

Abstract
STAT3 is a key element in many oncogenic pathways and, like other transcription factors, is an attractive target for development of novel anticancer drugs. However, interfering with STAT3 functions has been a difficult task and very few small molecule inhibitors have made their way to the clinic. OPB‐31121, an anticancer compound currently in clinical trials, has been reported to affect STAT3 signaling, although its mechanism of action has not been unequivocally demonstrated. In this study, we used a combined computational and experimental approach to investigate the molecular target and the mode of interaction of OPB‐31121 with STAT3. In parallel, similar studies were performed with known STAT3 inhibitors (STAT3i) to validate our approach. Computational docking and molecular dynamics simulation (MDS) showed that OPB‐31121 interacted with high affinity with the SH2 domain of STAT3. Interestingly, there was no overlap of the OPB‐31121 binding site with those of the other STAT3i. Computational predictions were confirmed by in vitro binding assays and competition experiments along with site‐directed mutagenesis of critical residues in the STAT3 SH2 domain. Isothermal titration calorimetry experiments demonstrated the remarkably high affinity of OPB‐31121 for STAT3 with Kd (10 nM) 2–3 orders lower than other STAT3i. Notably, a similar ranking of the potency of the compounds was observed in terms of inhibition of STAT3 phosphorylation, cancer cell proliferation and clonogenicity. These results suggest that the high affinity and efficacy of OPB‐31121 might be related to the unique features and mode of interaction of OPB‐31121 with STAT3. These unique characteristics make OPB‐31121 a promising candidate for further development and an interesting lead for designing new, more effective STAT3i.
Keywords: STAT3, Transcription factors, Small molecule inhibitors, Anticancer drugs, Cancer therapy, Mechanism of action, Computational modeling, Molecular dynamic simulation, Site-directed mutagenesis
Highlights
STAT3 is an attractive target for development of novel anticancer drugs.
OPB‐31121 is a putatively STAT3 inhibitor currently in clinical trials.
OPB‐31121 binds with high affinity to the SH2 domain of STAT3.
OPB‐31121 binds at a novel site in the SH2 domain distinct from other inhibitors.
1. Introduction
Signal Transducers and Activators of Transcription (STATs) are a family of latent cytoplasmic proteins that once activated regulate many aspects of cell growth, survival and differentiation (Levy and Darnell, 2002; Yu et al., 2009). The main function attributed to STAT proteins is to act as signal transducers and transcription factors with the ability to transmit signals from the cell membrane to the nucleus (Levy and Darnell, 2002; Yu et al., 2009). However, recent studies have revealed a far more complex picture with a range of novel and diverse functions associated with STAT signaling both in the nucleus and other cell compartments (Sehgal, 2008; Xu et al., 2007; Yu et al., 2014). The STAT family includes seven members (STAT1, 2, 3, 4, 5a, 5b, and 6) that share extensive structural homology (Yu et al., 2009). The main structural motifs of STAT proteins are the N‐terminal domain (NTD), coiled‐coil domain (CCD), DNA‐binding domain (DBD), Src Homology 2 domain (SH2) and C‐terminal domain (CTD). The NTD and CCD are required for nuclear translocation and protein–protein interaction, respectively (Levy and Darnell, 2002; Lim and Cao, 2006). The DBD is necessary for the recognition of specific DNA sequence elements and binding to gene promoters. The SH2 domain is the most conserved domain of the family and is required for formation of STAT3 dimers upon phosphorylation of specific tyrosine residues in the CTD of STAT proteins (Lim and Cao, 2006; Zhong et al., 1994). In the case of STAT3 the key event is the phosphorylation of tyrosine 705 (pY705). This promotes the interaction between the SH2 domains of distinct monomers and has been considered the main pathway of activation of STAT3 signaling to the nucleus. pY705 is induced by binding of cytokines and growth factors to the respective receptors and consequent activation of the receptor‐associated tyrosine kinases, like Janus Kinases (JAK) (Yu et al., 2009). Other non‐receptor associated kinases, such as Src, also activate nuclear STAT3 signaling through the phosphorylation of Y705. Furthermore, in addition to Y705 phosphorylation, STAT3 is phosphorylated at serine 727 (pS727) by various serine protein kinases (Zhang et al., 1995). This modification has been reported to enhance the STAT3 transcriptional activity (Wen et al., 1995) and, more recently, to control mitochondrial localization and function of STAT3 (Gough et al., 2009; Wegrzyn et al., 2009). Acetylation and methylation by protein acetyltransferases and methyltransferases play also relevant roles in controlling STAT3 functions in normal and pathological conditions (Kim et al., 2013a; Lee et al., 2012; Yu et al., 2014; Yuan et al., 2005). Furthermore, un‐phosphorylated STAT3, present both in the cytoplasm and nucleus, form dimers and has biological activity as transcription factor and signal transducer independent of its phosphorylation status (Liu et al., 2005; Sehgal, 2008; Timofeeva et al., 2012; Yang et al., 2007).
Alterations of the STAT3 signaling are associated with different human diseases (O'Shea and Plenge, 2012). STAT3 is over‐expressed and activated in many human cancers and promotes cell proliferation, survival, tumor angiogenesis and immune‐evasion (Sansone and Bromberg, 2012; Yu et al., 2009). Activation of the JAK/STAT3 pathway has been shown to contribute to tumor initiation and progression in various cancer models (Yu et al., 2014, 2009). Recently, activation of STAT3 has been associated with promotion and maintenance of cancer stem‐like cells (CSC), tumorigenicity and metastatic capability in many human cancers, including prostate cancer (Kroon et al., 2013; Marotta et al., 2011; Schroeder et al., 2014; Yu et al., 2014). In many cancers activation of STAT3 is associated with advanced disease, metastasis and clinical progression (Sansone and Bromberg, 2012; Yu et al., 2009). The JAK/STAT3 pathway contributes also to reduced response to treatment promoting survival and development of resistance after treatment with kinase inhibitors or, in prostate cancer, after androgen deprivation therapy (Lee et al., 2014; Schroeder et al., 2014; Sos et al., 2014). We have shown recently that activation of the JAK/STAT3 pathway contributes the establishment of immune‐tolerance and chemoresistance in a prostate cancer mouse model through the secretion of immunosuppressive cytokines in the tumor microenvironment (Toso et al., 2014).
Over‐activity of STAT3 in human cancers is frequently the result of deregulation of upstream pathways leading to activation of cytokine and growth factor receptor associated tyrosine kinases, like JAK family kinases (Grivennikov and Karin, 2008; Sansone and Bromberg, 2012; Yu et al., 2014). Alternative pathways controlling transcriptional and non‐transcriptional functions of STAT3 may have also important roles in abnormal activation of STAT3 signaling in cancer (Meier and Larner, 2014; Yu et al., 2014). In prostate cancer STAT3 has been reported to induce cell transformation and tumor development in the absence of pY705 (Qin et al., 2008). The oncogenic effect of STAT3 in this system depended on pS727 and transcriptional dependent and independent functions of STAT3 (Qin et al., 2008). Acetylation and methylation are also crucial for the role of STAT3 in the acquisition of cancer stem cell‐like phenotype and tumor progression (Kim et al., 2013a; Su et al., 2011).
Because of its central role in multiple oncogenic pathways and its diverse functions, STAT3 is an attractive target for development of anticancer drugs and great effort has been devoted over the last decade to the discovery of small molecule inhibitors (Debnath et al., 2012; Miklossy et al., 2013; Yu et al., 2009). Inhibitors of STAT3 can be classified as direct and indirect inhibitors (Benekli et al., 2009; Debnath et al., 2012). Indirect inhibitors interfere with cytokine and growth factor receptors or upstream kinases that phosphorylate STAT3. Conversely, direct inhibitors interact with the STAT3 protein and are expected to interfere with its multiple functions (Debnath et al., 2012). Direct inhibitors can be divided based on targeted protein domain, e.g. the NTD, DBD or SH2 domain. Due to its critical involvement in STAT3 activation, the SH2 domain has been seen as the most attractive site and SH2‐targeting compounds constitute the largest class of direct STAT3i (Debnath et al., 2012).
Genetic knockout, knockdown and small molecule inhibitors of STAT3 have been shown to prevent tumor development and growth in preclinical models (Chan et al., 2004; Kortylewski et al., 2005; Yu et al., 2009). However, despite the preclinical evidence that STAT3 would be an ideal target for cancer therapy, effective strategies to inhibit STAT3 in the clinic are still lacking (Debnath et al., 2012). This is largely due to the intrinsic difficulty of targeting directly transcription factors like STAT3. Consequently, few direct STAT3i have shown relevant activity in preclinical models in vivo and have been tested in clinical trials (Debnath et al., 2012). OPB‐31121 is a small molecule compound that has been recently reported to interfere with STAT3 signaling, although the underlying mechanism has not been clarified yet (Hayakawa et al., 2013; Kim et al., 2013b). OPB‐31121 exhibits potent anticancer activity in vitro and in tumor xenografts (Hayakawa et al., 2013; Kim et al., 2013b) and is currently investigated in clinical trials (https://clinicaltrials.gov). Understanding how OPB‐31121 interacts with STAT3 and the molecular basis of its potent anticancer effect would be highly relevant for further development of this class of compounds. In this study, we combined in silico and in vitro experiments to investigate how OPB‐31121 and other small molecule inhibitors interact with STAT3 and the functional consequences of the drug–target interaction. Importantly, our study reveals a unique mode of interaction of OPB‐31121 with the STAT3 SH2 domain not shared by any of the other STAT3i tested. These unique features might be at the basis of this compound's efficacy and make OPB‐31121 an interesting lead for further development and design of new, more effective STAT3i.
2. Materials and methods
2.1. Computational studies
The crystal structures of STAT3 protein was obtained from the available pdb file 1BG1 in the Protein Data Bank repository (Becker et al., 1998). All compounds structures were designed and optimized using Discovery Studio (DS, v. 2.5, Accelrys Inc., San Diego, CA, USA) (Laurini et al., 2011). All docking experiments were performed with Autodock 4.3 (Morris et al., 2009), with Autodock Tools 1.4.6 on a win64 platform following a consolidated procedure (Giliberti et al., 2010). The binding free energy, ΔGbind, between each drug and the protein was estimated resorting to the MM/PBSA (Molecular Mechanics/Poisson‐Boltzmann Surface Area) approach. According to this well‐validated methodology (Laurini et al., 2012), the binding free energy was obtained as the sum of the interaction energy between the receptor and the ligand (ΔEMM), the solvation free energy (ΔGsol), and the conformational entropy contribution (−TΔS), averaged over a series of snapshots from the corresponding MDS trajectories. The free energy of binding ΔGbind and the concentration of ligand that inhibits the protein activity by 50% (i.e., IC50) are related by the following fundamental equation: ΔGbind = −RT ln 1/IC50, where R is the gas constant and T is the temperature. Thus, once ΔGbind for a given protein/inhibitor couple is estimated by MM‐PBSA simulations, the relative IC50 value is also known by virtue of this relationship. The role of the key residues identified by PRBFED was further studied by performing computational alanine scanning (CAS) experiments (Guo et al., 2012). Accordingly, the absolute binding free energy of each mutant protein, in which one of the key residue was replaced with alanine, was calculated with the MM/PBSA method and corresponded to the difference in the binding free energy between the wild‐type (wt) and its alanine mutant (mut) counterpart.
2.2. Cell lines, plasmids, chemicals and antibodies
Human prostate cancer DU145 and LNCaP cell lines were purchased from American Type Culture Collection and maintained in RPMI supplemented with 10% (FBS) (PAA, Brunschwig, Basel, CH). STAT3 SH2 domain (amino acid residues 586–685) was subcloned into pGEX‐2T vector (GE Healthcare Europe GmbH) from pET28a‐STAT3‐SH2 domain (GenScript USA Inc) using BamHI and EcoRI restriction sites. Mutant constructs were generated using GENEART® Site‐Directed Mutagenesis System (Life Technologies). OPB‐31121 (Otsuka Pharmaceutical, Tokyo, Japan), STA‐21, and Stattic (ENZO LIFE SCIENCES AG, Lausen, CH), S31.201 and Cryptotanshinone (Merck KGaA, VWR, Dietikon, CH) were dissolved in DMSO. IL‐6 (10 ng/ml, R&D Systems Europe Ltd., Abingdon, UK), ampicillin (50 μg/ml, Eurobio) and IPTG (isopropyl‐β‐d‐thiogalactopyranoside, 1 mM, Promega, Dübendorf, CH) were dissolved in sterile water. Antibodies against STAT3, pSTAT3 Tyr705, pSTAT3 Ser727, were purchased from Cell Signaling Technology (BIOCONCEPT, Allschwil, CH), and GAPDH from Millipore (Zug, CH).
2.3. Western blotting
Cells were washed once in PBS and lysed in lysis buffer (25 mM Tris–HCl pH = 7.4, 50 mM KCl, 5 mM EDTA, 1% NP‐40, 0.5% Sodium deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitors cocktail (Roche Diagnostics (Schweiz) AG, Rotkreuz, CH), sodium orthovanadate (Na3VO4, Acros Organics) and phenylmethanesulfonylfluoride (PMSF, Sigma–Aldrich). After 20 min of incubation on ice samples were centrifuged for 15 min at 4 °C and proteins were quantified using BCA Protein Assay Kit (Pierce, Perbio Science Switzerland SA, Lausanne, CH). Proteins were loaded on 10–12% Sprint Next Gel (Amresco, Bioconcept, Allschwil CH) and analyzed by immunoblotting. Membranes were blocked for 1 h with 0.2% of I‐Block (Life Technologies) and then probed overnight at 4 °C with primary antibodies and for 1 h with horseradish peroxidase (HRP)‐conjugated secondary antibodies. Western Bright ECL detection system (WITEC AG, Littau, CH) was used for detection.
2.4. Cell viability
DU145 and LNCaP cells were plated in 96‐well plates in phenol red‐free RPMI supplemented with 10% serum. After 24 h cells were treated with the indicated STAT3 inhibitors. Cell viability was determined using MTT assay after 72 h (Genini et al., 2012). All assays were performed in triplicate and repeated in at least three independent experiments.
2.5. Colony forming assay
Cells were plated in triplicate in 6‐well plates. Drugs were added to the medium at increasing concentrations. After 10 days cells were fixed and stained with 1% crystal violet in 20% ethanol. Colonies were counted with an automated colony counter Alphaimager 3400 (Napoli et al., 2009). Results are represented as mean ± SD from 3 independent experiments.
2.6. Expression and purification of GST‐STAT3 SH2 domain
Escherichia coli strain BL21(DE3) (Life Technologies) transformed with the pGEX‐2T‐GST‐STAT3‐SH2 domain plasmids (WT, S636A, and V637A mutants) or pGEX‐2T‐GST (100 ng of DNA) was grown at 37 °C in LB medium containing ampicillin (50 μg/ml) to an OD 600 of 0.6–0.7. Cells were then induced with 1 mM IPTG for 4 h at 37 °C and subsequently harvested by centrifugation at 4000 × g. The bacterial pellet was resuspended in cold PBS containing protease inhibitors plus 1 mg/ml of lysozyme (Sigma–Aldrich) and sonicated (30 s of pulsing/30 s of pause for 6 times). Triton X‐100 (Sigma–Aldrich) was then added at a final concentration of 1% and the lysate was centrifuged for 20 min at 4 °C. Supernatant was filtered (0.45 μm), diluted 1:1 with cold PBS and purified by affinity chromatography using GSTrap HP column (GE Healthcare). Fusion proteins were eluted with 10 mM of glutathione, reduced, desalted in PBS and concentrated to 1 mg/ml.
2.7. Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments of STAT3i binding to the STAT3 SH2 domain were conducted with a Nano ITC Technology (TA Instruments) at 25 °C. After temperature equilibration, GST‐SH2‐WT, GST‐SH2/S636A or GS‐SH2/V637A mutant protein solutions (10μ M) were titrated with each inhibitor (100 μM in 1% v/v DMSO) by adding 1 μL of compound solution to the protein solution at intervals of 4 min. The titration of a GST‐SH2 domain in PBS solution containing 1% DMSO v/v with the same inhibitor solutions was used as blank test and to determine the heat of dilution of ligand. This reference experiment, carried out in the same way as the titration with protein sample, was subtracted from the sample data. The corrected binding isotherms were fitted to yield the values of the binding constant (Kd), the stoichiometry (n), and the binding enthalpy (ΔH) of each STAT3 SH2 domain/inhibitor binding event. Once the Kd for each inhibitor/protein was determined, the corresponding free energy of binding ΔGbind and the IC50 values were obtained via the above mentioned relationship: ΔGbind = −RT ln Kd = −RT ln 1/IC50.
2.8. Circular dichroism
CD spectra from GSH‐SH2‐WT, GST‐SH2/S636A or GST‐SH2/V637A mutants (0.1 mg/ml in 10 mM NaPO4, pH 7.4) were recorded on a Chirascan spectropolarimeter (Applied Photophysics) over the wavelength range from 195 to 260 nm at a band width of 1 nm, step size of 0.5 nm and 1s per step. The spectra in the far‐ultraviolet region required an average of five scans and were subtracted from blank spectra performed with GST in buffer.
3. Results
3.1. In silico analysis of the binding of OPB‐31121 to STAT3
We used various computational approaches to examine in silico the binding of OPB‐31121 (Hayakawa et al., 2013; Kim et al., 2013b) to STAT3 (Figure 1A). For comparison we used in our analyses selected STAT3i, like STA‐21 (Song et al., 2005), Stattic (Schust et al., 2006), S3I.201 (Siddiquee et al., 2007) and Cryptotanshinone (Shin et al., 2009), for which there was previous evidence of interaction with the STAT3 SH2 domain. OPB‐31121 was docked onto the SH2 domain and then the relevant drug/protein affinities were scored by molecular dynamics simulation (MDS) (Figure 1B). Using the same approach these parameters were determined for all the other STAT3i (Figure S1). In the case of Stattic, which is able to form covalent crosslinks with STAT3 (Schust et al., 2006), we considered only the initial step of non‐covalent interaction. Table 1 and Table S1 show the values of the calculated IC50, free energy of binding ΔGbind and the enthalpic and entropic components predicted for the interaction of each compound with the STAT3 SH2 domain obtained from these in silico analyses. The calculated IC50 value for OPB‐31121 was in the low nanomolar range (IC50, ∼18 nM). Notably, this value was about 2–3 orders of magnitude lower than the IC50 estimated for the other STAT3i (ranging from 1.4 to 27.2 μM).
Figure 1.
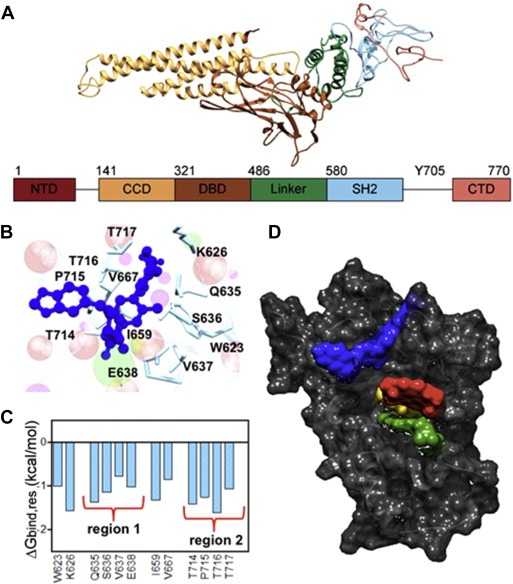
In silico binding of OPB‐31121 to STAT3. (A) Three‐dimensional structure of the STAT3 protein. The different domains of STAT3 are indicated in different colors indicated both in the structure and diagram. (B) Details of the binding site of OPB‐31121 in the STAT3 SH2 domain obtained from equilibrated MDS snapshots. The protein backbone is portrayed as a transparent sky blue ribbon; the main residues involved in drug interactions are shown as labeled colored sticks. OPB‐31121 is portrayed as atom‐colored sticks‐and‐balls. (C) Interaction spectrum for STAT3 in complex with OPB‐31121. Only residues for which ΔGbind is ≥ 0.75 kcal/mol are shown. (D) Binding pockets of different inhibitors on the STAT3 SH2 domain highlighted by their respective van der Waals surfaces. Dark gray, SH2 domain; blue, OPB‐31121; yellow, STA‐21; red, cryptotanshinone; green, S3I.201. Stattic is hidden by cryptotanshinone that binds to an overlapping site.
Table 1.
Predicted free energy of binding (ΔGbind) and IC50 values for OPB‐31121, Cryptotanshinone, STA‐21, S3I.201, and Stattic in complex with STAT3.
| OPB‐31121 | STA‐21 | Stattic | Crypto | S3I.201 | |
|---|---|---|---|---|---|
| ΔGbind (kcal/mol) | −10.54 ± 0.77 | −6.47 ± 0.88 | −6.99 ± 0.79 | −8.01 ± 0.61 | −6.23 ± 0.89 |
| IC50 (μM)a | 0.0187 | 17.900 | 7.400 | 1.400 | 27.200 |
ΔGbind and IC50 of ligand are related by the following fundamental equation: ΔGbind = −RT ln 1/IC50, where R is the gas constant and T is the temperature. Once ΔGbind for a given protein/ligand couple is estimated by MM‐PBSA simulations, the relative IC50 value is determined by virtue of this relationship.
To understand the basis of the remarkable high affinity of OPB‐31121 for STAT3 we performed a per‐residue deconvolution analysis of the free energy of binding (Figure 1C). The resulting interaction spectrum showed that the residues mostly involved in the binding of the drug clustered in two regions of the SH2 domain. Region 1 included residues from Q635 to E638 and region 2 included residues from T714 to T717. In addition, other four residues (i.e., W623, K626, I659, and V667) were found to be engaged in major stabilizing interactions with OPB‐31121. The same procedure was applied to the other STAT3i leading to the definition of the STAT3 interaction spectrum for each of the compounds (Figure S1E–H). Interestingly, the interaction spectra were compound‐specific with very little, if any, overlap between them. Moreover, the interaction region defined for OPB‐31121 was clearly distinct from those of all the other STAT3i. A visual representation of these results is given in Figure 1D, where each drug/STAT3 interaction surface is represented in a different color. Thus, our in silico data indicated that OPB‐31121 bound with remarkably high affinity to STAT3 and interacted with a distinct pocket in the SH2 domain with different residue specificity compared to other STAT3i.
3.2. In vitro assessment of the binding of OPB‐31121 to the STAT3 SH2 domain
The binding of OPB‐31121 to the SH2 domain of STAT3 was next investigated using recombinant GST‐tagged STAT3 SH2 domain and isothermal titration calorimetry (ITC). Consistent with the in silico data, ITC demonstrated high affinity binding of OPB‐31121 to the STAT3 SH2 domain yielding an experimental Kd of 10 nM (Figure 2A). We assessed the binding of other STAT3i using the same experimental approach. As predicted by the in silico analysis, S3I.201 also bound to the SH2 domain but with a substantially lower affinity compared to OPB‐31121 (Kd = 8 μM) (Figure 2B). All other STAT3i showed similar low binding affinities with experimental Kd in the micromolar range (Figure S2). Notably, the data from in vitro binding assays were in good agreement with the estimated IC50 values determined by MDS (Table 1). Control ITC experiments were conducted with recombinant GST to rule out non‐specific binding of the compounds. None of tested compounds showed any interaction with GST (Figure S3). Hence, the in vitro binding assays supported the computational chemistry prediction of high affinity binding of OPB‐31121 to the STAT3 SH2 domain.
Figure 2.
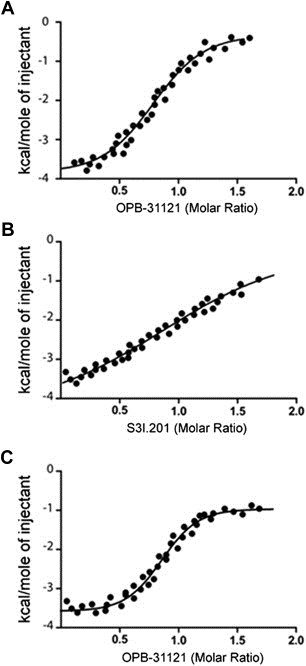
In vitro binding of OPB‐31221 to the STAT3 SH2 domain. (A) Isothermal titration calorimetry (ITC) data for the STAT3 SH2 domain/OPB‐31121 system. (B) ITC data for the STAT3 SH2 domain/S3I.201 system. (C) ITC analysis of OPB‐31121 interaction with the STAT3 SH2 domain after pre‐incubation with S3I.201.
The in silico analyses predicted also a distinct binding site for OPB‐31121 in the STAT3 SH2 domain compared to other STAT3i. In order to test the reliability of this prediction we performed competition assays with OPB‐31121 and S3I.201. The recombinant GST‐tagged STAT3 SH2 domain was incubated first with a saturating concentration of S3I.201 and then titrated with increasing concentrations of OPB‐31121 (Figure 2C). OPB‐31121 binding was similar in the presence and absence of S3I.201 yielding similar Kd values in both conditions. Thus, these data confirmed the in silico prediction of the existence of independent, non‐overlapping binding pockets for OPB‐31121 and other STAT3i in the STAT3 SH2 domain.
3.3. In silico alanine scanning and in vitro site‐directed mutagenesis analysis of the OPB‐31121 binding site
To further validate the predicted binding site of OPB‐31121 we selected two residues (S636 and V637) in the drug–target interaction region of the STAT3 SH2 domain defined by binding energy deconvolution analysis. The role of these two residues was first tested in silico by alanine scanning mutagenesis (Figure 3A–B). Turning either the S636 or V637 residue into alanine affected the positioning of OPB‐31121 in the binding pocket and greatly reduced the binding affinity resulting in a dramatic increase in the estimated IC50 values from 18 nM to 5 μM and 1.1 μM for S636A and V637A, respectively (Table 2). As proof of the specificity, we applied the same approach to S3I.201. Consistent with a distinct interaction site, neither the S636A nor V637A mutation affected significantly the binding mode and the estimated binding affinity of S3I.201 (Figure 3C–D and Table 2).
Figure 3.
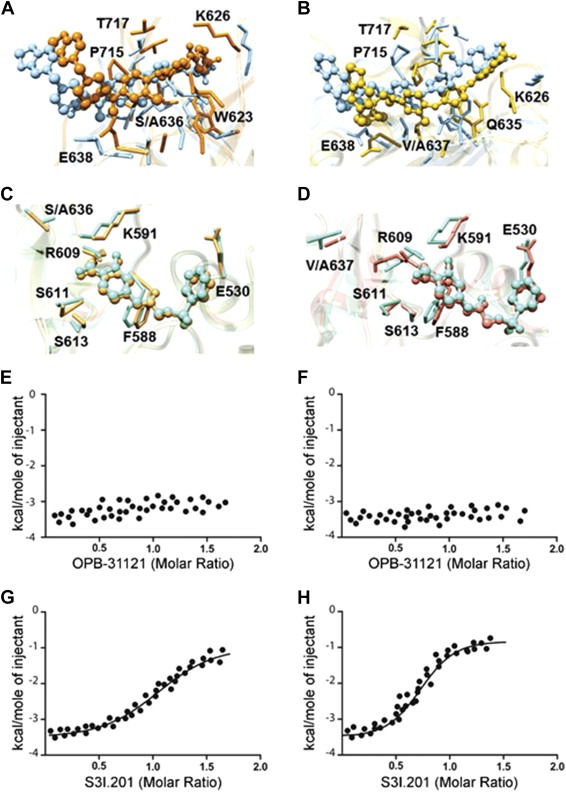
Mutational analysis of OPB‐31121 binding site in the STAT3 SH2 domain. (A) Superposition of the binding site of wild type (light blue) and S636A STAT3 mutant (orange) in complex with OPB‐31221. (B) Superposition of the binding site of wild type (light blue) and V637A STAT3 mutant (golden rod) in complex with OPB‐31221. (C) Superposition of the binding site of wild type (aquamarine) and S636A STAT3 mutant (sandy brown) in complex with S3I.201. (D) Superposition of the binding site of wild type (aquamarine) and V637A STAT3 mutant (salmon) in complex with S3I.201. In all panels drugs are depicted as colored sticks‐and‐balls, while main residues involved in the interactions are labeled and shown as colored sticks. Hydrogen atoms, water molecules, ions and counterions are omitted for clarity. (E) ITC data for S636A mutant STAT3 SH2 domain in complex with OPB‐31121. (F) ITC data for V637A mutant STAT3 SH2 domain in complex with OPB‐31121; (G) ITC data for S636A mutant STAT3 SH2 domain in complex with S3I.201; (H) ITC data for V637A mutant STAT3 SH2 domain in complex with S3I.201.
Table 2.
Predicted free energy of binding (ΔGbind), binding energy difference ΔΔGbind = ΔGbind(wild type) – ΔGbind(mutant), and IC50 values for OPB‐31121 and S3I.201 with S636A and V637A STAT3 mutants.
| S636A | V637A | |||
|---|---|---|---|---|
| OPB‐31121 | S3I.201 | OPB‐31121 | S3I.201 | |
| ΔGbind (kcal/mol) | −7.23 ± 0.64 | −6.15 ± 0.67 | −8.11 ± 0.69 | −6.26 ± 0.78 |
| ΔΔGbind (kcal/mol) | −3.31 | −0.08 | −2.43 | +0.03 |
| IC50 (μM) | 5 | 31.2 | 1.1 | 25.9 |
In parallel with the in silico studies, we performed in vitro site‐directed mutagenesis for the same residues and assessed binding to wild type and mutated GST‐tagged STAT3‐SH2 domain by ITC. Correct folding of the mutated SH2 domains was determined by comparing circular dichroism (CD) spectra of the wild‐type and mutant protein (Figure S4). Both wild type and mutant SH2 domains displayed the typical SH2 spectra indicating that the mutations did not affect the native conformation of the protein. However, the presence of the S636A or V637A mutation abrogated binding of OPB‐31121 in ITC experiments, sustaining the validity of the computational model (Figure 3E–F). Interestingly, the binding of the reference compound S3I.201 to the STAT3 SH2 domain was not affected by either mutation, showing affinities similar to that for the wild‐type protein (Figure 3G–H).
3.4. Inhibition of Y705 and S727 STAT3 phosphorylation by OPB‐31121
To assess the biological activity of OPB‐31121 we assessed its ability to interfere with STAT3 phosphorylation in human prostate cancer cells. Direct STAT3i may expect to block the interaction of STAT3 with protein kinases and likely prevent phosphorylation at Y705. In these assays we used two prostate cancer cell lines that exhibited constitutive (DU145) and IL‐6 inducible (LNCaP) Y705 phosphorylation, respectively. Cells were treated with increasing concentrations of OPB‐31121 for 16 h. LNCaP cells were stimulated with IL‐6 during the last 30 min of the incubation to induce pY705. OPB‐31121 at doses of 5–10 nM almost completely blocked pY705 in both cell lines (Figure 4A–B). We assessed the kinetics of pY705 inhibition using a dose of 10 nM of OPB‐31121. Incubation with OPB31121 for 4–8 h completely abrogated pY705 in both cell lines (Figure 4C–D). We assessed next the effect of OPB‐31121 on pS727, which in both DU145 and LNCaP cells is constitutively phosphorylated. Interestingly, OPB‐31121 reduced pS727 with dose dependence and kinetics similar to those observed for pY705 inhibition in both cell lines (Figure 4A–D). We noticed also a decrease of total STAT3 protein level in cells treated with OPB‐31121 at high concentrations (≥50 nM) and for longer incubation times (≥16 h).
Figure 4.
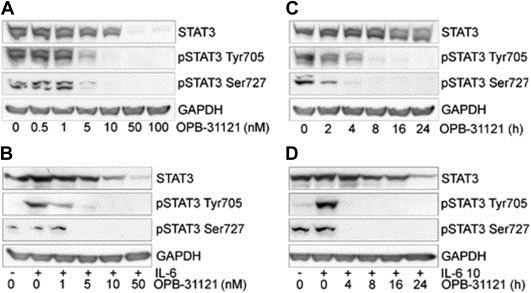
Inhibition of STAT3 phosphorylation at Y705 and S727 by OPB‐31121. (A–B) STAT3 phosphorylation in DU145 (A) and LNCaP (B) cells treated with the indicated concentrations of OPB‐31121 for 16 h (C–D) STAT3 phosphorylation in DU145 (C) and LNCaP (D) cells incubated with OPB‐31121 (10 nM) and analyzed at the indicated times. IL‐6 was added for 30 min at the end of the treatment with OPB‐31121 to induce pY705.
We performed similar experiments with the other STAT3i. All the compounds inhibited pY705 (Figure 5A). However, even for the most potent of these compounds (Cryptotanshinone) doses ≥ 5 μM were needed to significantly affect pY705. S3I.201, STA‐21 and Stattic were active at doses ≥20 μM to inhibit pY705 to a comparable level. Notably, these differences in potency reflected closely the differences in the binding affinity between OPB‐31121 and the other STAT3i. Interestingly, when we examined the kinetics of inhibition of pY705 and pS727 by cryptotanshinone and S3I.201 a reduction of pY705 was seen within 4 h (Figure S5A–B). However, significant inhibition of pS727 required longer incubation time (8–16 h). As seen with OPB‐31121, most of these compounds induced also a decrease of total STAT3 level at the highest doses tested. Although the cause of the reduction of total STAT3 needs to be investigated, this could be a consequence of the continuous presence of high concentrations of the inhibitors in cells interfering the synthesis or degradation of STAT3 protein at the transcriptional or post‐transcriptional level.
Figure 5.
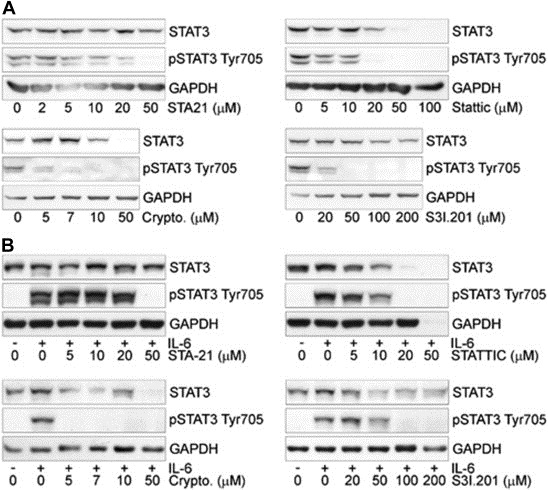
Inhibition of STAT3 phosphorylation at Y705 by STA‐21, Stattic, Cryptotanshinone, and S3I.201 for 16 h in DU145 (A) and LNCaP (B) cells.
Collectively, these experiments showed that OPB‐31121 reduced effectively both pY705 and pS727. OPB‐31121 acted at low doses and within few hours of incubation on both phosphorylation events. The activity of OPB‐31121 was not influenced by the preexisting phosphorylation status of Y705 and similar effects were seen in cells with constitutive and inducible phosphorylation at this site. Notably, in these cellular assays OPB‐31121 was about 100–1000 fold more potent than the other STAT3i tested here, in line with the high binding affinity for STAT3 demonstrated in vitro by this compound.
3.5. Antiproliferative activity of OPB‐31121 in prostate cancer cells
Our data showed that OPB‐31121 was highly effective in blocking both pY705 and pS727 in DU145 and LNCaP prostate cancer cell lines. Increased STAT3 levels and higher Y705 and S727 phosphorylation are frequent in human prostate cancer at the early (androgen‐dependent) and late (castration‐resistant) stages of the disease (Culig et al., 2005; Dhir et al., 2002; Mora et al., 2002). Activation of STAT3 signaling in prostate cancer is generally associated with poor clinical outcome (Culig et al., 2005; Dhir et al., 2002; Mora et al., 2002). Thus, the availability of a compound that could directly and effectively block STAT3 signaling through multiple downstream pathways could be highly advantageous in prostate cancer. OPB‐31121 has been reported to be active in preclinical models of various human cancers (Hayakawa et al., 2013; Kim et al., 2013b). However, the compound has never been tested in prostate cancer cells. Hence, we assessed the effects of OPB‐31121 on the viability and proliferation of LNCaP and DU145 cells, which are common models of androgen‐dependent and castration‐resistant prostate cancer, respectively. OPB‐31121 inhibited proliferation of both LNCaP and DU145 cells with IC50 values in the nanomolar range (18 and 25 nM) (Figure 6A). Colony formation was also strongly inhibited by OPB‐31121 at doses of 10–50 nM (Figure 6B). Interestingly, OPB‐31121 was effective in unstimulated LNCaP cells in line with the notion that the drug's activity was independent of the Tyr705 phosphorylation status. For comparison we tested the effects of the other STAT3i in the same cell lines. All the compounds affected cell proliferation, but the doses required to achieve significant levels of inhibition were considerably higher than those of OPB‐31121 (Figure 6A). Higher doses of these STAT3i were also required in the clonogenic assays (Figure 6B). Thus, in line with the higher binding affinity, OPB‐31121 was substantially more potent in suppressing cell proliferation and colony formation compared to other STAT3i.
Figure 6.
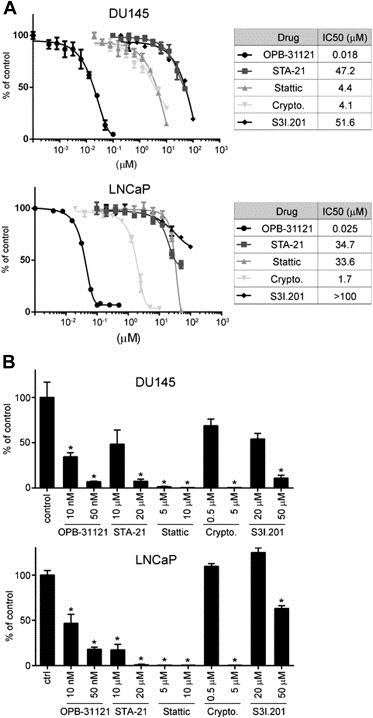
Inhibition of cell proliferation and colony formation by STAT3 inhibitors. (A) Cell viability determined by MTT assay in DU145 and LNCaP cells incubated with the indicated compounds. Left, IC50 values for each compound in the two cell lines. (B) Anchorage‐dependent clonal growth of DU145 and LNCaP cells treated with the indicated doses of OPB31121, STA‐21, Stattic, cryptotanshinone, and S3I.201. *P < 0.01.
4. Discussion
STAT3 is a latent cytoplasmic protein whose multiple functions are controlled by various post‐translational modifications (Yu et al., 2014, 2009). Phosphorylation at Y705 and S727 have been reported to enhance nuclear localization and transcriptional activity of STAT3. pS727 controls also mitochondrial functions of STAT3 (Levy and Darnell, 2002, 2014, 2009). Furthermore, un‐phosphorylated STAT3 is not devoid of biological activity and exerts both transcriptional and non‐transcriptional functions (Timofeeva et al., 2012; Yang et al., 2007). Because of its involvement in multiple biological pathways, STAT3 has an important role in human cancers sustaining neoplastic transformation and promoting tumor progression (Yu et al., 2014, 2009). Therefore, there is high interest in developing direct STAT3i that might interfere with the multiple and diverse functions of this pleiotropic transcription factor (Debnath et al., 2012). OPB‐31121 has been recently reported to inhibit STAT3 signaling and has relevant anticancer activity in preclinical models in vitro and in vivo (Hayakawa et al., 2013; Kim et al., 2013b). Based on its efficacy in preclinical models, phase I/II clinical trials have been initiated with this compound (Hayakawa et al., 2013; Kim et al., 2013b). Results from a first phase I study in patients with advanced solid tumors indicate that the drug orally administered was well tolerated and had a favorable toxicity profile, but low bioavailability and unfavorable pharmacokinetics impair its use in the clinic (Bendell et al., 2014). Moreover, despite the proven efficacy in preclinical models, questions remain about the intracellular target and mechanism of action of OPB‐31121 (Hayakawa et al., 2013; Kim et al., 2013b). In light of the recent preclinical and clinical data, this information would be highly valuable for continuing its development and generating new inhibitors with improved activity and pharmacological profile. In this study, we combined computational and experimental approaches to define the mode of interaction of OPB‐31121 with STAT3. For comparison, we performed similar studies with a series of structurally distinct STAT3i. To our knowledge, a detailed study of how different small molecules interact with the SH2 domain of STAT3 and how their binding mode impact on the biological activity of the compounds is missing. Indeed, even slight differences in the interaction site and binding affinity might be highly relevant in terms of biological activity and potency of the compounds. Interestingly, we found that OPB‐31121 has a remarkably high affinity for STAT3 and unique mode of interaction with the SH2 domain compared to other STAT3i.
In this study, we used computational docking and MDS to examine the potential binding site of OPB‐31121 in the SH2 domain of STAT3. The residues in the SH2 domain lining the putative binding site were identified and those affording major stabilizing contributions were investigated by free energy deconvolution and in silico alanine scanning mutagenesis. The same computational procedure was applied to the other STAT3i with the purpose of a direct comparison of binding modes and sites of interaction. Importantly, the computational predictions were validated by in vitro binding assays using ITC and purified STAT3 SH2 domain. Both series of experiments concurred to show that OPB‐31121 binds to STAT3 in the SH2 domain with very high affinity. Notably, both the computationally and experimentally estimated Kd values for OPB‐31121 were 2–3 orders of magnitude lower than those of the other STAT3i tested in this study. A similar ranking of the compounds was obtained in the cellular assays based on their efficacy on STAT3 phosphorylation and cell proliferation. All the data confirmed the substantially higher potency of OPB‐31121 compared to the other STAT3i.
In greater details, our in silico analysis identified two distinct binding pockets for small molecule inhibitors in the SH2 domain of STAT3: the first was occupied by OPB‐31121 and the second was common to all the other inhibitors (Figure 1D). The crystal structure of the STAT3‐SH2 domain reveals the existence of one hydrophilic and two hydrophobic sub‐pockets (Becker et al., 1998). Most STAT3i are predicted to bind either to the hydrophilic site, lined by the side chains of the K591, R609, S611, and S613 residues, or to the partially hydrophobic region composed by the K592, R595, I597, and I634 residues (Fletcher et al., 2008). Our computational analyses confirmed that all four STAT3i considered here (i.e., cryptotanshinone, STA‐21, Stattic, and S3I‐201) fit in these two sub‐pockets (Figure 1D and Figure S1). In contrast, OPB‐31121 was found to bind to a distinct region that included the third, hydrophobic sub‐pocket (Figure 1B–C). Furthermore, OPB‐31121 interacted with a consistently larger number of residues in the SH2 domain compared to the other compounds; this in turn contributed to the higher affinity of OPB‐31121 for STAT3, as indicated by the extremely favorable comparison of estimated IC50 (Table 1) and Kd values (Figure 2A). The ITC experiments concurred to support the in silico model of interaction of OPB‐31121 with the STAT3 SH2 domain. Competition experiments and site‐directed mutagenesis showed the specificity of the identified interaction site in the SH2 domain for OPB‐31121 (2, 3).
The presence of a distinct sub‐pocket and the high binding affinity of OPB‐31121 explain in part the high efficacy of the compound in inhibiting STAT3 phosphorylation in cells. Furthermore, in the case of OPB‐31121 inhibition of pY705 and pS727 occurred at similar doses and within the same time scale (∼4 h) (Figure 4). This was not the case with other STAT3i, like cryptotanshinone and S3I.201, for which the inhibition of pS727 was delayed with respect to pY705 inhibition (Figure S5). Thus, occupying a wider and distinct area in the SH2 domain, OPB‐31121 could impair more effectively the interaction of STAT3 with kinases and other proteins and prevent simultaneously and with higher efficiency phosphorylation of these residues compared to other STAT3i. Collectively, our results demonstrate that OPB‐31121 binds to the SH2 domain and interferes directly with STAT3 activation and signaling. Higher affinity for the target likely leads to higher potency in cellular assays and in vivo, although the compound's propensity to be internalized in cells and metabolized could influence its efficacy in biological systems.
Interfering with JAK/STAT3 signaling has been recently proposed as a valid option for treatment of cancer, including prostate cancer (Hedvat et al., 2009; Kroon et al., 2013; Schroeder et al., 2014). However, based on the current understanding of the multiple functions and diverse activation modes of STAT3, blocking pY705 alone may not be sufficient. Alternatively post‐translationally modified as well as un‐phosphorylated STAT3 are emerging as important mediators of STAT3 signaling in normal and cancer cells (Yu et al., 2014), emphasizing the need of compounds that could interact and interfere directly with STAT3. For instance, pS727 is frequently increased in prostate cancer and has been shown to be sufficient to drive prostate tumorigenesis and progression independently of pY705 (Qin et al., 2008). Furthermore, in preclinical models of prostate cancer inactivation of pS727 is sufficient to substantially reduce tumorigenicity (Qin et al., 2008). Therefore, in line with the prominent activation of STAT3 signaling in prostate cancer (Culig et al., 2005; Dhir et al., 2002; Mora et al., 2002), we tested the activity of OPB‐31121 in LNCaP and DU145 prostate cancer cell lines representative of androgen‐dependent and castration‐resistant tumors, respectively. We found that OPB‐31121 was a potent inhibitor of proliferation and clonogenicity in both cell models (Figure 6). Interestingly, the antiproliferative effect of OPB‐31121 was independent of the pY705 status and related to its high affinity for the target and ability to block effectively and concomitantly both pY705 and pS727. This raises the possibility that the efficacy of OPB‐31121 may not depend exclusively on the Y705 phosphorylation status and additional factors should be taken in consideration to identify potentially sensitive tumor types. Together, these findings suggest that the use of direct STAT3i like OPB‐31121 could be expanded to tumors that harbor not only constitutive pY705 but additional biomarkers (e.g., total and pS727 STAT3 level) should be considered.
Supporting information
The following are the supplementary data related to this article:
Figure S1 In silico binding of small molecule inhibitors to STAT3 SH2 domain. (A‐D) Models of STA‐21 (A), Stattic (B), Cryptotanshinone (C), and S3I.201 (D) bound to the SH2 domain of STAT3 as obtained from equilibrated MD simulation snapshots. Inhibitors are portrayed as atom‐colored balls‐and‐sticks. Main residues of the protein involved in the interaction with the drugs are shown as colored sticks and labeled. Hydrogen atoms, water molecules, ions and counterions are omitted for clarity. (E‐H) Interaction spectra of STA‐21 (E), Stattic (F), Cryptotanshinone (G), and S3I.201 (H) with the SH2 domain of STAT3 as obtained from the per‐residue deconvolution of the corresponding binding free energy.
Figure S2 Isothermal titration calorimetry (ITC) data for the STAT3 SH2 domain with STA‐21 (A), Stattic (B) and Cryptotanshinone (C).
Figure S3 Isothermal titration calorimetry (ITC) data for the GST protein/OPB‐31121 (A) and GST protein/S3I.201 (B) systems.
Figure S4 Circular dichroism spectra for wild type (A), S636A (B) and V637A mutant (C) STAT3 proteins.
Figure S5 Inhibition of STAT3 phosphorylation at Y705 and S727 by cryptotanshinone (7 μM) and S3I.201 (50 μM) in DU‐145 cells incubated for the indicated time.
Supplementary data
Acknowledgments
We thank Edwin Rock and Dusan Kostic (Otsuka Pharmaceuticals) for their continuous support and helpful comments. This work was supported by a grant from Otsuka Pharmaceuticals, Ticino Foundation for Cancer Research and Foundation Virginia Boeger to C.V.C.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.02.012.
Brambilla Lara, Genini Davide, Laurini Erik, Merulla Jessica, Perez Laurent, Fermeglia Maurizio, Carbone Giuseppina M., Pricl Sabrina, Catapano Carlo V., (2015), Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3), Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.02.012.
Contributor Information
Sabrina Pricl, Email: sabrina.pricl@di3.units.it.
Carlo V. Catapano, Email: carlo.catapano@ior.iosi.ch
References
- Becker, S. , Groner, B. , Muller, C.W. , 1998. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 394, 145–151. [DOI] [PubMed] [Google Scholar]
- Bendell, J.C. , Hong, D.S. , Burris, H.A. , Naing, A. , Jones, S.F. , Falchook, G. , Bricmont, P. , Elekes, A. , Rock, E.P. , Kurzrock, R. , 2014. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharmacol. 74, 125–130. [DOI] [PubMed] [Google Scholar]
- Benekli, M. , Baumann, H. , Wetzler, M. , 2009. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J. Clin. Oncol. 27, 4422–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, K.S. , Sano, S. , Kiguchi, K. , Anders, J. , Komazawa, N. , Takeda, J. , DiGiovanni, J. , 2004. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Invest. 114, 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culig, Z. , Steiner, H. , Bartsch, G. , Hobisch, A. , 2005. Interleukin-6 regulation of prostate cancer cell growth. J. Cell Biochem. 95, 497–505. [DOI] [PubMed] [Google Scholar]
- Debnath, B. , Xu, S. , Neamati, N. , 2012. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 55, 6645–6668. [DOI] [PubMed] [Google Scholar]
- Dhir, R. , Ni, Z. , Lou, W. , DeMiguel, F. , Grandis, J.R. , Gao, A.C. , 2002. Stat3 activation in prostatic carcinomas. Prostate 51, 241–246. [DOI] [PubMed] [Google Scholar]
- Fletcher, S. , Turkson, J. , Gunning, P.T. , 2008. Molecular approaches towards the inhibition of the signal transducer and activator of transcription 3 (Stat3) protein. ChemMedChem 3, 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genini, D. , Garcia-Escudero, R. , Carbone, G.M. , Catapano, C.V. , 2012. Transcriptional and non-transcriptional functions of PPARbeta/delta in non-small cell lung cancer. PLoS One 7, e46009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giliberti, G. , Ibba, C. , Marongiu, E. , Loddo, R. , Tonelli, M. , Boido, V. , Laurini, E. , Posocco, P. , Fermeglia, M. , Pricl, S. , 2010. Synergistic experimental/computational studies on arylazoenamine derivatives that target the bovine viral diarrhea virus RNA-dependent RNA polymerase. Bioorg. Med. Chem. 18, 6055–6068. [DOI] [PubMed] [Google Scholar]
- Gough, D.J. , Corlett, A. , Schlessinger, K. , Wegrzyn, J. , Larner, A.C. , Levy, D.E. , 2009. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324, 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov, S. , Karin, M. , 2008. Autocrine IL-6 signaling: a key event in tumorigenesis?. Cancer Cell 13, 7–9. [DOI] [PubMed] [Google Scholar]
- Guo, J. , Wang, X. , Sun, H. , Liu, H. , Yao, X. , 2012. The molecular basis of IGF-II/IGF2R recognition: a combined molecular dynamics simulation, free-energy calculation and computational alanine scanning study. J. Mol. Model. 18, 1421–1430. [DOI] [PubMed] [Google Scholar]
- Hayakawa, F. , Sugimoto, K. , Harada, Y. , Hashimoto, N. , Ohi, N. , Kurahashi, S. , Naoe, T. , 2013. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 3, e166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedvat, M. , Huszar, D. , Herrmann, A. , Gozgit, J.M. , Schroeder, A. , Sheehy, A. , Buettner, R. , Proia, D. , Kowolik, C.M. , Xin, H. , Armstrong, B. , Bebernitz, G. , Weng, S. , Wang, L. , Ye, M. , McEachern, K. , Chen, H. , Morosini, D. , Bell, K. , Alimzhanov, M. , Ioannidis, S. , McCoon, P. , Cao, Z.A. , Yu, H. , Jove, R. , Zinda, M. , 2009. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 16, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E. , Kim, M. , Woo, D.H. , Shin, Y. , Shin, J. , Chang, N. , Oh, Y.T. , Kim, H. , Rheey, J. , Nakano, I. , Lee, C. , Joo, K.M. , Rich, J.N. , Nam, D.H. , Lee, J. , 2013. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M.J. , Nam, H.J. , Kim, H.P. , Han, S.W. , Im, S.A. , Kim, T.Y. , Oh, D.Y. , Bang, Y.J. , 2013. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 335, 145–152. [DOI] [PubMed] [Google Scholar]
- Kortylewski, M. , Kujawski, M. , Wang, T. , Wei, S. , Zhang, S. , Pilon-Thomas, S. , Niu, G. , Kay, H. , Mule, J. , Kerr, W.G. , Jove, R. , Pardoll, D. , Yu, H. , 2005. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321. [DOI] [PubMed] [Google Scholar]
- Kroon, P. , Berry, P.A. , Stower, M.J. , Rodrigues, G. , Mann, V.M. , Simms, M. , Bhasin, D. , Chettiar, S. , Li, C. , Li, P.K. , Maitland, N.J. , Collins, A.T. , 2013. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 73, 5288–5298. [DOI] [PubMed] [Google Scholar]
- Laurini, E. , Col, V.D. , Mamolo, M.G. , Zampieri, D. , Posocco, P. , Fermeglia, M. , Vio, L. , Pricl, S. , 2011. Homology model and docking-based virtual screening for ligands of the sigma1 receptor. ACS Med. Chem. Lett. 2, 834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurini, E. , Marson, D. , Dal Col, V. , Fermeglia, M. , Mamolo, M.G. , Zampieri, D. , Vio, L. , Pricl, S. , 2012. Another brick in the wall. Validation of the sigma1 receptor 3D model by computer-assisted design, synthesis, and activity of new sigma1 ligands. Mol. Pharm. 9, 3107–3126. [DOI] [PubMed] [Google Scholar]
- Lee, H. , Zhang, P. , Herrmann, A. , Yang, C. , Xin, H. , Wang, Z. , Hoon, D.S. , Forman, S.J. , Jove, R. , Riggs, A.D. , Yu, H. , 2012. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. U. S. A. 109, 7765–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H.J. , Zhuang, G. , Cao, Y. , Du, P. , Kim, H.J. , Settleman, J. , 2014. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 26, 207–221. [DOI] [PubMed] [Google Scholar]
- Levy, D.E. , Darnell, J.E. , 2002. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662. [DOI] [PubMed] [Google Scholar]
- Lim, C.P. , Cao, X. , 2006. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2, 536–550. [DOI] [PubMed] [Google Scholar]
- Liu, L. , McBride, K.M. , Reich, N.C. , 2005. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. U. S. A. 102, 8150–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta, L.L. , Almendro, V. , Marusyk, A. , Shipitsin, M. , Schemme, J. , Walker, S.R. , Bloushtain-Qimron, N. , Kim, J.J. , Choudhury, S.A. , Maruyama, R. , Wu, Z. , Gonen, M. , Mulvey, L.A. , Bessarabova, M.O. , Huh, S.J. , Silver, S.J. , Kim, S.Y. , Park, S.Y. , Lee, H.E. , Anderson, K.S. , Richardson, A.L. , Nikolskaya, T. , Nikolsky, Y. , Liu, X.S. , Root, D.E. , Hahn, W.C. , Frank, D.A. , Polyak, K. , 2011. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 121, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, J.A. , Larner, A.C. , 2014. Toward a new STATe: the role of STATs in mitochondrial function. Semin. Immunol. 26, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy, G. , Hilliard, T.S. , Turkson, J. , 2013. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 12, 611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora, L.B. , Buettner, R. , Seigne, J. , Diaz, J. , Ahmad, N. , Garcia, R. , Bowman, T. , Falcone, R. , Fairclough, R. , Cantor, A. , Muro-Cacho, C. , Livingston, S. , Karras, J. , Pow-Sang, J. , Jove, R. , 2002. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 62, 6659–6666. [PubMed] [Google Scholar]
- Morris, G.M. , Huey, R. , Lindstrom, W. , Sanner, M.F. , Belew, R.K. , Goodsell, D.S. , Olson, A.J. , 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli, S. , Pastori, C. , Magistri, M. , Carbone, G.M. , Catapano, C.V. , 2009. Promoter-specific transcriptional interference and c-myc gene silencing by siRNAs in human cells. EMBO J. 28, 1708–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea, J.J. , Plenge, R. , 2012. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36, 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, H.R. , Kim, H.J. , Kim, J.Y. , Hurt, E.M. , Klarmann, G.J. , Kawasaki, B.T. , Duhagon Serrat, M.A. , Farrar, W.L. , 2008. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 68, 7736–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone, P. , Bromberg, J. , 2012. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J. Clin. Oncol. 30, 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder, A. , Herrmann, A. , Cherryholmes, G. , Kowolik, C. , Buettner, R. , Pal, S. , Yu, H. , Muller-Newen, G. , Jove, R. , 2014. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 74, 1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schust, J. , Sperl, B. , Hollis, A. , Mayer, T.U. , Berg, T. , 2006. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Sehgal, P.B. , 2008. Paradigm shifts in the cell biology of STAT signaling. Semin. Cell Dev. Biol. 19, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, D.S. , Kim, H.N. , Shin, K.D. , Yoon, Y.J. , Kim, S.J. , Han, D.C. , Kwon, B.M. , 2009. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 69, 193–202. [DOI] [PubMed] [Google Scholar]
- Siddiquee, K. , Zhang, S. , Guida, W.C. , Blaskovich, M.A. , Greedy, B. , Lawrence, H.R. , Yip, M.L. , Jove, R. , McLaughlin, M.M. , Lawrence, N.J. , Sebti, S.M. , Turkson, J. , 2007. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. U. S. A. 104, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, H. , Wang, R. , Wang, S. , Lin, J. , 2005. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 102, 4700–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos, M.L. , Levin, R.S. , Gordan, J.D. , Oses-Prieto, J.A. , Webber, J.T. , Salt, M. , Hann, B. , Burlingame, A.L. , McCormick, F. , Bandyopadhyay, S. , Shokat, K.M. , 2014. Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 8, 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, Y.J. , Lai, H.M. , Chang, Y.W. , Chen, G.Y. , Lee, J.L. , 2011. Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 30, 3186–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeeva, O.A. , Chasovskikh, S. , Lonskaya, I. , Tarasova, N.I. , Khavrutskii, L. , Tarasov, S.G. , Zhang, X. , Korostyshevskiy, V.R. , Cheema, A. , Zhang, L. , Dakshanamurthy, S. , Brown, M.L. , Dritschilo, A. , 2012. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J. Biol. Chem. 287, 14192–14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toso, A. , Revandkar, A. , Di Mitri, D. , Guccini, I. , Proietti, M. , Sarti, M. , Pinton, S. , Zhang, J. , Kalathur, M. , Civenni, G. , Jarrossay, D. , Montani, E. , Marini, C. , Garcia-Escudero, R. , Scanziani, E. , Grassi, F. , Pandolfi, P.P. , Catapano, C.V. , Alimonti, A. , 2014. Enhancing Chemotherapy efficacy in Pten-Deficient prostate tumors by activating the Senescence-associated antitumor immunity. Cell Rep. 9, 75–89. [DOI] [PubMed] [Google Scholar]
- Wegrzyn, J. , Potla, R. , Chwae, Y.J. , Sepuri, N.B. , Zhang, Q. , Koeck, T. , Derecka, M. , Szczepanek, K. , Szelag, M. , Gornicka, A. , Moh, A. , Moghaddas, S. , Chen, Q. , Bobbili, S. , Cichy, J. , Dulak, J. , Baker, D.P. , Wolfman, A. , Stuehr, D. , Hassan, M.O. , Fu, X.Y. , Avadhani, N. , Drake, J.I. , Fawcett, P. , Lesnefsky, E.J. , Larner, A.C. , 2009. Function of mitochondrial Stat3 in cellular respiration. Science 323, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, Z. , Zhong, Z. , Darnell, J.E. , 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250. [DOI] [PubMed] [Google Scholar]
- Xu, F. , Mukhopadhyay, S. , Sehgal, P.B. , 2007. Live cell imaging of interleukin-6-induced targeting of “transcription factor” STAT3 to sequestering endosomes in the cytoplasm. Am. J. Physiol. Cell Physiol. 293, C1374–C1382. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Liao, X. , Agarwal, M.K. , Barnes, L. , Auron, P.E. , Stark, G.R. , 2007. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 21, 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Lee, H. , Herrmann, A. , Buettner, R. , Jove, R. , 2014. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Pardoll, D. , Jove, R. , 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Z.L. , Guan, Y.J. , Chatterjee, D. , Chin, Y.E. , 2005. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Blenis, J. , Li, H.C. , Schindler, C. , Chen-Kiang, S. , 1995. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science 267, 1990–1994. [DOI] [PubMed] [Google Scholar]
- Zhong, Z. , Wen, Z. , Darnell, J.E. , 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Figure S1 In silico binding of small molecule inhibitors to STAT3 SH2 domain. (A‐D) Models of STA‐21 (A), Stattic (B), Cryptotanshinone (C), and S3I.201 (D) bound to the SH2 domain of STAT3 as obtained from equilibrated MD simulation snapshots. Inhibitors are portrayed as atom‐colored balls‐and‐sticks. Main residues of the protein involved in the interaction with the drugs are shown as colored sticks and labeled. Hydrogen atoms, water molecules, ions and counterions are omitted for clarity. (E‐H) Interaction spectra of STA‐21 (E), Stattic (F), Cryptotanshinone (G), and S3I.201 (H) with the SH2 domain of STAT3 as obtained from the per‐residue deconvolution of the corresponding binding free energy.
Figure S2 Isothermal titration calorimetry (ITC) data for the STAT3 SH2 domain with STA‐21 (A), Stattic (B) and Cryptotanshinone (C).
Figure S3 Isothermal titration calorimetry (ITC) data for the GST protein/OPB‐31121 (A) and GST protein/S3I.201 (B) systems.
Figure S4 Circular dichroism spectra for wild type (A), S636A (B) and V637A mutant (C) STAT3 proteins.
Figure S5 Inhibition of STAT3 phosphorylation at Y705 and S727 by cryptotanshinone (7 μM) and S3I.201 (50 μM) in DU‐145 cells incubated for the indicated time.
Supplementary data