

Abstract
The new age of Precision Cancer Medicine, with specific biomarkers being used to direct targeted agents, generally concerns only a subset of patients within a certain histopathologically defined tumor type. This paradigm is challenged by the need to perform widespread molecular screening in certified laboratories, with results available to clinicians within reasonable timeframe. Tumor heterogeneity and clonal evolution must be considered in the decision making process. Adaptive and innovative clinical trial designs exploring predictive algorithms and reconsideration of traditional efficacy endpoints are required to rapidly translate scientific discoveries into patient care. Furthermore, international collaboration in cancer research and open discussions on the availability of investigational agents will likely redefine the drug development and approval process in the coming years.
Keywords: Clinical trial design, Precision Cancer Medicine, Targeted therapy
Highlights
Co‐developing biomarkers and drugs is crucial in genomically‐guided therapies.
Innovative trials have adaptive designs for patient accrual and drug selection.
Trials with novel efficacy endpoints in non‐advanced settings are anticipated.
Circulating biomarkers may help individualize therapy in acquired resistance setting.
Abbreviations
- CSC
Cancer stem cell
- COSMIC
Catalogue of Somatic Mutations in Cancer
- CTC
Circulating tumor cell
- ctDNA
Circulating tumor DNA
- CRC
Colorectal cancer
- DCR
Disease control rate
- EMA
European Medicines Agency
- FDA
Food and Drug Administration
- GIST
Gastrointestinal stromal cell tumor
- ICGC
International Cancer Genome Consortium
- IDE
Investigational Device Exemption
- NCI
National Cancer Institute
- NSCLC
Non-small cell lung cancer
- OS
Overall survival
- PCM
Precision Cancer Medicine
- PFS
Progression-free survival
- RECIST
Response Evaluation Criteria In Solid Tumors
- TCGA
The Cancer Genome Atlas
- TTG
Time to tumor growth
- TGR
Tumor growth rate
- WIN
Worldwide Innovative Networking
1. Introduction
With the development and clinical use of molecularly targeted agents, it soon became clear that only selected patient populations derive benefit from such therapies. Precision Cancer Medicine (PCM) has emerged from the accumulated evidence on matching targeted agents with tumor molecular aberrations (Hoelder et al., 2012). Drugs designed to interact with a specific target, and especially those that used predictive biomarkers, were able to produce the highest relative improvement in response rate and survival (Ocana et al., 2013). Current knowledge gathered from large‐scale collaborative sequencing projects such as the Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC), in addition to publicly available resources such as the cBioPortal for Cancer Genomics and the Catalogue of Somatic Mutations in Cancer (COSMIC) have facilitated our understanding of the genetic interpatient tumor heterogeneity in multiple cancers subtypes (Dienstmann et al., 2013a).
However, recent studies have also described striking intrapatient intratumor heterogeneity and how clonal evolution under treatment pressure may represent major challenges to PCM, questioning the value of a single needle biopsy or surgical excision to accurately capture the complete genomic landscape of a patient's cancer (Bedard et al., 2013; Gerlinger et al., 2014). Moreover, with few exceptions, most druggable genomic aberrations are present only in small to moderate proportions of patients, further emphasizing multicenter collaboration in early drug development as critical for successful clinical trial enrollment. Nevertheless, we believe that the described heterogeneity in genomic profiles, in particular, applies to bystander mutations and that true tumor‐driving events are usually present in the majority of subclones from the primary tumor as well as the metastatic lesions (Yap et al., 2012). Therefore, regimens that target genomic alterations with high variant frequencies are expected to provide substantial tumor responses. As clinical responses to targeted agents are consistently abrogated by the development of drug resistance, we see repeated tumor biopsies of progressing lesions and/or characterization of circulating markers (tumor cells, tumor DNA) as a key component of patient's care, allowing identification of mechanisms of resistance and potentially guiding alternative treatment options with investigational agents (Dienstmann et al., 2013a). Clinical trial designs for cancer diagnostics and therapeutics must take into consideration these rate‐limiting steps in order to efficiently and dynamically incorporate genomic data and assess the value of matching profiled patients to specific interventions or targeted therapies. Here, we discuss some of the challenges to rapidly translate scientific discoveries to effective drug development programs and present clinical trial frameworks to test PCM with novel efficacy endpoints. Of note, our objective is to present key concepts on this topic, knowing that most innovative trial designs in fact combine these ideas and take advantage of adaptive flexible models for successful proof‐of‐concept.
2. Biomarker – drug co‐development
Clinical trial design in the era of PCM is dictated by the type of biomarker being testing or developed (Yap et al., 2010). Predictive biomarkers inform the investigator of potential anti‐tumor activity of a given therapy. Prognostic biomarkers provide information on the risk of relapse, disease progression or death. Pharmacogenomic biomarkers inform how patients respond to a drug with respect to toxicity or efficacy. Analytical validity, clinical validity and clinical utility of biomarkers need to be established during the development process. Analytical validation means confirming that the test measures with adequate sensitivity and specificity what it claims to measure. Clinical validity of a biomarker refers to how well the test works in identifying patients who will or will not respond or present toxicity to a given therapy. Finally, clinical utility means that measuring the biomarker and using it for decision‐making is beneficial to patients relative to the standard of care (Simon and Roychowdhury, 2013).
Co‐development of biomarkers and drugs is essential for the success of genomically‐guided therapies, but this strategy raises many technical and sometimes ethical issues. First, the main objectives of the trial are not only to assess the safety and efficacy of the drug, but also to investigate the performance of the diagnostic in that specific therapeutic context. Therefore, timing and alignment of the development processes, which rely on a coordinated preclinical assessment of potential biomarkers, are crucial steps for effective clinical translation. For example, this knowledge guides the decision of whether to recruit marker‐negative patients (i.e., those that are not expected to benefit from the drug) in the trial. In addition to robust and validated diagnostic assays, the pharmacological properties of the drug should be assessed before clinical testing. These include pharmacokinetics/pharmacodynamics modeling, definition of readouts of pathway inhibition and the most appropriate drug scheduling for achievement of biological effects. Importantly, specific genomic variants that are expected to predict sensitivity should be functionally validated. For non‐hotspot gene alterations it may be difficult to know whether they are involved in deregulating a particular pathway and what is the potency of the drug in this context. Systems biology and experimental models relating genomic events to drug effectiveness are needed before variants of unproven biological significance are utilized for clinical decision‐making regarding therapies. Of note, even when the diagnostic assay is validated, the gene alteration is a known driver event in a particular tumor type and a potent selective drug is available, there is no guarantee of success in a different context – the higher efficacy of vemurafenib and dabrafenib in BRAF V600E melanoma as compared to colorectal cancer (CRC) is a clear example (Dienstmann et al., 2013a).
The turnaround time for results of biomarker tests, particularly clinical next‐generation sequencing, is an important consideration for patients undergoing molecular profiling, especially in the metastatic setting when treatment decisions have to be made in a short timeframe. As an alternative to the traditional approach of centralized biomarker analysis just before considering the inclusion of the patient in a trial, we favor the alternative strategy of local prescreening at academic institutions while patients are still receiving standard treatment for advanced disease. This approach is time and tissue saving, increasing the chances of patient recruitment in early clinical trials, although the financial burden of prescreening tests is transferred from trial sponsors to health care providers (Rodon et al., 2012). All these issues have to be taken into consideration during the design of clinical trials that incorporate biomarkers.
3. Genomic and clinical databases – longitudinal cohort studies with nested clinical trials
One framework currently used by many large cancer institutions and national cancer cooperative groups is to prospectively profile a large number of patients to establish a longitudinal cohort with molecular characterization and clinical annotation (Bedard et al., 2013). This is an inclusive approach, with actionable targets being screened for all individual patients regardless of cancer type. Patients become “genomic information donors” and these databases, as opposed to TCGA, have valuable outcome data. In addition, as most patients enrolled in these programs have refractory disease, they can give “real world” information on relevant targets in this setting. As shown in Figure 1, patients with specific molecular aberrations are often “opportunistically” enrolled into Phase 1, 2 or 3 clinical trials of matched targeted agents. The coupling of a molecular characterization strategy with a drug development program has been widely embraced, although the clinical utility of this approach is still unproven. These initiatives are classified as “protocolized” or “particular” depending on whether profiling and drug matching are central parts of an umbrella protocol or not. Furthermore, they can either offer a limited gene aberration profile with a priori defined matched agents or propose a comprehensive genomic analysis irrespective of targeted drug availability. Examples of particular comprehensive programs include the Michigan Oncology Sequencing Project (MI‐ONCOSEQ), which integrates whole genome sequencing, exome capture sequencing and transcriptome analysis of tumor aberrations with multiple molecular‐driven clinical trials (Roychowdhury et al., 2011); and the Canadian IMPACT trial, with systematic use of massively parallel sequencing data for PCM in a timely fashion (Tran et al., 2013). The SAFIR and MOSCATO French trials are protocolized initiatives of targeted gene sequencing and comparative genomic hybridization guiding recruitment of breast cancer patients or Phase 1 trial candidates in matched trials, with all eligible patients under closer follow‐up (Andre et al., 2014). Another example is the Molecular Analysis for Therapy Choice (MATCH) NCI trial, an umbrella protocol for multiple single‐arm Phase 2 trials that plans to assign over 1000 patients that progress after 1 line of standard therapy to matched agents based on next generation sequencing, with at least 1/4 enrollment restricted to “rare” tumors. Interestingly, NCI plans to genotype 100 “exceptional responders” (trial participants who show noticeable improvements after treatment with cancer drugs that did not provide much benefit to others), in the hope of elucidating sensitivity mechanisms.
Figure 1.
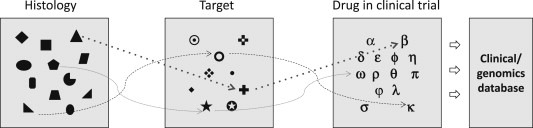
Graphical representation of longitudinal cohort studies with nested trials and clinical‐genomics databases.
Importantly, as many gene aberrations are detected in a limited number of patients, the prognostic and/or predictive value derived from analyses of individual mutations should be considered only hypothesis‐generating. Additionally, patient attrition rates, i.e., the number of patients with actionable alterations who were not treated as recommended, have not been systematically reported. There are other caveats of these initiatives, such as the need of a portfolio of drugs under investigation, on‐purpose fresh tumor biopsies, the complexity involved in interpreting sequencing results and dealing with incidental genomic findings (Dienstmann et al., 2013b).
4. Early clinical trials
An increasingly large number of putative biomarkers assessed by sophisticated technologies are being used in Phase 1 trials. The lack of fully validated and reproducible diagnostic assays is a major concern in this scenario. To prevent the stifling of innovative clinical and translational research, clearer guidelines are needed to support the use of what is best described as biomarkers that are “fit for the intended purpose” (Garcia et al., 2011). Current Food and Drug Administration (FDA) regulations require that biomarker tests used to assign therapy to patients must be studied under an Investigational Device Exemption (IDE). In early clinical trials, pharmacodynamic biomarkers must follow very rigorous standards in order to accurately define “proof‐of‐mechanism”. However, predictive biomarkers for selecting patients may be explored according to less strict validation criteria, as they may be considered the “best guess” for clinical efficacy (Yap et al., 2010). Predictive biomarkers can be either part of inclusion criteria of the trial or simply be used by investigators for “enrichment” strategies. Successfully enriching early clinical trials with patients whose tumors harbor specific molecular aberrations that may predict response can demonstrate “proof‐of‐concept” and encourage further research with a given drug or target. Lack of anticancer effect in the “best‐case scenario”, providing that sufficient target inhibition is achieved, may ultimately redefine drug development strategies. Importantly, delineating a selected patient population does not restrict the late development of a specific drug to that subpopulation. If predictive biomarkers prove to be robust and useful in early clinical trials, they are further clinically validated in larger trials. When the predictive value is not clear, and therefore the clinical utility of the biomarker is not obvious, a randomized or stratified strategy is needed to properly assess both drug and the biomarker in Phase 2 or 3 trials, as discussed later.
There is considerable current interest in defining more precise measurements of experimental drug efficacy. Analysis of response magnitude can offer insights into disease biology that go beyond tumor genotyping and matched targeted therapies. With the plethora of investigational drugs entering genomically‐driven clinical trials, several with overlapping targets but exhibiting different pharmacokinetic and pharmacodynamic properties, the magnitude of anti‐tumor effects for guiding “go‐versus‐no go” decisions is further emphasized. In this context, Response Evaluation Criteria In Solid Tumors (RECIST) criteria represent the gold standard method for assessment of the tumor response to antineoplastic agents. Of note, the thresholds that dictate the decision‐making for patients are somewhat arbitrary cut‐offs on the continuous response scale: −30% for partial response, +20% or occurrence of new lesions for progressive disease, and between these two values for stable disease. Furthermore, individual responses are pooled and the subtleties of treatment effect in individual patients and single tumor lesions are diluted or obfuscated by aggregate data. To overcome these inadequacies, a number of alternative methods exploring tumor metabolism/perfusion changes have been proposed (Levy et al., 2013). In addition, the immune component of the response (Nishino et al., 2013) and potential value of tumor kinetics with quantitative evaluation of tumor growth over time (Ferte et al., 2014) are under investigation, as discussed later.
With the interest in understanding tumor area as a continuous measure, waterfall and spider plots are increasingly being used as representative displays of individual patient tumor responses (Figure 2). Waterfall plots only show the best on‐study change in tumor burden relative to baseline, which may lead to misrepresentation of the kinetics of tumor growth over time by focusing on a single static measurement relative to a single point of reference. An alternative that addresses this limitation is the spider plot that shows a change in tumor volume over time longitudinally from a normalized baseline of zero (LoRusso et al., 2010). Spider plots allow visualization of time‐on‐treatment/time‐to‐progression for each individual patient. This information is also depicted in swim plots, well‐known horizontal barplots of time‐to‐event that frequently complement waterfall plots in many publications of early clinical trials.
Figure 2.
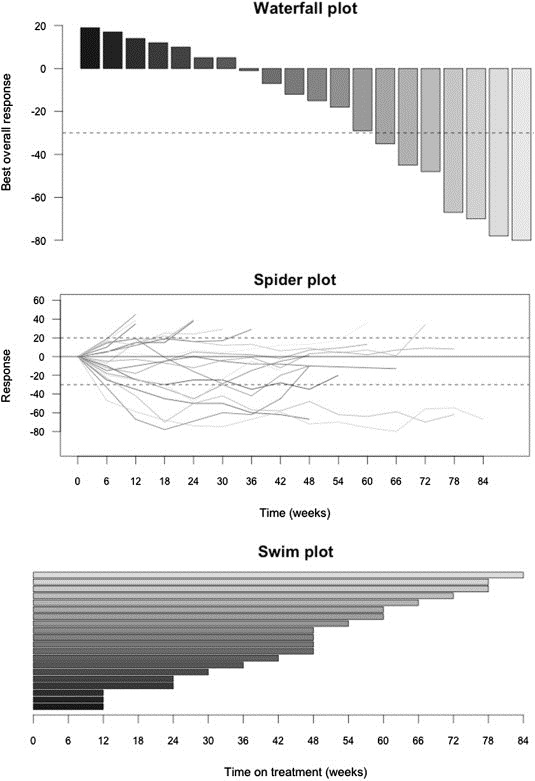
Illustrative plots to summarize response and clinical benefit in genomically‐guided clinical trials. All plots represent the same fictitious database with RECIST response rate and time on treatment information.
4.1. Trial design
4.1.1. Histology‐agnostic, aberration‐specific trials
In this framework, seen in Figure 3A, patients with different tumor histologies but who harbor the same molecular aberration receive a matched targeted in the context of expansion cohorts of a Phase 1 trial or as a separate Phase 2 trial, with efficacy as the primary endpoint. An example is the inclusion of different cancer types, either solid tumors or hematological malignancies that harbor BRAF V600 mutations, in a basket trial that evaluates vemurafenib (NCT01524978). The caveat is the possibility of insufficient representation of patients with tumor types that harbor the aberration of interest, leading to false‐negative conclusions. Therefore, one can stratify by histology, taking into consideration the reported frequencies of the genomic event (Bedard et al., 2013). This strategy may be adapted to increase enrollment of patients with tumor types that demonstrate early signals of antitumor activity while excluding those who lack preliminary response. Furthermore, additional cohorts with different tumor types can be created, and patients with related molecular aberrations can also be enrolled – those with newly identified fusion genes known to activate the pathway and sensitize to the agent under investigation similarly to gene copy number alterations or mutations, for example. Although this framework will not directly lead to regulatory approval, given its exploratory nature, it does provide a platform to determine the differences in functionality of the same molecular alteration across multiple cancer types.
Figure 3.
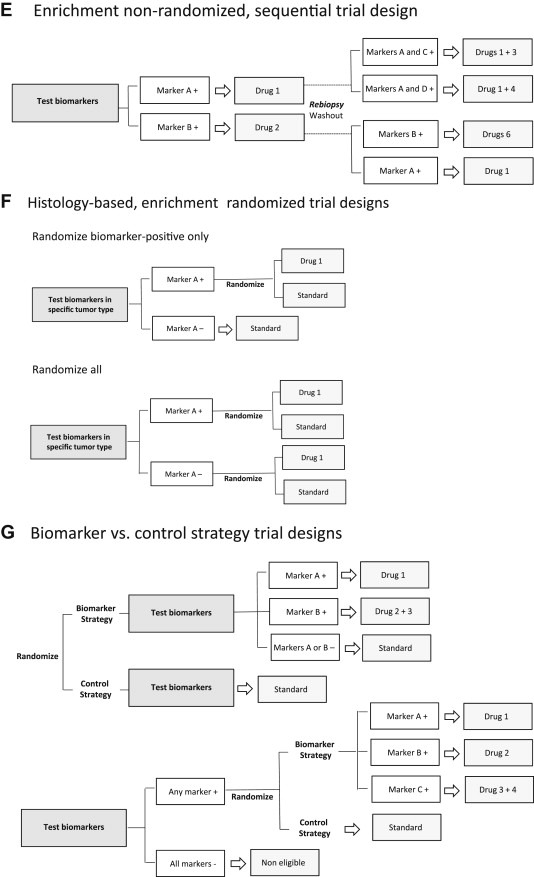
A. Histology‐agnostic, aberration‐specific trials (“Basket” design), patients receive a matched targeted therapy according to genomic profile irrespective of cancer type; B. Original “N‐of‐1” design with randomization, with analysis of the average effects of each drug across individuals; C: Modified “N‐of‐1” sequential design with each patient as his/her own control, comparing progression‐free survival on therapy guided by molecular aberration with that achieved on the regimen immediately preceding trial enrollment; D: Histology‐based, enrichment non‐randomized, hypothesis‐generating design; E: Enrichment non‐randomized sequential approach, with rebiopsies in the refractory setting guiding a second‐generation drug or a combination of targeted agents; F: Different randomized frameworks – to assess the predictive capacity of biomarkers with unknown clinical validity one needs to have the outcome of biomarker‐positive and ‐negative patients separately after experimental and control treatments, and to evaluate if a biomarker is prognostic, one needs to assess the outcome of biomarker‐positive and ‐negative patients on control treatment; G: Biomarker versus control strategy, with direct comparison of the outcomes of patients treated according to a biomarker‐based strategy with those assigned a control treatment.
4.1.2. “N‐of‐1” clinical trial design
“N‐of‐1” or single subject clinical trials, which evaluate an individual patient for efficacy or toxicity of different sequential interventions, have been successfully performed in non‐oncological settings. According to the original “N‐of‐1” trial design, depicted in Figure 3B, patients can be randomly assigned to different agents in different sequential orders, with washout periods between drugs to minimize crossover effects. At completion, the individual effect of each drug and the average effects of each drug across individuals can be analyzed. However, a modified “N‐of‐1” framework represents a promising strategy to investigate the value of individualized therapy in cancer, particularly for patients who harbor rare molecular aberrations, irrespective of tumor type. As seen in Figure 3C, each patient is used as his or her own control and the treatment effect of the current matched drug is compared with that of the most recent earlier drug. The WINTHER trial (NCT01856296), led by the Worldwide Innovative Networking (WIN) Consortium, is an example of a modified “N‐of‐1” design that is using a variety of advanced profiling technologies to comprehensively characterize oncogenic events in over 200 patients with different cancers. The trial compares patients' progression‐free survival (PFS) on therapy guided by profiling results with that achieved on the regimen immediately preceding trial enrollment.
4.1.3. Histology‐based trials
Enrichment is increasingly being used as a strategy to improve study efficiency in trials enrolling patients with a specific tumor type. In the traditional non‐randomized enrichment design, seen in Figure 3D, all potentially eligible patients are first tested for multiple genomic aberrations and biomarker‐positives receive a matched targeted therapy. Endpoints are objective tumor response, magnitude and durability of response. Results should be considered hypothesis‐generating because of the non‐randomized setting, but when significant antitumor activity is observed it may justify Phase 3 testing. The CUSTOM National Cancer Institute (NCI) trial (NCT01306045) evaluating five different matched targeted agents in molecularly‐defined subpopulations of non‐small cell lung cancer (NSCLC) is one example. In the sequential non‐randomized enrichment design, depicted in Figure 3E, one of the main objectives is the understanding of mechanism of acquired drug resistance. The second stage utilizes the putative biomarkers identified in repeated tumor biopsies to direct therapy, which can be a second‐generation drug or a combination of targeted agents. These regimens are predicted to have increased efficacy, additive or synergistic effects, what requires prior knowledge on candidate mechanisms of acquired drug resistance to the initial therapy. One example is the LOGIC trial in BRAF V600 mutated melanoma, assessing multiple rationale combinations of targeted agents in patients progressing to first‐line BRAF inhibitor therapy (NCT01820364). Of note, this approach to identify and tackle resistance can also be integrated into clinical trials using histology‐agnostic frameworks.
When the predictive value of a biomarker is unclear after early clinical development, a randomization strategy with control arm in recommended. Figure 3F describes trial designs that incorporate biomarkers in randomized frameworks. Recently, the NCI began enrolling patients with a variety of rare and common solid tumors in a prospective study called the Molecular Profiling based Assignment of Cancer Therapeutics (M‐PACT), which aims to gauge whether drugs that target certain mutations and pathways implicated but seldom studied in certain cancers, will benefit patients. Patients in M‐PACT will be randomized to either receive a treatment targeted to their specific mutation or pathway, or they will receive an agent in the comparator arm not pharmacogentically determined. If patients progress on the comparator arm, they will have the option to cross over to matched targeted treatment.
It is important to understand that in order to properly assess the predictive capacity of biomarkers with unknown clinical validity one needs to have the outcome of biomarker‐positive and ‐negative patients separately after experimental and control treatments, at the cost of a large sample size (see “randomized all” design in Figure 3F). Furthermore, to evaluate if a biomarker is prognostic, such as BRAF V600E in CRC, one needs to assess the outcome of biomarker‐positive and ‐negative patients on control treatment, a comparison that is possible in “randomize biomarker‐positive only” and “randomize all” study designs but not in single‐arm or non‐randomized enrichment trials (Tajik et al., 2013). In addition to evaluation of the predictive and/or prognostic effects of a biomarker, other objectives of the randomized part of Phase 2 trials include: (i) to evaluate surrogate endpoints such as PFS; (ii) to identify the magnitude of benefit for Phase 3 design; (iii) to select best dose if included in any arm; and (iv) allow early stopping rules for ineffective or toxic arms.
4.1.4. Biomarker versus control strategy
An interesting design for randomized Phase 2 or 3 trials, which applies to both histology‐agnostic and histology‐specific frameworks, compares the overall benefit of therapy based on tumor molecular profiling versus conventional therapy in patients with refractory cancer. In the “biomarker versus control strategy” trials, eligible patients are randomized to treatment strategies instead of specific drugs, as illustrated in Figure 3G. Randomization can take place either upfront, before biomarker testing, or only if a molecular aberration is identified for which an approved/experimental targeted agent is available – this framework is being explored in the SHIVA trial (NCT01771458) (Le Tourneau et al., 2012).
4.1.5. Trials with adaptive design
An adaptive trial uses information obtained while the trial is ongoing (i.e., interim analyses) to modify the course of the trial. The objectives are to make studies more efficient, more likely to demonstrate an effect of the drug if one exists, or more informative (Sleijfer et al., 2013). As suggested by the FDA and European Medicines Agency (EMA), potential adaptations should be preplanned and registered before the trial is initiated (Ocana et al., 2013).
Well‐known examples of adaptive measures in clinical trials include early stopping rules in case of lack of efficacy or unacceptable toxicity and changing doses or schedules of drugs in order to improve the benefit–toxicity profile. More recently, novel adaptation strategies have been proposed. In the adaptive accrual design, after the initial “learning phase”, the ratio of patients randomly assigned to the experimental arm versus the control arm changes from the standard 1:1 to increase the proportion of patients randomized to the arm that is doing better, what augments the statistical power to detect a relevant magnitude of clinical benefit. One can envision a trial that begins with a biomarker‐stratified first stage until a pre‐defined accrual is reached – if the results of the interim analysis comparing the outcome of the experimental versus control treatment in biomarker negatives are not promising, recruitment in this arm is terminated and the second stage continues as an enrichment trial in biomarker‐positive patients until the planned total sample size is recruited. The BATTLE‐2 study (Biomarker‐integrated Approaches of Targeted Therapy for Lung Cancer Elimination 2), for example, is a biomarker‐based and biopsy‐mandatory prospective trial to guide treatment of heavily pre‐treated metastatic NSCLC patients (NCT01248247). In the “adaptive phase”, randomization to different drugs or combinations is weighted based on mutation profile results generated in real time. A similar framework can be applied to studies assessing the predictive value of gene expression signatures. Instead of using a fixed model – built on the training data only – throughout the entire study, adaptive strategies use the information on patients enrolled earlier in the testing set to continuously update the model and refine accrual (Xiao et al., 2014). Adaptive models increase the weights of good predictors and decrease the weights of unstable predictors, improving the overall performance of the classifier and selecting the “best” matched therapy based on current patients' characteristics. These algorithms may facilitate the use of molecular signatures to predict the clinical outcomes of patients in prospective clinical studies.
Finally, we envision enrichment trials with adaptive designs where predictive algorithms incorporating prior knowledge (based on in silico models of drug sensitivity, ex vivo experiments, preclinical or early clinical data) are used to guide the best matched targeted therapy in particular settings: (i) when multiple druggable aberrations are identified in a patient's tumor sample and more than one agent is available for testing; and (ii) when one driver genomic event is identified and the investigator has to choose amongst various drugs with overlapping mechanisms of action (targeting the same driver event) but with different potency/activity according to coexisting genomic aberrations. These “machine‐learning predictive models” can complement molecular tumor boards efforts to identify the “best guess”. We refer to a recent publication as an illustrative example that development of such algorithms is of great interest (Pemovska et al., 2013).
Adaptive clinical trials can be large and expensive because of the number of treatment arms, but their ability to halt further clinical studies with unfavorable agents can reduce the total cost of drug development (Sleijfer et al., 2013).
4.2. Innovative endpoints
The limitations of RECIST criteria in measuring the impact of treatment on the kinetics of tumor growth can be particularly challenging in the context of Phase 2 trials, as previously discussed. Alternative endpoints that incorporate time between radiological evaluations can provide clinically useful information to investigators:
-
a.
Dynamic assessment of tumor growth rate (TGR) allows subtle and quantitative characterization of drug activity, especially of targeted agents expected to produce disease stabilization. This efficacy measure was very informative in a retrospective analysis of large Phase 3 trials evaluating sorafenib and everolimus in advanced renal cell carcinoma, for example (Ferte et al., 2013);
-
b.
Early tumor shrinkage (more than 20% response rate in the first 8 weeks), as suggested by a recent study that analyzed the added benefit from cetuximab therapy compared with chemotherapy alone in the first‐line setting of advanced KRAS wild‐type CRC (Piessevaux et al., 2013);
-
c.
Time to tumor growth (TTG), which was able to capture the benefit of bevacizumab in first‐line treatment of CRC and accurately predicted overall survival in the Phase 3 setting (Claret et al., 2013);
-
d.
Disease control rate (DCR) at 8 weeks, the efficacy endpoint in the BATTLE program, revealed substantial activity of sorafenib treatment among KRAS mutated NSCLC patients (Kim et al., 2011);
-
e.
Progression‐free survival ratio (PFS‐2/PFS‐1), as previously described in the WINTHER trial. This efficacy assessment is based on the timeframe that patients are on the former standard therapy (period 1) compared with the time frame of the genomically‐guided therapy (period 2), with patients acting as their own controls. The validity of this approach is unknown, given the uncertain correlation in PFS between sequential inactive therapies.
In addition, intermediate endpoints of clinical response are also being incorporated to the decision‐making process of adaptive clinical trials. One example of this approach is the multi‐arm STAMPEDE trial in prostate cancer: an increased benefit in biochemical (i.e., PSA) response or PFS as an interim endpoint supports increased accrual to the selected arms while maintaining overall survival (OS) as the major final endpoint. Enumeration and profiling of circulating tumor cells (CTCs) and dynamic changes in circulating tumor DNA (ctDNA) are also promising intermediate endpoints. The DETECT III trial (NCT01619111) is evaluating the potential benefit of lapatinib plus standard therapy compared with standard therapy alone in patients with HER2‐positive CTCs and HER2‐negative metastatic breast cancer, and is addressing the role of dynamic changes in HER2‐positive CTC levels as an early biomarker of treatment response/resistance. Importantly, it should be noted that the innovative endpoints presented here still need further validation in order to be considered predictive of clinical benefit.
4.3. Neoadjuvant setting
The testing of novel targeted agents in the preoperative (neoadjuvant) setting offers a potentially rapid and efficient strategy for drug development utilizing pathologic complete response, a surrogate marker for survival, as the primary endpoint. In addition, neoadjuvant studies allow the assessment of drug effects on the target (pharmacodynamic response) and the clinical validation of predictive biomarkers. Molecular profiling of the residual tumor in the surgical specimen may also provide insights into actionable mechanisms of resistance. A classical example, the I‐SPY 2 trial (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging and Molecular Analysis 2) employs an adaptive trial design for locally advanced breast cancer patients in the neoadjuvant setting, with different targeted agents being added to standard chemotherapy according to molecular profile. Of note, the FDA recently announced consideration of neoadjuvant trials for accelerated drug approval in early breast cancer, particularly for tumors with high risk of recurrence and unfavorable prognosis (FDA, 2012).
A particularly challenging clinical trial framework concerns efficacy assessment of drugs targeting the cancer stem cell (CSC) population (Vermeulen et al., 2012). In the advanced setting, short‐term decrease in tumor volume can be unrelated to the effect of the drug on the CSC population. Conversely, therapeutic interventions that show no evidence of inducing radiological response or that show short‐term progression of the disease, might be very successful in eliminating CSCs and preventing metastasis. An indirect assessment of the drug's efficacy in targeting the CSC fraction could be measurement of the time to development of new metastatic lesions. However, determining post‐treatment surrogate markers of “clonogenicity” in residual cancer cells in settings where neoadjuvant therapy is given appears to be the ideal framework to test whether effectively targeting cells with self‐renewal capacity is able to reduce therapeutic failure and recurrence. We envision that quantification of the fraction of CSC‐marker‐positive CTCs before, during, and after treatment could also be applied to monitor treatment responses.
5. Approval process of genomically‐guided therapies
Reviewing the design of Phase 3 clinical trials that investigate targeted agents goes beyond the scope of this manuscript and we refer the reader to a recent publication (Ocana et al., 2013). Randomized controlled trials will likely remain necessary for the registration of many agents. On the other hand, for targeted drugs that manifest spectacular early efficacy signals, a randomized trial may not be required to proceed with registration – even for ethical reasons – and/or may not be feasible – in cases of rare genomic aberrations. However, confirmatory experience to evaluate the therapeutic indexes of promising drugs in sufficient numbers of patients whose tumors harbor the molecular target should be available. In fact, drug/tumor biomarker pairs with strong relationships have been suggested to warrant an alternative, accelerated path to regulatory approval (Chabner, 2011; McClellan et al., 2011). One example is the approval of ponatinib in chronic myeloid leukemia refractory to dasatinib or nilotinib or who had the BCR‐ABL T315I mutation based on results of a large single‐arm Phase 2 trial (Cortes et al., 2013).
Important considerations in this setting include the size of the safety database that will be required for initial approval, knowing that genomic‐based therapies are usually indicated to relatively small patient populations. In addition, how combinations of investigational agents should be developed so that the activity and safety as required by regulatory agencies are properly documented. FDA has recently released guidelines recognizing these challenges (FDA, 2013). There is a clear need for collaboration among regulatory agencies, industry, and academics at the forefront of genomic technology in order to develop new approaches for comprehensive genomic testing in the drug and test approval processes. Publicly sponsored trials can give relevant information by conducting head‐to‐head comparisons of different treatment regimens, exploring combinations of treatments, and investigating whether targeted drugs approved for one type of cancer are effective against other types – trials unlikely to be performed by pharmaceutical companies once a drug has received approval from the FDA or EMA for a particular indication.
6. Off‐label use of matched targeted therapies – rare aberrations/rare populations
As discussed above, clinical trials of agents targeting a molecular aberration that has a low prevalence in a less common type of cancer may not be feasible in the context of histology‐based studies. In addition, in the setting of histology‐agnostic trials, access to oncologic therapies can be problematic as it depends on geographical issues, availability of slots to treat patients in a timely manner, strict inclusion criteria, and therapeutic dose levels – it can be unclear whether a given patient who does not respond is a true non‐responder or has simply not received the right dosage. Access to the compassionate use of specific agents and the use of drugs outside of their regulatory agency approved indication impose additional challenges, especially with regards to reimbursement issues. One example is a rare patient with gastrointestinal stromal cell tumor (GIST) that has an activating BRAF V600E mutation. Physicians may want to offer off‐label use of an existing therapy that targets BRAF, such as vemurafenib or dabrafenib, both approved in melanoma. Case reports of spectacular responses to matched targeted agents are becoming ubiquitous, but there is an inherent bias to publish positive results, making it difficult to aggregate information from lots of “N‐of‐1” experiments. Mechanisms to annotate lack of response in this setting are missing. As proposed by Dr. Richard Schilsky, the ideal solution is a national formulary of targeted agents against common aberrations, so that every patient receiving a matched therapy in the off‐label setting can be tracked (Schilsky, 2014). These pharmacy exchange programs could generate ever‐growing databanks integrating the genomic information with therapeutic response and outcome. The American Society of Clinical Oncology rapid learning systems and cognitive computing platforms called CancerLinQ is an alternative. We envision that information derived from these registries should be added to knowledge databases such as My Cancer Genome (http://www.mycancergenome.org/) and become readily available to oncologists worldwide, providing annotated predictive genomic markers in cancer and potentially changing the paradigm of drug approval process.
7. Conclusions
In this review manuscript we present the complexities of research in PCM. As previously documented (Tajik et al., 2013), we observed substantial variability in the labeling of clinical trial designs for evaluating biomarkers for treatment selection. The research community, as proposed here, should adopt a unified nomenclature. In addition, these trials break the traditional differences of Phase 1, 2 and 3 designs, introducing new concepts such as Phase 1 expansion cohorts that “replace” Phase 2 testing and regulatory approval based on non‐randomized trials. Other challenges include technical limitations of molecular tests, logistical issues for patient accrual in clinical trials and critical unsolved regulatory issues. Importantly, genomics knowledge is ahead of our ability to therapeutically target tumors, given that many mutations identified by sequencing are either linked to unapproved drugs or are not druggable by currently available therapies. Genetic heterogeneity at intratumoral and inter‐patient levels and clonal evolution of tumors over time are among the major obstacles for PCM. Given the potentials of genomic characterization of CTCs and ctDNA, these circulating blood biomarkers are expected to be important for monitoring the emergence of treatment‐resistant clones under selective pressures and to provide an efficient model of individualized therapy. Hopefully, in the near future, novel molecular imaging tools will be standardized and validated to help overcome the drawbacks associated with relying on tumor tissue alone. Clinical trial strategies to interrogate tumor heterogeneity are particularly difficult to implement and will require not only the active participation of patients who are willing to undergo repeated investigations, but also the collaborative engagement of clinicians and scientists (Bedard et al., 2013). We strongly believe that clinical trials with innovative endpoints, exploring matched targeted agents in settings other than advanced refractory disease, and using adaptive strategies will represent a major advance for PCM. Researchers will be able to use data from a set of patient's treatments to tailor enrollment of additional subjects, more quickly eliminating ineffective or toxic drugs, and allowing the knowledge learned throughout the course of the trial to be used for individualizing therapy.
Conflicts of interest
Authors declare no competing interests in relation to the work described.
Funding
R. Dienstmann is a recipient of “La Caixa International Program for Cancer Research & Education”.
Dienstmann Rodrigo, Rodon Jordi, Tabernero Josep, (2015), Optimal design of trials to demonstrate the utility of genomically‐guided therapy: Putting Precision Cancer Medicine to the test, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.06.014.
References
- Andre, F. , Bachelot, T. , Commo, F. , 2014. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 15, 267–274. [DOI] [PubMed] [Google Scholar]
- Bedard, P.L. , Hansen, A.R. , Ratain, M.J. , Siu, L.L. , 2013. Tumour heterogeneity in the clinic. Nature. 501, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabner, B.A. , 2011. Early accelerated approval for highly targeted cancer drugs. N. Engl. J. Med. 364, 1087–1089. [DOI] [PubMed] [Google Scholar]
- Claret, L. , Gupta, M. , Han, K. , 2013. Evaluation of tumor-size response metrics to predict overall survival in Western and Chinese patients with first-line metastatic colorectal cancer. J. Clin. Oncol. 31, 2110–2114. [DOI] [PubMed] [Google Scholar]
- Cortes, J.E. , Kim, D.W. , Pinilla-Ibarz, J. , 2013. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 369, 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann, R. , Rodon, J. , Barretina, J. , 2013. Genomic medicine frontier in human solid tumors: prospects and challenges. J. Clin. Oncol. 31, 1874–1884. [DOI] [PubMed] [Google Scholar]
- Dienstmann, R. , Rodon, J. , Tabernero, J. , 2013. Biomarker-driven patient selection for early clinical trials. Curr. Opin. Oncol. 25, 305–312. [DOI] [PubMed] [Google Scholar]
- FDA, 2012. Draft Guidance for Industry. Pathologic Complete Response in Neoadjuvant Treatment of High-risk Early-stage Breast Cancer: Use as an Endpoint to Support Accelerated Approval http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM305501.pdf [Google Scholar]
- FDA, 2013. Paving the Way for Personalized Medicine: FDA's Role in a New Era of Medical Product Development http://www.fda.gov/downloads/scienceresearch/specialtopics/personalizedmedicine/ucm372421.pdf [Google Scholar]
- Ferte, C. , Fernandez, M. , Hollebecque, A. , 2014. Tumor growth rate is an early indicator of antitumor drug activity in phase I clinical trials. Clin. Cancer Res. 20, 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferte, C. , Koscielny, S. , Albiges, L. , 2013. Tumor growth rate provides useful information to evaluate sorafenib and everolimus treatment in metastatic renal cell carcinoma patients: an integrated analysis of the TARGET and RECORD phase 3 trial data. Eur. Urol. 65, 713–720. [DOI] [PubMed] [Google Scholar]
- Garcia, V.M. , Cassier, P.A. , de Bono, J. , 2011. Parallel anticancer drug development and molecular stratification to qualify predictive biomarkers: dealing with obstacles hindering progress. Cancer Discov. 1, 207–212. [DOI] [PubMed] [Google Scholar]
- Gerlinger, M. , Horswell, S. , Larkin, J. , 2014. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 46, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelder, S. , Clarke, P.A. , Workman, P. , 2012. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol. Oncol. 6, 155–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E.S. , Herbst, R.S. , Wistuba, I.I. , Lee, J.J. , 2011. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 1, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tourneau, C. , Kamal, M. , Tredan, O. , 2012. Designs and challenges for personalized medicine studies in oncology: focus on the SHIVA trial. Target Oncol. 7, 253–265. [DOI] [PubMed] [Google Scholar]
- Levy, A. , Hollebecque, A. , Ferte, C. , 2013. Tumor assessment criteria in phase I trials: beyond RECIST. J. Clin. Oncol. 31, 395 [DOI] [PubMed] [Google Scholar]
- LoRusso, P.M. , Anderson, A.B. , Boerner, S.A. , 2010. Making the investigational oncology pipeline more efficient and effective: are we headed in the right direction?. Clin. Cancer Res. 16, 5956–5962. [DOI] [PubMed] [Google Scholar]
- McClellan, M. , Benner, J. , Schilsky, R. , 2011. An accelerated pathway for targeted cancer therapies. Nat. Rev. Drug Discov. 10, 79–80. [DOI] [PubMed] [Google Scholar]
- Nishino, M. , Giobbie-Hurder, A. , Gargano, M. , 2013. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin. Cancer Res. 19, 3936–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocana, A. , Amir, E. , Vera-Badillo, F. , 2013. Phase III trials of targeted anticancer therapies: redesigning the concept. Clin. Cancer Res. 19, 4931–4940. [DOI] [PubMed] [Google Scholar]
- Pemovska, T. , Kontro, M. , Yadav, B. , 2013. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 3, 1416–1429. [DOI] [PubMed] [Google Scholar]
- Piessevaux, H. , Buyse, M. , Schlichting, M. , 2013. Use of early tumor shrinkage to predict long-term outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 31, 3764–3775. [DOI] [PubMed] [Google Scholar]
- Rodon, J. , Saura, C. , Dienstmann, R. , 2012. Molecular prescreening to select patient population in early clinical trials. Nat. Rev. Clin. Oncol. 9, 359–366. [DOI] [PubMed] [Google Scholar]
- Roychowdhury, S. , Iyer, M.K. , Robinson, D.R. , 2011. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci. Transl. Med. 3, 111ra121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilsky, R.L. , 2014. Implementing personalized cancer care. Nat. Rev. Clin. Oncol. 11, 432–438. [DOI] [PubMed] [Google Scholar]
- Simon, R. , Roychowdhury, S. , 2013. Implementing personalized cancer genomics in clinical trials. Nat. Rev. Drug Discov. 12, 358–369. [DOI] [PubMed] [Google Scholar]
- Sleijfer, S. , Bogaerts, J. , Siu, L.L. , 2013. Designing transformative clinical trials in the cancer genome era. J. Clin. Oncol. 31, 1834–1841. [DOI] [PubMed] [Google Scholar]
- Tajik, P. , Zwinderman, A.H. , Mol, B.W. , 2013. Trial designs for personalizing cancer care: a systematic review and classification. Clin. Cancer Res. 19, 4578–4588. [DOI] [PubMed] [Google Scholar]
- Tran, B. , Brown, A.M. , Bedard, P.L. , 2013. Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. Int. J. Cancer. 132, 1547–1555. [DOI] [PubMed] [Google Scholar]
- Vermeulen, L. , de Sousa e Melo, F. , Richel, D.J. , 2012. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 13, e83–89. [DOI] [PubMed] [Google Scholar]
- Xiao, G. , Ma, S. , Minna, J. , 2014. Adaptive prediction model in prospective molecular signature-based clinical studies. Clin. Cancer Res. 20, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap, T.A. , Gerlinger, M. , Futreal, P.A. , 2012. Intratumor heterogeneity: seeing the wood for the trees. Sci. Transl. Med. 4, 127ps10 [DOI] [PubMed] [Google Scholar]
- Yap, T.A. , Sandhu, S.K. , Workman, P. , 2010. Envisioning the future of early anticancer drug development. Nat. Rev. Cancer. 10, 514–523. [DOI] [PubMed] [Google Scholar]