et al., 2013). In some cases, macrophage activation syndrome accompanies CRS. This is noteworthy since pre‐clinical models of CAR T‐cell induced CRS implicate macrophage activation as a key component of disease pathogenesis (van der Stegen et al., 2013).
Emerging evidence suggests that severity of CRS depends upon disease burden (Brentjens et al., 2013; Maude et al., 2014b). Since CRS can progress rapidly, close monitoring of patients is required. This is particularly the case in the 24–48 h period after the infusion of cells. However, delayed occurrence of CRS may also be seen, particularly when cells undergo substantial expansion in‐vivo from small starting numbers. Management of CRS poses a challenging dilemma since some degree of cytokine release accompanies T‐cell activation and effector activity, while therapeutic blockade of this process may entail the use of one or more immunosuppressive agents. On the other hand, severe CRS can be rapidly lethal, as has occurred in one patient treated with HER2 re‐targeted CAR T‐cells (see section 4.2) (Morgan et al., 2010). Recently, both diagnostic and grading systems have been proposed, in addition to treatment algorithms for this syndrome (Davila et al., 2014; Lee et al., 2014). Serum C‐reactive protein (CRP) has been identified a potential biomarker for CRS, supplementing clinical parameters to facilitate the stratification of patients that are likely to need more intensive treatment. Depending upon severity, management can involve symptomatic treatment, fluid replacement, oxygen and vasopressor support, and immunosuppression with agents such as the IL‐6 receptor α‐blocking antibody, tocilizumab and/or corticosteroids.
5.2. Neurotoxicity
Neurotoxicity is another serious potential toxicity arising from CAR T‐cell therapy and has been observed in several patients treated with CD19‐targeted CAR T‐cells (Davila et al., 2014; Lee et al., 2015; Maude et al., 2014b) and in a patient with glioblastoma treated locally with IL13Rα2‐targeted CAR T‐cells (Brown et al., 2015). Symptoms of neurotoxicity include visual hallucination, delirium, dysphasia and epilepsy or seizures and the cause of this toxicity is not yet known. Although CD19 CAR T‐cells have been found in the cerebral spinal fluid (CSF) of most patients treated with CD19 CAR T‐cells in one trial at UPenn (regardless of encephalopathy), all 6/21 patients who had neurotoxicity had higher concentrations of CSF CAR T‐cells (Lee et al., 2015). This was irrespective of whether there CNS leukaemic blasts were present. In contrast, not all patients demonstrating neurotoxicity had evidence of CAR T‐cells in the CSF in another trial (Davila et al., 2014), despite clinically evident delirium at the time of CSF collection. As neurotoxicity is also observed in patients treated with blinatumomab, a T‐cell activating bispecific antibody that engages both CD3 on T‐cells and CD19 on tumour cells (Topp et al., 2014), it is speculated that toxicity arises from generalized T‐cell mediated inflammation rather than targeted CAR T‐cell attack of CNS tissue. Whilst neurotoxicity has been fully reversible and self‐limiting in two large trials to date (Davila et al., 2014; Lee et al., 2015) it is a clear concern for CAR T‐cell therapy, particularly as it does not correlate with the severity of CRS and so is harder to predict. Understanding the mechanisms behind neurological toxicities will be critical for the development of safer CAR T‐cell therapy and for more effective management of these adverse effects.
5.3. On‐target off‐tumour toxicity
On‐target toxicity is best illustrated by the propensity of CD19‐targeted CAR T‐cells to cause B‐cell aplasia, with resultant hypogammaglobulinaemia. In the context of otherwise untreatable B‐cell malignancy, such toxicity is deemed acceptable since it can be mitigated by implementation of intravenous or subcutaneous immunoglobulin replacement therapy (Maher, 2012). Another example is the hepatotoxicity induced by carbonic anhydrase IX re‐targeted CAR T‐cells (Lamers et al., 2006), which is discussed more fully in section 4.2 above. On‐target off‐tumour toxicity is a particular concern with CAR T‐cell immunotherapy since most targets are self‐antigens that are expressed to some degree in normal tissues. Furthermore, CAR T‐cells are activated by a far lower level of antigen than is required for such a response to a monoclonal antibody‐based therapy (Ahmed et al., 2009; Watanabe et al., 2015).
5.4. The use of suicide genes in CAR T‐cell immunotherapy
In light of the significant risk of toxicity associated with CAR‐based immunotherapy, it is logical to consider the co‐expression of a suicide gene in such engineered T‐cells. Most clinical experience has been obtained with the herpes simplex virus thymidine kinase gene, which renders target cells susceptible to ganciclovir‐mediated elimination. However, this approach is limited by the immunogenicity of this enzyme (Traversari et al., 2007). More recently cell depletion systems include truncated EGFR (Wang et al., 2011), CD20 (Philip et al., 2014) or a CD34/CD20 epitope tag (RQR8) (Philip et al., 2014), proteins that render CAR T‐cells susceptible to elimination by cetuximab or rituximab, respectively.
Perhaps the most elegant and effective solution is the inducible caspase 9 (iCasp9) system, comprising a fusion of human caspase 9 to a 12kD human FK506 binding protein moiety (Straathof et al., 2005). This fusion protein is activated by the small molecule dimerizer AP1903, leading to rapid apoptosis and clearance of T‐cells (Di Stasi et al., 2011). Efficacy and safety of iCasp9 has been demonstrated in patients who received donor‐derived T‐cells to promote immune reconstitution following haploidentical HSCT. In the event that GvHD occurred, AP1903 administration resulted in elimination of iCasp9‐expressing T‐cells within 30 min (Di Stasi et al., 2011, 2014, 2015), accompanied by decline in cytokinaemia. This response has been replicated in pre‐clinical models using CAR T‐cells (Budde et al., 2013).
6. Particular challenges hindering CAR T‐cell immunotherapy of solid tumours
Much of the excitement surrounding CAR T‐cell immunotherapy stems from its profound therapeutic efficacy in B‐cell malignancy, notably ALL. By contrast, solid tumours have proved much more challenging to treat. The paucity of tumour‐specific targets represents a fundamental hurdle in this regard, meaning that there is a significant risk of on‐target toxicity accompanying tumour regression. Moreover, tumours create a hostile microenvironment characterized by abnormal vasculature, areas of hypoxia and dysregulated metabolism that can hinder T‐cell migration, survival and effector function. The desmoplastic stromal reaction induced by many cancers imposes an additional barrier to T‐cell infiltration and can actively inhibit the anti‐tumour immune response (Watt and Kocher, 2013). These issues, coupled with the panoply of immunosuppressive factors within the tumour microenvironment (Wu et al., 2015) can render T‐cells unresponsive. A number of these challenges are discussed further below.
6.1. Improving the precision of tumour targeting using CAR T‐cells
Very few targets found in solid tumours are only expressed by transformed or dispensable cell types. Consequently, target selection is a key challenge to safe targeting. Dual targeting of two or more tumour‐associated targets represents one approach that may be used to enhance discrimination by CAR T‐cells between abnormal and healthy tissue. One alternative approach that may assist with this entails the co‐expression of inhibitory CARs (iCARs) together with their activating counterparts. Inhibitory CARs may be directed against molecules that are found in healthy organs but which are down‐regulated or lost in tumours. Inhibitory signalling may be provided by naturally occurring checkpoint molecule, such as CTLA‐4, PD‐1 (Fedorov et al., 2013), BTLA‐4 or LAG‐3 (Chicaybam and Bonamino, 2015) or a variety of phosphatases that naturally regulate T‐cell signalling. There is considerable interest in the development of systems to balance activating and inhibitory signalling so that T‐cells are not activated outside of the tumour microenvironment.
6.2. The need to increase tumour‐specific T‐cell homing
A second major limitation to the effectiveness of adoptive T‐cell immunotherapy is poor trafficking of CAR T‐cells to tumour deposits. In some malignancies however, loco‐regional disease is the primary cause of disease‐related morbidity and mortality, providing a strong rationale for regional T‐cell delivery. An example of this principle is squamous cell carcinoma of head and neck. Intra‐tumoural delivery of ErbB re‐targeted CAR T‐cells is currently under Phase 1 evaluation in this tumour type (clinicaltrials.gov; NCT01818323) (van Schalkwyk et al., 2013). In a similar vein, epithelial ovarian cancer (EOC) and malignant pleural mesothelioma (MPM) may both be amenable to regional delivery of CAR T‐cells, owing to their propensity for localized dissemination within the peritoneal and pleural cavities, respectively (Adusumilli et al., 2014; Parente‐Pereira et al., 2013). A Phase I trial evaluating intra‐pleural delivery (Adusumilli et al., 2014) is currently ongoing at MSKCC (clinicaltrials.gov; NCT02414269) while Phase II studies using intra‐peritoneal/intra‐pleural delivery of CAR T‐cells are planned for MPM and EOC/MPM, respectively (Papa et al., 2015). Recently, three patients with recurrent glioblastoma were locally treated with IL13Rα2 targeted CAR T‐cells (Brown et al., 2015). Intracranial delivery was well tolerated over a maximum of 12 infusions, with evidence of a transient anti‐tumour response, as determined by a reduction in IL13Rα2 positive tumour cells following T‐cell delivery and an increase in necrotic tumour volume by MRI.
In general however, metastatic disease is the main cause of morbidity and death from solid tumours, mandating the systemic (intravenous) delivery of T‐cells. When administered in this manner, T‐cells become physically stuck in the lungs for several hours, after which they re‐distribute mainly to the liver and spleen (Kershaw et al., 2006; Parente‐Pereira et al., 2011). Whilst some migration of CAR T‐cells to tumour sites has been observed in both haematological (Brentjens et al., 2011; Kalos et al., 2011; Savoldo et al., 2011) and solid tumours (e.g. ovarian, pancreatic and mesothelioma) (Ahmed et al., 2015; Tanyi et al., 2015), there is a clear need to improve the efficiency of T‐cell trafficking to tumour deposits.
Several experimental strategies are under investigation to address this need. Many rely on the fact that chemokine over‐production is prevalent in many solid tumours. To exploit, this, CAR T‐cells may be engineered to express a chemokine receptor tailor designed to match the chemokine expression profile of a specific cancer type. Illustrating this, many malignant melanomas secrete CXCL1 and CXCL8. Both of these chemokines bind to the CXCR2 receptor, primarily found on neutrophils and monocytes and which mediates migration to sites of inflammation. While CXCR2 is not widely expressed by T‐cells, transgenic pmel‐1 T‐cells (specific for the gp100 antigen) have been engineered to express this receptor and consequently achieve enhanced localization within melanoma tumours (Peng et al., 2010). Similarly, over‐expression of the chemokine (C–C motif) receptor 2 (CCR2) confers responsiveness of CAR T‐cells to CCL2, a chemokine that is over‐produced in several tumours including neuroblastoma, lung cancer and mesothelioma (Asai et al., 2013; Craddock et al., 2010; Moon et al., 2011). To achieve improved intra‐tumoural trafficking, it is also possible to take advantage of endogenously expressed chemokine receptors by CAR T‐cells. For example, CCL5 (also known as RANTES) is a potent T‐cell chemoattractant that binds to CCR1, 3 and 5. When expressed in neuroblastoma cells using an oncolytic adenovirus and delivered in combination with CAR T‐cells, this approach also promotes enhanced intra‐tumoural T‐cell trafficking (Nishio et al., 2014). Low‐dose irradiation treatment or cyclophosphamide has also been shown to increase migration of T‐cells to prostate cancer bone metastases in mice through induction of the potent T‐cell chemoattractant stromal‐derived factor‐1 (CXCL‐12), which binds to CXCR4 that is naturally expressed in some T‐cell subsets (Pinthus et al., 2004).
An additional strategy to increase T‐cell infiltration into tumours relies on normalizing of tumour vasculature, through the use of anti‐angiogenic drugs. The angiogenic switch within solid tumours is regulated by vascular endothelial growth factor (VEGF), which stimulates blood vessel growth by binding to VEGF‐receptor 2 (VEGFR2) on endothelial cells but which also has immunosuppressive properties (Mulligan et al., 2009). Levels of VEGF correlate negatively with the presence of tumour‐infiltrating lymphocytes (TILs) in ovarian cancer (Zhang et al., 2003). However, infiltration of adoptively transferred T‐cells into melanoma tumours may be increased using anti‐VEGF therapy (Shrimali et al., 2010) or B‐Raf inhibition, which leads to down‐regulation of VEGF in both murine and human tumour biopsies (Liu et al., 2013).
6.3. Dealing with the tumour stroma problem
Tumour‐related stroma comprises extracellular matrix (ECM), fibroblasts, stellate cells, pericytes, immune cells and endothelial cells that co‐conspire to provide a pro‐tumourigenic environment (Hanahan and Weinberg, 2011). Dense stroma can constitute a physical barrier to T‐cell infiltration and loosening of the stroma with either collagenase or heparanase can increase T‐cell distribution within the tumour (Salmon et al., 2012) and enhance anti‐tumour activity of CAR T‐cells (Caruana et al., 2015).
Several reports demonstrate that the stroma may sequester T‐cells away from tumour cells (Ene‐Obong et al., 2013; Molon et al., 2011; Salmon et al., 2012; Weishaupt et al., 2007), and this has been linked to the actions of CXCL12, a chemokine released by cancer‐associated fibroblasts (CAFs) in colorectal (Akishima‐Fukasawa et al., 2009) and ovarian cancer (Scotton et al., 2002), and by stellate cells in pancreatic cancer (Ene‐Obong et al., 2013). Numerous approaches have been used to block the CXCL12/CXCR4 pathway in order to increase intra‐tumoural accumulation of T‐cells, such as (i) administration of the CXCR4 inhibitor AMD3100 (Feig et al., 2013; Righi et al., 2011); (ii) expression of a CXCR4 antagonist from an oncolytic vaccinia virus (Gil et al., 2014) and (iii) targeting the source of CXCL12, either through agents to render stellate cells quiescent (Ene‐Obong et al., 2013) or by targeting CAFs directly. Fibroblast‐activation protein α (FAP) is expressed in CAFs found in many malignancies, including breast, colorectal, ovarian and lung cancers (Garin‐Chesa et al., 1990). In addition, it is also expressed by some tumour cells themselves, including by sarcomas (Dohi et al., 2009) and mesothelioma (Schuberth et al., 2013). Selective ablation of FAP+ CAFs in transgenic mice allows immunological control of PDAC tumour growth (Kraman et al., 2010), suggesting that targeting of these cells could also augment CAR T‐cell therapy. Second generation CAR T‐cells targeted against FAP inhibited tumour growth in both MPM and lung cancer models (Schuberth et al., 2013; Wang et al., 2014a). In a further study using A549 lung adenocarcinoma xenografts, FAP‐targeted CAR T‐cells exhibited anti‐tumour activity, in a manner that was enhanced by combination with EphA2 CAR T‐cells (Kakarla et al., 2013). Whilst no severe toxicity was observed in these animal models, adoptive transfer of FAP‐specific CAR T‐cells induced cachexia and lethal bone toxicity in mice bearing subcutaneous xenografts owing to antigen expression on multi‐potent bone marrow stromal cells (BMSCs) (Tran et al., 2013). Since FAP is also expressed in human BMSCs (Tran et al., 2013), caution must be applied in the clinical application of FAP‐targeted CAR T‐cells. A Phase I trial (clinicaltrials.gov; NCT01722149) is currently investigating the use of FAP CAR T‐cells administered directly in the pleural cavity for the treatment of MPM (Petrausch et al., 2012). As FAP is expressed both in all subtypes of mesothelioma cells and in CAFs, this approach can target both cell types to maximize therapeutic efficacy (Schuberth et al., 2013).
6.4. Addressing the intra‐tumoural metabolic challenge
Intra‐tumoural acidosis and metabolic impairment both inhibit T‐cell cytotoxicity and thereby may hamper the success of CAR T‐cell immunotherapy for solid tumours. It was discovered by Otto Warburg in 1927 that tumour cells metabolize glucose in a distinctive manner compared to non‐malignant cells (Warburg et al., 1927). Normal cells rely on mitochondrial oxidative phosphorylation (OXPHOS) to generate energy whereas tumour cells have a higher rate of aerobic glycolysis, resulting in increased lactate production even in the presence of plentiful oxygen. This is known as the “Warburg effect” and leads to a drop in extracellular pH (Gatenby and Gillies, 2004). In normal tissues, pH is around 7.4 whereas in tumours it is not uncommon to find a pH of 6.5 or less (Calcinotto et al., 2012; Gerweck and Seetharaman, 1996). This acidic environment impairs T‐cell metabolism and effector function.
Activated T‐cells also rely on glycolysis as a source of energy; naïve T‐cells use fatty acid oxidation whereas, upon antigen encounter, T‐cells shift to glycolysis to sustain effector function (Sukumar et al., 2013). The resultant lactic acid produced by glycolysis is exported from the T‐cell, a process that is dependent on a gradient between cytoplasmic and extracellular lactic acid concentrations. A lowered pH in the tumour microenvironment disturbs this gradient, hindering lactic acid export and thus disturbing T‐cell metabolism resulting in a hypo‐responsive state (Calcinotto et al., 2012; Fischer et al., 2007; Nakagawa et al., 2015).
In contrast to the substantial amount of work that has focused on optimizing CAR T‐cell signalling, very little attention has been paid to date on metabolic manipulations to improve therapeutic efficacy of CAR T‐cells. Inhibition of acid secretion by proton pump inhibitors (PPIs) such as esomeprazole has been shown to enhance efficacy of both active and adoptive immunotherapy (Calcinotto et al., 2012). Other strategies aim to promote memory T‐cell formation by inhibiting glycolytic metabolism, as memory T‐cells do not rely heavily on this and instead preferentially use fatty acid OXPHOS. Rapamycin blocks mTOR signalling and has been shown to increase memory CD8+ T‐cell precursors (Araki et al., 2009). Similarly, metformin is an activator of AMPK that restores fatty acid oxidation and promotes CD8+ memory T‐cell formation (Pearce et al., 2009). A third approach inhibits glycolysis with 2‐deoxyglucose (2DG) to prevent terminal differentiation and enhance memory T‐cell formation. Treatment of CD8+ T‐cells with 2DG led to enhanced migration to lymphoid tissues. Although migration of adoptively transferred T‐cells to melanoma tumours was not initially increased, 2DG treated T‐cells accumulated more efficiently over time, resulting in improved anti‐tumour activity (Sukumar et al., 2013).
6.5. The challenge of intra‐tumoural hypoxia
Low oxygen availability is a hallmark of many solid tumours and there is evidence that hypoxic conditions within the tumour microenvironment can negatively affect cytotoxic T‐cell (CTL) function. Accumulation of hypoxia‐inducible factor (HIF)1‐α suppresses T‐cell effector function (Schlie et al., 2011) and furthermore can render tumour cells more resistant to CTL‐mediated lysis by HIF1‐α dependent induction of the microRNA miR‐210 (Noman et al., 2012). An immunosuppressive hypoxic environment is further encouraged by the recruitment of regulatory T‐cells (Tregs) to hypoxic regions of the tumour. This may be achieved by hypoxia‐driven production of cytokines and chemokines such as transforming growth factor (TGF)‐β and CCL28 by tumour cells (Facciabene et al., 2011; Hasmim et al., 2013). Furthermore, hypoxic Tregs are more effective than their normoxic counterparts in suppressing the proliferation of effector T‐cells (Ben‐Shoshan et al., 2008). As IL‐2 expression is reduced under hypoxic conditions, Kim et al. sought to redress this by generating hypoxia‐resistant T‐cells that up‐regulate IL‐2 when oxygen content is low (Kim et al., 2008). In a model of B cell lymphoma, genetically modified T‐cells sustained effector function and proliferative capacity even when oxygen content was 1%, leading to enhanced anti‐tumour activity and overall survival compared to parental T‐cells (Kim et al., 2008).
A further mechanism of hypoxia‐induced immunosuppression is elicited by the release of ATP from dying tumour cells. Released ATP is metabolized to adenosine by the ecto‐enzymes CD39 and CD73, both of which are upregulated by hypoxia (Sitkovsky et al., 2014). Adenosine binds to the A2A‐adenosine receptor (A2AR) on T‐cells, leading to augmented levels of intracellular cyclic AMP (cAMP) and inhibition of T‐cell receptor triggered signalling. In addition, genes that contain the cAMP response element (CRE) or the hypoxia responsive element (HRE) are upregulated, leading to expression of immunosuppressive proteins such as TGF‐β, IL‐10, CTLA‐4 and PD‐1 (Sitkovsky et al., 2014). There is a clear rationale to target both hypoxia and the hypoxia driven A2‐adenosinergic pathway in order to improve T‐cell effector function.
Agents that target various stages of the A2‐adenosinergic pathway include: i) inhibitors of CD39 and CD73; ii) adenosine‐degrading enzymes and iii) A2AR antagonists. Anti‐CD73 antibody therapy and antagonists of A2AR synergize with anti‐PD1 antibody treatment to enhance therapeutic activity in several pre‐clinical models (Allard et al., 2013; Beavis et al., 2015). Similarly, knockdown or antagonism of A2AR in adoptively transferred T‐cells increases anti‐tumour activity (Darcy et al., 2014; Ohta et al., 2006). HER2 CAR T‐cells up‐regulate expression of A2AR after antigen activation and combination treatment with SCH58621, a small molecule A2AR antagonist enhances survival, which is improved even further in combination with anti‐PD‐1 treatment (Darcy et al., 2014).
Modifications to the way that CAR T‐cells are cultured prior to infusion may also render them more able to function in a hypoxic environment, albeit transiently. Culture of CTLs in the presence of the adenosine receptor agonist NECA (5′‐N‐Ethylcarboxamidoadenosine) during the expansion phase leads to selection of more resistant cells that are able to proliferate even in the presence of NECA (Ohta et al., 2009). Although this hypo‐responsiveness was transient, it is possible that temporarily decreasing A2ARhi populations of T‐cells may improve their in‐vivo function in the first few days after infusion.
Finally, it was recently reported that respiratory hyperoxia (60% oxygen) decreases intra‐tumoural hypoxia and concentrations of extracellular adenosine, leading to reversal of hypoxia‐adenosinergic immunosuppression. This led to increased infiltration of adoptively transferred T‐cells and NK cells, resulting in improved tumour regression (Hatfield et al., 2015), supporting the idea that supplemental oxygen can be given as a co‐adjuvant therapy.
6.6. Miscellaneous approaches to address the tumour microenvironment problem
An alternative strategy to improve T‐cell survival and effector function within the hostile tumour microenvironment entails the engineering of CAR T‐cells to release one or more cytokines. Several groups have shown that CAR T‐cells that express IL‐12 achieve improved anti‐tumour activity (known variously as “armoured CARs” or “Trucks”) (Chmielewski et al., 2011; Pegram et al., 2012; Zhang et al., 2011). Alternatively, additional survival signals may be provided by engineering CAR T‐cells to secrete a common gamma cytokine such as IL‐2, IL‐7, IL‐15 or IL‐21 (Markley and Sadelain, 2010; Quintarelli et al., 2007) or to acquire heightened responsiveness to a tumour‐derived cytokine such as IL‐4 (Wilkie et al., 2010) or colony‐stimulating factor‐1 (Lo et al., 2008).
7. Combination therapy
Targeted therapies and cytotoxic agents can modulate the immune response, which raises the possibility that these can be combined with immunotherapy to improve clinical outcomes (reviewed by Melero et al., 2015; Vanneman and Dranoff, 2012).
7.1. Combination with checkpoint inhibitors
A major impediment to the efficacy of CAR T‐cells is the immunosuppressive tumour microenvironment, where increased expression of inhibitory signals for activated T‐cells play a role in hampering the immune response. These inhibitory checkpoint receptors include cytotoxic T lymphocyte‐associated antigen 4 (CTLA4) and programmed death‐1 (PD‐1), which are expressed on activated T‐cells and act to inhibit effector function. Whilst these signalling pathways limit excessive inflammation and therefore damage to healthy tissue during a normal immune response, tumours have evolved to hijack these mechanisms to evade immune attack. For example, the ligand for PD‐1 (programmed death‐ligand 1 (PDL‐1)) is overexpressed on tumour cells and macrophages and is associated with poorer prognosis in several cancers including renal cell carcinoma (RCC) and ovarian cancer (Hamanishi et al., 2007; Maine et al., 2014; Thompson et al., 2006). Monoclonal antibodies that block checkpoint receptors and their ligands such as CTLA4, PD‐1 and PDL‐1 have already demonstrated remarkable activity in the clinic, even in refractory solid tumours, with responses seen in patients with melanoma, RCC, NSCLC and ovarian cancer (Brahmer et al., 2012; Hodi et al., 2010; Topalian et al., 2012). A logical evolution of CAR immunotherapy therefore is to combine CAR T‐cells with checkpoint inhibitors, an approach which has already proved advantageous in several pre‐clinical models (John et al., 2013; Peng et al., 2012; Teo et al., 2015). The concern with this combination is the potential for uncontrolled proliferation and activation of CAR T‐cells, although it would be possible to mitigate against this with the use of suicide genes such as the iCasp9 system (see Section 5.4).
7.2. Combination with targeted therapies
Some targeted therapies manifest important immunomodulatory properties in addition to their effects on tumour cells. Inhibitors of the mTOR pathway can enhance CD8+ T‐cell activation and augment differentiation into memory T cells (Vanneman and Dranoff, 2012) and have already been discussed in section 6.4. In addition, sunitinib is an inhibitor of multiple tumour‐associated tyrosine kinases but also decreases the numbers of myeloid‐derived suppressor cells (MDSCs) and Tregs. Furthermore, treatment with sunitinib decreases expression of CTLA4 and PD‐1 on T‐cells and PDL‐1 on MDSCs (Ozao‐Choy et al., 2009) and blocks STAT3 signalling. Ablation of Stat3 in T‐cells prior to adoptive transfer allows proliferation in vivo and tumour growth inhibition, a result that is duplicated with combination therapy of sunitinib and adoptively transferred T‐cells in pre‐clinical models of RCC and melanoma (Kujawski et al., 2010).
7.3. Combination with cytotoxic therapies
The immunological aspects of chemotherapy agents have been well documented elsewhere (Galluzzi et al., 2012; Zitvogel et al., 2008) and there is increasing interest in combining adoptive immunotherapy with conventional therapy to improve clinical outcome. Some chemotherapies such as cyclophosphamide, mitoxantrone, doxorubicin and oxaliplatin are known to induce an immunogenic cell death (ICD), which is characterized by exposure of calreticulin at the cell surface and release of ATP and the chromatin‐binding high mobility group B1 (HMGB1) and can trigger an adaptive immune response (Pol et al., 2015). Cyclophosphamide is the best studied alongside CAR T‐cell therapy owing to its use as a lymphodepleting agent. The importance of pre‐conditioning was demonstrated by enhanced CD19 CAR T‐cell persistence in patients who received cyclophosphamide (Brentjens et al., 2011), which not only reduces numbers of immunosuppressive T‐cells and makes room for transferred CAR T‐cells but may also allow spontaneous expansion of transferred T‐cells to maintain homeostasis in a phenomenon known as rebound overshoot (Proietti et al., 2012).
Other chemotherapies do not lead to ICD yet are able to induce immunogenic modulation that can sensitize tumour cells to CAR T‐cells. This has been observed in several models using various drugs. For example, pre‐treatment of prostate, breast and colorectal cancer cells with docetaxel sensitizes tumour cells to PSA, MUC1 and CEA specific T‐cells, respectively (Hodge et al., 2013). Modulation of the immune system by low dose chemotherapy can reduce mechanisms of immunosuppression and trigger recruitment of CD8+ lymphocytes to tumours, with patients receiving dose dense cisplatin and paclitaxel showing elevated serum levels of IL2 and IFNγ indicative of cytotoxic T‐cell activity (Chang et al., 2013). Combination of low dose chemotherapy with CAR T‐cells can therefore be used to enhance therapeutic efficacy, as demonstrated in a pre‐clinical model of ovarian cancer where low dose carboplatin had no anti‐tumour activity alone but improved the efficacy of ErbB targeted CAR T‐cells (Parente‐Pereira et al., 2013). Immune modulating activity of other chemotherapeutic drugs has also been described which warrants their investigation in combination with CAR T‐cells. For example, platinum‐based chemotherapeutics reduce expression of PDL2 on tumour cells and dendritic cells (Lesterhuis et al., 2011) and several commonly used drugs are able to increase the permeability of tumour cells to granzyme B (Ramakrishnan et al., 2010). In the latter, granzyme B uptake is perforin independent and leads to the death of neighbouring cells that do not express an antigen to which cytotoxic T‐lymphocytes are specific. This bystander effect can enhance the overall efficacy of CAR T‐cells, particularly in heterogenous tumours where the target antigen may not be expressed on all cells.
8. The development of “off‐the‐shelf” CAR T‐cells
Currently, the personalized nature of CAR T‐cell therapy restricts their use to specialized facilities that are able to manufacture genetically modified autologous T‐cells under good manufacturing practice (GMP). Collection, purification and activation of T‐cells are all subject to variability based on the natural variation, disease stage and prior treatment of the patient. Additionally, transduction efficiency of T‐cells can vary and poor transduction/expansion can restrict certain patients from receiving the planned dose of CAR T‐cells (Lee et al., 2015). Furthermore, the expansion phase required to generate large numbers of CAR T‐cells required can delay treatment to patients.
Attempts to generate allogeneic “off the shelf” CAR T‐cells are thus in development. This is not possible without genetic engineering, as the endogenous TCR in donor T‐cells recognizes allogeneic HLA molecules and minor antigens in the host to react against non‐autologous tissue, leading to GvHD. It is possible to ablate TCR expression through gene disruption using zinc finger nucleases (ZFNs), transcription activator‐like effector nucleases (TALENs) or CRISP/Cas9 technology (reviewed in Themeli et al., 2015). This approach has been used to generate transgenic TCR (Berdien et al., 2014; Provasi et al., 2012) and CAR T‐cells (Poirot et al., 2015; Torikai et al., 2012; Valton et al., 2015). Poirot et al. developed a CD19‐targeted CAR T‐cell population that is deficient in expression of both the TCR and CD52, the target of the lymphodepleting monoclonal antibody alemtuzumab. Knockout of these two genes using TALEN technology allows the prospect of allogeneic delivery without GvHD and confers resistance to alemtuzumab to allow CAR T‐cell engraftment in the presence of host T‐cell depletion (Poirot et al., 2015). However, patients would be profoundly immune compromized in response to alemtuzumab therapy. A similar approach using TALEN‐mediated gene editing generated TCRαβ deficient CAR T‐cells that are also resistant to clofradine, cytarabine and fludaribine, three commonly used lymphodepleting chemotherapeutic agents (Valton et al., 2015). As an additional safety feature, allogeneic CAR T‐cells can be engineered to co‐express the RQR8 gene to render them sensitive to rituximab (Smith et al., 2015). In this manner, CAR T‐cells can be depleted in the event of GvHD.
A second approach to create allogeneic CAR T‐cells relies on the use of progenitor cells as a cell source (reviewed extensively in Themeli et al., 2015). Unlike T‐cells, their precursors cannot cause GvHD as they complete their differentiation in the recipient's thymus and thus become tolerant to the host (Themeli et al., 2015). Transduction of induced pluripotent stem cell (iPSC) derived T‐cells with a CD19‐targeted CAR gives rise to an innate γδ T‐cell‐like population, despite expression of the endogenous TCR. In a proof of principle study, these CAR‐TiPSC T‐cells generated in‐vitro showed anti‐tumour efficacy against a CD19+ lymphoma in‐vivo that was comparable to that of T‐cells derived from peripheral blood (Themeli et al., 2013). However, this research is at an early stage and more studies are needed to assess the expansion and functional capabilities of iPSC‐derived T‐cells. To date, autologous T‐cells represent the most developed source of CAR T‐cell therapy.
9. Conclusions
First developed over 20 years ago, CAR T‐cells are now an exciting and realistic treatment option for haematological malignancies. The need for more targeted and personalized medicine has become clearer over time with regards to developing novel therapies and adoptive T‐cell therapy is no longer considered an experimental and risky approach. Indeed, the clinical successes seen with CD19‐targeted CAR T‐cells pave the way for broader clinical application, particularly in the arena of solid cancers. The safety of CAR T‐cells remains a concern and emphasizes the need to identify safe antigen targets. However, huge developments have been made in this area as clinical experience increases and now there are grading systems for CRS, with management of toxicity becoming more advanced. In particular, the use of the IL6Rα antagonist antibody tocilizumab and suicide systems has the potential to greatly reduce serious adverse effects.
Several challenges have been identified that have hampered success, especially against solid tumours. Extensive research is ongoing to address these issues, including strategies to enhance trafficking to the tumour, in‐vivo persistence and the ability of CAR T‐cells to thrive in the hostile tumour microenvironment in order to maximize therapeutic efficacy. In addition, there is potential to combine CAR T‐cell therapy with conventional chemotherapeutics or other exciting emerging immunotherapeutics such as checkpoint inhibitors.
As technologies develop, the ability to develop “off the shelf” universal CAR T‐cells generated from healthy donors also provides an exciting opportunity. Coupled with increasing interest in the field from commercial entities, this may accelerate clinical implementation and the advance of CAR T‐cell therapy as a first line therapy.
Acknowledgements
Research in the author's laboratory is supported by Breast Cancer Now (SP296 and 2013PhD145), Worldwide Cancer Research (13‐1017), the Wellcome Trust, Cancer Research UK and Bayer Healthcare (co‐funded drug discovery award), Leukemia and Lymphoma Research (13007), the June Hancock Mesothelioma Research Fund (JH/18/2014), The Jon Moulton Charitable Foundation (clinical trial award), the Medical Research Council (MR/M024733/1), the Experimental Cancer Medicine Centre at King's College London and by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Whilding Lynsey M., Maher John, (2015), CAR T‐cell immunotherapy: The path from the by‐road to the freeway?, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.10.012.
This is a contribution to the special issue edited by Johanna Olweus, Cancer Immunotherapy.
References
- Achkova, D. , Spicer, J. , Maher, J. , 2015. Development of immunotherapy for anaplastic large cell lymphoma (ALCL) using CSF1R retargeted human T lymphocytes. Proceedings of the American Association for Cancer Research Meeting, Philadelphia, Pennsylvania Abstract 3144 [Google Scholar]
- Adusumilli, P.S. , Cherkassky, L. , Villena-Vargas, J. , Colovos, C. , Servais, E. , Plotkin, J. , Jones, D.R. , Sadelain, M. , 2014. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 6, 261ra151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, A.S. , Ormiston-Smith, N. , Sasieni, P.D. , 2015. Trends in the lifetime risk of developing cancer in Great Britain: comparison of risk for those born from 1930 to 1960. Br. J. Cancer. 112, 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, N. , Brawley, V.S. , Hegde, M. , Robertson, C. , Ghazi, A. , Gerken, C. , Liu, E. , Dakhova, O. , Ashoori, A. , Corder, A. , Gray, T. , Wu, M.F. , Liu, H. , Hicks, J. , Rainusso, N. , Dotti, G. , Mei, Z. , Grilley, B. , Gee, A. , Rooney, C.M. , Brenner, M.K. , Heslop, H.E. , Wels, W.S. , Wang, L.L. , Anderson, P. , Gottschalk, S. , 2015. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 33, 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, N. , Salsman, V.S. , Kew, Y. , Shaffer, D. , Powell, S. , Zhang, Y.J. , Grossman, R.G. , Heslop, H.E. , Gottschalk, S. , 2010. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 16, 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, N. , Salsman, V.S. , Yvon, E. , Louis, C.U. , Perlaky, L. , Wels, W.S. , Dishop, M.K. , Kleinerman, E.E. , Pule, M. , Rooney, C.M. , Heslop, H.E. , Gottschalk, S. , 2009. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther.: J. Am. Soc. Gene Ther. 17, 1779–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akishima-Fukasawa, Y. , Nakanishi, Y. , Ino, Y. , Moriya, Y. , Kanai, Y. , Hirohashi, S. , 2009. Prognostic significance of CXCL12 expression in patients with colorectal carcinoma. Am. J. Clin. Pathol. 132, 202–210. quiz 307 [DOI] [PubMed] [Google Scholar]
- Allard, B. , Pommey, S. , Smyth, M.J. , Stagg, J. , 2013. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 19, 5626–5635. [DOI] [PubMed] [Google Scholar]
- Almasbak, H. , Walseng, E. , Kristian, A. , Myhre, M.R. , Suso, E.M. , Munthe, L.A. , Andersen, J.T. , Wang, M.Y. , Kvalheim, G. , Gaudernack, G. , Kyte, J.A. , 2015. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 22, 391–403. [DOI] [PubMed] [Google Scholar]
- Altenschmidt, U. , Kahl, R. , Moritz, D. , Schnierle, B.S. , Gerstmayer, B. , Wels, W. , Groner, B. , 1996. Cytolysis of tumor cells expressing the Neu/erbB-2, erbB-3, and erbB-4 receptors by genetically targeted naive T lymphocytes. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2, 1001–1008. [PubMed] [Google Scholar]
- Altvater, B. , Landmeier, S. , Pscherer, S. , Temme, J. , Juergens, H. , Pule, M. , Rossig, C. , 2009. 2B4 (CD244) signaling via chimeric receptors costimulates tumor-antigen specific proliferation and in vitro expansion of human T cells. Cancer Immunol. Immunother. 58, 1991–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anurathapan, U. , Chan, R.C. , Hindi, H.F. , Mucharla, R. , Bajgain, P. , Hayes, B.C. , Fisher, W.E. , Heslop, H.E. , Rooney, C.M. , Brenner, M.K. , Leen, A.M. , Vera, J.F. , 2014. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther.: J. Am. Soc. Gene Ther. 22, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, K. , Turner, A.P. , Shaffer, V.O. , Gangappa, S. , Keller, S.A. , Bachmann, M.F. , Larsen, C.P. , Ahmed, R. , 2009. mTOR regulates memory CD8 T-cell differentiation. Nature. 460, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai, H. , Fujiwara, H. , An, J. , Ochi, T. , Miyazaki, Y. , Nagai, K. , Okamoto, S. , Mineno, J. , Kuzushima, K. , Shiku, H. , Inoue, H. , Yasukawa, M. , 2013. Co-introduced functional CCR2 potentiates in vivo anti-lung cancer functionality mediated by T cells double gene-modified to express WT1-specific T-cell receptor. PLoS One. 8, e56820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala, A. , Chung, C.S. , Grutkoski, P.S. , Song, G.Y. , 2003. Mechanisms of immune resolution. Crit. Care Med. 31, S558–S571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty, G.L. , Haas, A.R. , Maus, M.V. , Torigian, D.A. , Soulen, M.C. , Plesa, G. , Chew, A. , Zhao, Y. , Levine, B.L. , Albelda, S.M. , Kalos, M. , June, C.H. , 2014. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavis, P.A. , Milenkovski, N. , Henderson, M.A. , John, L.B. , Allard, B. , Loi, S. , Kershaw, M.H. , Stagg, J. , Darcy, P.K. , 2015. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 3, 506–517. [DOI] [PubMed] [Google Scholar]
- Ben-Shoshan, J. , Maysel-Auslender, S. , Mor, A. , Keren, G. , George, J. , 2008. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 38, 2412–2418. [DOI] [PubMed] [Google Scholar]
- Berdien, B. , Mock, U. , Atanackovic, D. , Fehse, B. , 2014. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 21, 539–548. [DOI] [PubMed] [Google Scholar]
- Brahmer, J.R. , Tykodi, S.S. , Chow, L.Q. , Hwu, W.J. , Topalian, S.L. , Hwu, P. , Drake, C.G. , Camacho, L.H. , Kauh, J. , Odunsi, K. , Pitot, H.C. , Hamid, O. , Bhatia, S. , Martins, R. , Eaton, K. , Chen, S. , Salay, T.M. , Alaparthy, S. , Grosso, J.F. , Korman, A.J. , Parker, S.M. , Agrawal, S. , Goldberg, S.M. , Pardoll, D.M. , Gupta, A. , Wigginton, J.M. , 2012. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. New Engl. J. Med. 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, M.K. , 2013. CAR T cells for acute myeloid leukemia: the LeY of the land. Mol. Ther. 21, 1983–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, R.J. , Davila, M.L. , Riviere, I. , Park, J. , Wang, X. , Cowell, L.G. , Bartido, S. , Stefanski, J. , Taylor, C. , Olszewska, M. , Borquez-Ojeda, O. , Qu, J. , Wasielewska, T. , He, Q. , Bernal, Y. , Rijo, I.V. , Hedvat, C. , Kobos, R. , Curran, K. , Steinherz, P. , Jurcic, J. , Rosenblat, T. , Maslak, P. , Frattini, M. , Sadelain, M. , 2013. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, R.J. , Latouche, J.B. , Santos, E. , Marti, F. , Gong, M.C. , Lyddane, C. , King, P.D. , Larson, S. , Weiss, M. , Riviere, I. , Sadelain, M. , 2003. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 9, 279–286. [DOI] [PubMed] [Google Scholar]
- Brentjens, R.J. , Riviere, I. , Park, J.H. , Davila, M.L. , Wang, X. , Stefanski, J. , Taylor, C. , Yeh, R. , Bartido, S. , Borquez-Ojeda, O. , Olszewska, M. , Bernal, Y. , Pegram, H. , Przybylowski, M. , Hollyman, D. , Usachenko, Y. , Pirraglia, D. , Hosey, J. , Santos, E. , Halton, E. , Maslak, P. , Scheinberg, D. , Jurcic, J. , Heaney, M. , Heller, G. , Frattini, M. , Sadelain, M. , 2011. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 118, 4817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, R.J. , Santos, E. , Nikhamin, Y. , Yeh, R. , Matsushita, M. , La Perle, K. , Quintas-Cardama, A. , Larson, S.M. , Sadelain, M. , 2007. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 13, 5426–5435. [DOI] [PubMed] [Google Scholar]
- Bridgeman, J.S. , Hawkins, R.E. , Bagley, S. , Blaylock, M. , Holland, M. , Gilham, D.E. , 2010. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 184, 6938–6949. [DOI] [PubMed] [Google Scholar]
- Brown, C.E. , Badie, B. , Barish, M.E. , Weng, L. , Ostberg, J.R. , Chang, W.C. , Naranjo, A. , Starr, R. , Wagner, J. , Wright, C. , Zhai, Y. , Bading, J.R. , Ressler, J.A. , Portnow, J. , D'Apuzzo, M. , Forman, S.J. , Jensen, M.C. , 2015. Bioactivity and safety of IL13Ralpha2-Redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 21, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde, L.E. , Berger, C. , Lin, Y. , Wang, J. , Lin, X. , Frayo, S.E. , Brouns, S.A. , Spencer, D.M. , Till, B.G. , Jensen, M.C. , Riddell, S.R. , Press, O.W. , 2013. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 8, e82742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcinotto, A. , Filipazzi, P. , Grioni, M. , Iero, M. , De Milito, A. , Ricupito, A. , Cova, A. , Canese, R. , Jachetti, E. , Rossetti, M. , Huber, V. , Parmiani, G. , Generoso, L. , Santinami, M. , Borghi, M. , Fais, S. , Bellone, M. , Rivoltini, L. , 2012. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 72, 2746–2756. [DOI] [PubMed] [Google Scholar]
- Carpenito, C. , Milone, M.C. , Hassan, R. , Simonet, J.C. , Lakhal, M. , Suhoski, M.M. , Varela-Rohena, A. , Haines, K.M. , Heitjan, D.F. , Albelda, S.M. , Carroll, R.G. , Riley, J.L. , Pastan, I. , June, C.H. , 2009. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. U. S. A. 106, 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana, I. , Savoldo, B. , Hoyos, V. , Weber, G. , Liu, H. , Kim, E.S. , Ittmann, M.M. , Marchetti, D. , Dotti, G. , 2015. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 21, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C.L. , Hsu, Y.T. , Wu, C.C. , Lai, Y.Z. , Wang, C. , Yang, Y.C. , Wu, T.C. , Hung, C.F. , 2013. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res. 73, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicaybam, L. , Bonamino, M.H. , 2015. Construction and validation of an activating and inhibitory chimeric antigen receptor (CAR) system. Proceedings of the American Association for Cancer Research Meeting, Philadelphia, Pennsylvania Abstract number 3156 [Google Scholar]
- Chmielewski, M. , Kopecky, C. , Hombach, A.A. , Abken, H. , 2011. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71, 5697–5706. [DOI] [PubMed] [Google Scholar]
- Chow, K.K. , Naik, S. , Kakarla, S. , Brawley, V.S. , Shaffer, D.R. , Yi, Z. , Rainusso, N. , Wu, M.F. , Liu, H. , Kew, Y. , Grossman, R.G. , Powell, S. , Lee, D. , Ahmed, N. , Gottschalk, S. , 2013. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther.: J. Am. Soc. Gene Ther. 21, 629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri, N. , Camisa, B. , Cocchiarella, F. , Forcato, M. , Oliveira, G. , Provasi, E. , Bondanza, A. , Bordignon, C. , Peccatori, J. , Ciceri, F. , Lupo-Stanghellini, M.T. , Mavilio, F. , Mondino, A. , Bicciato, S. , Recchia, A. , Bonini, C. , 2013. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 121, 573–584. [DOI] [PubMed] [Google Scholar]
- Craddock, J.A. , Lu, A. , Bear, A. , Pule, M. , Brenner, M.K. , Rooney, C.M. , Foster, A.E. , 2010. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 33, 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz, C.R. , Micklethwaite, K.P. , Savoldo, B. , Ramos, C.A. , Lam, S. , Ku, S. , Diouf, O. , Liu, E. , Barrett, A.J. , Ito, S. , Shpall, E.J. , Krance, R.A. , Kamble, R.T. , Carrum, G. , Hosing, C.M. , Gee, A.P. , Mei, Z. , Grilley, B.J. , Heslop, H.E. , Rooney, C.M. , Brenner, M.K. , Bollard, C.M. , Dotti, G. , 2013. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 122, 2965–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcy, P.K. , John, L.B. , Neeson, P. , Henderson, M.A. , Kershaw, M.H. , Beavis, P.A. , 2014. Targeting tumor-induced immunosuppression leads to potent anti-tumor responses by CAR T cells. Australasian Society for Immunology Annual Scientific Meeting 2014 [Google Scholar]
- Davies, D.M. , Maher, J. , 2015. TSLPR: a new CAR in the showroom for B-ALL. Blood. 126, 567–569. [DOI] [PubMed] [Google Scholar]
- Davies, D.M. , Foster, J. , Van Der Stegen, S.J. , Parente-Pereira, A.C. , Chiapero-Stanke, L. , Delinassios, G.J. , Burbridge, S.E. , Kao, V. , Liu, Z. , Bosshard-Carter, L. , Van Schalkwyk, M.C. , Box, C. , Eccles, S.A. , Mather, S.J. , Wilkie, S. , Maher, J. , 2012. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 18, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila, M.L. , Riviere, I. , Wang, X. , Bartido, S. , Park, J. , Curran, K. , Chung, S.S. , Stefanski, J. , Borquez-Ojeda, O. , Olszewska, M. , Qu, J. , Wasielewska, T. , He, Q. , Fink, M. , Shinglot, H. , Youssif, M. , Satter, M. , Wang, Y. , Hosey, J. , Quintanilla, H. , Halton, E. , Bernal, Y. , Bouhassira, D.C. , Arcila, M.E. , Gonen, M. , Roboz, G.J. , Maslak, P. , Douer, D. , Frattini, M.G. , Giralt, S. , Sadelain, M. , Brentjens, R. , 2014. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 6, 224ra225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi, A. , De Angelis, B. , Rooney, C.M. , Zhang, L. , Mahendravada, A. , Foster, A.E. , Heslop, H.E. , Brenner, M.K. , Dotti, G. , Savoldo, B. , 2009. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 113, 6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi, A. , Tey, S.K. , Dotti, G. , Fujita, Y. , Kennedy-Nasser, A. , Martinez, C. , Straathof, K. , Liu, E. , Durett, A.G. , Grilley, B. , Liu, H. , Cruz, C.R. , Savoldo, B. , Gee, A.P. , Schindler, J. , Krance, R.A. , Heslop, H.E. , Spencer, D.M. , Rooney, C.M. , Brenner, M.K. , 2011. Inducible apoptosis as a safety switch for adoptive cell therapy. New Engl. J. Med. 365, 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi, O. , Ohtani, H. , Hatori, M. , Sato, E. , Hosaka, M. , Nagura, H. , Itoi, E. , Kokubun, S. , 2009. Histogenesis-specific expression of fibroblast activation protein and dipeptidylpeptidase-IV in human bone and soft tissue tumours. Histopathology. 55, 432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong, C.P. , Westwood, J.A. , Yong, C.S. , Murphy, A. , Devaud, C. , John, L.B. , Darcy, P.K. , Kershaw, M.H. , 2013. Engineering T cell function using chimeric antigen receptors identified using a DNA library approach. PLoS One. 8, e63037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak, H.F. , 1986. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. New Engl. J. Med. 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- Ene-Obong, A. , Clear, A.J. , Watt, J. , Wang, J. , Fatah, R. , Riches, J.C. , Marshall, J.F. , Chin-Aleong, J. , Chelala, C. , Gribben, J.G. , Ramsay, A.G. , Kocher, H.M. , 2013. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 145, 1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshhar, Z. , Waks, T. , Gross, G. , Schindler, D.G. , 1993. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. U. S. A. 90, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facciabene, A. , Peng, X. , Hagemann, I.S. , Balint, K. , Barchetti, A. , Wang, L.P. , Gimotty, P.A. , Gilks, C.B. , Lal, P. , Zhang, L. , Coukos, G. , 2011. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 475, 226–230. [DOI] [PubMed] [Google Scholar]
- Fedorov, V.D. , Themeli, M. , Sadelain, M. , 2013. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 5, 215ra172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig, C. , Jones, J.O. , Kraman, M. , Wells, R.J. , Deonarine, A. , Chan, D.S. , Connell, C.M. , Roberts, E.W. , Zhao, Q. , Caballero, O.L. , Teichmann, S.A. , Janowitz, T. , Jodrell, D.I. , Tuveson, D.A. , Fearon, D.T. , 2013. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A. 110, 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney, H.M. , Akbar, A.N. , Lawson, A.D. , 2004. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 172, 104–113. [DOI] [PubMed] [Google Scholar]
- Finney, H.M. , Lawson, A.D. , Bebbington, C.R. , Weir, A.N. , 1998. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 161, 2791–2797. [PubMed] [Google Scholar]
- Fischer, K. , Hoffmann, P. , Voelkl, S. , Meidenbauer, N. , Ammer, J. , Edinger, M. , Gottfried, E. , Schwarz, S. , Rothe, G. , Hoves, S. , Renner, K. , Timischl, B. , Mackensen, A. , Kunz-Schughart, L. , Andreesen, R. , Krause, S.W. , Kreutz, M. , 2007. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 109, 3812–3819. [DOI] [PubMed] [Google Scholar]
- G.B, B. , O'Hara, M.H. , Nelson, A.M. , McGarvey, M. , Torigian, D.A. , Lacey, S.F. , Melenhorst, J.J. , Levine, B. , Plesa, G. , June, C.H. , 2015. Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. 2015 ASCO Annual Meeting J. Clin. Oncol. Chicago, Illinois, p. suppl; abstr 3007 [Google Scholar]
- Galluzzi, L. , Senovilla, L. , Zitvogel, L. , Kroemer, G. , 2012. The secret ally: immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 11, 215–233. [DOI] [PubMed] [Google Scholar]
- Garin-Chesa, P. , Old, L.J. , Rettig, W.J. , 1990. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. U. S. A. 87, 7235–7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby, R.A. , Gillies, R.J. , 2004. Why do cancers have high aerobic glycolysis?. Nat. Rev. Cancer. 4, 891–899. [DOI] [PubMed] [Google Scholar]
- Gattinoni, L. , Restifo, N.P. , 2013. Moving T memory stem cells to the clinic. Blood. 121, 567–568. [DOI] [PubMed] [Google Scholar]
- Geiger, T.L. , Nguyen, P. , Leitenberg, D. , Flavell, R.A. , 2001. Integrated src kinase and costimulatory activity enhances signal transduction through single-chain chimeric receptors in T lymphocytes. Blood. 98, 2364–2371. [DOI] [PubMed] [Google Scholar]
- Gerweck, L.E. , Seetharaman, K. , 1996. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 56, 1194–1198. [PubMed] [Google Scholar]
- Gil, M. , Komorowski, M.P. , Seshadri, M. , Rokita, H. , McGray, A.J. , Opyrchal, M. , Odunsi, K.O. , Kozbor, D. , 2014. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J. Immunol. 193, 5327–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, S. , Tasian, S.K. , Ruella, M. , Shestova, O. , Li, Y. , Porter, D.L. , Carroll, M. , Danet-Desnoyers, G. , Scholler, J. , Grupp, S.A. , June, C.H. , Kalos, M. , 2014. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 123, 2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano Attianese, G.M. , Marin, V. , Hoyos, V. , Savoldo, B. , Pizzitola, I. , Tettamanti, S. , Agostoni, V. , Parma, M. , Ponzoni, M. , Bertilaccio, M.T. , Ghia, P. , Biondi, A. , Dotti, G. , Biagi, E. , 2011. In vitro and in vivo model of a novel immunotherapy approach for chronic lymphocytic leukemia by anti-CD23 chimeric antigen receptor. Blood. 117, 4736–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, M.C. , Latouche, J.B. , Krause, A. , Heston, W.D. , Bander, N.H. , Sadelain, M. , 1999. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grada, Z. , Hegde, M. , Byrd, T. , Shaffer, D.R. , Ghazi, A. , Brawley, V.S. , Corder, A. , Schonfeld, K. , Koch, J. , Dotti, G. , Heslop, H.E. , Gottschalk, S. , Wels, W.S. , Baker, M.L. , Ahmed, N. , 2013. TanCAR: a novel bispecific chimeric antigen receptor for Cancer immunotherapy. Mol. Ther. Nucleic Acids. 2, e105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, G. , Waks, T. , Eshhar, Z. , 1989. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. U. S. A. 86, 10024–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, S.A. , Kalos, M. , Barrett, D. , Aplenc, R. , Porter, D.L. , Rheingold, S.R. , Teachey, D.T. , Chew, A. , Hauck, B. , Wright, J.F. , Milone, M.C. , Levine, B.L. , June, C.H. , 2013. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedan, S. , Carpenito, C. , McGettigan, S.E. , Frigault, M.J. , Scholler, J. , Zhao, Y. , June, C.H. , 2012. Redirection of TH17 cells with a car containing the ICOS costimulatory domain enhances function, antitumor activity and persistence of TH17 cells. Mol. Ther. 20, S130 Abstract 329 [Google Scholar]
- Guest, R.D. , Hawkins, R.E. , Kirillova, N. , Cheadle, E.J. , Arnold, J. , O'Neill, A. , Irlam, J. , Chester, K.A. , Kemshead, J.T. , Shaw, D.M. , Embleton, M.J. , Stern, P.L. , Gilham, D.E. , 2005. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J. Immunother. 28, 203–211. [DOI] [PubMed] [Google Scholar]
- Hamanishi, J. , Mandai, M. , Iwasaki, M. , Okazaki, T. , Tanaka, Y. , Yamaguchi, K. , Higuchi, T. , Yagi, H. , Takakura, K. , Minato, N. , Honjo, T. , Fujii, S. , 2007. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. U. S. A. 104, 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2011. Hallmarks of cancer: the next generation. Cell. 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hasmim, M. , Noman, M.Z. , Messai, Y. , Bordereaux, D. , Gros, G. , Baud, V. , Chouaib, S. , 2013. Cutting edge: hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-beta1. J. Immunol. 191, 5802–5806. [DOI] [PubMed] [Google Scholar]
- Hatfield, S.M. , Kjaergaard, J. , Lukashev, D. , Schreiber, T.H. , Belikoff, B. , Abbott, R. , Sethumadhavan, S. , Philbrook, P. , Ko, K. , Cannici, R. , Thayer, M. , Rodig, S. , Kutok, J.L. , Jackson, E.K. , Karger, B. , Podack, E.R. , Ohta, A. , Sitkovsky, M.V. , 2015. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 7, 277ra230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes, N.M. , Snook, M.B. , Trapani, J.A. , Cerruti, L. , Jane, S.M. , Smyth, M.J. , Darcy, P.K. , 2001. Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J. Immunol. 166, 182–187. [DOI] [PubMed] [Google Scholar]
- Hegde, M. , Corder, A. , Chow, K.K. , Mukherjee, M. , Ashoori, A. , Kew, Y. , Zhang, Y.J. , Baskin, D.S. , Merchant, F.A. , Brawley, V.S. , Byrd, T.T. , Krebs, S. , Wu, M.F. , Liu, H. , Heslop, H.E. , Gottschalk, S. , Yvon, E. , Ahmed, N. , 2013. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther.: J. Am. Soc. Gene Ther. 21, 2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimberger, A.B. , Hlatky, R. , Suki, D. , Yang, D. , Weinberg, J. , Gilbert, M. , Sawaya, R. , Aldape, K. , 2005. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 11, 1462–1466. [DOI] [PubMed] [Google Scholar]
- Heslop, H.E. , 2010. Safer CARS. Mol. Ther.: J. Am. Soc. Gene Ther. 18, 661–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge, J.W. , Garnett, C.T. , Farsaci, B. , Palena, C. , Tsang, K.Y. , Ferrone, S. , Gameiro, S.R. , 2013. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer. 133, 624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi, F.S. , O'Day, S.J. , McDermott, D.F. , Weber, R.W. , Sosman, J.A. , Haanen, J.B. , Gonzalez, R. , Robert, C. , Schadendorf, D. , Hassel, J.C. , Akerley, W. , van den Eertwegh, A.J. , Lutzky, J. , Lorigan, P. , Vaubel, J.M. , Linette, G.P. , Hogg, D. , Ottensmeier, C.H. , Lebbe, C. , Peschel, C. , Quirt, I. , Clark, J.I. , Wolchok, J.D. , Weber, J.S. , Tian, J. , Yellin, M.J. , Nichol, G.M. , Hoos, A. , Urba, W.J. , 2010. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl. J. Med. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach, A. , Heuser, C. , Sircar, R. , Tillmann, T. , Diehl, V. , Kruis, W. , Pohl, C. , Abken, H. , 1997. T cell targeting of TAG72+ tumor cells by a chimeric receptor with antibody-like specificity for a carbohydrate epitope. Gastroenterology. 113, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Hombach, A. , Koch, D. , Sircar, R. , Heuser, C. , Diehl, V. , Kruis, W. , Pohl, C. , Abken, H. , 1999. A chimeric receptor that selectively targets membrane-bound carcinoembryonic antigen (mCEA) in the presence of soluble CEA. Gene Ther. 6, 300–304. [DOI] [PubMed] [Google Scholar]
- Hudecek, M. , Sommermeyer, D. , Kosasih, P.L. , Silva-Benedict, A. , Liu, L. , Rader, C. , Jensen, M.C. , Riddell, S.R. , 2015. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 3, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai, C. , Mihara, K. , Andreansky, M. , Nicholson, I.C. , Pui, C.H. , Geiger, T.L. , Campana, D. , 2004. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 18, 676–684. [DOI] [PubMed] [Google Scholar]
- Irving, B.A. , Weiss, A. , 1991. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 64, 891–901. [DOI] [PubMed] [Google Scholar]
- John, L.B. , Devaud, C. , Duong, C.P. , Yong, C.S. , Beavis, P.A. , Haynes, N.M. , Chow, M.T. , Smyth, M.J. , Kershaw, M.H. , Darcy, P.K. , 2013. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 19, 5636–5646. [DOI] [PubMed] [Google Scholar]
- Johnson, L.A. , Scholler, J. , Ohkuri, T. , Kosaka, A. , Patel, P.R. , McGettigan, S.E. , Nace, A.K. , Dentchev, T. , Thekkat, P. , Loew, A. , Boesteanu, A.C. , Cogdill, A.P. , Chen, T. , Fraietta, J.A. , Kloss, C.C. , Posey, A.D. , Engels, B. , Singh, R. , Ezell, T. , Idamakanti, N. , Ramones, M.H. , Li, N. , Zhou, L. , Plesa, G. , Seykora, J.T. , Okada, H. , June, C.H. , Brogdon, J.L. , Maus, M.V. , 2015. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 7, 275ra222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonnalagadda, M. , Mardiros, A. , Urak, R. , Wang, X. , Hoffman, L.J. , Bernanke, A. , Chang, W.C. , Bretzlaff, W. , Starr, R. , Priceman, S. , Ostberg, J.R. , Forman, S.J. , Brown, C.E. , 2015. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol. Ther.: J. Am. Soc. Gene Ther. 23, 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlon, K.S. , Brown, C. , Cooper, L.J. , Raubitschek, A. , Forman, S.J. , Jensen, M.C. , 2004. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 64, 9160–9166. [DOI] [PubMed] [Google Scholar]
- Kakarla, S. , Chow, K.K. , Mata, M. , Shaffer, D.R. , Song, X.T. , Wu, M.F. , Liu, H. , Wang, L.L. , Rowley, D.R. , Pfizenmaier, K. , Gottschalk, S. , 2013. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther.: J. Am. Soc. Gene Ther. 21, 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos, M. , Levine, B.L. , Porter, D.L. , Katz, S. , Grupp, S.A. , Bagg, A. , June, C.H. , 2011. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandalaft, L.E. , Powell, D.J. , Coukos, G. , 2012. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J. Transl. Med. 10, 157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katari, U.L. , Keirnan, J.M. , Worth, A.C. , Hodges, S.E. , Leen, A.M. , Fisher, W.E. , Vera, J.F. , 2011. Engineered T cells for pancreatic cancer treatment. HPB (Oxford). 13, 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz, S.C. , Burga, R.A. , McCormack, E. , Wang, L.J. , Mooring, W. , Point, G.R. , Khare, P.D. , Thorn, M. , Ma, Q. , Stainken, B.F. , Assanah, E.O. , Davies, R. , Espat, N.J. , Junghans, R.P. , 2015. Phase I hepatic immunotherapy for metastases study of intra-Arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 21, 3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenderian, S.S. , Ruella, M. , Shestova, O. , Klichinsky, M. , Aikawa, V. , Morrissette, J.J. , Scholler, J. , Song, D. , Porter, D.L. , Carroll, M. , June, C.H. , Gill, S. , 2015. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 29, 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw, M.H. , Westwood, J.A. , Parker, L.L. , Wang, G. , Eshhar, Z. , Mavroukakis, S.A. , White, D.E. , Wunderlich, J.R. , Canevari, S. , Rogers-Freezer, L. , Chen, C.C. , Yang, J.C. , Rosenberg, S.A. , Hwu, P. , 2006. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 12, 6106–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. , Peng, G. , Hicks, J.M. , Weiss, H.L. , Van Meir, E.G. , Brenner, M.K. , Yotnda, P. , 2008. Engineering human tumor-specific cytotoxic T cells to function in a hypoxic environment. Mol. Ther.: J. Am. Soc. Gene Ther. 16, 599–606. [DOI] [PubMed] [Google Scholar]
- Kloss, C.C. , Condomines, M. , Cartellieri, M. , Bachmann, M. , Sadelain, M. , 2013. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 31, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, J.N. , Dudley, M.E. , Carpenter, R.O. , Kassim, S.H. , Rose, J.J. , Telford, W.G. , Hakim, F.T. , Halverson, D.C. , Fowler, D.H. , Hardy, N.M. , Mato, A.R. , Hickstein, D.D. , Gea-Banacloche, J.C. , Pavletic, S.Z. , Sportes, C. , Maric, I. , Feldman, S.A. , Hansen, B.G. , Wilder, J.S. , Blacklock-Schuver, B. , Jena, B. , Bishop, M.R. , Gress, R.E. , Rosenberg, S.A. , 2013. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 122, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, J.N. , Dudley, M.E. , Feldman, S.A. , Wilson, W.H. , Spaner, D.E. , Maric, I. , Stetler-Stevenson, M. , Phan, G.Q. , Hughes, M.S. , Sherry, R.M. , Yang, J.C. , Kammula, U.S. , Devillier, L. , Carpenter, R. , Nathan, D.A. , Morgan, R.A. , Laurencot, C. , Rosenberg, S.A. , 2012. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 119, 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, J.N. , Dudley, M.E. , Kassim, S.H. , Somerville, R.P. , Carpenter, R.O. , Stetler-Stevenson, M. , Yang, J.C. , Phan, G.Q. , Hughes, M.S. , Sherry, R.M. , Raffeld, M. , Feldman, S. , Lu, L. , Li, Y.F. , Ngo, L.T. , Goy, A. , Feldman, T. , Spaner, D.E. , Wang, M.L. , Chen, C.C. , Kranick, S.M. , Nath, A. , Nathan, D.A. , Morton, K.E. , Toomey, M.A. , Rosenberg, S.A. , 2015. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 33, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler, D. , Chmielewski, M. , Rappl, G. , Hombach, A. , Riet, T. , Schmidt, A. , Hombach, A. , Wendtner, C. , Abken, H. , 2011. CD28 costimulation impairs the efficacy of a redirected T-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing Lck activation. Mol. Ther.: J. Am. Soc. Gene Ther. 19, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, S. , Sengupta, S. , Tyler, B. , Bais, A.J. , Ma, Q. , Doucette, S. , Zhou, J. , Sahin, A. , Carter, B.S. , Brem, H. , Junghans, R.P. , Sampath, P. , 2012. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 18, 5949–5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraman, M. , Bambrough, P.J. , Arnold, J.N. , Roberts, E.W. , Magiera, L. , Jones, J.O. , Gopinathan, A. , Tuveson, D.A. , Fearon, D.T. , 2010. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 330, 827–830. [DOI] [PubMed] [Google Scholar]
- Krause, A. , Guo, H.F. , Latouche, J.B. , Tan, C. , Cheung, N.K. , Sadelain, M. , 1998. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 188, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo, K. , Imai, C. , Lorenzini, P. , Kamiya, T. , Kono, K. , Davidoff, A.M. , Chng, W.J. , Campana, D. , 2014. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 74, 93–103. [DOI] [PubMed] [Google Scholar]
- Kujawski, M. , Zhang, C. , Herrmann, A. , Reckamp, K. , Scuto, A. , Jensen, M. , Deng, J. , Forman, S. , Figlin, R. , Yu, H. , 2010. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 70, 9599–9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana, Y. , Asakura, Y. , Utsunomiya, N. , Nakanishi, M. , Arata, Y. , Itoh, S. , Nagase, F. , Kurosawa, Y. , 1987. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 149, 960–968. [DOI] [PubMed] [Google Scholar]
- Lamers, C.H. , Sleijfer, S. , van Steenbergen, S. , van Elzakker, P. , van Krimpen, B. , Groot, C. , Vulto, A. , den Bakker, M. , Oosterwijk, E. , Debets, R. , Gratama, J.W. , 2013. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther.: J. Am. Soc. Gene Ther. 21, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers, C.H. , Sleijfer, S. , Vulto, A.G. , Kruit, W.H. , Kliffen, M. , Debets, R. , Gratama, J.W. , Stoter, G. , Oosterwijk, E. , 2006. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 24, e20–22. [DOI] [PubMed] [Google Scholar]
- Lamers, C.H. , Willemsen, R. , van Elzakker, P. , van Steenbergen-Langeveld, S. , Broertjes, M. , Oosterwijk-Wakka, J. , Oosterwijk, E. , Sleijfer, S. , Debets, R. , Gratama, J.W. , 2011. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. 117, 72–82. [DOI] [PubMed] [Google Scholar]
- Lanitis, E. , Poussin, M. , Hagemann, I.S. , Coukos, G. , Sandaltzopoulos, R. , Scholler, N. , Powell, D.J. , 2012. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol. Ther.: J. Am. Soc. Gene Ther. 20, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanitis, E. , Poussin, M. , Klattenhoff, A.W. , Song, D. , Sandaltzopoulos, R. , June, C.H. , Powell, D.J. , 2013. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 1, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D.W. , Gardner, R. , Porter, D.L. , Louis, C.U. , Ahmed, N. , Jensen, M. , Grupp, S.A. , Mackall, C.L. , 2014. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 124, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D.W. , Kochenderfer, J.N. , Stetler-Stevenson, M. , Cui, Y.K. , Delbrook, C. , Feldman, S.A. , Fry, T.J. , Orentas, R. , Sabatino, M. , Shah, N.N. , Steinberg, S.M. , Stroncek, D. , Tschernia, N. , Yuan, C. , Zhang, H. , Zhang, L. , Rosenberg, S.A. , Wayne, A.S. , Mackall, C.L. , 2015. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 385, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesterhuis, W.J. , Punt, C.J. , Hato, S.V. , Eleveld-Trancikova, D. , Jansen, B.J. , Nierkens, S. , Schreibelt, G. , de Boer, A. , Van Herpen, C.M. , Kaanders, J.H. , van Krieken, J.H. , Adema, G.J. , Figdor, C.G. , de Vries, I.J. , 2011. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J. Clin. Invest. 121, 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Peng, W. , Xu, C. , Lou, Y. , Zhang, M. , Wargo, J.A. , Chen, J.Q. , Li, H.S. , Watowich, S.S. , Yang, Y. , Tompers Frederick, D. , Cooper, Z.A. , Mbofung, R.M. , Whittington, M. , Flaherty, K.T. , Woodman, S.E. , Davies, M.A. , Radvanyi, L.G. , Overwijk, W.W. , Lizee, G. , Hwu, P. , 2013. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 19, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, A.S. , Taylor, J.R. , Farzaneh, F. , Kemeny, D.M. , Dibb, N.J. , Maher, J. , 2008. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol. Immunol. 45, 1276–1287. [DOI] [PubMed] [Google Scholar]
- Louis, C.U. , Savoldo, B. , Dotti, G. , Pule, M. , Yvon, E. , Myers, G.D. , Rossig, C. , Russell, H.V. , Diouf, O. , Liu, E. , Liu, H. , Wu, M.F. , Gee, A.P. , Mei, Z. , Rooney, C.M. , Heslop, H.E. , Brenner, M.K. , 2011. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 118, 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher, J. , 2012. Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol. 2012, 278093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher, J. , 2014. Clinical immunotherapy of B-cell malignancy using CD19-targeted CAR T-cells. Curr. Gene Ther. 14, 35–43. [DOI] [PubMed] [Google Scholar]
- Maher, J. , Brentjens, R.J. , Gunset, G. , Riviere, I. , Sadelain, M. , 2002. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat. Biotechnol. 20, 70–75. [DOI] [PubMed] [Google Scholar]
- Maine, C.J. , Aziz, N.H. , Chatterjee, J. , Hayford, C. , Brewig, N. , Whilding, L. , George, A.J. , Ghaem-Maghami, S. , 2014. Programmed death ligand-1 over-expression correlates with malignancy and contributes to immune regulation in ovarian cancer. Cancer Immunol. Immunother. 63, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley, J.C. , Sadelain, M. , 2010. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 115, 3508–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude, S.L. , Barrett, D. , Teachey, D.T. , Grupp, S.A. , 2014. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 20, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude, S.L. , Frey, N. , Shaw, P.A. , Aplenc, R. , Barrett, D.M. , Bunin, N.J. , Chew, A. , Gonzalez, V.E. , Zheng, Z. , Lacey, S.F. , Mahnke, Y.D. , Melenhorst, J.J. , Rheingold, S.R. , Shen, A. , Teachey, D.T. , Levine, B.L. , June, C.H. , Porter, D.L. , Grupp, S.A. , 2014. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 371, 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude, S.L. , Teachey, D.T. , Porter, D.L. , Grupp, S.A. , 2015. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 125, 4017–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus, M.V. , Haas, A.R. , Beatty, G.L. , Albelda, S.M. , Levine, B.L. , Liu, X. , Zhao, Y. , Kalos, M. , June, C.H. , 2013. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 1, 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero, I. , Berman, D.M. , Aznar, M.A. , Korman, A.J. , Gracia, J.L. , Haanen, J. , 2015. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer. 15, 457–472. [DOI] [PubMed] [Google Scholar]
- Miao, H. , Choi, B.D. , Suryadevara, C.M. , Sanchez-Perez, L. , Yang, S. , De Leon, G. , Sayour, E.J. , McLendon, R. , Herndon, J.E. , Healy, P. , Archer, G.E. , Bigner, D.D. , Johnson, L.A. , Sampson, J.H. , 2014. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One. 9, e94281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone, M.C. , Fish, J.D. , Carpenito, C. , Carroll, R.G. , Binder, G.K. , Teachey, D. , Samanta, M. , Lakhal, M. , Gloss, B. , Danet-Desnoyers, G. , Campana, D. , Riley, J.L. , Grupp, S.A. , June, C.H. , 2009. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther.: J. Am. Soc. Gene Ther. 17, 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molon, B. , Ugel, S. , Del Pozzo, F. , Soldani, C. , Zilio, S. , Avella, D. , De Palma, A. , Mauri, P. , Monegal, A. , Rescigno, M. , Savino, B. , Colombo, P. , Jonjic, N. , Pecanic, S. , Lazzarato, L. , Fruttero, R. , Gasco, A. , Bronte, V. , Viola, A. , 2011. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 208, 1949–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon, E.K. , Carpenito, C. , Sun, J. , Wang, L.C. , Kapoor, V. , Predina, J. , Powell, D.J. , Riley, J.L. , June, C.H. , Albelda, S.M. , 2011. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 17, 4719–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, R.A. , Johnson, L.A. , Davis, J.L. , Zheng, Z. , Woolard, K.D. , Reap, E.A. , Feldman, S.A. , Chinnasamy, N. , Kuan, C.T. , Song, H. , Zhang, W. , Fine, H.A. , Rosenberg, S.A. , 2012. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 23, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, R.A. , Yang, J.C. , Kitano, M. , Dudley, M.E. , Laurencot, C.M. , Rosenberg, S.A. , 2010. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther.: J. Am. Soc. Gene Ther. 18, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenroth, A. , Cartellieri, M. , Schmitz, M. , Gunes, S. , Weigle, B. , Bachmann, M. , Abken, H. , Rieber, E.P. , Temme, A. , 2007. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate. 67, 1121–1131. [DOI] [PubMed] [Google Scholar]
- Mulligan, J.K. , Day, T.A. , Gillespie, M.B. , Rosenzweig, S.A. , Young, M.R. , 2009. Secretion of vascular endothelial growth factor by oral squamous cell carcinoma cells skews endothelial cells to suppress T-cell functions. Hum. Immunol. 70, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniappan, A. , Banapour, B. , Lebkowski, J. , Talib, S. , 2000. Ligand-mediated cytolysis of tumor cells: use of heregulin-zeta chimeras to redirect cytotoxic T lymphocytes. Cancer Gene Ther. 7, 128–134. [DOI] [PubMed] [Google Scholar]
- Nakagawa, Y. , Negishi, Y. , Shimizu, M. , Takahashi, M. , Ichikawa, M. , Takahashi, H. , 2015. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol. Lett. 167, (2) 72–86. [DOI] [PubMed] [Google Scholar]
- Nishio, N. , Diaconu, I. , Liu, H. , Cerullo, V. , Caruana, I. , Hoyos, V. , Bouchier-Hayes, L. , Savoldo, B. , Dotti, G. , 2014. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 74, 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman, M.Z. , Buart, S. , Romero, P. , Ketari, S. , Janji, B. , Mari, B. , Mami-Chouaib, F. , Chouaib, S. , 2012. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 72, 4629–4641. [DOI] [PubMed] [Google Scholar]
- Ohta, A. , Gorelik, E. , Prasad, S.J. , Ronchese, F. , Lukashev, D. , Wong, M.K. , Huang, X. , Caldwell, S. , Liu, K. , Smith, P. , Chen, J.F. , Jackson, E.K. , Apasov, S. , Abrams, S. , Sitkovsky, M. , 2006. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. U. S. A. 103, 13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, A. , Kjaergaard, J. , Sharma, S. , Mohsin, M. , Goel, N. , Madasu, M. , Fradkov, E. , Ohta, A. , Sitkovsky, M. , 2009. In vitro induction of T cells that are resistant to A2 adenosine receptor-mediated immunosuppression. Br. J. Pharmacol. 156, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozao-Choy, J. , Ma, G. , Kao, J. , Wang, G.X. , Meseck, M. , Sung, M. , Schwartz, M. , Divino, C.M. , Pan, P.Y. , Chen, S.H. , 2009. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 69, 2514–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pameijer, C.R. , Navanjo, A. , Meechoovet, B. , Wagner, J.R. , Aguilar, B. , Wright, C.L. , Chang, W.C. , Brown, C.E. , Jensen, M.C. , 2007. Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T cell antigen receptor. Cancer Gene Ther. 14, 91–97. [DOI] [PubMed] [Google Scholar]
- Papa, S. , van Schalkwyk, M. , Maher, J. , 2015. Clinical evaluation of ErbB-targeted CAR T-cells, following intracavity delivery in patients with ErbB-expressing solid tumors. Methods Mol. Biol. 1317, 365–382. [DOI] [PubMed] [Google Scholar]
- Parente-Pereira, A.C. , Burnet, J. , Ellison, D. , Foster, J. , Davies, D.M. , van der Stegen, S. , Burbridge, S. , Chiapero-Stanke, L. , Wilkie, S. , Mather, S. , Maher, J. , 2011. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J. Clin. Immunol. 31, 710–718. [DOI] [PubMed] [Google Scholar]
- Parente-Pereira, A.C. , Whilding, L.M. , Brewig, N. , van der Stegen, S.J. , Davies, D.M. , Wilkie, S. , van Schalkwyk, M.C. , Ghaem-Maghami, S. , Maher, J. , 2013. Synergistic chemoimmunotherapy of epithelial ovarian Cancer using ErbB-retargeted T cells combined with carboplatin. J. Immunol. 191, 2437–2445. [DOI] [PubMed] [Google Scholar]
- Park, J.H. , Riviere, I. , Wang, X. , Bernal, Y. , Purdon, T. , Halton, E. , Curran, K.J. , Sauter, C.S. , Sadelain, M. , Brentjens, R.J. , 2015. Efficacy and safety of CD19-targeted 19-28z CAR modified T cells in adult patients with relapsed or refractory B-ALL. 2015 ASCO Annual Meeting J. Clin. Oncol. Chicago, Illinois, p. suppl; abstr 7010 [Google Scholar]
- Park, J.R. , Digiusto, D.L. , Slovak, M. , Wright, C. , Naranjo, A. , Wagner, J. , Meechoovet, H.B. , Bautista, C. , Chang, W.C. , Ostberg, J.R. , Jensen, M.C. , 2007. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther.: J. Am. Soc. Gene Ther. 15, 825–833. [DOI] [PubMed] [Google Scholar]
- Pearce, E.L. , Walsh, M.C. , Cejas, P.J. , Harms, G.M. , Shen, H. , Wang, L.S. , Jones, R.G. , Choi, Y. , 2009. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram, H.J. , Lee, J.C. , Hayman, E.G. , Imperato, G.H. , Tedder, T.F. , Sadelain, M. , Brentjens, R.J. , 2012. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 119, 4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, W. , Liu, C. , Xu, C. , Lou, Y. , Chen, J. , Yang, Y. , Yagita, H. , Overwijk, W.W. , Lizee, G. , Radvanyi, L. , Hwu, P. , 2012. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 72, 5209–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, W. , Ye, Y. , Rabinovich, B.A. , Liu, C. , Lou, Y. , Zhang, M. , Whittington, M. , Yang, Y. , Overwijk, W.W. , Lizee, G. , Hwu, P. , 2010. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 16, 5458–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrausch, U. , Schuberth, P.C. , Hagedorn, C. , Soltermann, A. , Tomaszek, S. , Stahel, R. , Weder, W. , Renner, C. , 2012. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer. 12, 615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip, B. , Kokalaki, E. , Mekkaoui, L. , Thomas, S. , Straathof, K. , Flutter, B. , Marin, V. , Marafioti, T. , Chakraverty, R. , Linch, D. , Quezada, S.A. , Peggs, K.S. , Pule, M. , 2014. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 124, 1277–1287. [DOI] [PubMed] [Google Scholar]
- Pinthus, J.H. , Waks, T. , Malina, V. , Kaufman-Francis, K. , Harmelin, A. , Aizenberg, I. , Kanety, H. , Ramon, J. , Eshhar, Z. , 2004. Adoptive immunotherapy of prostate cancer bone lesions using redirected effector lymphocytes. J. Clin. Invest. 114, 1774–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzitola, I. , Anjos-Afonso, F. , Rouault-Pierre, K. , Lassailly, F. , Tettamanti, S. , Spinelli, O. , Biondi, A. , Biagi, E. , Bonnet, D. , 2014. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 28, 1596–1605. [DOI] [PubMed] [Google Scholar]
- Poirot, L. , Philip, B. , Schiffer-Mannioui, C. , Le Clerre, D. , Chion-Sotinel, I. , Derniame, S. , Bas, C. , Potrel, P. , Lemaire, L. , Duclert, A. , Galetto, R. , Lebuhotel, C. , Eyquem, J. , Cheung, G.W. , Gouble, A. , Arnould, S. , Peggs, K.S. , Pule, M. , Scharenberg, A. , Smith, J. , 2015. Multiplex genome edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 75, (18) 3853–3864. [DOI] [PubMed] [Google Scholar]
- Pol, J. , Vacchelli, E. , Aranda, F. , Castoldi, F. , Eggermont, A. , Cremer, I. , Sautes-Fridman, C. , Fucikova, J. , Galon, J. , Spisek, R. , Tartour, E. , Zitvogel, L. , Kroemer, G. , Galluzzi, L. , 2015. Trial Watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology. 4, e1008866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, D.L. , Levine, B.L. , Kalos, M. , Bagg, A. , June, C.H. , 2011. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 365, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proietti, E. , Moschella, F. , Capone, I. , Belardelli, F. , 2012. Exploitation of the propulsive force of chemotherapy for improving the response to cancer immunotherapy. Mol. Oncol. 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi, E. , Genovese, P. , Lombardo, A. , Magnani, Z. , Liu, P.Q. , Reik, A. , Chu, V. , Paschon, D.E. , Zhang, L. , Kuball, J. , Camisa, B. , Bondanza, A. , Casorati, G. , Ponzoni, M. , Ciceri, F. , Bordignon, C. , Greenberg, P.D. , Holmes, M.C. , Gregory, P.D. , Naldini, L. , Bonini, C. , 2012. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 18, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule, M.A. , Savoldo, B. , Myers, G.D. , Rossig, C. , Russell, H.V. , Dotti, G. , Huls, M.H. , Liu, E. , Gee, A.P. , Mei, Z. , Yvon, E. , Weiss, H.L. , Liu, H. , Rooney, C.M. , Heslop, H.E. , Brenner, M.K. , 2008. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 14, 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule, M.A. , Straathof, K.C. , Dotti, G. , Heslop, H.E. , Rooney, C.M. , Brenner, M.K. , 2005. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 12, 933–941. [DOI] [PubMed] [Google Scholar]
- Qin, H. , Cho, M. , Haso, W. , Zhang, L. , Tasian, S.K. , Oo, H.Z. , Negri, G.L. , Lin, Y. , Zou, J. , Mallon, B.S. , Maude, S. , Teachey, D.T. , Barrett, D.M. , Orentas, R.J. , Daugaard, M. , Sorensen, P.H. , Grupp, S.A. , Fry, T.J. , 2015. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood. 126, 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintarelli, C. , Vera, J.F. , Savoldo, B. , Giordano Attianese, G.M. , Pule, M. , Foster, A.E. , Heslop, H.E. , Rooney, C.M. , Brenner, M.K. , Dotti, G. , 2007. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 110, 2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainusso, N. , Brawley, V.S. , Ghazi, A. , Hicks, M.J. , Gottschalk, S. , Rosen, J.M. , Ahmed, N. , 2012. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 19, 212–217. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan, R. , Assudani, D. , Nagaraj, S. , Hunter, T. , Cho, H.I. , Antonia, S. , Altiok, S. , Celis, E. , Gabrilovich, D.I. , 2010. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J. Clin. Invest. 120, 1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell, S.R. , Sommermeyer, D. , Berger, C. , Liu, L.S. , Balakrishnan, A. , Salter, A. , Hudecek, M. , Maloney, D.G. , Turtle, C.J. , 2014. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 20, 141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righi, E. , Kashiwagi, S. , Yuan, J. , Santosuosso, M. , Leblanc, P. , Ingraham, R. , Forbes, B. , Edelblute, B. , Collette, B. , Xing, D. , Kowalski, M. , Mingari, M.C. , Vianello, F. , Birrer, M. , Orsulic, S. , Dranoff, G. , Poznansky, M.C. , 2011. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 71, 5522–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, D.S. , Neeson, P.J. , Khot, A. , Peinert, S. , Tai, T. , Tainton, K. , Chen, K. , Shin, M. , Wall, D.M. , Honemann, D. , Gambell, P. , Westerman, D.A. , Haurat, J. , Westwood, J.A. , Scott, A.M. , Kravets, L. , Dickinson, M. , Trapani, J.A. , Smyth, M.J. , Darcy, P.K. , Kershaw, M.H. , Prince, H.M. , 2013. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther.: J. Am. Soc. Gene Ther. 21, 2122–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saied, A. , Licata, L. , Burga, R.A. , Thorn, M. , McCormack, E. , Stainken, B.F. , Assanah, E.O. , Khare, P.D. , Davies, R. , Espat, N.J. , Junghans, R.P. , Katz, S.C. , 2014. Neutrophil:lymphocyte ratios and serum cytokine changes after hepatic artery chimeric antigen receptor-modified T-cell infusions for liver metastases. Cancer Gene Ther. 21, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon, H. , Franciszkiewicz, K. , Damotte, D. , Dieu-Nosjean, M.C. , Validire, P. , Trautmann, A. , Mami-Chouaib, F. , Donnadieu, E. , 2012. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 122, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson, J.H. , Choi, B.D. , Sanchez-Perez, L. , Suryadevara, C.M. , Snyder, D.J. , Flores, C.T. , Schmittling, R.J. , Nair, S.K. , Reap, E.A. , Norberg, P.K. , Herndon, J.E. , Kuan, C.T. , Morgan, R.A. , Rosenberg, S.A. , Johnson, L.A. , 2014. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 20, 972–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo, B. , Ramos, C.A. , Liu, E. , Mims, M.P. , Keating, M.J. , Carrum, G. , Kamble, R.T. , Bollard, C.M. , Gee, A.P. , Mei, Z. , Liu, H. , Grilley, B. , Rooney, C.M. , Heslop, H.E. , Brenner, M.K. , Dotti, G. , 2011. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 121, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlie, K. , Spowart, J.E. , Hughson, L.R. , Townsend, K.N. , Lum, J.J. , 2011. When cells suffocate: autophagy in cancer and immune cells under low oxygen. Int. J. Cel. Biol. 2011, 470597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth, P.C. , Hagedorn, C. , Jensen, S.M. , Gulati, P. , van den Broek, M. , Mischo, A. , Soltermann, A. , Jungel, A. , Marroquin Belaunzaran, O. , Stahel, R. , Renner, C. , Petrausch, U. , 2013. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 11, 187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster, S.J. , Svoboda, J. , Nasta, S. , Porter, D.L. , Mato, A. , Shah, G.D. , Landsburg, D.J. , Chong, E.A. , Lacey, S.F. , Melenhorst, J.J. , Chew, A. , Hasskarl, J. , Shah, N.N. , Wasik, M.A. , Marcucci, K. , Zheng, Z. , Levine, B. , June, C.H. , 2015. Phase IIa trial of chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with relapsed or refractory CD19+ lymphomas. 2015 ASCO Annual Meeting J. Clin. Oncol. Chicag, Illinois, p. suppl; abstr 8516 [Google Scholar]
- Scotton, C.J. , Wilson, J.L. , Scott, K. , Stamp, G. , Wilbanks, G.D. , Fricker, S. , Bridger, G. , Balkwill, F.R. , 2002. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 62, 5930–5938. [PubMed] [Google Scholar]
- Sharifzadeh, Z. , Rahbarizadeh, F. , Shokrgozar, M.A. , Ahmadvand, D. , Mahboudi, F. , Jamnani, F.R. , Moghimi, S.M. , 2013. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett. 334, 237–244. [DOI] [PubMed] [Google Scholar]
- Sheen, A.J. , Sherlock, D.J. , Irlam, J. , Hawkins, R.E. , Gilham, D.E. , 2003. T lymphocytes isolated from patients with advanced colorectal cancer are suitable for gene immunotherapy approaches. Br. J. Cancer. 88, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimali, R.K. , Yu, Z. , Theoret, M.R. , Chinnasamy, D. , Restifo, N.P. , Rosenberg, S.A. , 2010. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 70, 6171–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitkovsky, M.V. , Hatfield, S. , Abbott, R. , Belikoff, B. , Lukashev, D. , Ohta, A. , 2014. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol. Res. 2, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. , Poirot, L. , Philip, B. , Schiffer-Mannoui, C. , Derniame, S. , Zhan, H. , Stafford, S. , Reynier, S. , Arnould, S. , Gouble, A. , Qasim, W. , Peggs, K. , Pule, M. , Sourdive, D. , 2015. UCART19, an allogeneic “off-the-shelf” adoptive T-cell immunotherapy against CD19+ B-cell leukemias. ASCO Annual Meeting J. Clin. Oncol. p. (suppl; abstr 3069) [Google Scholar]
- Song, D.G. , Ye, Q. , Carpenito, C. , Poussin, M. , Wang, L.P. , Ji, C. , Figini, M. , June, C.H. , Coukos, G. , Powell, D.J. , 2011. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 71, 4617–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D.G. , Ye, Q. , Poussin, M. , Harms, G.M. , Figini, M. , Powell, D.J. , 2012. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 119, 696–706. [DOI] [PubMed] [Google Scholar]
- Stephan, M.T. , Ponomarev, V. , Brentjens, R.J. , Chang, A.H. , Dobrenkov, K.V. , Heller, G. , Sadelain, M. , 2007. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat. Med. 13, 1440–1449. [DOI] [PubMed] [Google Scholar]
- Straathof, K.C. , Pule, M.A. , Yotnda, P. , Dotti, G. , Vanin, E.F. , Brenner, M.K. , Heslop, H.E. , Spencer, D.M. , Rooney, C.M. , 2005. An inducible caspase 9 safety switch for T-cell therapy. Blood. 105, 4247–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar, M. , Liu, J. , Ji, Y. , Subramanian, M. , Crompton, J.G. , Yu, Z. , Roychoudhuri, R. , Palmer, D.C. , Muranski, P. , Karoly, E.D. , Mohney, R.P. , Klebanoff, C.A. , Lal, A. , Finkel, T. , Restifo, N.P. , Gattinoni, L. , 2013. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 123, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, M. , Shi, H. , Liu, C. , Liu, J. , Liu, X. , Sun, Y. , 2014. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res. 16, R61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann, J.B. , Smyth, M.J. , 2007. Immune surveillance of tumors. J. Clin. Invest. 117, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada, K. , Geng, D. , Sakoda, Y. , Bansal, N. , Srivastava, R. , Li, Z. , Davila, E. , 2012. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 18, 6436–6445. [DOI] [PubMed] [Google Scholar]
- Tammana, S. , Huang, X. , Wong, M. , Milone, M.C. , Ma, L. , Levine, B.L. , June, C.H. , Wagner, J.E. , Blazar, B.R. , Zhou, X. , 2010. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum. Gene Ther. 21, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanyi, J.L. , Haas, A.R. , Beatty, G.R. , Morgan, M.A. , Stashwick, C.J. , O'Hara, M.H. , Porter, D.L. , Maus, M.V. , Levine, B.L. , Lacey, S.F. , Nelson, A.M. , McGarvey, M. , Kerr, N.D.S. , Plesa, G. , June, C.H. , 2015. Safety and feasibility of chimeric antigen receptor modified T cells directed against mesothelin (CART-meso) in patients with mesothelin expressing cancers. Proceedings of the American Association for Cancer Research Meeting, Philadelphia, Pennsylvania Abstract number CT105 [Google Scholar]
- Teo, P.Y. , Yang, C. , Whilding, L.M. , Parente-Pereira, A.C. , Maher, J. , George, A.J. , Hedrick, J.L. , Yang, Y.Y. , Ghaem-Maghami, S. , 2015. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: strategies to enhance T cell killing. Adv. Healthc. Mater. 4, 1180–1189. [DOI] [PubMed] [Google Scholar]
- Themeli, M. , Kloss, C.C. , Ciriello, G. , Fedorov, V.D. , Perna, F. , Gonen, M. , Sadelain, M. , 2013. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 31, 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themeli, M. , Riviere, I. , Sadelain, M. , 2015. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 16, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, R.H. , Kuntz, S.M. , Leibovich, B.C. , Dong, H. , Lohse, C.M. , Webster, W.S. , Sengupta, S. , Frank, I. , Parker, A.S. , Zincke, H. , Blute, M.L. , Sebo, T.J. , Cheville, J.C. , Kwon, E.D. , 2006. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 66, 3381–3385. [DOI] [PubMed] [Google Scholar]
- Till, B.G. , Jensen, M.C. , Wang, J. , Chen, E.Y. , Wood, B.L. , Greisman, H.A. , Qian, X. , James, S.E. , Raubitschek, A. , Forman, S.J. , Gopal, A.K. , Pagel, J.M. , Lindgren, C.G. , Greenberg, P.D. , Riddell, S.R. , Press, O.W. , 2008. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 112, 2261–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till, B.G. , Jensen, M.C. , Wang, J. , Qian, X. , Gopal, A.K. , Maloney, D.G. , Lindgren, C.G. , Lin, Y. , Pagel, J.M. , Budde, L.E. , Raubitschek, A. , Forman, S.J. , Greenberg, P.D. , Riddell, S.R. , Press, O.W. , 2012. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 119, 3940–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, S.L. , Hodi, F.S. , Brahmer, J.R. , Gettinger, S.N. , Smith, D.C. , McDermott, D.F. , Powderly, J.D. , Carvajal, R.D. , Sosman, J.A. , Atkins, M.B. , Leming, P.D. , Spigel, D.R. , Antonia, S.J. , Horn, L. , Drake, C.G. , Pardoll, D.M. , Chen, L. , Sharfman, W.H. , Anders, R.A. , Taube, J.M. , McMiller, T.L. , Xu, H. , Korman, A.J. , Jure-Kunkel, M. , Agrawal, S. , McDonald, D. , Kollia, G.D. , Gupta, A. , Wigginton, J.M. , Sznol, M. , 2012. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. New Engl. J. Med. 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp, M.S. , Gokbuget, N. , Zugmaier, G. , Klappers, P. , Stelljes, M. , Neumann, S. , Viardot, A. , Marks, R. , Diedrich, H. , Faul, C. , Reichle, A. , Horst, H.A. , Bruggemann, M. , Wessiepe, D. , Holland, C. , Alekar, S. , Mergen, N. , Einsele, H. , Hoelzer, D. , Bargou, R.C. , 2014. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 32, 4134–4140. [DOI] [PubMed] [Google Scholar]
- Torikai, H. , Reik, A. , Liu, P.Q. , Zhou, Y. , Zhang, L. , Maiti, S. , Huls, H. , Miller, J.C. , Kebriaei, P. , Rabinovitch, B. , Lee, D.A. , Champlin, R.E. , Bonini, C. , Naldini, L. , Rebar, E.J. , Gregory, P.D. , Holmes, M.C. , Cooper, L.J. , 2012. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 119, 5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, E. , Chinnasamy, D. , Yu, Z. , Morgan, R.A. , Lee, C.C. , Restifo, N.P. , Rosenberg, S.A. , 2013. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 210, 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traversari, C. , Marktel, S. , Magnani, Z. , Mangia, P. , Russo, V. , Ciceri, F. , Bonini, C. , Bordignon, C. , 2007. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 109, 4708–4715. [DOI] [PubMed] [Google Scholar]
- Urbanska, K. , Lanitis, E. , Poussin, M. , Lynn, R.C. , Gavin, B.P. , Kelderman, S. , Yu, J. , Scholler, N. , Powell, D.J. , 2012. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 72, 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton, J. , Guyot, V. , Marechal, A. , Filhol, J.M. , Juillerat, A. , Duclert, A. , Duchateau, P. , Poirot, L. , 2015. A multidrug-resistant engineered CAR T cell for allogeneic combination immunotherapy. Mol. Ther.: J. Am. Soc. Gene Ther. 23, (9) 1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stegen, S.J. , Davies, D.M. , Wilkie, S. , Foster, J. , Sosabowski, J.K. , Burnet, J. , Whilding, L.M. , Petrovic, R.M. , Ghaem-Maghami, S. , Mather, S. , Jeannon, J.P. , Parente-Pereira, A.C. , Maher, J. , 2013. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity?. J. Immunol. 191, 4589–4598. [DOI] [PubMed] [Google Scholar]
- van der Stegen, S.J. , Hamieh, M. , Sadelain, M. , 2015. The pharmacology of second-generation chimeric antigen receptors. Nature reviews. Drug Discov. 14, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schalkwyk, M.C. , Papa, S.E. , Jeannon, J.P. , Guerrero Urbano, T. , Spicer, J.F. , Maher, J. , 2013. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum. Gene Ther. Clin. Dev. 24, 134–142. [DOI] [PubMed] [Google Scholar]
- Vanneman, M. , Dranoff, G. , 2012. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer. 12, 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinay, D.S. , Ryan, E.P. , Pawelec, G. , Talib, W.H. , Stagg, J. , Elkord, E. , Lichtor, T. , Decker, W.K. , Whelan, R.L. , Kumara, H.M. , Signori, E. , Honoki, K. , Georgakilas, A.G. , Amin, A. , Helferich, W.G. , Boosani, C.S. , Guha, G. , Ciriolo, M.R. , Chen, S. , Mohammed, S.I. , Azmi, A.S. , Keith, W.N. , Bilsland, A. , Bhakta, D. , Halicka, D. , Fujii, H. , Aquilano, K. , Ashraf, S.S. , Nowsheen, S. , Yang, X. , Choi, B.K. , Kwon, B.S. , 2015. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin. Cancer Biol. (in press). http://dx.doi.org/10.1016/j.semcancer.2015.03.004 [DOI] [PubMed] [Google Scholar]
- Wang, E. , Wang, L.C. , Tsai, C.Y. , Bhoj, V. , Gershenson, Z. , Moon, E. , Newick, K. , Sun, J. , Lo, A. , Baradet, T. , Feldman, M.D. , Barrett, D. , Pure, E. , Albelda, S. , Milone, M.C. , 2015. Generation of potent T-cell immunotherapy for Cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol. Res. 3, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L.C. , Lo, A. , Scholler, J. , Sun, J. , Majumdar, R.S. , Kapoor, V. , Antzis, M. , Cotner, C.E. , Johnson, L.A. , Durham, A.C. , Solomides, C.C. , June, C.H. , Pure, E. , Albelda, S.M. , 2014. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2, 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Chang, W.C. , Wong, C.W. , Colcher, D. , Sherman, M. , Ostberg, J.R. , Forman, S.J. , Riddell, S.R. , Jensen, M.C. , 2011. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 118, 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Zhang, W.Y. , Han, Q.W. , Liu, Y. , Dai, H.R. , Guo, Y.L. , Bo, J. , Fan, H. , Zhang, Y. , Zhang, Y.J. , Chen, M.X. , Feng, K.C. , Wang, Q.S. , Fu, X.B. , Han, W.D. , 2014. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin. Immunol. 155, 160–175. [DOI] [PubMed] [Google Scholar]
- Warburg, O. , Wind, F. , Negelein, E. , 1927. The metabolism of tumors in the Body. J. Gen. Physiol. 8, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, K. , Terakura, S. , Martens, A.C. , van Meerten, T. , Uchiyama, S. , Imai, M. , Sakemura, R. , Goto, T. , Hanajiri, R. , Imahashi, N. , Shimada, K. , Tomita, A. , Kiyoi, H. , Nishida, T. , Naoe, T. , Murata, M. , 2015. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 194, 911–920. [DOI] [PubMed] [Google Scholar]
- Watt, J. , Kocher, H.M. , 2013. The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration. Oncoimmunology. 2, e26788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weishaupt, C. , Munoz, K.N. , Buzney, E. , Kupper, T.S. , Fuhlbrigge, R.C. , 2007. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 13, 2549–2556. [DOI] [PubMed] [Google Scholar]
- Wilkie, S. , Burbridge, S.E. , Chiapero-Stanke, L. , Pereira, A.C. , Cleary, S. , van der Stegen, S.J. , Spicer, J.F. , Davies, D.M. , Maher, J. , 2010. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 285, 25538–25544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie, S. , Picco, G. , Foster, J. , Davies, D.M. , Julien, S. , Cooper, L. , Arif, S. , Mather, S.J. , Taylor-Papadimitriou, J. , Burchell, J.M. , Maher, J. , 2008. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J. Immunol. 180, 4901–4909. [DOI] [PubMed] [Google Scholar]
- Wilkie, S. , van Schalkwyk, M.C. , Hobbs, S. , Davies, D.M. , van der Stegen, S.J. , Pereira, A.C. , Burbridge, S.E. , Box, C. , Eccles, S.A. , Maher, J. , 2012. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 32, 1059–1070. [DOI] [PubMed] [Google Scholar]
- Wu, A.A. , Drake, V. , Huang, H.S. , Chiu, S. , Zheng, L. , 2015. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology. 4, e1016700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Conejo-Garcia, J.R. , Katsaros, D. , Gimotty, P.A. , Massobrio, M. , Regnani, G. , Makrigiannakis, A. , Gray, H. , Schlienger, K. , Liebman, M.N. , Rubin, S.C. , Coukos, G. , 2003. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. New Engl. J. Med. 348, 203–213. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Kerkar, S.P. , Yu, Z. , Zheng, Z. , Yang, S. , Restifo, N.P. , Rosenberg, S.A. , Morgan, R.A. , 2011. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther. 19, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Wang, Q.J. , Yang, S. , Kochenderfer, J.N. , Zheng, Z. , Zhong, X. , Sadelain, M. , Eshhar, Z. , Rosenberg, S.A. , Morgan, R.A. , 2009. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 183, 5563–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, X.S. , Matsushita, M. , Plotkin, J. , Riviere, I. , Sadelain, M. , 2010. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther.: J. Am. Soc. Gene Ther. 18, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Di Stasi, A. , Tey, S.K. , Krance, R.A. , Martinez, C. , Leung, K.S. , Durett, A.G. , Wu, M.F. , Liu, H. , Leen, A.M. , Savoldo, B. , Lin, Y.F. , Grilley, B.J. , Gee, A.P. , Spencer, D.M. , Rooney, C.M. , Heslop, H.E. , Brenner, M.K. , Dotti, G. , 2014. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 123, 3895–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Dotti, G. , Krance, R.A. , Martinez, C.A. , Naik, S. , Kamble, R.T. , Durett, A.G. , Dakhova, O. , Savoldo, B. , Di Stasi, A. , Spencer, D.M. , Lin, Y.F. , Liu, H. , Grilley, B.J. , Gee, A.P. , Rooney, C.M. , Heslop, H.E. , Brenner, M.K. , 2015. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 125, 4103–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Li, J. , Wang, Z. , Chen, Z. , Qiu, J. , Zhang, Y. , Wang, W. , Ma, Y. , Huang, N. , Cui, K. , Li, J. , Wei, Y.Q. , 2013. Cellular immunotherapy for carcinoma using genetically modified EGFR-specific T lymphocytes. Neoplasia. 15, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel, L. , Apetoh, L. , Ghiringhelli, F. , Kroemer, G. , 2008. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 8, 59–73. [DOI] [PubMed] [Google Scholar]