

Abstract
The Hippo pathway has emerged as a fundamental regulator in tissue growth, organ size and stem cell functions, and tumorigenesis when deregulated. However, its roles and associated molecular mechanisms underlying oral squamous cell carcinoma (OSCC) initiation and progression remain largely unknown. Here, we identified TAZ, the downstream effector of Hippo signaling, as a novel bona fide oncogene by promoting cell proliferation, migration/invasion and chemoresistance in OSCC. TAZ promoted epithelial‐to‐mesenchymal transition (EMT) and also was involved in TGF‐β1‐induced EMT in oral cancer cells. Furthermore, enriched TAZ sustained self‐renewal, maintenance, tumor‐seeding potential of oral cancer stem cells (CSCs). Remarkably, enforced TAZ overexpression conferred CSCs‐like properties on differentiated non‐CSCs and fueled phenotypic transition from non‐CSCs to CSCs‐like cells. Mechanistically, TAZ‐TEADs binding and subsequent transcriptional activation of EMT mediators and pluripotency factors are presumably responsible for TAZ‐mediated EMT and non‐CSCs‐to‐CSCs conversion. Importantly, aberrant TAZ overexpression was found to be associated with tumor size, pathological grade and cervical lymph node metastasis, as well as unfavorable prognosis. Pharmacological repression of TAZ by simvastatin resulted in potent anti‐cancer effects against OSCC. Taken together, our findings have revealed critical links between TAZ, EMT and CSCs in OSCC initiation and progression, and also established TAZ as a novel cancer biomarker and viable druggable target for OSCC therapeutics.
Keywords: Hippo, TAZ, Epithelial to mesenchymal transition, Cancer stem cell, Oral squamous cell carcinoma
Highlights
TAZ serves as a novel bona fide oncogene by promoting cell proliferation, migration and invasion in OSCC.
TAZ is capable of triggering EMT and required for TGF‐β1‐induced EMT in oral cancer cells.
TAZ sustains self‐renewal, maintenance, tumor‐seeding potential of oral cancer stem cells.
TAZ overexpression associated with higher pathological grade, lymph node metastasis, and unfavorable prognosis.
Pharmacological repression of TAZ by simvastatin resulted in potent anti‐cancer effects against OSCC.
1. Introduction
Metastatic spread and chemoresistance/radioresistance largely account for therapeutic failure and cancer‐related death in human cancers, including oral squamous cell carcinoma (OSCC) (Wan et al., 2013). The emergence of locoregional recurrence, metastatic dissemination and therapeutic resistance, alone or in combination, dictates OSCC patient's prognosis (Scully and Bagan, 2009). Mounting evidence has established that epithelial–mesenchymal transition (EMT) is intricately involved in cancer invasion‐metastasis cascade and therapeutic resistance by endowing cancer cells with enhanced motility, invasiveness and resistance to apoptosis/senescence (Polyak and Weinberg, 2009). Multiple master transcription factors (Snail, Twist, Zeb1/2 et al.) coupled with epigenetic regulators orchestrates EMT via converging on E‐cadherin repression during cancer progression. The contextual EMT‐inducing signals like TGF‐β1, primarily emanating from tumor‐associated microenvironment, activate EMT to fuel metastatic dissemination (Lamouille et al., 2014). However, the key mediators and molecular circuits underlying oral cancer EMT remain incompletely recognized thus far.
Oral cancer cells display prominent phenotypic and functional heterogeneities, likely contributing to cancer metastasis and therapeutic resistance (Sinha et al., 2013). To address these heterogeneities, the cancer stem cell (CSC) model has postulated that tumors contain a unique cell subpopulation termed as CSCs or tumor‐initiating cells (TICs) with self‐renewal and tumor‐seeding properties. Importantly, these CSCs have been proposed to sustain tumor overgrowth and drive metastatic spread, ultimately leading to disease relapse and mortality. With regard to the cellular origin of CSCs, several studies have revealed that these CSCs may arise from naturally residing stem/progenitor cells. However, others argued that they could be transformed or dedifferentiated from differentiated cells and then clonally expanded (Beck and Blanpain, 2013; Mani et al., 2008). Strikingly, some cancer cells, if not all, were able to re‐acquire stemness and exhibit CSCs‐like properties upon undergoing EMT, also termed CSCs plasticity (Chaffer et al., 2013; Mani et al., 2008; Polyak and Weinberg, 2009). Ectopic overexpression of Snail/Twist/Zeb1 in malignant mammary epithelial cells generated CSCs in vitro and in vivo, thus raising the possibility that CSCs can be generated de novo from differentiated non‐CSCs by EMT (Chaffer et al., 2013; Mani et al., 2008). Moreover, several EMT‐associated factors including TGF‐β1, HGF and IL‐6 potently induced an EMT‐CSCs program in differentiated cancer cells and reprogrammed them into CSCs‐like cells (Chaffer et al., 2013; Iliopoulos et al., 2011; Vermeulen et al., 2010). Furthermore, intravital lineage tracing provided compelling evidence that such non‐CSCs to CSCs conversion indeed occurred in vivo, thus substantiating the dynamic nature and plasticity of cancer cells (Zomer et al., 2013). Thus, these findings provide experimental support to the concept that CSC hierarchy may be considered flexible and interconvertible between CSCs and more differentiated non‐CSCs. Nonetheless, the molecular determinants to drive non‐CSCs to CSCs conversion and promote CSC expansion remain incompletely known.
The Hippo pathway has emerged as a major regulator in organ size control, stem cell homeostasis and tumorigenesis (Harvey et al., 2013; Zhao et al., 2011). In mammals, the Hippo pathway comprises a kinase cassette including MST1/2, LATS1/2, and downstream effectors: transcriptional co‐activator with PDZ‐binding motif (TAZ, also known as WWTR1) and yes‐associated protein (YAP). Once Hippo activation, MST1/2 phosphorylates LATS1/2 and in turn phosphorylates and inactivates TAZ/YAP by their cytoplasmic retention and proteasome‐mediated degradation. In contrast, inactivated Hippo results in a hypophosphorylated state of TAZ/YAP and facilitates their nuclear translocation where they drive downstream target transcriptions via forming complexes with TEAD1‐4 and Smads (Zhao et al., 2011). Notably, deregulated Hippo pathway has been tightly linked to cancer initiation and progression (Harvey et al., 2013). Aberrant TAZ/YAP overexpression and/or amplification have been implicated in fundamental cellular programs, such as cell proliferation, migration, invasion and EMT (Chan et al., 2008; Overholtzer et al., 2006; Zhang et al., 2009). Strikingly, TAZ has been identified as a potent determinant of CSCs self‐renewal and expansion, and holds unexpected capabilities to confer CSCs traits to non‐CSCs in breast cancer (Bartucci et al., 2015; Cordenonsi et al., 2011). However, the functional roles of TAZ responsible for OSCC EMT and CSCs maintenance have not yet been explored.
Here, we identified TAZ as a novel oncogenic driver involved in OSCC initiation and progression and revealed critical molecular links between TAZ and EMT as well as CSCs. Moreover, pharmacological repression of TAZ by simvastatin induced potent therapeutic effects against OSCC.
2. Materials and methods
Detailed experimental materials, methods and relevant references were described in supplementary experimental procedures, for example cell culture, vectors construction and virus preparation, cell transfection/infection, real‐time RT‐PCR, western blot, immunohistochemistry, tumorsphere formation, xenograft model et al. These experiments were performed as described previously with minor modifications (Cordenonsi et al., 2011; Li et al., 2013; Liu et al., 2010). All chemicals were purchased from Sigma–Aldrich. The siRNA sequences, antibodies and PCR primers were listed in Supplementary Tables 3–5. All experimental studies involving human and animals were approved by Research Ethic Committee and Animal Research Committee of Nanjing Medical University.
3. Results
3.1. Overexpressed TAZ in OSCC associates with aggressive clinicopathological features and patients' prognosis
To explore the expression of TAZ in OSCC, we initially evaluated the mRNA and protein abundance of TAZ in a panel of OSCC cell lines as compared to the immortalized oral epithelial cell line HIOEC. The qPCR data revealed significantly increased TAZ transcripts in all cancerous cells in relative to HIOCE (Figure 1A), and western blot results further indicated remarkably upregulated TAZ protein in most cell lines, except SCC9 cells with comparable abundance (Figure 1B). Additionally, TAZ was pronouncedly increased in oral cancerous tissues compared to the pair‐matched adjacent non‐cancerous tissues (n = 4) (Figure 1B). Then, we measured TAZ expression by immunohistochemistry in a retrospective cohort of 111 primary OSCC samples. As shown in Figure 1C, TAZ positive staining was identified in nucleus or both nucleus and cytoplasm in cancer specimens from diverse locations including tongue, buccal, mouth floor and gingiva, whereas weak or negative staining was detected in the normal counterparts. Based on the immunohistochemical staining scores, high TAZ expression was identified in approximately 50.4% (56/111) in cancer samples and 20.8% (5/24) in normal samples, thus indicating aberrant TAZ overexpression in a fraction of OSCC (P < 0.001, Figure 1D).
Figure 1.
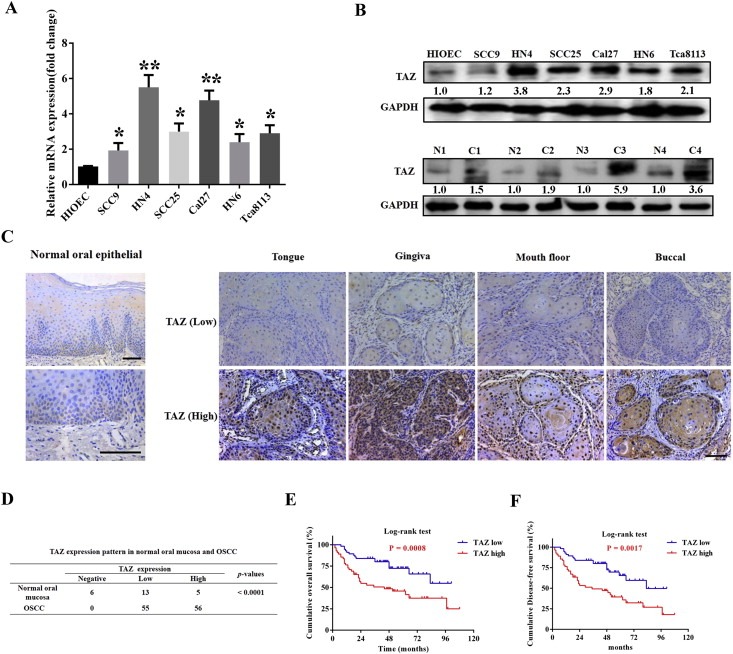
TAZ is overexpressed in OSCC and associates with patients survival A: TAZ mRNA levels were measured by real‐time RT‐PCR in six OSCC cell lines as compared with human immortalized oral epithelial (HIOEC). B: TAZ protein levels were determined by western blot (WB) in OSCC cell lines and paired tumor‐adjacent non‐tumor tissues (n = 4). Representative images of WB are shown. The “N” stands for non‐tumor tissue, and “C” stands for cancer in the lower panel. C: TAZ expression in human normal oral mucosa and OSCC specimens from diverse primary sites was evaluated by immunohistochemical staining. Rare TAZ positive staining was observed in basal cells from healthy oral epithelial. Scale bar: 100 μm. D: Expression patterns of TAZ in human OSCC samples and normal counterparts were statistically determined. E, F: Overall and disease‐free survival analyses of patients with high or low expression of TAZ were estimated by Kaplan–Meier method and compared with log‐rank test. Data shown here are mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ANOVA analysis.
To further understand the clinical significance of TAZ overexpression in OSCC, we set out to identify the potential associations between TAZ expression and patients' clinicopathological parameters. As shown in Supplementary Table 1, there were no significant correlations found between TAZ and patient age, gender, primary sites and clinical stage. Noticeably, TAZ abundance associated with tumor size, pathological grade and cervical nodal metastasis (P < 0.05). Moreover, patients with high TAZ had much lower overall and disease‐free survival rates compared to the counterparts with low TAZ as estimated by Kaplan–Meier analyses (Figure 1E and F, P = 0.0008 and 0.0017). Importantly, TAZ abundance was further identified as an independent predictor for patients' prognosis (P = 0.003 and 0.011, Cox proportional hazards regression model, Supplementary Table 2). Taken together, our data reveal that overexpressed TAZ associates with aggressive clinicopathological features and unfavorable patients' prognosis in OSCC.
3.2. TAZ promotes cell proliferation, migration and invasion, and EMT
Given the proposed oncogenic roles of TAZ in cancers (Bhat et al., 2011; Chan et al., 2008) and our initial findings of TAZ as an OSCC biomarker, we then sought to unravel the biological roles of TAZ in OSCC via loss‐of‐function and gain‐of‐function approaches. Two independent shRNA lentiviral vectors targeting human TAZ were exploited to infect cancer cells with relative high endogenous TAZ and yielded stable cells (Cal27‐shTAZ, HN4‐shTAZ). The knockdown efficiency and specificity were confirmed by the facts that endogenous TAZ and its downstream target CTGF were significantly inhibited, whereas no remarkable change of YAP, the TAZ homolog, was detected (Figure 2A). Impaired cell proliferation and G1/G2 cell cycle arrest were observed following TAZ knockdown as revealed by MTT assay and flow cytometry (Figure 2B–C). The proportions of apoptotic cells were significantly elevated approximately 2 folds relative to their corresponding controls (9.3% versus 4.3% for Cal27; 12.2% versus 5.1% for HN4) upon TAZ depletion (Figure 2D). Consistently, several cell cycle regulators including p16, p21 and p53, together with apoptosis marker cleaved‐PARP were upregulated (Figure 2E). Moreover, TAZ depletion induced impaired migration and invasion as detected via wound healing and transwell invasion assays, in line with increased E‐cadherin as well as reduced vimentin (Figure 2F–H).
Figure 2.
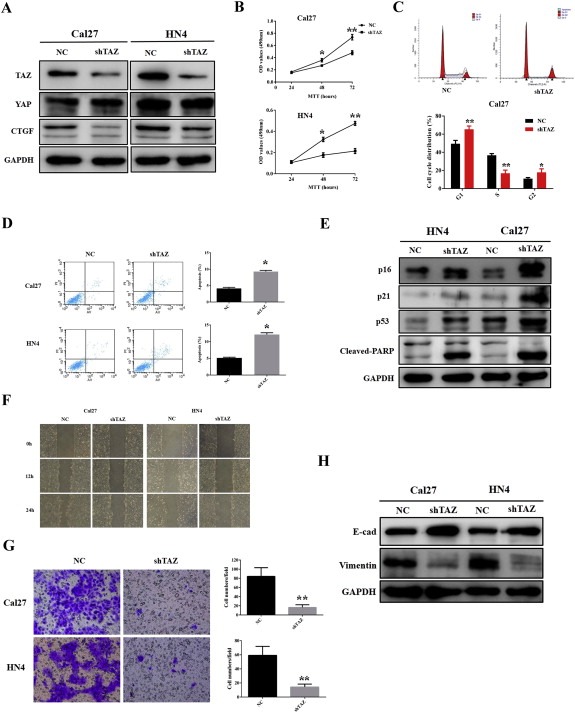
TAZ knockdown inhibits cell proliferation, migration and invasion, and triggers cell apoptosis in tongue cancer cells A: Endogenous TAZ was efficiently silenced by shTAZ lentiviral particles in Cal27 and HN4 cells. Representative images of WB are shown. B: Cell viability and proliferation were remarkably suppressed when endogenous TAZ was silenced as measured by MTT assay. C: TAZ depletion resulted in G1/2‐phase cell cycle arrest as measured by flow cytometry in Cal27 cells. D: Increased percentages of cell undergoing apoptosis were evident in shTAZ‐infected cells as assayed by Annexin V‐PI double staining. E: The expression of multiple cell cycle regulators including p16, p21 and p53, and cell apoptosis marker cleaved‐PARP was measured by western blot. Representative images are shown. F, G: The migration and invasion abilities were significantly reduced in shTAZ‐treated cells in 24 h wound healing and transwell assays. H: The abundances of E‐cadherin and Vimentin were measured by WB following endogenous TAZ knockdown. Representative images are shown. Data shown here are mean ± SD from three independent experiments, *p < 0.05, **p < 0.01, Student's t‐test.
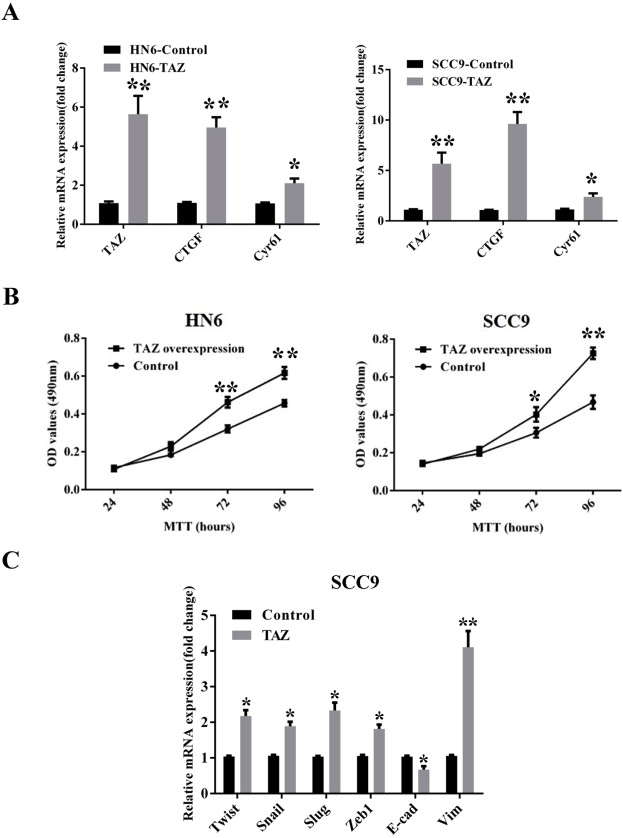
To further confirm the tumorigenic roles of TAZ in OSCC, we subcloned the full‐length human TAZ cDNA with an N‐terminal Flag tag into a lentiviral vector, and generated stable TAZ‐overexpressing cells in HN6 and SCC9 (relatively low endogenous TAZ). As expected, both TAZ mRNA and protein, as well as two downstream targets CTGF and Cyr61, were significantly increased following TAZ overexpression (Figure 3A and Supplementary Fig.S1A). Overexpression of TAZ induced more rapid cell proliferation in HN6 and SCC9 (Supplementary Fig.S1B), consistent with the well‐established pro‐proliferative function of TAZ. Interestingly, as shown in Figure 3B, TAZ‐overexpressed cells became elongated, scattering distributed with fibroblast‐like appearance, while control cells remained typical cobblestone morphology with tight adhesion. Remarkably enhanced cell migration and invasion were also observed in TAZ‐overexpressed cells (Figure 3C–D). These morphological and functional changes induced by TAZ were reminiscent of EMT triggered by Snail and MT1‐MMP in SCC9 cells as we reported previously (Yang et al., 2013; Zhu et al., 2012), suggesting that TAZ might be capable of promoting EMT in oral cancer cells. To confirm this notion, the abundance and locations of E‐cadherin and vimentin were further determined by immunofluorescence, real‐time PCR and western blot assays. The data revealed apparent E‐cadherin loss and vimentin gain in TAZ‐transduced cells compared with control cells (Figure 3E and Supplementary Fig.S1C). Notably, several EMT‐orchestrating transcription factors Twist, Snail and Slug were significantly upregulated upon TAZ overexpression (Figure 3F and Supplementary Fig.S1C).
Figure 3.
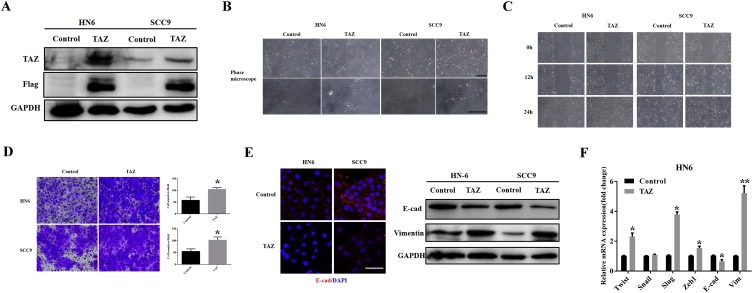
Ectopic TAZ overexpression induces EMT‐like changes A: TAZ overexpression was confirmed by western blot in cellular lysates from cells infected with TAZ cDNA lentivirus. Representative images are shown. B: Enforced TAZ overexpression resulted in EMT‐like morphological changes from cobble‐like to spindle‐like appearance under phase contrast microscopy. C, D: The cell motilities and invasion were remarkably enhanced after TAZ overexpression as gauged by wound healing assay(C) and transwell‐invasion assay(D). E: E‐cadherin expression was probed by immunoflurescent staining in TAZ‐overexpressing and control cells (left panel). Scale bar: 100 μm. The abundance of EMT markers E‐cadherin and vimentin following TAZ overexpression were assayed by western blot (right panel). Representative images are shown. F: The mRNA levels of EMT‐associated transcription factors and E‐cadherin/vimentin were determined by real‐time RT‐PCR. Data showed here are mean ± SD from three independent experiments, *p < 0.05, **p < 0.01, Student's t‐test.
We next developed a xenograft animal model to further substantiate the oncogenic roles of TAZ in OSCC. As displayed in Supplementary Fig.S2A‐D, these data from tumor volume and weight, Ki‐67/TUNEL staining revealed that tumor growth was substantially compromised when endogenous TAZ was inhibited, whereas TAZ overexpression resulted in significantly accelerated tumor growth. Additionally, much weaker or stronger E‐cadherin staining was observed in Cal27‐shTAZ or HN6‐TAZ xenograft, respectively (Supplementary Fig.S2C,D). To further verify the pro‐metastatic roles of TAZ, HN6‐TAZ and control cells were systemically injected by tail vein to assess lung metastasis. As shown in Supplementary Fig.S3A‐B, significantly more and larger metastatic lesions were identified in animals receiving HN6‐TAZ cells for 6 weeks than those inoculated with control cells. Collectively, our data demonstrate that TAZ has pleiotropic tumorigenic roles underlying oral cancer progression and acts as a bona fide oncogene by modulating cell proliferation, apoptosis, migration and invasion, as well as metastasis in oral cancers.
3.3. TAZ is involved in TGF‐β1‐induced EMT
Mounting evidence indicates that EMT‐mediated metastatic spread dictates patients survival (Valastyan and Weinberg, 2011). The above data have revealed key roles of TAZ in mediating invasion and metastasis presumably by triggering EMT. To further substantiate this, we determined the functions of TAZ using an in vitro EMT model induced by TGF‐β1. TGF‐β1 is a well‐established EMT inducer under diverse physical and pathological settings, and also promotes metastasis in advanced cancers (Zavadil and Bottinger, 2005). As shown in Supplementary Fig.S4A, typical morphological changes of cells including cell elongation and scattering upon rhTGF‐β1 exposure were observed, indicative of cell undergoing EMT. The immunoflurescent staining and PCR data of multiple EMT markers (E‐cadherin, vimentin) and mediators (Twist, Snail and Slug) further confirmed EMT induced by rhTGF‐β1 in HN6 cells (Supplementary Fig.S4B–C). Notably, TAZ, YAP and CTGF transcripts were significantly upregulated up rhTGF‐β1 treatment in HN6 cells (Supplementary Fig.S4D, similar data not shown for Cal27). As displayed in Figure 4A‐B, rhTGF‐β1 exposure resulted in dramatic reduction of E‐cadherin as well as vimentin gain in control cells, but failed in TAZ‐depleted cells. Moreover, TAZ knockdown significantly abrogated the upregulation of these EMT mediators following rhTGF‐β1 stimulation (Supplementary Fig.S4E).
Figure 4.
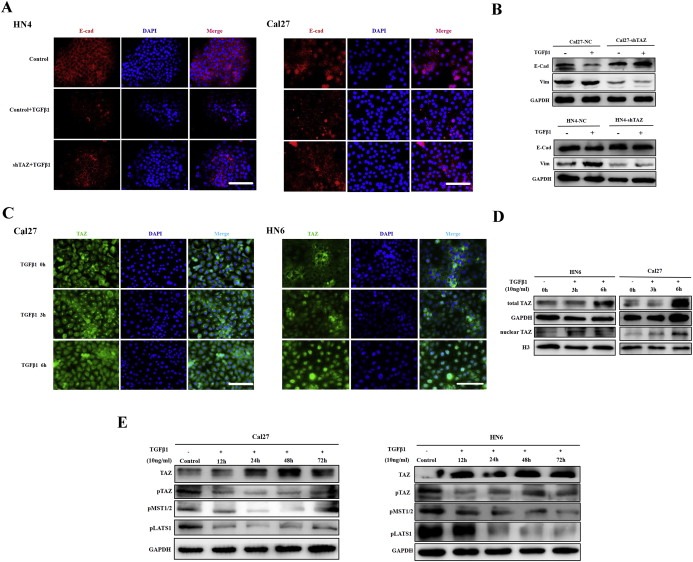
TAZ is required for TGF‐β1‐induced EMT A: Immunofluorescence staining of E‐cadherin in cells with stable TAZ silencing or control cells following treatment with rhTGF‐β1 (10μ/ml) for 48 h. Scale bar:100 μm. B: The protein abundances of E‐cadherin and Vimentin were determined by western blot after Cal27‐shTAZ and HN4‐shTAZ cells were treated with rhTGF‐β1 (10μ/ml) for 48 h, respectively. Representative images of WB are shown. C: rhTGF‐β1 treatment induced TAZ nuclear translocation in confluent Cal27 and HN6 cells as visualized by immunofluorescence staining during a 6‐h time course. D: rhTGF‐β1 exposure resulted in increased total and nuclear TAZ as gauged by western blot using total protein samples or nuclear fractions. Representative images are shown. E: TGF‐β1 induced total TAZ upregulation and dephosphorylation (TAZ‐Ser89) by inhibiting and inactivating Hippo kinases as measured by MST1/2 (MST‐Thr183/Mst2‐Thr180) and LATS1 (Lats1‐Thr1079) phosphorylation. Representative images are shown. Data shown here are from three independent experiments.
To further confirm the roles of TAZ in rhTGF‐β1‐triggered EMT, we determined the abundance, cytoplasmic/nuclear localization and phosphorylation status of TAZ following rhTGF‐β1 treatment. Considering the localization and phosphorylation of endogenous TAZ were tightly regulated by cell density (Supplementary Fig. S5), the nearly confluent cells were treated with rhTGF‐β1 during 6 h time course. Notably, nuclear translocation of TAZ was observed following rhTGF‐β1 exposure in both Cal27 and HN6 cells (Figure 4C), irrespective of their endogenous levels of TAZ. Complementary, both total and nuclear TAZ abundances were significantly increased upon rhTGF‐β1 treatment (Figure 4D). Given the intricate crosstalk between Hippo‐TAZ and TGF‐β signaling (Mauviel et al., 2012), we hypothesized that TGF‐β1‐induced TAZ upregulation and nuclear translocation might, at least in part, be mediated through affecting Hippo kinases. Indeed, as displayed in Figure 4E, the phosphorylated TAZ (pTAZ‐Ser89), MST1/2 (pMst1‐Thr183/Mst2‐Thr180) and Lats1 (pLats1‐Thr1079) were remarkably reduced following rhTGF‐β1 treatment, thus suggesting that rhTGF‐β1 inactivated these Hippo kinases and in turn lead to TAZ upregulation via dephosphorylation and nuclear translocation. Consistently, when endogenous TGF‐β1 pathway was pharmacologically inhibited by inhibitor SB431542, Hippo signaling was activated as evidenced by increased Lats1 and MST1/2 phosphorylation, and reduced TAZ, CTGF and Cyr61 (Supplementary Fig.S6). Taken together, our findings indicate that TAZ is critically involved in TGF‐β1‐mediated EMT in OSCC cells. TAZ upregulation induced by TGF‐β1 was partially due to dephosphorylation and nuclear translocation by inactivated Hippo signaling.
3.4. TAZ promotes cancer stem cell maintenance and expansion in OSCC
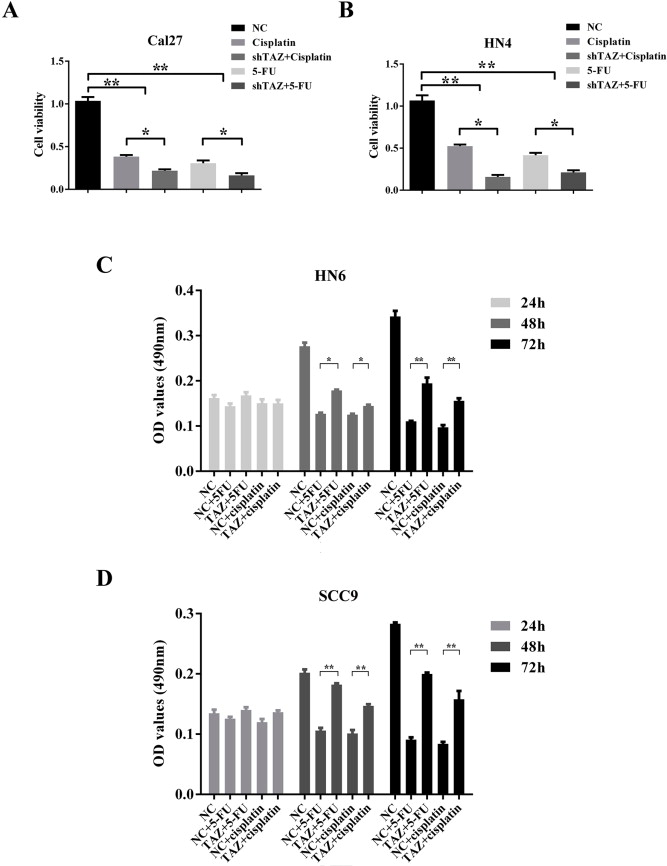
Hippo pathway has been identified as a pivotal mediator for normal and cancer stem cell self‐renewal and differentiation (Hao et al., 2014; Zhao et al., 2011). TAZ was capable of conferring CSCs‐like traits on non‐CSC cells in breast cancers (Cordenonsi et al., 2011). Intrigued by these finding, we next asked whether TAZ was involved in CSC maintenance and expansion in OSCC. To address this, the colony formation and tumorsphere assays as surrogate readouts for CSCs‐like properties were exploited in cells with TAZ depletion or overexpression. Colony formation was pronouncedly impaired following endogenous TAZ knockdown, while significantly enhanced in TAZ‐overexpressing cells (Figure 5A–B). Additionally, TAZ depletion resulted in significantly less and smaller tumorspheres, whereas enforced TAZ overexpression yielded much more and larger spheres. Similar findings were also replicated in the secondary round of tumorsphere formation using the single disassociated cell from primary tumorsphere (Figure 5C). Complementarily, 5‐FU or cisplatin induced significantly lower cell viability in TAZ‐knockdown cells as compared to control cells (Supplementary Fig. S7A–B). However, TAZ‐overexpressing cells displayed more resistance to these conventional cytotoxic drugs (Supplementary Fig. S7C–D). Thus, these findings offer important clues for potential TAZ involvement in oral CSCs.
Figure 5.
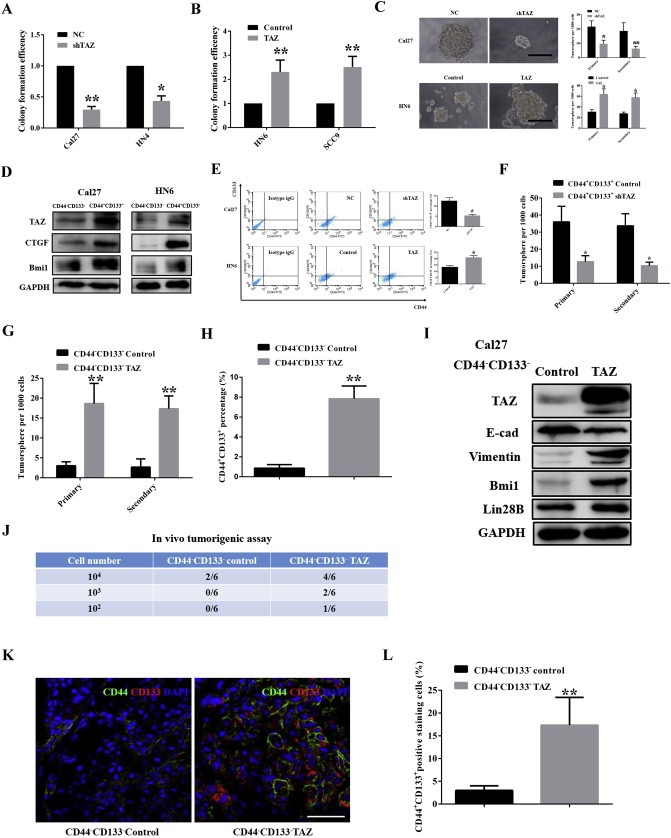
TAZ is essential for oral CSCs maintenance and endows non‐CSCs with some CSCs‐like traits A, B: TAZ knockdown resulted in reduced colony formation efficiencies, while TAZ overexpression significantly enhanced clonal formation in vitro. Colony formation efficiency was defined as the ratios of the numbers of colony generated from indicated cells, while the colony formation in controls was designed as 1. C: TAZ depletion led to fewer and smaller tumorspheres, whereas its overexpression resulted in more and larger tumorspheres. Scale bar:50 μm. D: TAZ, CTGF and Bmi1 were enriched in CD44+CD133+(oral CSC subpopulation) cells. E: The percentages of CD44+CD133+ subpopulation were increased upon TAZ overexpression, while reduced following TAZ knockdown as measured by FACS. F: Tumorsphere formation was impaired when freshly isolated CD44+CD133+ Cal27 cells infected with shTAZ lentivirus. G: Enhanced tumorsphere formation was evident in these freshly isolated CD44−CD133− Cal27 cells infected with TAZ cDNA lentivirus. H: The percentage of CD44+CD133+ subpopulation was significantly increased following TAZ overexpression in freshly sorted CD44−CD133− Cal27 cells (72 h). I: The abundances of TAZ, EMT markers (E‐cadherin, vimentin) and CSCs makers (Bmi1, Lin28B) were probed in CD44−CD133− Cal27 cells infected TAZ cDNA lentivirus (72 h) using western blot. J: The tumor‐initiating potentials of freshly sorted CD44−CD133−Cal27 cells following TAZ cDNA or control lentivirus infection were evaluated by in vivo tumorigenic assay. K, L: CD44CD133 double positive cells in tumor xenograft from CD44−CD133− Cal27 control and CD44−CD133− Cal27 with TAZ enforced overexpression were identified by immunofluorescence staining and quantified. Scale bar: 50 μm. Representative images are shown. Data shown here are mean ± SD from two or three independent experiments, *p < 0.05, **p < 0.01, Student's t‐test.
We next prospectively isolated putative CSCs fractions from cell lines by fluorescent‐activated cell sorting (FACS) using CD44 and CD133 as surface markers (Grosse‐Gehling et al., 2013; Prince et al., 2007), and then determined their phenotypes via in vitro tumorsphere and in vivo tumorigenic assays at limited dilution. To exclude the effects of varied endogenous TAZ on CSCs‐like subpopulation, both Cal27 and HN6 cells with high and low TAZ were selected. The FACS data indicated that CD44+CD133+ subpopulation accounted for approximate 12.04% and 14.63% in Cal27 and HN6 cells, respectively (Supplementary Fig.S8A). CD44+CD133+ cells displayed dramatically stronger capacities to generate tumorsphere in suspension, while most CD44−CD133− died and the survivors produced much smaller aggregates. Typical tumorsphere was extremely rare in CD44−CD133− cells (Supplementary Fig.S8B). Considering the serum as a potent differentiation inducer for cells cultured as tumorsphere, we added 10% FBS into tumorsphere cultures and observed that these floating sphere become attached and grown into monolayers with typical epithelial morphology and cell contact within 5–7 days. TAZ, along with multiple CSCs markers Bmi1 (Allegra et al., 2014), Lin28B (Zhou et al., 2013) and c‐myc (Wang et al., 2008) was pronouncedly reduced after serum addition, in line with serum‐induced CSCs differentiated into non‐CSCs progenies (Supplementary Fig. S8C). To further substantiate the unique properties of CD44+CD133+ CSCs, freshly sorted CD44+CD133+ and CD44−CD133− cells at limited dilutions combined with Matrigels were inoculated in flanks of nude mice. Data from in vivo tumorigenic assay indicated that CD44+CD133+ subpopulation had much stronger tumor‐initiating potentials as compared with CD44−CD133− subpopulation, suggesting that CD44+CD133+ subpopulation was enriched with CSCs (Supplementary Fig. S8D). H&E staining of tumor masses showed typical histomorphological manifestations reminiscent of squamous cell carcinoma (data not shown). Importantly, CD44+CD133+ cells generated CD44−CD133− counterparts in vivo as assessed by CD44 and CD133 double immunofluorescence staining, thus indicating that CD44+CD133+ CSCs gave rise to CD44−CD133− non‐CSCs progenies (Supplementary Fig. S8E). Together, these results indicate that CD44+CD133+ subpopulations in OSCC cell lines were enriched with CSCs with unique self‐renewal and tumor‐initiating properties.
Intrigued by TAZ, CTGF and Bmi1 enrichments in freshly sorted CD44+CD133+ CSCs from Cal27 and HN6 (Figure 5D) as well as their reductions in serum‐induced tumorsphere differentiation (Supplementary Fig. S8C), we hypothesized that TAZ was required for oral CSCs maintenance. To prove this, we monitored the percentages of CD44+CD133+ cells by manipulating endogenous TAZ. Indeed, TAZ knockdown or ectopic overexpression resulted in increased or reduced proportions of CD44+CD133+ subpopulations, respectively (Figure 5E). Moreover, as shown in Figure 5F, freshly‐sorted CD44+CD133+ cells generated significantly less tumorsphere following shTAZ lentivirus infection.
Growing evidence has suggest that CSC hierarchy might not be unidirectional as originally thought, but bi‐directional and interconvertible by evidenced that non‐CSCs were successfully reprogramed into CSCs spontaneously, induced by EMT or dedifferentiation in certain contexts (Chaffer et al., 2011; Mani et al., 2008; Vermeulen et al., 2010). Intrigued by increased proportions of CD44+CD133+ proportions followed TAZ overexpression, we reasoned that one possibility accounted for these changes, that is phenotypical transition from CD44−CD133− into CD44+CD133+ cells fueled by TAZ. To substantiate this, we overexpressed TAZ in freshly isolated CD44−CD133− cells and monitored their properties in vitro and in vivo. As shown in Figure 5G–I and Supplementary Fig.S9, remarkably more sphere formation, enhanced migration/invasion, increased CD44+CD133+ percentage as well as upregulated Bmi1 and lin28B were observed in CD44−CD133− cells infected with TAZ lentivirus. More importantly, when CD44−CD133− cells with or without forced TAZ overexpression at limited dilution were transplanted into immunodeficient mice, pronouncedly enhanced tumor‐initiating potentials were detected in TAZ‐overexpressed cells as indicated by more tumor formation and much shorter latency (Figure 5J and data not shown). Immunoflurescent staining data revealed increased CD44+CD133+ cells observed in samples derived from CD44−CD133− TAZ cells transplantation (Figure 5K–L). Collectively, our data indicate that TAZ is required for oral CSCs self‐renewal and maintenance. TAZ has capacities to endow non‐CSCs with CSCs‐like traits and facilitate the phenotypic transition from non‐CSCs to CSCs‐like cells.
3.5. TAZ‐TEADs interaction is involved in EMT and CSCs maintenance
TAZ primarily binds with TEADs and acts in concert to dictate transcriptional output of the downstream targets to regulate diverse biological processes (Chan et al., 2009; Zhang et al., 2009). We next wondered whether TAZ‐TEADs interaction was essential for oral cancer EMT and CSCs. To address this, we first knocked down four member of endogenous TEADs (TEAD1,2,3,4) via siRNAs in Cal27 and HN6 cells as measured by TEAD4 protein level and TEAD1‐4 transcripts (Figure 6A, and data not shown). Concomitantly, TEADs silencing led to E‐cadherin gain and vimentin loss, as well as Bmi1 and lin28B downregulation (Figure 6B). The efficiencies of colony formation and tumorsphere formation were remarkably compromised in TEADs‐depleted cells (Supplementary Figure 10A–C). Moreover, to testify whether TEADs knockdown can reverse the effects of TAZ, HN6 cells with stable TAZ overexpression were further treated with siTEADs and subjected to further analyses. Indeed, TEADs knockdown significantly attenuated the resultant expression changes of E‐cadherin, vimentin and Bmi1 induced by TAZ, whereas the control siRNAs failed. Moreover, TEADs silencing largely abolished the effects induced by TAZ as measured by colony formation, tumorsphere and migration assays (Figure 6C–D, Supplementary Fig.S10D and data not shown).
Figure 6.
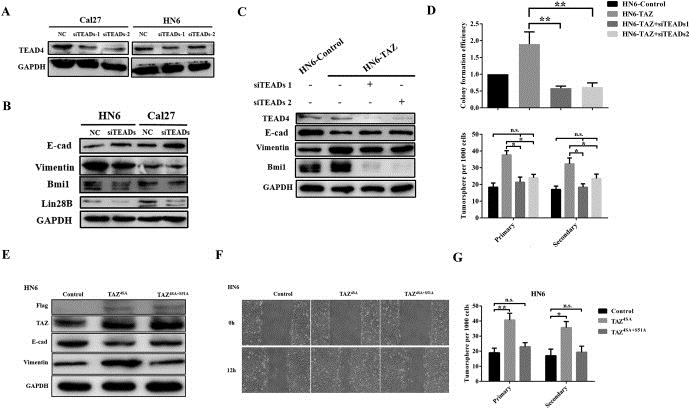
TAZ‐TEADs interaction mediates TAZ‐mediated EMT and CSCs maintenance A: Endogenous human TEADs were efficiently inhibited by siTEADs targeting human TEAD1‐4 as measured by TEAD4 protein as readout. B: The protein abundances of E‐cadherin, vimentin, Lin28B and Bmi1 were assessed when endogenous TEADs were repressed. C: The protein abundances of E‐cadherin, vimentin and Bmi1 were determined in TAZ overexpressing HN6 cells (HN6‐TAZ) when treated with siTEADAs (48 h). D: TEADs silencing attenuated the effects of TAZ overexpression in colony and tumorsphere formation. Colony formation efficiency was defined as the ratios of the numbers of colony generated from indicated cells, while the colony formation in controls was designed as 1. E,F: HN6 cells transiently transfected with constitutive active TAZ (TAZ4SA) induced EMT‐like changes, while cells transfected with TEADs binding‐deficient mutant (TAZ4SA+S51A) failed, as evidenced by EMT markers expression and migration assay. Controls cell received empty plasmids. G: HN6 cells with TAZ4SA transfection displayed significantly increased tumorsphere formation, while the efficiency of TAZ4SA+S51A transfected cells was comparable to control cells. Representative images are shown. Data shown here are mean ± SD from three independent experiments, *p < 0.05, **p < 0.01, ANOVA analyses.
To consolidate the critical roles of TAZ‐TEADs in EMT and CSCs, we transfected cells with TAZ mutant constructs (TAZ4SA, constitutive nuclear active TAZ; TAZ4SA+S51A, TEADs‐binding domain deleted mutant) (Lei et al., 2008; Zhang et al., 2009), generated stable clones and monitored their phenotypical changes. As shown in Figure 6E–F and Supplementary Fig.S10E, TAZ4SA overexpression resulted in typical EMT morphological changes and markers switch together with accelerated cell migration, while no similar alterations were detected in TAZ4SA + S51A cells. Moreover, TAZ4SA cells generated much more and larger tumorsphere as compared with TAZ4SA + S51A and control cells (Figure 6G). As the functional outputs of TAZ‐TEADs were largely mediated by transcriptional activation, we further found that these EMT transcription factors (Twist, Snail, Slug) and CSCs regulators (c‐myc, Sox2) were significantly decreased upon either TAZ or TEADs knockdown (Supplementary Fig.S10F‐G). In agreement with this, transcriptional activation of these factors were largely abrogated when cells were treated with TAZ mutant without TEAD binding as comparison with constitutive nuclear TAZ mutant (Supplementary Fig.S10H). Taken together, these data support the notion that TAZ‐TEADs interaction and transcriptional activation are essential for EMT and CSCs in OSCC.
3.6. Pharmacological inhibition of TAZ induces anti‐cancer therapeutic effects in vitro and in vivo
Having revealed the essential roles of TAZ in OSCC, we believed that TAZ might be therapeutically targeted to treat oral cancer, especially for those with TAZ hyperactivation. Recently, statins robustly inhibited Hippo‐TAZ/YAP via opposing TAZ/YAP nuclear translocation and in turn inactivated downstream effects (Sorrentino et al., 2014; Wang et al., 2014), thus raising the possibility that the statins might become as therapeutic agents against oral cancer. To address this, we first determined whether simvastatin was able to inhibit TAZ in vitro. Indeed, simvastatin remarkably reduced TAZ protein abundance in a time‐ and dose‐dependent manner (Figure 7A–B). In addition, the transcripts of TAZ, YAP and CTGF were also markedly downregulated (Supplementary Fig.S11A). Moreover, impaired cell proliferation and migration, tumorsphere formation as well as increased apoptosis were detected upon simvastatin treatment (Figure 7C–F and data not shown). Compared with single agent treatment, more pronounced effects were detected when cells were challenged with both simvastatin and 5‐FU/cisplatin than single agent treatment (Supplementary Fig.S11B). Not surprisingly, the expression changes of markers for cell proliferation, apoptosis and migration further confirmed the phenotypic changes upon simvastatin exposure (Figure 7G). As simvastatin is a chemical inhibitor of enzyme HMG‐CoA reductase by catalyzing mevalonic acid production, these aforementioned effects might be attributed to other targets beyond Hippo‐TAZ/YAP. To discern this possibility, we treated HN6 cells with stable TAZ overexpression (HN6‐TAZ) and control cells with empty vector (HN6‐Control) with simvastatin, respectively. As shown in Figure 7H and Supplementary Fig.S11C, HN6‐TAZ cells exhibited more resistance to simvastatin as measured by cell proliferation, apoptosis and migration in comparison with HN6‐Control cells. Furthermore, we performed an in vitro rescue experiment by TAZ overexpression following simvastatin treatment. Enforced TAZ overexpression, in part although not all, abolished the inhibitory effects exerted by simvastatin (Figure 7I–J). Therefore, these findings indicate that therapeutic effects of simvastatin might, at least in part, be attributed to TAZ inhibition in oral cancer cells.
Figure 7.
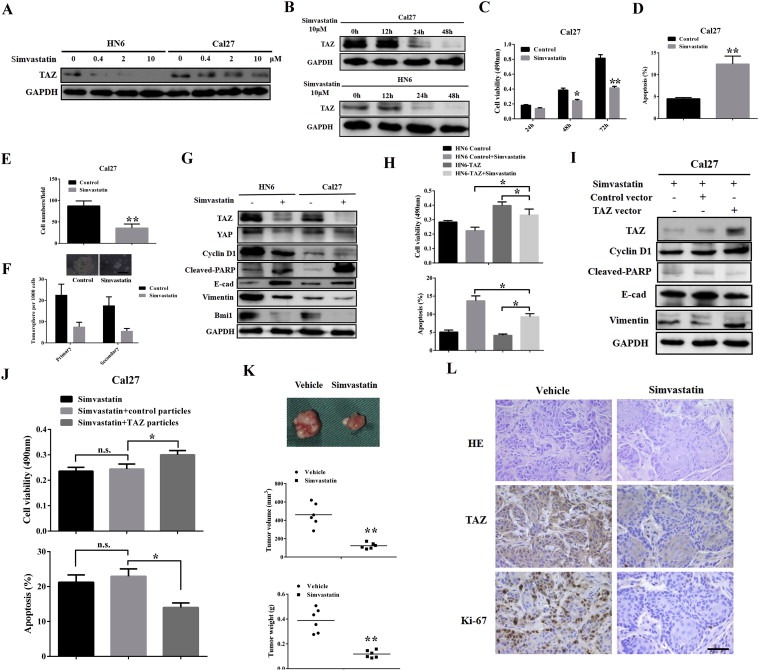
Pharmacological inhibition of TAZ by simvastatin induced anti‐cancer effects and inhibited tumor growth in vivo A,B: Simvastatin induced TAZ inhibition in a dose‐ (chemical treatment for 48 h) and time‐dependent manner in HN6 and Cal27 cells. C–F: Simvastatin treatment resulted in impaired cell proliferation (C), cell apoptosis (D), reduced migration (E) and tumorsphere (F) in Cal27 cells. Scale bar:50 μm. G: The protein abundances of TAZ, YAP, cleaved‐PARP, E‐cadherin, Vimentin and Bmi1 were assayed by western blot following simvastatin exposure. H: TAZ‐overexpressing cells were more resistant to simvastatin challenge as gauged by cell viability (upper panel) and apoptosis assays (lower panel). I–J: TAZ overexpression was able to attenuate the effects induced by simvastatin in Cal27 cells as measured by markers expression and cell viability/apoptosis assays. K: Simvastatin treatment by oral gavage inhibited tumor growth in a xenograft animal model. Upper panel: representative images of tumor masses from animals received simvastatin or vehicle treatment. Middle and lower panel: final tumor volume and weight. L: Representative images of H&E staining and TAZ, Ki‐67 immunohistochemical staining in tumor xenograft samples. Scale bar: 50 μm. Data shown here are mean ± SD from three independent experiments, *p < 0.05, **p < 0.01, ANOVA analyses and Student's t‐test.
To further confirm the therapeutic effects of simvastatin against oral cancer, we generated a xenograft animal model and treated animals with simvastatin. After tumor masses were reached approximately 200 mm3 in volume, these animals were randomly distributed into two groups and received simvastatin (150 mg/kg/day) or vehicle by oral gavage every day for consecutive 2 weeks. The simvastatin treatment was well tolerated in animals. As indicated in Figure 7K, tumor volume and weight in mice received simvastatin were much lower than those in control animals (P = 0.01). Most tumors exhibited retarded growth or regression after simvastatin treatment, which was also evidenced by remarkable downregulation of TAZ and Ki‐67, and increased TUNEL staining (Figure 7L and Supplementary Fig.S12) in simvastatin‐treated samples. To address whether tumor will regrow following simvastatin withdrawal, we treated animals bearing tumor xenograft with simvastatin for two weeks, then withdrew the drug and monitored tumor growth for another two weeks (Supplementary Fig.S13A). Our data showed some tumor regrowth after simvastatin discontinued (Supplementary Fig.S13B), thus suggesting that long‐term administration of simvastatin or in combination with other chemotherapeutic agents might resulted in durable tumor regression. Taken together, our data reveal that simvastatin has therapeutic potency against oral cancer partially through inactivating TAZ.
4. Discussion
Here we have defined TAZ, one of Hippo signaling transducers, as a novel bona fide oncogene with pleiotropic roles underlying OSCC tumorigenesis. Our data reveal that TAZ is aberrantly overexpressed in a fraction of OSCC, and associates with aggressive clinicopathological features and unfavorable patients' prognosis. Notably, TAZ promotes proliferation, anti‐apoptosis, migration and invasion, as well as EMT and CSCs maintenance in oral cancer. Furthermore, simvastatin has been identified as a robust chemical inhibitor of TAZ and is therapeutically effective against oral cancer.
The Hippo pathway is an evolutionarily conserved signaling for tissue growth, whereas its perturbation links to tumorigenesis. Mutation and deregulated expression of Hippo members including TAZ have been revealed in multiple human cancers (Harvey et al., 2013). For example, aberrant TAZ overexpression has been observed and associated with aggressive features and unfavorable survival in papillary thyroid carcinoma, breast cancer, and malignant glioma (Bhat et al., 2011; Chan et al., 2008; de Cristofaro et al., 2011). Our data extend these previous findings and further reveal that TAZ is aberrantly overexpressed in human oral cancers and associates with aggressiveness and adverse patient prognosis. These data support the notion that TAZ is a novel diagnostic and prognostic biomarker for oral cancer and holds promise as one of essential clinicopathological factors when select appropriate treatment and predict prognosis for patients.
Our findings from both gain and loss‐of‐function assays indicate that TAZ has multiple tumorigenic roles by regulating cell proliferation, apoptosis, migration and invasion in oral cancer cells, which is in agreement with the pro‐proliferative and anti‐apoptosis functions of Hippo‐TAZ in physical and pathological settings (Chan et al., 2008; Dong et al., 2007; Zhang et al., 2009). Of note, typical morphological changes, markers switch and induction of EMT inducers like Twist and Snail upon TAZ overexpression, as well as the pro‐metastatic functions of TAZ in vitro and in vivo provided strong evidence that TAZ is capable of promoting EMT in oral cancer cells. Complementary, we also unraveled the pivotal roles of TAZ required for TGF‐β1‐induced EMT. In the TGF‐β1‐triggered EMT model, rhTGF‐β1 not only promoted TAZ transcription, but also reduced its phosphorylation and fuelled its nuclear translocation by inactivating Hippo kinases. These findings are consistent with recent findings that in response to TGF‐β1, TAZ binds heteromeric Smad complexes and is recruited to TGF‐β response elements in nucleus, and in turn activates downstream targets in embryonic stem cells (Varelas et al., 2008). Furthermore, TAZ/YAP is necessary to promote and maintain TGF‐β‐induced tumorigenic phenotypes such as increased cell migration and invasion by coordinating TAZ/YAP‐TEAD‐Smad2/3‐mediated transcriptional program in breast cancer cells (Hiemer et al., 2014). Together, these findings indicate that TAZ is a potent EMT inducer, and may act downstream of TGF‐β/Smads pathway and mediate some, if not all, its functional outputs like EMT in cancer. Further studies are still needed to unravel the intricate crosstalk between Hippo‐TAZ and TGF‐β/Smads pathway behind oral tumorigenesis.
Previous reports have established an important role of TAZ in regulating self‐renewal and differentiation in both normal and malignant stem cells (Bartucci et al., 2015; Cordenonsi et al., 2011; Hong et al., 2005). To extend this, we provide evidence that TAZ mediates self‐renewal and maintenance of oral CSCs, largely based on the following findings: (1) TAZ enrichment in oral CSCs subpopulation and decreased during serum‐induced tumorsphere differentiation; (2) impaired tumorsphere formation, reduced CSCs percentages and increased chemosensitivity upon endogenous TAZ knockdown, and opposite results following TAZ overexpression; (3) positive associations between TAZ and high pathological grade or metastasis in clinical samples. Thus, TAZ is a new regulatory governor of CSCs in human oral cancer.
Accumulating evidence supports the existence of a dynamic equilibrium and bidirectional interconvertibility between CSCs and non‐CSCs in some, if not all, human cancers (Chaffer et al., 2013; Iliopoulos et al., 2011; Vermeulen et al., 2010). The conversion from non‐CSCs to CSCs, also term CSCs plasticity, has increasingly been recognized as a novel plausible source of cellular origins for CSCs besides normal stem cells or progenitors (Scheel and Weinberg, 2011). This plasticity also might account for tumor relapse, metastatic dissemination and treatment resistance following therapeutic interventions (Beck and Blanpain, 2013; Gupta et al., 2009; Scheel and Weinberg, 2011). A seminal finding uncovered that TAZ conferred CSCs‐related properties to non‐CSCs and allowed them to regain stemness by EMT in breast cancer, thus raising an intriguing possibility that TAZ functioned as a molecular switcher between CSCs and non‐CSCs (Cordenonsi et al., 2011). Our data reveal that enforced overexpression of TAZ in freshly purified non‐CSCs significantly enhanced their self‐renewal, increased CSCs proportions as well as in vivo tumorigenic‐initiating properties, thus suggesting that TAZ is able to confer CSCs traits onto more differentiated oral cancer cells. Similarly, De Maria R et al. identified TAZ as a central mediator of breast CSCs properties and revealed that TAZ overexpression in differentiated cancer cells induced cell transformation and conferred tumorigenicity and migratory activities (Bartucci et al., 2015). However, the key issues still remain that whether EMT induced by ectopic TAZ is functionally coupled with oral CSCs plasticity, although we and other reports support that EMT and CSCs plasticity are tightly interconnected, while other findings favor the their independence (Beck et al., 2015; Mani et al., 2008; Morel et al., 2008; Schwitalla et al., 2013; Yang et al., 2013). Therefore, our findings together with others (Bartucci et al., 2015; Bhat et al., 2011; Cordenonsi et al., 2011) support that TAZ could act as a functional phenotypic switcher by fuelling novel CSCs generation from non‐CSCs in certain cancers.
Paracrine signals from tumor‐associated microenvironment induced and maintained stem cell traits in both normal and transformed breast cells (Scheel et al., 2011). These contextual signals from tumor stroma, for example TGF‐β1 and HGF, are sufficient to convert non‐CSCs to CSCs and restore CSCs traits in more differentiated cancer cells (Scheel et al., 2011; Vermeulen et al., 2010). We reason that, in addition to genomic amplification and transcriptional activation of TAZ, signals from tumor‐associated stroma might be also involved in TAZ hyperactivation which in turn promotes oral CSCs maintenance and plasticity. This was supported by the findings that several mitogenic growth factors usually emanated from tumor microenvironment, for example EGF and TGF‐β1, inactivated the Hippo signaling and in turn activated TAZ/YAP by dephosphorylation and nuclear accumulation (Fan et al., 2013; Varelas et al., 2008). On the other hand, the induction signal for TAZ hyperactivation might be from increased stiffness of tumor stroma, since TAZ/YAP serves as key sensors and mediators of mechanical cues instructed by cellular microenvironment, and mechanical stress regulates stem cell behaviors and cell fate decision by affecting Hippo‐TAZ/YAP signaling (Halder et al., 2012). It will be interesting to unravel the roles of TAZ driving CSCs plasticity from the perspective of tumor–tumor microenvironment interaction.
TAZ functions as a transcriptional coactivator in complex with TEADs to mediate downstream transcriptional events. Disruption of TAZ‐TEADs binding blocked the major effects mediated by TAZ including oncogenic transformation, pro‐proliferation and EMT (Chan et al., 2009; Zhang et al., 2009). In agreement with this, our data indicate that TAZ‐TEADs binding is critically involved in TAZ‐induced EMT in oral cancer cells as evidenced by the facts that EMT phenotypic changes were largely abrogated in TEADs knockdown and TAZ mutant experiments. A line of evidence suggest that TAZ promotes EMT by inducing EMT‐associated factors like Snail and Foxc2 in breast cancer (Lei et al., 2008). Similarly, our data reveal that TAZ activates transcription of EMT inducers such as Twist, Snail and Slug, which are presumably responsible for EMT in oral cancer cells. This is also supported by the identification and verification of TEAD1 binding sites in promoter regions of Twist, Snail and Slug in embryonal rhabdomyosarcoma cell (Tremblay et al., 2014).
Growing evidence suggests that complex transcriptional regulatory networks including transcription factors and signaling pathways converges on CSCs plasticity (Vermeulen et al., 2010; Wong et al., 2008). Our data suggest that transcriptional program mediated by pluripotent mediators such as c‐myc, Sox2, lin28B and Bmi1 induced by TAZ is presumably responsible for oral CSCs plasticity. Because several factors important for pluripotency and self‐renewal such as Sox2 were found to contain putative TEAD‐binding sites and were bound by TEADs complex during stem cell renewal (Lian et al., 2010). Moreover, Sox2 is a novel functional marker for skin CSCs by directly regulating target genes controlling cancer stemness (Boumahdi et al., 2014). Indeed, these transcription factors were the key reprogramming mediators for iPS generation from differentiated cells and also critical for CSCs self‐renewal and expansion (Takahashi et al., 2007; Yang et al., 2010). It's plausible for us to reason that similar reprogramming process induced by these transcription factors exists for both normal and malignant stem cell generation, which requires further experimental substantiations.
Verteporfin was the firstly identified chemical to inhibit YAP‐induced liver oncogenic overgrowth by disturbing YAP‐TEAD binding, thus providing proof‐of‐principle that targeting Hippo‐TAZ/YAP was pharmacologically viable in cancer therapeutics (Liu‐Chittenden et al., 2012). Intriguingly, statins inhibited proliferation and self‐renewal of breast cancer cells via reducing TAZ/YAP abundance and nuclear localization (Sorrentino et al., 2014; Wang et al., 2014). Here our data reveal that TAZ and its oncogenic functions in OSCC could be inhibited both in vitro and in vivo by simvastatin. Thus, we propose that, simvastatin, alone or in combinations with current chemotherapeutic agents, may have additional beneficial effects for oral cancer patients. In addition, the CSC model emphasizes the crucial importance to target CSCs in cancer therapy. However, more plasticity exists in cancer cells than conventionally anticipated, offering the rationale for novel therapeutic strategies to block such phenotypic transition. The optimal therapeutic regimens may comprise agents that target CSCs, non‐CSCs and their interconversion, ultimately achieving long‐term and truly curative outcomes (Medema, 2013).
In conclusion, we have elucidated the tumorigenic roles of TAZ responsible for oral cancer initiation and progression and revealed TAZ as a novel biomarker with diagnostic and prognostic significance. Our findings identify TAZ as an important mediator of EMT and oral CSCs, and also a viable therapeutic target against oral cancer.
Conflict of interests
The authors declare no interests of conflict.
Author contributions
Jie Cheng, Jianrong Yang and Zhongwu Li conceived and supervised the whole project, designed experiments, analyzed the data and wrote the paper. Zhongwu Li, Yanling Wang, Yumin Zhu, Chunping Yuan carried out the most experiments. Dongmiao Wang, Wei Zhang, Bin Qi and Jin Qiu performed histological and statistical analyses. Xiaomeng Song, Jinhai Ye, Hemin Wu, Hongbing Jiang, Laikui Liu and Yuan Zhang provided clinical samples and follow‐up data. Liang‐Nian Song offered helpful discussions and contributed to manuscript writing.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
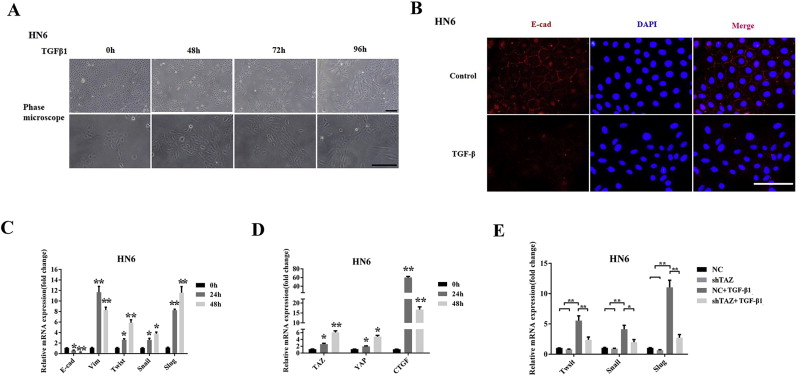
Figure. 4 A: Morphological changes were monitored under phase‐contrast microscope after HN6 cells were treated by rhTGF‐β1(10μ/ml, 96h). Scale bar:50μm. B: E‐cadherin location and abundance was visualized by immunoflurescent staining in HN6 cells with or without rhTGF‐β1 exposure. C: The expression of EMT associated markers and transcriptional regulators was assessed by real‐time RT‐PCR in HN6 cells with or without rhTGF‐β1 treatment. D: The transcripts of TAZ, YAP and CTGF were also determined in HN6 cells with or without rhTGF‐β1 treatment. E: The transcripts of Twist, Snail and Slug were measured in indicated cells treated with or without rhTGF‐β1. Data shown here are mean±SD from three independent experiments. *p<0.05, **p<0.01, ANOVA and Student's t‐test.
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
We are grateful to Prof. Guan Kunliang and Chen Wantao for their kind sharing with experimental materials. This work was supported, in whole or in part, by National Natural Science Foundation of China (Grant No. 81100737), Natural Science Foundation of Jiangsu Province (Grant No. BK2011762, BK20130898), Specialized Research Fund for the Doctoral Program of Higher Education (Grant No. 20113234120003), China Postdoctoral Science Foundation (2014M560436), and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (Grant No.2014‐37, JX10531802).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.01.007.
Li Zhongwu, Wang Yanling, Zhu Yumin, Yuan Chunping, Wang Dongmiao, Zhang Wei, Qi Bin, Qiu Jin, Song Xiaomeng, Ye Jinhai, Wu Heming, Jiang Hongbing, Liu Laikui, Zhang Yuan, Song Liang-Nian, Yang Jianrong, Cheng Jie, (2015), The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.01.007.
Contributor Information
Jianrong Yang, Email: cj992118@yahoo.com.
Jie Cheng, Email: leonardo_cheng@163.com.
References
- Allegra, E. , Trapasso, S. , Pisani, D. , Puzzo, L. , 2014. The role of BMI1 as a biomarker of cancer stem cells in head and neck cancer: a review. Oncology 86, 199–205. [DOI] [PubMed] [Google Scholar]
- Bartucci, M. , Dattilo, R. , Moriconi, C. , Pagliuca, A. , Mottolese, M. , Federici, G. , Benedetto, A.D. , Todaro, M. , Stassi, G. , Sperati, F. , Amabile, M.I. , Pilozzi, E. , Patrizii, M. , Biffoni, M. , Maugeri-Sacca, M. , Piccolo, S. , De Maria, R. , 2015. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 34, (6) 681–690. [DOI] [PubMed] [Google Scholar]
- Beck, B. , Blanpain, C. , 2013. Unravelling cancer stem cell potential. Nat. Rev. Cancer 13, 727–738. [DOI] [PubMed] [Google Scholar]
- Beck, B. , Lapouge, G. , Rorive, S. , Drogat, B. , Desaedelaere, K. , Delafaille, S. , Dubois, C. , Salmon, I. , Willekens, K. , Marine, J.C. , Blanpain, C. , 2015. Different levels of twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 16, 67–79. [DOI] [PubMed] [Google Scholar]
- Bhat, K.P. , Salazar, K.L. , Balasubramaniyan, V. , Wani, K. , Heathcock, L. , Hollingsworth, F. , James, J.D. , Gumin, J. , Diefes, K.L. , Kim, S.H. , Turski, A. , Azodi, Y. , Yang, Y. , Doucette, T. , Colman, H. , Sulman, E.P. , Lang, F.F. , Rao, G. , Copray, S. , Vaillant, B.D. , Aldape, K.D. , 2011. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 25, 2594–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumahdi, S. , Driessens, G. , Lapouge, G. , Rorive, S. , Nassar, D. , Le Mercier, M. , Delatte, B. , Caauwe, A. , Lenglez, S. , Nkusi, E. , Brohee, S. , Salmon, I. , Dubois, C. , del Marmol, V. , Fuks, F. , Beck, B. , Blanpain, C. , 2014. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250. [DOI] [PubMed] [Google Scholar]
- Chaffer, C.L. , Brueckmann, I. , Scheel, C. , Kaestli, A.J. , Wiggins, P.A. , Rodrigues, L.O. , Brooks, M. , Reinhardt, F. , Su, Y. , Polyak, K. , Arendt, L.M. , Kuperwasser, C. , Bierie, B. , Weinberg, R.A. , 2011. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. U S A 108, 7950–7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer, C.L. , Marjanovic, N.D. , Lee, T. , Bell, G. , Kleer, C.G. , Reinhardt, F. , D'Alessio, A.C. , Young, R.A. , Weinberg, R.A. , 2013. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, S.W. , Lim, C.J. , Guo, K. , Ng, C.P. , Lee, I. , Hunziker, W. , Zeng, Q. , Hong, W. , 2008. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 68, 2592–2598. [DOI] [PubMed] [Google Scholar]
- Chan, S.W. , Lim, C.J. , Loo, L.S. , Chong, Y.F. , Huang, C. , Hong, W. , 2009. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J. Biol. Chem. 284, 14347–14358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi, M. , Zanconato, F. , Azzolin, L. , Forcato, M. , Rosato, A. , Frasson, C. , Inui, M. , Montagner, M. , Parenti, A.R. , Poletti, A. , Daidone, M.G. , Dupont, S. , Basso, G. , Bicciato, S. , Piccolo, S. , 2011. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772. [DOI] [PubMed] [Google Scholar]
- de Cristofaro, T. , Di Palma, T. , Ferraro, A. , Corrado, A. , Lucci, V. , Franco, R. , Fusco, A. , Zannini, M. , 2011. TAZ/WWTR1 is overexpressed in papillary thyroid carcinoma. Eur. J. Cancer 47, 926–933. [DOI] [PubMed] [Google Scholar]
- Dong, J. , Feldmann, G. , Huang, J. , Wu, S. , Zhang, N. , Comerford, S.A. , Gayyed, M.F. , Anders, R.A. , Maitra, A. , Pan, D. , 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, R. , Kim, N.G. , Gumbiner, B.M. , 2013. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. U S A 110, 2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Gehling, P. , Fargeas, C.A. , Dittfeld, C. , Garbe, Y. , Alison, M.R. , Corbeil, D. , Kunz-Schughart, L.A. , 2013. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J. Pathol. 229, 355–378. [DOI] [PubMed] [Google Scholar]
- Gupta, P.B. , Chaffer, C.L. , Weinberg, R.A. , 2009. Cancer stem cells: mirage or reality?. Nat. Med. 15, 1010–1012. [DOI] [PubMed] [Google Scholar]
- Halder, G. , Dupont, S. , Piccolo, S. , 2012. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol 13, 591–600. [DOI] [PubMed] [Google Scholar]
- Hao, J. , Zhang, Y. , Jing, D. , Li, Y. , Li, J. , Zhao, Z. , 2014. Role of Hippo signaling in cancer stem cells. J. Cell Physiol 229, 266–270. [DOI] [PubMed] [Google Scholar]
- Harvey, K.F. , Zhang, X. , Thomas, D.M. , 2013. The Hippo pathway and human cancer. Nat. Rev. Cancer 13, 246–257. [DOI] [PubMed] [Google Scholar]
- Hiemer, S.E. , Szymaniak, A.D. , Varelas, X. , 2014. The transcriptional regulators TAZ and YAP direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J. Biol. Chem. 289, 13461–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, J.H. , Hwang, E.S. , McManus, M.T. , Amsterdam, A. , Tian, Y. , Kalmukova, R. , Mueller, E. , Benjamin, T. , Spiegelman, B.M. , Sharp, P.A. , Hopkins, N. , Yaffe, M.B. , 2005. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078. [DOI] [PubMed] [Google Scholar]
- Iliopoulos, D. , Hirsch, H.A. , Wang, G. , Struhl, K. , 2011. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. U S A 108, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille, S. , Xu, J. , Derynck, R. , 2014. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, Q.Y. , Zhang, H. , Zhao, B. , Zha, Z.Y. , Bai, F. , Pei, X.H. , Zhao, S. , Xiong, Y. , Guan, K.L. , 2008. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell Biol 28, 2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Wang, Y. , Qiu, J. , Li, Q. , Yuan, C. , Zhang, W. , Wang, D. , Ye, J. , Jiang, H. , Yang, J. , Cheng, J. , 2013. The polycomb group protein EZH2 is a novel therapeutic target in tongue cancer. Oncotarget 4, 2532–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, I. , Kim, J. , Okazawa, H. , Zhao, J. , Zhao, B. , Yu, J. , Chinnaiyan, A. , Israel, M.A. , Goldstein, L.S. , Abujarour, R. , Ding, S. , Guan, K.L. , 2010. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 24, 1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden, Y. , Huang, B. , Shim, J.S. , Chen, Q. , Lee, S.J. , Anders, R.A. , Liu, J.O. , Pan, D. , 2012. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L.K. , Jiang, X.Y. , Zhou, X.X. , Wang, D.M. , Song, X.L. , Jiang, H.B. , 2010. Upregulation of vimentin and aberrant expression of E-cadherin/beta-catenin complex in oral squamous cell carcinomas: correlation with the clinicopathological features and patient outcome. Mod. Pathol. 23, 213–224. [DOI] [PubMed] [Google Scholar]
- Mani, S.A. , Guo, W. , Liao, M.J. , Eaton, E.N. , Ayyanan, A. , Zhou, A.Y. , Brooks, M. , Reinhard, F. , Zhang, C.C. , Shipitsin, M. , Campbell, L.L. , Polyak, K. , Brisken, C. , Yang, J. , Weinberg, R.A. , 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauviel, A. , Nallet-Staub, F. , Varelas, X. , 2012. Integrating developmental signals: a Hippo in the (path)way. Oncogene 31, 1743–1756. [DOI] [PubMed] [Google Scholar]
- Medema, J.P. , 2013. Cancer stem cells: the challenges ahead. Nat. Cell Biol 15, 338–344. [DOI] [PubMed] [Google Scholar]
- Morel, A.P. , Lievre, M. , Thomas, C. , Hinkal, G. , Ansieau, S. , Puisieux, A. , 2008. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3, e2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer, M. , Zhang, J. , Smolen, G.A. , Muir, B. , Li, W. , Sgroi, D.C. , Deng, C.X. , Brugge, J.S. , Haber, D.A. , 2006. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. U S A 103, 12405–12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak, K. , Weinberg, R.A. , 2009. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer 9, 265–273. [DOI] [PubMed] [Google Scholar]
- Prince, M.E. , Sivanandan, R. , Kaczorowski, A. , Wolf, G.T. , Kaplan, M.J. , Dalerba, P. , Weissman, I.L. , Clarke, M.F. , Ailles, L.E. , 2007. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. U S A 104, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel, C. , Eaton, E.N. , Li, S.H. , Chaffer, C.L. , Reinhardt, F. , Kah, K.J. , Bell, G. , Guo, W. , Rubin, J. , Richardson, A.L. , Weinberg, R.A. , 2011. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel, C. , Weinberg, R.A. , 2011. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells?. Int. J. Cancer 129, 2310–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalla, S. , Fingerle, A.A. , Cammareri, P. , Nebelsiek, T. , Goktuna, S.I. , Ziegler, P.K. , Canli, O. , Heijmans, J. , Huels, D.J. , Moreaux, G. , Rupec, R.A. , Gerhard, M. , Schmid, R. , Barker, N. , Clevers, H. , Lang, R. , Neumann, J. , Kirchner, T. , Taketo, M.M. , van den Brink, G.R. , Sansom, O.J. , Arkan, M.C. , Greten, F.R. , 2013. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38. [DOI] [PubMed] [Google Scholar]
- Scully, C. , Bagan, J.V. , 2009. Recent advances in oral oncology 2008; squamous cell carcinoma imaging, treatment, prognostication and treatment outcomes. Oral Oncol. 45, e25–30. [DOI] [PubMed] [Google Scholar]
- Sinha, N. , Mukhopadhyay, S. , Das, D.N. , Panda, P.K. , Bhutia, S.K. , 2013. Relevance of cancer initiating/stem cells in carcinogenesis and therapy resistance in oral cancer. Oral Oncol. 49, 854–862. [DOI] [PubMed] [Google Scholar]
- Sorrentino, G. , Ruggeri, N. , Specchia, V. , Cordenonsi, M. , Mano, M. , Dupont, S. , Manfrin, A. , Ingallina, E. , Sommaggio, R. , Piazza, S. , Rosato, A. , Piccolo, S. , Del Sal, G. , 2014. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol 16, 357–366. [DOI] [PubMed] [Google Scholar]
- Takahashi, K. , Tanabe, K. , Ohnuki, M. , Narita, M. , Ichisaka, T. , Tomoda, K. , Yamanaka, S. , 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Tremblay, A.M. , Missiaglia, E. , Galli, G.G. , Hettmer, S. , Urcia, R. , Carrara, M. , Judson, R.N. , Thway, K. , Nadal, G. , Selfe, J.L. , Murray, G. , Calogero, R.A. , De Bari, C. , Zammit, P.S. , Delorenzi, M. , Wagers, A.J. , Shipley, J. , Wackerhage, H. , Camargo, F.D. , 2014. The Hippo transducer YAP1 Transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 26, (2) 273–287. [DOI] [PubMed] [Google Scholar]
- Valastyan, S. , Weinberg, R.A. , 2011. Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas, X. , Sakuma, R. , Samavarchi-Tehrani, P. , Peerani, R. , Rao, B.M. , Dembowy, J. , Yaffe, M.B. , Zandstra, P.W. , Wrana, J.L. , 2008. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol 10, 837–848. [DOI] [PubMed] [Google Scholar]
- Vermeulen, L. , De Sousa, E.M.F. , van der Heijden, M. , Cameron, K. , de Jong, J.H. , Borovski, T. , Tuynman, J.B. , Todaro, M. , Merz, C. , Rodermond, H. , Sprick, M.R. , Kemper, K. , Richel, D.J. , Stassi, G. , Medema, J.P. , 2010. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol 12, 468–476. [DOI] [PubMed] [Google Scholar]
- Wan, L. , Pantel, K. , Kang, Y. , 2013. Tumor metastasis: moving new biological insights into the clinic. Nat. Med. 19, 1450–1464. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Wang, H. , Li, Z. , Wu, Q. , Lathia, J.D. , McLendon, R.E. , Hjelmeland, A.B. , Rich, J.N. , 2008. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 3, e3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Wu, Y. , Wang, H. , Zhang, Y. , Mei, L. , Fang, X. , Zhang, X. , Zhang, F. , Chen, H. , Liu, Y. , Jiang, Y. , Sun, S. , Zheng, Y. , Li, N. , Huang, L. , 2014. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. U S A 111, E89–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, D.J. , Liu, H. , Ridky, T.W. , Cassarino, D. , Segal, E. , Chang, H.Y. , 2008. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C.C. , Zhu, L.F. , Xu, X.H. , Ning, T.Y. , Ye, J.H. , Liu, L.K. , 2013. Membrane Type 1 Matrix Metalloproteinase induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. BMC Cancer 13, 171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M.H. , Hsu, D.S. , Wang, H.W. , Wang, H.J. , Lan, H.Y. , Yang, W.H. , Huang, C.H. , Kao, S.Y. , Tzeng, C.H. , Tai, S.K. , Chang, S.Y. , Lee, O.K. , Wu, K.J. , 2010. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol 12, 982–992. [DOI] [PubMed] [Google Scholar]
- Zavadil, J. , Bottinger, E.P. , 2005. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Liu, C.Y. , Zha, Z.Y. , Zhao, B. , Yao, J. , Zhao, S. , Xiong, Y. , Lei, Q.Y. , Guan, K.L. , 2009. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 284, 13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, B. , Tumaneng, K. , Guan, K.L. , 2011. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol 13, 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Ng, S.B. , Chng, W.J. , 2013. LIN28/LIN28B: an emerging oncogenic driver in cancer stem cells. Int. J. Biochem. Cell Biol 45, 973–978. [DOI] [PubMed] [Google Scholar]
- Zhu, L.F. , Hu, Y. , Yang, C.C. , Xu, X.H. , Ning, T.Y. , Wang, Z.L. , Ye, J.H. , Liu, L.K. , 2012. Snail overexpression induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. Lab Invest 92, 744–752. [DOI] [PubMed] [Google Scholar]
- Zomer, A. , Ellenbroek, S.I. , Ritsma, L. , Beerling, E. , Vrisekoop, N. , Van Rheenen, J. , 2013. Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells 31, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Figure. 4 A: Morphological changes were monitored under phase‐contrast microscope after HN6 cells were treated by rhTGF‐β1(10μ/ml, 96h). Scale bar:50μm. B: E‐cadherin location and abundance was visualized by immunoflurescent staining in HN6 cells with or without rhTGF‐β1 exposure. C: The expression of EMT associated markers and transcriptional regulators was assessed by real‐time RT‐PCR in HN6 cells with or without rhTGF‐β1 treatment. D: The transcripts of TAZ, YAP and CTGF were also determined in HN6 cells with or without rhTGF‐β1 treatment. E: The transcripts of Twist, Snail and Slug were measured in indicated cells treated with or without rhTGF‐β1. Data shown here are mean±SD from three independent experiments. *p<0.05, **p<0.01, ANOVA and Student's t‐test.
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data