

Abstract
Introduction
The molecular characterization of circulating tumor cells (CTCs) is a promising tool for the repeated and non‐invasive evaluation of predictive and prognostic factors. Challenges associated with CTC characterization using the only FDA approved method for CTC enumeration, the CellSearch technique, include the presence of an excess of leukocytes in CTC‐enriched blood fractions. Here we aimed to identify colorectal tumor‐specific gene expression levels in the blood of patients with and without detectable CTCs according to CellSearch criteria.
Materials and methods
Blood of 30 healthy donors (HDs) and 142 metastatic colorectal cancer (mCRC) patients was subjected to CellSearch CTC enumeration and isolation. In all samples, 95 mRNAs were measured by reverse transcriptase quantitative PCR (RT‐qPCR). HD blood samples and patient samples with three or more CTCs were compared to identify CTC‐specific mRNAs. Patient samples without detectable CTCs were separately analyzed.
Results
Thirty‐four CTC‐specific mRNAs were higher expressed in patients with ≥3 CTCs compared with HDs (Mann–Whitney U‐test P < 0.05). Among patients without detectable CTCs, a HD‐unlike subgroup was identified which could be distinguished from HDs by the expression of epithelial genes such as KRT19, KRT20 and AGR2. Also, in an independent patient set, a similar HD‐unlike group could be identified among the patients without detectable CTCs according to the CellSearch system.
Conclusion
Extensive molecular characterization of colorectal CTCs is feasible and a subgroup of patients without detectable CTCs according to CellSearch criteria bears circulating tumor load, which may have clinical consequences. This CTC‐specific gene panel for mCRC patients may enable the exploration of CTC characterization as a novel means to further individualize cancer treatment.
Keywords: Circulating tumor cells, Metastatic colorectal cancer, Liver resection, Gene expression, Real-time PCR
Highlights
We measure the expression of 95 mRNAs in enriched circulating tumor cells (CTCs).
We identify CTC‐specific mRNAs based on which three patient clusters are identified.
A subgroup of patients without detectable CTCs differed from healthy donors.
This distinction was driven by the expression of various epithelial genes.
A similar HD‐unlike subgroup was identified in an independent patient set.
Abbreviations
- CTC
circulating tumor cell
- CK
cytokeratin
- Ct
threshold cycle
- DFS
disease-free survival
- HD
healthy donor
- mCRC
metastatic colorectal cancer
- OS
overall survival
- QC
quality control
- RT-qPCR
reverse transcriptase quantitative PCR
1. Introduction
Colorectal cancer (CRC) is a highly heterogeneous disease, in its presentation as well as its prognosis. The liver is a predominant site of metastases; approximately 25% of CRC patients present with synchronous hepatic metastases, while ultimately more than 50% of patients initially presenting with non‐metastatic CRC will develop liver metastases in the course of their disease (Taylor, 1996). When the metastases are confined to the liver and are deemed resectable, patients are increasingly undergoing partial liver resection aiming for curation (van der Pool et al., 2010; Verhoef et al., 2009). Recent data suggest that a selected patient group undergoing this procedure achieves long‐term survival (Brouquet et al., 2011). Nevertheless, up to half of patients undergoing such major abdominal surgery will develop disease relapse in the liver or at other distant sites within one year (Abdalla et al., 2004; Adam et al., 2008; Ito et al., 2008; Zakaria et al., 2007) prompting the need for prognostic factors discriminating patients benefiting from this approach versus those who do not. In primary CRC, several factors have been shown to be prognostic such as microsatellite instability, mRNAs profiles and KRAS and BRAF gene mutations (Imamura et al., 2012, 2012, 2013, 2013, 2013, 2014, 2012). However, at the time of metastatic disease, clonal selection and genomic instability can lead to important discrepancies between primary tumor and metastases, which can be augmented by the passing of time and administration of systemic therapy (Campbell et al., 2010). Heterogeneity between primary tumor and metastases has been described for clinically highly relevant predictive factors such as KRAS (Baldus et al., 2010; Jiang et al., ASCO Molecular Markers 2010 (abstr 49)). As a result, when attempting to identify factors prognostic or predictive for the behavior of metastatic disease, such discrepancies between primary tumor and metastases can be crucial. As such, predictive and prognostic models should be established in metastatic tissue rather than in the primary tumor. Unfortunately, metastatic tissue is often hard to obtain for diagnostic purposes, and only through invasive procedures.
A potential alternative approach for characterizing metastatic solid lesions is the characterization of circulating tumor cells (CTCs) (Alix‐Panabieres and Pantel, 2013; van de Stolpe et al., 2011). A CTC count has been identified as a powerful prognostic marker in metastatic colorectal (Cohen et al., 2008), breast (Cristofanilli et al., 2004) and prostate cancer (de Bono et al., 2008). Additionally, CTC characterization for drug target expression (Attard et al., 2009; Riethdorf et al., 2010), mutations (Holdhoff et al., 2009) and gene expression by reverse transcriptase quantitative PCR (RT‐qPCR) (Cohen et al., 2006; Sieuwerts et al., 2011; Smirnov et al., 2005; Xi et al., 2007) could have the potential to greatly improve treatment decision making.
However, significant challenges remain, such as the low frequency of CTCs in the circulation, and the additional presence of up to a thousand remaining leukocytes, even after CellSearch EpCAM‐based enrichment (Sieuwerts et al., 2009a). To enable CTC characterization by RT‐qPCR, we have minimized the potential confounding contribution contaminating leukocytes by focusing solely on genes expressed in tumor tissue that are not, or at a much lower level, expressed by leukocytes. Using stringent selection methods combined with a sensitive but robust cDNA pre‐amplification, we were able to reliably quantify a CTC‐specific gene panel in blood of metastatic breast cancer patients (Sieuwerts et al., 2011).
In the current study, we set out to identify such a CTC‐specific gene panel by qualifying a large panel of mRNAs in CellSearch‐enriched CTCs of metastatic CRC (mCRC) patients prior to partial liver resection. To this end, we compared the expression of this mRNA panel between healthy donors (HDs) and patients without detectable CTCs as defined by CellSearch. By measuring these CTC‐specific genes in patients without detectable CTCs, a HD‐unlike group was identified, and the prognosis of these patients was compared with HD‐like patients without evidence of circulating tumor load according to their CTC‐specific gene expression.
2. Materials and methods
2.1. Patients and blood samples
As part of a prospective study, which included patients with mCRC about to undergo resection of hepatic metastases, 142 patients were included from June 2008 until June 2011. The first (primary) cohort of 107 patients was used to perform all our initial analyses. A second independent cohort of 35 patients, who were included in the same study under the same in‐ and exclusion criteria but at a later time point, was subsequently analyzed to substantiate our findings obtained in the primary cohort. From all these patients, 30 mL blood samples were taken for CTC enumeration and characterization (for details see next paragraph) by way of venipuncture before liver metastasis resection and prior to surgical tumor manipulation. This study was approved by the Leiden University Medical Center and Erasmus University Medical Center Institutional Review Boards (METC P05.182) and all patients were included in the Erasmus University Medical Center, Rotterdam, Netherlands, after written informed consent was obtained. Additionally, 30 mL blood samples were drawn from 30 healthy volunteers (age 21–58) to evaluate gene expression in HDs. The same HDs were used in the analysis of the primary and the secondary independent patient set (Figure 1).
Figure 1.
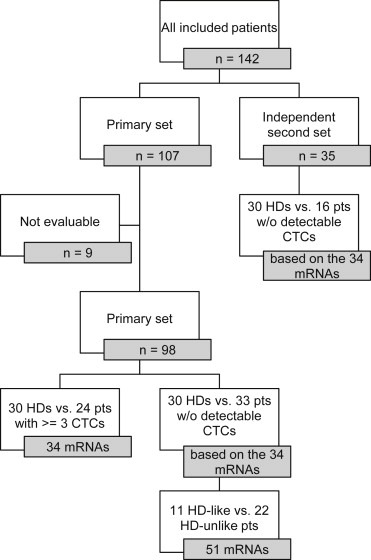
Flowchart of patient sets for all analyses. Flowchart depicting the patient set including the primary set and the independent second set as well as the number of genes identified in the analysis of healthy donors (HDs) and patients (pts) without (w/o) detectable CTCs (left bottom branch) and in the comparison of HDs and patients with ≥3 CTCs (right bottom branch). Nine patients were excluded from the primary patient set because of insufficient mRNA quality according to our QC standards (see Materials and Methods).
2.2. Isolation and enumeration of CTCs
Two samples of 30 mL blood from the 142 mCRC patients prior to liver metastasis resection were drawn in CellSave™ tubes (Veridex LLC, Raritan, NJ) for enumeration or in EDTA tubes for isolation. Prior to CTC enumeration and isolation, a density gradient‐based enrichment step was applied as described before (Lalmahomed et al., 2010). Briefly, immediately before subsequent CTC enumeration or isolation, 30 mL blood was pooled and centrifuged for 10 min at 800 × g. After removal of plasma, 15 mL CTC buffer was added and mixed, and pelleting the total supernatant volume was carefully placed onto 6 mL of Lymphoprep (Axis‐Shield, Dundee, Scotland), a Ficoll density‐gradient medium. After centrifugation at 400 × g for 10 min, the top buffer layer was discarded. Then, 7.5 mL of suspension including the buffy coat was aspirated with a reversed 10 mL pipette, allowing optimal isolation of the mononuclear cell layer, and pipetted into a regular CellTracks™ tube (Veridex). For CTC enumeration, samples were processed on the CellTracks™ AutoPrep System (Veridex) using the CellSearch™ Epithelial Cell Kit (Veridex) within 96 h after collection and CTC counts were determined on the CellTracks™ Analyzer (Veridex) according to the manufacturer's instructions and as described before (Mostert et al., 2011; Sieuwerts et al., 2009a). A cell was counted as a CTC if it was an intact, nucleated DAPI+ cell, lacking CD45 and expressing cytokeratin (CK)8/9/19, according to the manufacturer's instructions.
2.3. mRNA isolation from CTCs, RT‐qPCR and quality control
For gene expression studies, in parallel with the enumeration studies, 30 mL of blood from patients and HDs was drawn in EDTA tubes, subjected to Ficoll enrichment as described above and enriched for CTCs on the CellTracks™ AutoPrep System using the CellSearch™ Profile Kit (Veridex) within 24 h after collection. RNA isolation was performed with the AllPrep DNA/RNA Micro Kit (Qiagen, Valencia, CA), and cDNA synthesis, pre‐amplification, real time PCR and normalization procedures to quantify gene expression levels were performed as described in detail before (Sieuwerts et al., 2009a). The quality control (QC) measures that were taken to ensure the linear and homogeneous nature of pre‐amplification, adequate PCR efficiency and reproducibility of each assay have also been described in detail before (Sieuwerts et al., 2009a). Only patients with sufficient QC mRNA signal for 3 reference genes (GUSB, HMBS and HPRT1 with an average value ≤26 Ct) were included in the final analysis, based on which material of 9 from the originally 142 patients (6.3%) were excluded from the final mRNA analysis.
Besides the 3 reference genes, 92 mRNAs (Supplementary Table 1) were selected to be putatively CTC‐specific, i.e., were described in silico to be relatively low expressed in leukocytes compared with CRC (SAGE Genie Database of the Cancer Genome Anatomy Project (http://cgap.nci.nih.gov/SAGE/AnatomicViewer), and could be reliably and linearly measured based on our previously described QC measures (Sieuwerts et al., 2009, 2011). For each mRNA assay, a first cutoff to eliminate false‐positive mRNA expression signals due to leukocyte contamination was established at the median of the expression in HDs. As a result, H19 and MMP3 were excluded from the original panel of 92 mRNAs, leading to a total of 90 mRNAs, which were used for identification of CTC‐specific mRNAs and comparison of HDs to patients without detectable CTCs (see below and Figure 1).
2.4. Statistical analysis
Stata v13 and Analyse‐it v2.26 were used for statistical analyses and generation of box‐plots. The strength of the associations between continuous variables was assessed with non‐parametric Spearman rank correlation. Differences in the expression levels in various groups were tested with the non‐parametric Mann–Whitney U test or the Kruskal–Wallis test, when appropriate, and differences in baseline patient and tumor characteristics by the Fisher's exact test. CTC‐specific profiles were identified by Class Comparison in Biometric Research Branch ArrayTools (http://linus.nci.nih.gov/BRB‐ArrayTools.html), using a permutation P‐value cut‐off of <0.05 (two‐sample t‐test). Hierarchical clustering analysis was performed using Cluster and Treeview (Eisen et al., 1998) and a custom Perl script to visualize the gene expression values. DAVID (Database for Annotation, Visualization, and Integrated Discovery (Dennis et al., 2003; Huang da et al., 2009)) was used to functionally annotate genes and identify the over‐represented functions, with P‐values corrected for multiple testing via Benjamini–Hochberg's procedure. Unless stated otherwise, all statistical tests are 2‐sided with P < 0.05 considered statistically significant.
3. Results
3.1. Patient characteristics
Among 107 included mCRC patients about to undergo hepatic metastasis resection in the primary patient set, 98 patients were evaluable for mRNA gene expression (Figure 1). The 35 patients in the independent patient set, who were included in the same study under the same inclusion criteria but at a later time point and used to further characterize samples without detectable CTCs, were all evaluable for mRNA gene expression. Detailed clinicopathological information for the primary set of 98 patients and the independent second set is available in Table 1. Patient and tumor characteristics were well balanced between the primary and independent second set and no significant differences in the proportions of any of the patient or tumor characteristics between the two groups were observed.
Table 1.
Patient characteristics of the primary patient set, according to CTC count and mRNA cluster, and the second independent patient set.
| Patient characteristics | Primary patient set | Second independent patient set | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of pts | % | mRNA cluster | P‐value | CTC count | P‐value | No. of pts | % | P‐value | |||
| 1 | 2 | <3 | ≥3 | ||||||||
| All patients | 98 | 100% | 36 | 62 | 74 | 24 | 35 | ||||
| Sex | |||||||||||
| Female | 40 | 41% | 10 | 30 | 0.060 | 24 | 16 | 0.004 | 12 | 34% | 0.550 |
| Male | 58 | 59% | 26 | 32 | 50 | 8 | 23 | 66% | |||
| Age at time of surgery | |||||||||||
| <60 | 35 | 36% | 14 | 21 | 0.665 | 25 | 10 | 0.625 | 12 | 34% | 1.000 |
| ≥60 | 63 | 64% | 22 | 41 | 49 | 14 | 23 | 66% | |||
| Presentation of metastasis | |||||||||||
| synchronous | 62 | 63% | 19 | 43 | 0.129 | 48 | 14 | 0.629 | 24 | 69% | 0.682 |
| metachronous | 36 | 37% | 17 | 19 | 26 | 10 | 11 | 31% | |||
| Primary tumor in situ at time of surgery | |||||||||||
| yes | 26 | 27% | 8 | 18 | 0.636 | 21 | 5 | 0.598 | 7 | 20% | 0.502 |
| no | 72 | 73% | 28 | 44 | 53 | 19 | 28 | 80% | |||
| Elapsed time between primary tumor and metastasis resectiona | |||||||||||
| <6 months | 22 | 22% | 7 | 15 | 0.612 | 19 | 3 | 0.257 | 6 | 17% | 0.802 |
| ≥6 months | 64 | 65% | 26 | 38 | 47 | 17 | 22 | 63% | |||
| CTC count | |||||||||||
| <3 | 74 | 76% | 29 | 45 | 0.486 | 29 | 83% | 0.482 | |||
| ≥3 | 24 | 24% | 7 | 17 | 6 | 17% | |||||
| Primary tumor characteristics | |||||||||||
| Locationb | |||||||||||
| right hemicolon | 16 | 16% | 4 | 12 | 0.299 | 12 | 4 | 1.000 | 7 | 20% | 0.716 |
| left hemicolon | 50 | 51% | 17 | 33 | 38 | 12 | 18 | 51% | |||
| rectum | 32 | 33% | 15 | 17 | 24 | 8 | 9 | 26% | |||
| Tumor sizeb | |||||||||||
| T0 | 1 | 1% | 1 | 0 | 1.000 | 1 | 0 | 1.000 | 0 | 0% | 0.243 |
| T1 | 0 | 0% | 0 | 0 | 0 | 0 | 0 | 0% | |||
| T2 | 13 | 13% | 4 | 9 | 10 | 3 | 1 | 3% | |||
| T3 | 64 | 65% | 25 | 39 | 50 | 14 | 24 | 69% | |||
| T4 | 9 | 9% | 3 | 6 | 7 | 2 | 7 | 20% | |||
| Lymph node involvementb | |||||||||||
| N0 | 33 | 34% | 18 | 15 | 0.091 | 25 | 8 | 0.545 | 10 | 29% | 0.6746 |
| N1 | 29 | 30% | 8 | 21 | 25 | 4 | 13 | 37% | |||
| N2 | 20 | 20% | 7 | 13 | 15 | 5 | 9 | 26% | |||
| Dukes classificationb | |||||||||||
| A | 1 | 1% | 0 | 1 | 0.11 | 0 | 1 | 0.303 | 0 | 0% | 0.8964 |
| B | 16 | 16% | 10 | 6 | 11 | 5 | 5 | 14% | |||
| C | 16 | 16% | 6 | 10 | 14 | 2 | 5 | 14% | |||
| D | 62 | 63% | 19 | 43 | 48 | 14 | 24 | 69% | |||
| Gradeb | |||||||||||
| 1 (well differentiated) | 3 | 3% | 1 | 2 | 1000 | 3 | 0 | 0.179 | 3 | 9% | 0.2871 |
| 2 (moderately differentiated) | 58 | 59% | 23 | 35 | 41 | 17 | 18 | 51% | |||
| 3 (poorly differentiated) | 7 | 7% | 3 | 4 | 7 | 0 | 3 | 9% | |||
| Neoadjuvant chemotherapyb | |||||||||||
| yes | 10 | 10% | 6 | 4 | 0.086 | 9 | 1 | 0.444 | 4 | 11% | 0.5049 |
| no | 71 | 72% | 22 | 49 | 54 | 17 | 19 | 54% | |||
| Adjuvant chemotherapyb | |||||||||||
| yes | 9 | 9% | 3 | 6 | 1000 | 8 | 1 | 0.676 | 4 | 11% | 0.4646 |
| no | 74 | 76% | 25 | 49 | 57 | 17 | 18 | 51% | |||
| Induction chemotherapy before liver resectionb | |||||||||||
| yes | 52 | 53% | 15 | 37 | 0.017 | 38 | 14 | 0.813 | 22 | 63% | 0.4291 |
| no | 44 | 45% | 21 | 23 | 34 | 10 | 13 | 37% | |||
| Site of metastasis | |||||||||||
| liver‐only | 89 | 91% | 33 | 56 | 1000 | 68 | 21 | 0.684 | 32 | 91% | 1000 |
| liver and other sites | 9 | 9% | 3 | 6 | 6 | 3 | 3 | 9% | |||
Detailed clinicopathologic characteristics of the 98 patients from the primary patient set with pathology‐confirmed colorectal liver metastases and mRNA data available, depicted according to CTC‐count (<3 vs. ≥3, the clinically relevant prognostic cut‐off level and according to mRNA (cluster 1 vs. 2). The last column depicts the clinicopathologic characteristics of the independent second patients set, which were included in the same study at a later time point and used to validate some of our findings in the primary patient set. Significant P‐values as obtained by the Fisher's exact test are depicted in italics and P‐values are depicted in bold if the value is <0.05.
Numbers do not add up to 100% because patients undergoing primary tumor resection after liver resection, and patients who have not undergone primary tumor resection at all, were left out.
Numbers do not add up to 100% because of missing data.
3.2. CTC counts
Among all 98 patients from the primary set with QC mRNA, 24 patients (24%) had ≥3 CTCs per 30 mL blood, 41 patients (42%) had 1 or 2 detectable CTCs, while in 33 patients (34%) no CTCs were identified. The number of patients below and above the cut‐off of 3 CTCs was not equally distributed among males and females; relatively more female than male patients had CTC counts above 3 cells per 30 mL blood (Fisher's exact, P 0.004, Table 1). In the second set, six patients had ≥3 CTCs per 30 mL blood (17%); while in 16 patients (46%), no CTCs were identified. The unequal distribution in CTC counts among males and females was not observed in the smaller second set of 35 patients.
3.3. CTC‐specific mRNAs
As described, besides the 3 reference genes, 90 mRNAs could be reliably and linearly measured in mCRC CTCs. To select CTC‐specific mRNAs, we compared the mRNA expression between samples with presumed substantial circulating tumor gene expression and samples without any circulating tumor load. We therefore compared the 24 patients with ≥3 CTCs in the primary set with the 30 HDs, and thus identified a panel of 34 mRNAs of which the transcripts were at a permutation P < 0.05 higher expressed in the patients with ≥3 CTCs compared with HDs (Class comparison BRB‐array tools, Supplementary Table 2).
3.4. Unsupervised hierarchical clustering of CTC‐specific mRNAs
After identifying CTC‐specific genes, we wished to study possible heterogeneity in their expression between patients. To this end, we used the identified 34 CTC‐specific mRNAs to perform unsupervised 2‐dimensional average linking hierarchical clustering analysis of all 98 patients from the primary set with available QC mRNA data (Figure 2). This clustering analysis revealed two main clusters, cluster 1 and 2. Cluster 2 could be further divided into two patient clusters, which, contrarily to patient cluster 1, were both characterized by a relatively high expression of epithelial genes such as KRT19 and KRT20. Patient cluster 2a was characterized by the high expression of genes such as FABP1, CDX1 and CDH17. These genes are marked by the blue rectangle at the right in Figure 2. No specific common category was significantly enriched in this gene cluster by DAVID functional gene annotation analysis. Patient cluster 2b largely lacked expression of these genes, but did express REG1A, IGFBP5 and AGR2 (genes are marked by the red rectangle at the right in Figure 2). DAVID analysis identified “secreted” as the most significant category for seven genes (PRSS8, RARRES2, COL4A1, LAD1, AGR2, IGFBP3, IGFBP5) in this 15‐gene cluster (5.3‐fold enrichment, Benjamini P 0.04). No significant association was found between the different patient clusters and CTC counts, neither the continuous CTC counts (dark green bars at the bottom of Figure 2) nor a dichotomized CTC count <3 versus ≥3 per 30 mL blood (green [<3] and red bars [≥3] at the bottom of Figure 2). The patients with ≥3 CTCs do seem to be enriched in patient cluster 2a, but differences were not statistically significant. Comparing the patients in the two respective mRNA clusters (1 and 2a & b) for their baseline clinicopathological characteristics showed that patients belonging to clusters 2a and 2b had more frequently had induction chemotherapy administered than those in cluster 1 (P 0.017, Table 1).
Figure 2.
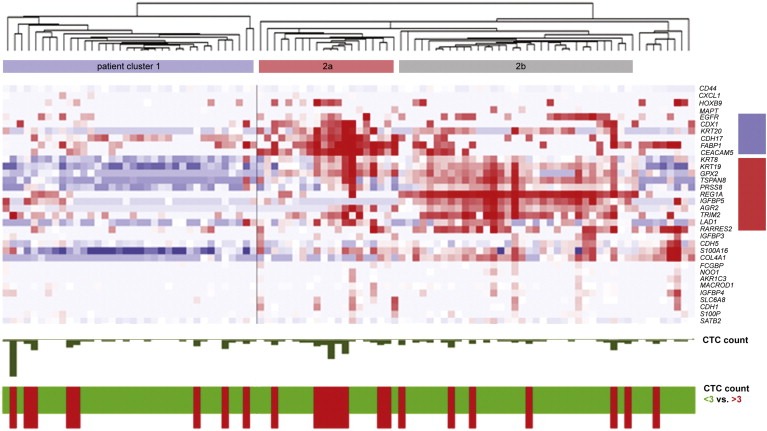
Cluster analysis of mRNA data. Unsupervised hierarchical clustering analysis comparing mRNA expression profiles in CellSearch‐enriched blood samples of 98 mCRC patients with available mRNA data. Each horizontal line corresponds to a gene; each vertical line represents a patient. Red color signifies a transcript level above the median level, white color represents an intermediate level and blue color a transcript level below the median level of the particular mRNA in all samples. Three patient clusters (1 and 2a and b) can be distinguished. Clusters 2a and b were characterized by two gene clusters (depicted by the blue and red rectangles, respectively). CTC count as enumerated by CellSearch is depicted on the bottom of the graph; in the ‘CTC count’ panel, the number of CTCs is represented by the size of the green bars (range 0–35), the red line signifies the cut‐off of 3 CTCs. In the “CTC count 0 vs. <3 vs. ≥3” panel, a green bar to a CTC count <3 and a red bar to a CTC count ≥3 per 30 mL blood.
3.5. CTC‐specific mRNAs and prognosis
For those patients evaluable for relapse‐rate, disease‐free (DFS) and overall survival (OS), we assessed the correlation of the 91 patients making up the three distinct patient clusters (cluster 1, 2a and 2b) based on the 34 CTC‐specific mRNAs with prognosis. Seventy patients were evaluable for disease recurrence rate and survival. Of the other 21, two patients still had the primary tumor in situ at the time of liver resection, eight patients underwent liver resection as part of a two‐stage approach and thus still had residual liver metastases in situ after the liver resection and 11 did not undergo liver resection because extensive and/or progressive disease was seen during surgery. Of 70 evaluable patients, 49 experienced a relapse. Comparing the relapse‐rate within 1 year after liver resection, no difference was seen between the clusters (Pearson's X2 statistic 1.22, P 0.5442). Comparing overall survival (OS), a trend was seen towards shorter OS (HR 1.96 [95%CI 0.88–4.33] Logrank trend P 0.098) for patients in cluster 2.
3.6. Circulating tumor load in patients without detectable CTCs: mRNA analysis
Based on the results of our previous study in breast cancer (Sieuwerts et al., 2009, 2011), we hypothesized that patients without detectable CTCs, i.e., as defined by the CellSearch enumeration definition, could harbor tumor‐specific gene expression in the CellSearch‐enriched fractions of their blood. This circulating tumor load would nonetheless not be identified and counted as CTCs by the CellSearch enumeration procedure. To this end, we assessed whether or not the blood of the 33 patients without detectable CTCs contained gene transcripts related to circulating tumor load. For this analysis we took an unbiased approach and employed an unsupervised hierarchical clustering analysis (Figure 3) using the top 75% variably expressed genes (n = 69, Supplementary Table 2) in this cohort of the 33 patients without detectable CTCs according to CellSearch criteria (marked in red at the bottom bar) and 30 HDs (marked in green at the bottom bar). This analysis revealed 2 clear subgroups. Interestingly, a subgroup of patients without detectable CTCs according to the CellSearch system (the HD‐unlike cluster; marked in red at the top bar) was clearly distinct from HDs and other patients without CTCs (the HD‐alike cluster; marked in blue at the top bar). These patients had high expression of epithelial genes such as KRT19 and KRT20, but also of the genes IGFBP5, AGR2, S100A16 and LAD1, which were previously established to identify epithelial tumor load in breast cancer patients (Sieuwerts et al., 2011). This result strongly suggested that the blood of the HD‐unlike patients indeed contained CTCs or fragments thereof, which were not detected by the CellSearch enumeration procedure, but nevertheless were captured by the CellSearch Profile Kit. To gain further insight into the possible effect of stochastic variation between the CTC enrichment and enumeration tubes, we also performed this analysis with patients with 1 enumerated CTC. While some of these patients cluster together with healthy donors, probably due to stochastic variation, the majority of these patients (23 out of 31) end up in the HD‐unlike cluster (Supplementary Figure 1).
Figure 3.
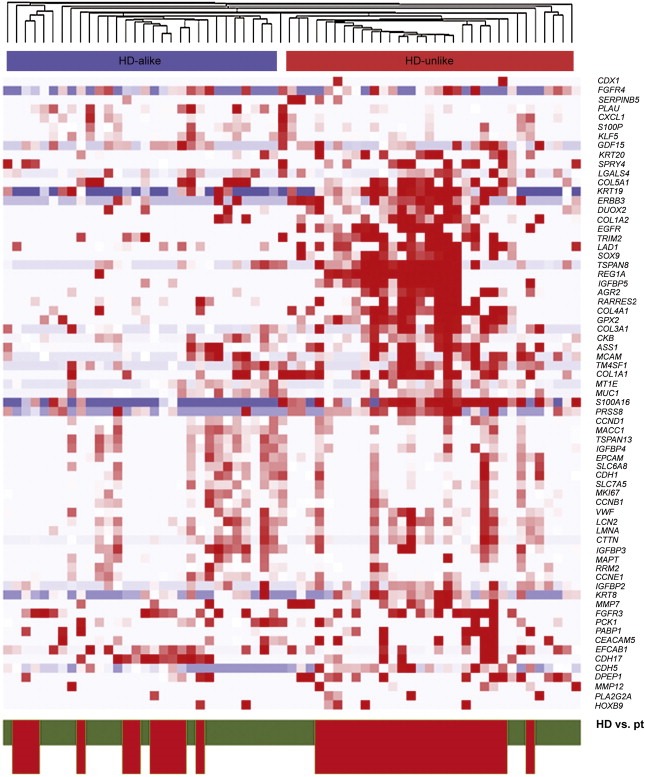
Clustering of HDs and patients without detectable CTCs. Unsupervised hierarchical cluster analysis comparing mRNA gene expression profiles in CTC‐enriched fractions from 33 mCRC patients without detectable CTCs and mRNA data available for 30 HDs. Data shown have been subjected to median normalization of each individual sample across all genes followed by median normalization of each individual gene across all samples. Columns represent patient samples, rows represent the 69 genes that were the top 75% variably expressed between the patients without detectable CTCs and the HDs. Red color indicates a transcript level above the median level, white color indicates a median transcript level, and blue color indicates a transcript level below the median level of the particular mRNA in all samples. Depicted gene clusters were identified at an average linkage correlation greater than 0.2. At the top, the HD‐unlike patient cluster is indicated in red; the HD‐alike patient cluster in blue. Healthy donors (HD, green) and patients (pt, red) are depicted on the bottom of the graph.
While we analyzed genes that are not or at a very low level expressed by leukocytes, we wished to exclude the possibility that the mRNA expression from the leukocyte background, that is still present after EpCAM enrichment, accounted for the clustering of HD‐unlike patients with patients with detectable CTCs. For this, we compared the expression of PTPRC (CD45, a leukocyte marker) between HDs, HD‐unlike and HD‐like patients. We did see a higher PTPRC expression in healthy donors compared to patients (Supplementary Figure 1), but no difference between HD‐like and HD‐unlike patients (data not shown).
3.7. Identification of HD‐unlike patients in an independent patient set
To assess whether a HD‐unlike patient group was also present in an independent patient set, we analyzed the 34 genes that were differentially expressed between HDs and patients with ≥3 CTCs in the primary patient set and thus CTC‐specific (see above) also in the independent secondary set consisting of 35 patients. We chose to do this analysis based on the 34 CTC‐specific genes, as we hypothesized that the HD‐unlike patients were distinct from HDs and HD‐like patients due to presence of circulating tumor load. By using the 34 CTC‐specific genes, we anticipated to be able to identify a subgroup of patients without detectable CTCs but with evidence of circulating tumor load in the independent patient set. As described above, characteristics of the HD‐unlike patients did not differ significantly from the original set of 98 patients (Table 1). We clustered our 16 patients with zero CTCs from the secondary set as well as the 33 patients with zero CTCs from the primary set with the 30 HDs to be able to view whether patients with evidence of circulating tumor load indeed clustered with the previously identified HD‐unlike patients. Indeed, we again observed a distinction into HD‐alike and HD‐unlike patients (Figure 4), and the previously identified HD‐unlike patients from the primary set were again distinguished as such. Of the 16 patients without detectable CTCs that were added from the second patient set, three grouped in the HD‐unlike cluster independently confirming the existence of HD‐unlike patients amongst those without detectable CTCs.
Figure 4.
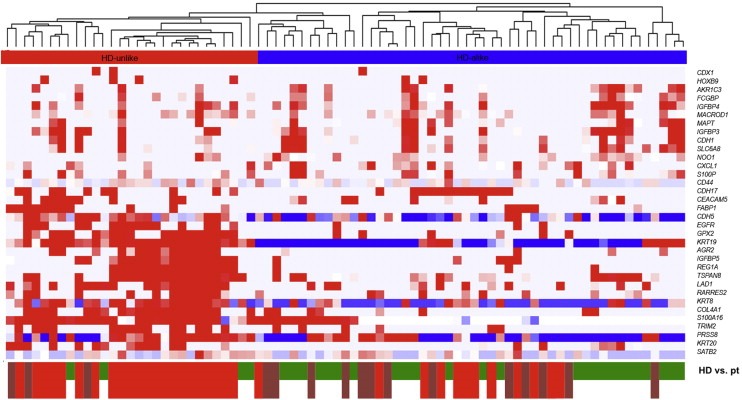
Clustering of patients from the primary and secondary sets without enumerated CTCs with healthy donors (HDs). Unsupervised hierarchical clustering analysis comparing mRNA gene expression profiles in mCRC CTC‐enriched fractions from 49 patients without detectable CTCs and QC mRNA available (33 from the primary set and 16 from the independent second set) and 30 HDs. Data shown have been subjected to median normalization of each individual sample across all genes followed by median normalization of each individual gene across all samples. Columns represent patient samples, rows represent genes. Red color indicates a transcript level above the median level, white color indicates a median transcript level, and blue color indicates a transcript level below the median level of the particular mRNA in all samples. Depicted gene clusters were identified at an average linkage correlation greater than 0.2. At the top, the HD‐unlike patient cluster is indicated in red; the HD‐alike patient cluster in blue. Healthy donors (HD, green), primary patients (pt, bright red) and secondary patients (dark red) are depicted on the bottom of the graph.
3.8. Prognosis in HD‐unlike and HD‐like patients
Thus, we had identified a HD‐unlike patient group without detectable CTCs according to CellSearch criteria, but with evidence of circulating tumor load by CTC‐specific gene expression. Next, we wished to conduct an exploratory analysis to assess whether this circulating tumor load also conferred to a poorer prognosis compared with HD‐like patients. Only 11 HD‐like and 14 HD‐unlike patients were evaluable (two patients still had the primary tumor in situ at the time of liver resection, two patients underwent liver resection as part of a two‐stage approach and thus still had residual liver metastases in situ after the liver resection and four did not undergo liver resection because extensive and/or progressive disease was seen during surgery). The 1‐year recurrence rate in the HD‐unlike group and the HD‐like group were both 36%. Likewise, no differences were seen in overall survival (HR; 1.54, P 0.53).
3.9. CTC detection markers in HDs and patients without detectable CTCs
Above we showed that a group of HD‐unlike patients in whose blood no CTCs were detected with CellSearch CTC enumeration nevertheless showed evidence of circulating tumor load. A possible explanation for the inability to enumerate CTCs in these patients could be an insufficient expression of epithelial markers needed for CTCs to be enumerated according to the CellSearch criteria (the cytokeratins KRT8, KRT18 and KRT19), while sufficient EpCAM expression did enable their capture for subsequent gene expression profiling. To explore this hypothesis, we focused on the expression levels of the epithelial markers EPCAM and KRT8, KRT18 and KRT19 in the RNA fractions isolated from CellSearch‐enriched blood of 30 HDs, 49 patients without detectable CTCs (23 HD‐alike, 26 HD‐unlike), and 30 patients with ≥3 CTCs (Figure 5). The expression levels of cytokeratins 8 and 19 significantly differed between HDs and patients with ≥3 CTCs (Bonferroni contrast; KRT8, P < 0.0001; KRT19, P < 0.0001), whilst those of cytokeratin 18 (KRT18, P 1.000) and EpCAM (P 0.2849) did not (Figure 5). When we tested for differences in expression between HD‐alike and HD‐unlike patients, those of the cytokeratins 8 and 19 were again significantly different, whereas EPCAM and cytokeratin 18 were not (KRT8, P < 0.0001; KRT19, P < 0.0001; KRT18, P 0.1411; EPCAM, P 1.0000).
Figure 5.
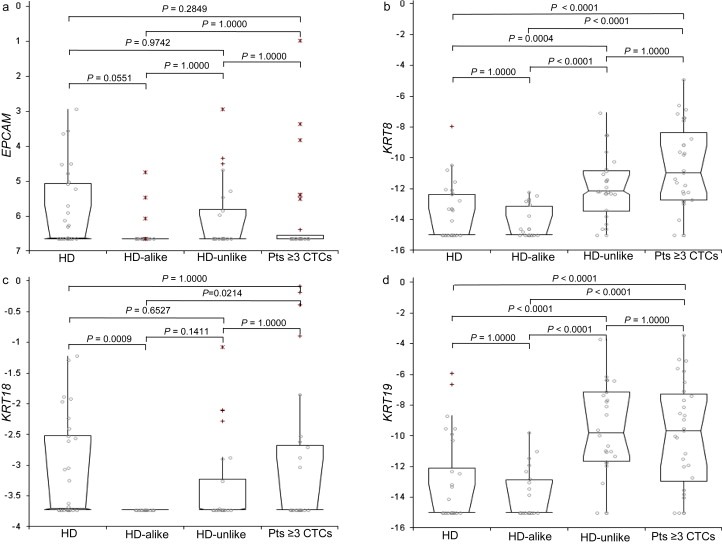
Expression of CTC‐markers in CellSearch‐enriched blood from 30 HDs and 49 patients without detectable CTCs compared with patients with three or more CTCs. Expression levels of EPCAM, KRT8, KRT18 and KRT19 are depicted for 30 HDs, 23 HD‐alike, 26 HD‐unlike and 30 patients with three or more CTCs from the whole patient set. Expression levels were measured in RNA isolated from the EpCAM‐enriched (CellSearch) blood fractions and are depicted as deltaCt to the median of the reference genes (GUSB, HMBS and HPRT1). Box plots represent median, 95% confidence interval (box) and outlier (+; >1.5 and <3 interquartile range,*; >3 interquartile range) expression levels. Level of significance tested with Kruskal–Wallis test followed by post‐hoc contrast analysis. HD, healthy donor; pts, patients.
4. Discussion
CTCs offer an exciting new opportunity to assess prognostic and predictive markers repeatedly during the course of cancer. CTCs are presumed to represent actual metastatic tumor load, and may thus provide more accurate information to guide treatment decisions than the primary tumor. After having previously shown the feasibility of measuring a CTC‐specific gene panel in the CTCs of metastatic breast cancer patients (Sieuwerts et al., 2011), we show here that, using genes clinically relevant in CRC, a similar approach can be successfully applied to CTCs from mCRC patients with overall a much lower CTC count. Moreover, we show the presence of circulating tumor load as suggested by epithelial gene expression in a large subgroup of patients without detectable CTCs.
While several studies have described the expression levels of up to 10 different genes in the blood of CRC patients (Koyanagi et al., 2008; Terrin et al., 2008; Xi et al., 2007; Yie et al., 2008), no work has been published on the broad‐scale molecular characterization of CTCs of mCRC patients after CellSearch enrichment. This EpCAM‐based CellSearch enrichment has the significant advantage of being semi‐automated, but does not result in a pure CTC population. To still enable reliable measurement of clinically relevant genes, we therefore selected, in silico, genes that are not or at a very low level expressed by leukocytes.
By comparing HDs and patients with ≥3 CTCs, we identified 34 CTC‐specific mRNAs. Based on the unsupervised clustering analysis with the 34 CTC‐specific mRNAs, we could distinguish three patient clusters, one of them –patient cluster 1‐ characterized by the absence of epithelial gene expression. Patient cluster 2a was characterized by the high expression of genes such as FABP1, CDX1, CDH17 and KRT20. FABP1 was previously shown to be higher expressed in good‐prognosis CRC primary tumors and metastases (Yamazaki et al., 1999) and a marker of differentiation (Lawrie et al., 2004) but has also been described as a useful CTC detection marker (Smirnov et al., 2005). CDX1 has been described as a tumor suppressor gene (Guo et al., 2004) reducing proliferation through Cyclin D1 (Lynch et al., 2003), and is frequently rearranged in relation to rearrangements at the APC locus (Domon‐Dell et al., 2003). CDH17 mediates cell–cell adhesion in intestinal epithelial cells (Berndorff et al., 1994), is specific to cancers of the digestive system (Su et al., 2008) and has previously been associated with poor prognosis (Wang et al., 2004). KRT20 is an epithelial marker, which has also frequently been used to identify circulating tumor load in CRC patients (Iinuma et al., 2011; Vlems et al., 2003), but which is not included in the CellSearch CTC enumeration kit. Patients in cluster 2b expressed REG1A, IGFBP5 and AGR2, of which the latter were previously determined to be epithelial‐specific in our breast cancer studies (Sieuwerts et al., 2011), while AGR2 is also up regulated in microsatellite instability‐high (MSI‐H) tumors (Kim et al., 2004; Mori et al., 2004). REG1A is a gene correlated with poor prognosis (Astrosini et al., 2008) and microsatellite instability (Lee et al., 2008). Patients in this cluster have a lower expression of KRT20, making its individual use as a CTC detection marker doubtful. Noteworthy is that we did not see a clear relation of the different clusters with CTC‐count, for which the aforementioned issues resulting in discrepancies between CTC enumeration and gene expression are the most probable explanation. Correlation with disease free‐survival and overall survival showed a trend towards poorer prognosis in patients in cluster 2 as a whole. It should be noted that this was an exploratory analysis and because of the small patient groups, no correction was done for the administration of induction chemotherapy before liver resection and clinical risk score, both known important risk factors for DFS and OS in this patient population (Ayez et al., 2011).
Remarkably, when comparing patients without detectable CTCs with HDs, a cluster of patients had a gene expression profile strongly differing from that of HDs. These HD‐unlike patients were characterized by the expression of known epithelial markers such as cytokeratins and EPCAM, but also by the expression of four other genes previously determined to be epithelial‐specific (S100A16, AGR2, IGFBP5 and LAD1 (Sieuwerts et al., 2011)). Others have also described two of these genes, S100A16 and AGR2, as expressed in CTCs (Smirnov et al., 2005). Among the 30 other genes were genes involved in focal adhesion (the collagens and Cyclin D1) and extracellular matrix (ECM)‐receptor interaction (the collagens and VWF), among others. Also when looking at patients with zero or one enumerated CTCs as well as in our independent secondary patient set, a subgroup of patients was identified with expression of epithelial marker genes in the absence of counted CTCs. Together, the gene expression pattern of these patients strongly suggests the presence of circulating tumor load, CTCs or cell fragments like exosomes, which are not being detected or recognized as such by the CellSearch system in the blood drawn in parallel for CTC enumeration. A few factors could cause this discrepancy between CTC count and CTC molecular profile. Literature suggests that CRC cells can lack cytokeratin 8, 18 or 19 expression (Joosse et al., 2012; Moll et al., 1992), the markers by which a CTC is defined in CellSearch. In breast cancer as well, we have described a lack of cytokeratin expression for certain breast cancer subtypes (Mostert et al., 2015), reflecting the epithelial‐to‐mesenchymal (EMT)‐like phenotype of these cells. A lack of cytokeratin 8/18/19 expression would not affect the enrichment of CTCs by immunomagnetic enrichment, which is based on their EpCAM‐expression. However, the lack of cytokeratin expression would result in these cells not being identified as epithelial cells and thus not counted as CTCs in the subsequent enumeration step. Indeed, KRT8 and KRT19 mRNAs were significantly higher expressed in HD‐unlike than in HD‐alike patients without detectable CTCs, while this difference was not apparent for EPCAM or KRT9. EpCAM‐based immunomagnetic isolation leads to capture of about a thousand leukocytes per 7,5 mL blood in both HDs and patients, which would explain the lack of difference in EPCAM expression. KRT8 and KRT19, however, are only expressed by epithelial cells, and the expression of KRT8 and KRT19 in HD‐unlike patients alludes to the presence of circulating tumor load. The presence of this KRT mRNA expression apparently does not always lead to the identification of CTCs by the CellSearch enumeration method, possibly because these cells or cell fragments do not meet the morphologic criteria for CTCs. To further study the possible influence of EMT on the detection of circulating tumor load by RT‐qPCR in the absence of enumerated CTCs, it would be of great interest to measure EMT‐associated genes such as vimentin (VIM). However, because these genes are also abundantly expressed by leukocytes (Sieuwerts et al., 2009a), we could not measure these genes reliably in our current samples, which still contained a leukocyte background despite the CellSearch enrichment. Single‐cell CTCs or a pure population of CTCs could circumvent these problems, and should thus be a major focus of research in this field.
Other reasons for the discrepancies between CTC counts and gene expression could be an imperfect correlation of mRNA and protein expression and the well‐known issue of stochastic variation (Allan and Keeney, 2010). Stochastic variation leads to differences in CTC content in two blood tubes drawn in parallel, and becomes particularly important when highly sensitive techniques such as PCR are applied to samples containing low CTC numbers such as in this study. If we assume the CTC enrichment and enumeration tubes to be paired tests, the chance of the enumeration tube to contain zero CTCs while the expected value is one (as occurs in the CTC enrichment tube), is 0.37. The chance of finding circulating tumor load in the enriched CTCs of 21 patients out of 35 patients with zero enumerated CTCs, is 0.013, making it unlikely that stochastic variation is an important factor in our findings. In addition, the strict morphological criteria that have to be met for cells to be counted as CTCs exclude the counting of small CTCs or CTC fragments (Coumans et al., 2010), both of which nonetheless can confer an mRNA signal. The method of cell fixation is also a variable differing between the CTC enumeration and CTC isolation method. The blood for CTC enumeration is kept in CellSave tubes containing a preservative enabling their storage for up to 96 h before processing, while the blood for CTC isolation is kept in EDTA blood and processed within 24 h. The stability of the CTC enumeration sample has been tested and shown by the manufacturer. We have tested the stability of mRNA in EDTA, showing stability for at least 24 h (Supplementary Figure 2). Nonetheless, the difference in fixation and storage time could theoretically confer to a difference in CTC enrichment efficiency. We did not see a difference in leukocyte‐specific mRNA expression between HD‐like and HD‐unlike patients (Kruskal–Wallis, P 0.27), and while we measure genes that are not or at a low level expressed in leukocytes, we cannot completely exclude an effect of the number of background leukocytes after CTC enrichment on the measured gene expression.
Whatever the cause, measurement of epithelial gene expression seems to be able to detect circulating tumor load among patients without detectable CTCs. The prognostic value of small CTCs or CTC fragments not meeting the criteria of CTC according to the CellSearch procedure, has recently been described for prostate cancer (Coumans et al., 2010). In this small study, an exploratory analysis did not show a correlation of the HD‐unlike profile with poor prognosis, but this will have to be repeated in a larger study group. Exosomes have gained the interest of researchers as they are thought to function as crosstalk between tumor cells and the microenvironment, and as such could be directly involved in the process of metastasis. No broad consensus has been reached on the optimal method for isolation of exosomes from blood plasma, and no data are available on possible co‐isolation of exosomes when performing CellSearch EpCAM‐based enrichment of whole blood. Because CTC isolation in this study was preceded by a Ficoll‐based density gradient enrichment, it seems unlikely that exosomes would still be present in our samples. However, comparison of the gene expression in isolated single or purified CTCs versus exosomes would be of great interest to further increase our understanding of the mechanism of tumor metastasis, and could provide more insight into the source of the measured HD‐unlike mRNA expression in our patient samples.
Our CRC CTC‐specific 34‐gene panel enables the large‐scale characterization of CTCs despite their presence among an excess of contaminating leukocytes. This method might increase the sensitivity of CTC detection, making it more suitable to use in mCRC and possibly localized CRC as well. We can also explore the value of CTCs as a surrogate biopsy of metastases and improve our insight into metastasis biology. Moreover, CTC characterization by the CTC‐specific profile permits studies assessing the use of CTC gene transcripts as a predictive tool to guide treatment decisions.
In conclusion, the generation of this CRC CTC‐specific gene panel enables the exploration of CTC characterization as a novel means to further individualize cancer treatment based on better prognostic and predictive factors.
Financial support
This study was in part financially supported by Cancer Genomics Netherlands (CGC.nl)/Netherlands Organization for Scientific Research (NWO), and the Stichting Coolsingel (Coolsingel Foundation). The sponsors had no involvement in the collection, analysis and interpretation data; in the writing of the report; or in the decision to submit the paper for publication.
Supporting information
The following are the supplementary data related to this article:
Supplementary Table 1 Specifics of the Taqman assays used for detection of mRNA transcripts. Official gene symbol, reason for inclusion in our gene expression panel, Assay ID and Gene description of 95 mRNAs included in initial selection for measurement in CTCs. CTCM, well‐known CTC marker; DT, drug target; EM, epithelial marker (as discovered in our previous breast cancer studies (Sieuwerts et al., 2011)); ENM, endothelial marker; LM, leukocyte marker; REF, reference gene marker; SCM, stem cell marker.
Supplementary Table 2 Top 75% variably expressed between HDs and patients without detectable CTCs (a) and between HDs and patients with ≥3 CTCs (b). Panel a: The 34 mRNAs, which were at a Permutation P‐value <0.05 (two‐sample t‐test) differentially expressed between HDs and patients with ≥3 CTCs are depicted. All mRNAs were higher expressed in patients than in HDs. For reference purposes, the parametric P‐value and false discovery rate (FDR) are also given. Panel b: The 69 mRNAs, which were the top 75% variably, expressed genes between 30 HDs and 33 patients without detectable CTCs are depicted.
Supplementary Figure 2 Stability and detectability of RNA in CellSave preservative versus EDTA blood at different time points. Ct‐values of reference genes and percentages of 93 target genes that were detectable of MCF7 breast cancer cell line cells by RTq‐PCR. Both spiked and unspiked samples were analyzed, at 0, 24, 48, 72 and 96 hours after blood draw, and compared between EDTA versus CellSave preservative tubes.
Supplementary Figure 1 Clustering of patients (pat) from the primary set with 0–1 enumerated CTC and healthy donors (HDs). Unsupervised hierarchical clustering analysis comparing mRNA gene expression profiles in mCRC CTC‐enriched fractions from 66 patients with 0–1 detectable CTC and QC mRNA available and 30 HDs. Data shown have been subjected to median normalization of each individual sample across all genes followed by median normalization of each individual gene across all samples. Columns represent patient samples, rows represent genes. Red color indicates a transcript level above the median level, white color indicates a median transcript level, and blue color indicates a transcript level below the median level of the particular mRNA in all samples. Depicted gene clusters were identified at an average linkage correlation greater than 0.2.
{kind=link}
Acknowledgments
The authors would like to thank Ms. Maxime Look for her help with the statistical analyses, the surgeons, anaesthesiologists and operating room personnel for their assistance in collecting the blood samples, and the patients for their willingness to participate in this study. We would also like to thank Cancer Genomics Netherlands for their support.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.molonc.2015.01.001.
Mostert Bianca, Sieuwerts Anieta M., Bolt-de Vries Joan, Kraan Jaco, Lalmahomed Zarina, van Galen Anne, van der Spoel Petra, de Weerd Vanja, Ramírez-Moreno Raquel, Smid Marcel, Verhoef Cornelis, IJzermans Jan N.M., Gratama Jan W., Sleijfer Stefan, Foekens John A., Martens John W.M., (2015), mRNA expression profiles in circulating tumor cells of metastatic colorectal cancer patients, Molecular Oncology 9, doi: 10.1016/j.molonc.2015.01.001.
References
- Abdalla, E.K. , Vauthey, J.N. , Ellis, L.M. , Ellis, V. , Pollock, R. , Broglio, K.R. , Hess, K. , Curley, S.A. , 2004. Recurrence and outcomes following hepatic resection, radiofrequency ablation, and combined resection/ablation for colorectal liver metastases. Ann. Surg. 239, 818–825. (discussion 825–817) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam, R. , de Haas, R.J. , Wicherts, D.A. , Aloia, T.A. , Delvart, V. , Azoulay, D. , Bismuth, H. , Castaing, D. , 2008. Is hepatic resection justified after chemotherapy in patients with colorectal liver metastases and lymph node involvement?. J. Clin. Oncol. 26, 3672–3680. [DOI] [PubMed] [Google Scholar]
- Alix-Panabieres, C. , Pantel, K. , 2013. Circulating tumor cells: liquid biopsy of cancer. Clin. Chem. 59, 110–118. [DOI] [PubMed] [Google Scholar]
- Allan, A.L. , Keeney, M. , 2010. Circulating tumor cell analysis: technical and statistical considerations for application to the clinic. J. Oncol. 2010, 426218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrosini, C. , Roeefzaad, C. , Dai, Y.Y. , Dieckgraefe, B.K. , Jons, T. , Kemmner, W. , 2008. REG1A expression is a prognostic marker in colorectal cancer and associated with peritoneal carcinomatosis. Int. J. Cancer 123, 409–413. [DOI] [PubMed] [Google Scholar]
- Attard, G. , Swennenhuis, J.F. , Olmos, D. , Reid, A.H. , Vickers, E. , A'Hern, R. , Levink, R. , Coumans, F. , Moreira, J. , Riisnaes, R. , Oommen, N.B. , Hawche, G. , Jameson, C. , Thompson, E. , Sipkema, R. , Carden, C.P. , Parker, C. , Dearnaley, D. , Kaye, S.B. , Cooper, C.S. , Molina, A. , Cox, M.E. , Terstappen, L.W. , de Bono, J.S. , 2009. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 69, 2912–2918. [DOI] [PubMed] [Google Scholar]
- Ayez, N. , Lalmahomed, Z.S. , van der Pool, A.E. , Vergouwe, Y. , van Montfort, K. , de Jonge, J. , Eggermont, A.M. , Ijzermans, J.N. , Verhoef, C. , 2011. Is the clinical risk score for patients with colorectal liver metastases still useable in the era of effective neoadjuvant chemotherapy?. Ann. Surg. Oncol. 18, 2757–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldus, S.E. , Schaefer, K.L. , Engers, R. , Hartleb, D. , Stoecklein, N.H. , Gabbert, H.E. , 2010. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin. Cancer Res. 16, 790–799. [DOI] [PubMed] [Google Scholar]
- Berndorff, D. , Gessner, R. , Kreft, B. , Schnoy, N. , Lajous-Petter, A.M. , Loch, N. , Reutter, W. , Hortsch, M. , Tauber, R. , 1994. Liver-intestine cadherin: molecular cloning and characterization of a novel Ca(2+)-dependent cell adhesion molecule expressed in liver and intestine. J. Cell Biol. 125, 1353–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouquet, A. , Abdalla, E.K. , Kopetz, S. , Garrett, C.R. , Overman, M.J. , Eng, C. , Andreou, A. , Loyer, E.M. , Madoff, D.C. , Curley, S.A. , Vauthey, J.N. , 2011. High survival rate after two-stage resection of advanced colorectal liver metastases: response-based selection and complete resection define outcome. J. Clin. Oncol. 29, 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, P.J. , Yachida, S. , Mudie, L.J. , Stephens, P.J. , Pleasance, E.D. , Stebbings, L.A. , Morsberger, L.A. , Latimer, C. , McLaren, S. , Lin, M.L. , McBride, D.J. , Varela, I. , Nik-Zainal, S.A. , Leroy, C. , Jia, M. , Menzies, A. , Butler, A.P. , Teague, J.W. , Griffin, C.A. , Burton, J. , Swerdlow, H. , Quail, M.A. , Stratton, M.R. , Iacobuzio-Donahue, C. , Futreal, P.A. , 2010. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, S.J. , Alpaugh, R.K. , Gross, S. , O'Hara, S.M. , Smirnov, D.A. , Terstappen, L.W. , Allard, W.J. , Bilbee, M. , Cheng, J.D. , Hoffman, J.P. , Lewis, N.L. , Pellegrino, A. , Rogatko, A. , Sigurdson, E. , Wang, H. , Watson, J.C. , Weiner, L.M. , Meropol, N.J. , 2006. Isolation and characterization of circulating tumor cells in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 6, 125–132. [DOI] [PubMed] [Google Scholar]
- Cohen, S.J. , Punt, C.J. , Iannotti, N. , Saidman, B.H. , Sabbath, K.D. , Gabrail, N.Y. , Picus, J. , Morse, M. , Mitchell, E. , Miller, M.C. , Doyle, G.V. , Tissing, H. , Terstappen, L.W. , Meropol, N.J. , 2008. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 3213–3221. [DOI] [PubMed] [Google Scholar]
- Coumans, F.A. , Doggen, C.J. , Attard, G. , de Bono, J.S. , Terstappen, L.W. , 2010. All circulating EpCAM+CK+CD45- objects predict overall survival in castration-resistant prostate cancer. Ann. Oncol. 21, 1851–1857. [DOI] [PubMed] [Google Scholar]
- Cristofanilli, M. , Budd, G.T. , Ellis, M.J. , Stopeck, A. , Matera, J. , Miller, M.C. , Reuben, J.M. , Doyle, G.V. , Allard, W.J. , Terstappen, L.W. , Hayes, D.F. , 2004. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. New Engl. J. Med. 351, 781–791. [DOI] [PubMed] [Google Scholar]
- de Bono, J.S. , Scher, H.I. , Montgomery, R.B. , Parker, C. , Miller, M.C. , Tissing, H. , Doyle, G.V. , Terstappen, L.W. , Pienta, K.J. , Raghavan, D. , 2008. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 14, 6302–6309. [DOI] [PubMed] [Google Scholar]
- Dennis, G. , Sherman, B.T. , Hosack, D.A. , Yang, J. , Gao, W. , Lane, H.C. , Lempicki, R.A. , 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3 [PubMed] [Google Scholar]
- Domon-Dell, C. , Schneider, A. , Moucadel, V. , Guerin, E. , Guenot, D. , Aguillon, S. , Duluc, I. , Martin, E. , Iovanna, J. , Launay, J.F. , Duclos, B. , Chenard, M.P. , Meyer, C. , Oudet, P. , Kedinger, M. , Gaub, M.P. , Freund, J.N. , 2003. Cdx1 homeobox gene during human colon cancer progression. Oncogene 22, 7913–7921. [DOI] [PubMed] [Google Scholar]
- Eisen, M.B. , Spellman, P.T. , Brown, P.O. , Botstein, D. , 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, R.J. , Suh, E.R. , Lynch, J.P. , 2004. The role of Cdx proteins in intestinal development and cancer. Cancer Biol. Ther. 3, 593–601. [DOI] [PubMed] [Google Scholar]
- Holdhoff, M. , Schmidt, K. , Donehower, R. , Diaz, L.A. , 2009. Analysis of circulating tumor DNA to confirm somatic KRAS mutations. J. Natl. Cancer Inst. 101, 1284–1285. [DOI] [PubMed] [Google Scholar]
- Huang da, W. , Sherman, B.T. , Lempicki, R.A. , 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Iinuma, H. , Watanabe, T. , Mimori, K. , Adachi, M. , Hayashi, N. , Tamura, J. , Matsuda, K. , Fukushima, R. , Okinaga, K. , Sasako, M. , Mori, M. , 2011. Clinical significance of circulating tumor cells, including Cancer stem-like cells, in peripheral blood for recurrence and prognosis in patients with Dukes' stage B and C Colorectal Cancer. J. Clin. Oncol. 29, 1547–1555. [DOI] [PubMed] [Google Scholar]
- Imamura, Y. , Morikawa, T. , Liao, X. , Lochhead, P. , Kuchiba, A. , Yamauchi, M. , Qian, Z.R. , Nishihara, R. , Meyerhardt, J.A. , Haigis, K.M. , Fuchs, C.S. , Ogino, S. , 2012. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res. 18, 4753–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, H. , Are, C. , Gonen, M. , D'Angelica, M. , Dematteo, R.P. , Kemeny, N.E. , Fong, Y. , Blumgart, L.H. , Jarnagin, W.R. , 2008. Effect of postoperative morbidity on long-term survival after hepatic resection for metastatic colorectal cancer. Ann. Surg. 247, 994–1002. [DOI] [PubMed] [Google Scholar]
- Jiang, J. , Mostert, B. , Lalmahomed, Z. , Ramírez-Moreno, R. , Sieuwerts, A. , Kraan, J. , IJzermans, J. , Verhoef, C. , Wang, Y. , Sleijfer, S. , 2010. KRAS and BRAF Mutation Status Analysis: Primary Tumor versus Metastasis and Circulating Tumor Cells in 42 Patients with Colorectal Cancer. ASCO Molecular Markers (abstr 49) [Google Scholar]
- Joosse, S.A. , Hannemann, J. , Spotter, J. , Bauche, A. , Andreas, A. , Muller, V. , Pantel, K. , 2012. Changes in keratin expression during metastatic progression of breast cancer: impact on the detection of circulating tumor cells. Clin. Cancer Res. 18, 993–1003. [DOI] [PubMed] [Google Scholar]
- Kim, H. , Nam, S.W. , Rhee, H. , Shan Li, L. , Ju Kang, H. , Hye Koh, K. , Kyu Kim, N. , Song, J. , Tak-Bun Liu, E. , Kim, H. , 2004. Different gene expression profiles between microsatellite instability-high and microsatellite stable colorectal carcinomas. Oncogene 23, 6218–6225. [DOI] [PubMed] [Google Scholar]
- Koyanagi, K. , Bilchik, A.J. , Saha, S. , Turner, R.R. , Wiese, D. , McCarter, M. , Shen, P. , Deacon, L. , Elashoff, D. , Hoon, D.S. , 2008. Prognostic relevance of occult nodal micrometastases and circulating tumor cells in colorectal cancer in a prospective multicenter trial. Clin. Cancer Res. 14, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalmahomed, Z.S. , Kraan, J. , Gratama, J.W. , Mostert, B. , Sleijfer, S. , Verhoef, C. , 2010. Circulating tumor cells and sample size: the more, the better. J. Clin. Oncol. 28, e288–e289. (author reply e290) [DOI] [PubMed] [Google Scholar]
- Lawrie, L.C. , Dundas, S.R. , Curran, S. , Murray, G.I. , 2004. Liver fatty acid binding protein expression in colorectal neoplasia. Br. J. Cancer 90, 1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W.S. , Seo, G. , Shin, H.J. , Yun, S.H. , Yun, H. , Choi, N. , Lee, J. , Son, D. , Cho, J. , Kim, J. , Cho, Y.B. , Chun, H.K. , Lee, W.Y. , 2008. Identification of differentially expressed genes in microsatellite stable HNPCC and sporadic colon cancer. J. Surg. Res. 144, 29–35. [DOI] [PubMed] [Google Scholar]
- Lochhead, P. , Imamura, Y. , Morikawa, T. , Kuchiba, A. , Yamauchi, M. , Liao, X. , Qian, Z.R. , Nishihara, R. , Wu, K. , Meyerhardt, J.A. , Fuchs, C.S. , Ogino, S. , 2012. Insulin-like growth factor 2 messenger RNA binding protein 3 (IGF2BP3) is a marker of unfavourable prognosis in colorectal cancer. Eur. J. Cancer 48, 3405–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead, P. , Kuchiba, A. , Imamura, Y. , Liao, X. , Yamauchi, M. , Nishihara, R. , Qian, Z.R. , Morikawa, T. , Shen, J. , Meyerhardt, J.A. , Fuchs, C.S. , Ogino, S. , 2013. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J. Natl. Cancer Inst. 105, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, J. , Keller, M. , Guo, R.J. , Yang, D. , Traber, P. , 2003. Cdx1 inhibits the proliferation of human colon cancer cells by reducing cyclin D1 gene expression. Oncogene 22, 6395–6407. [DOI] [PubMed] [Google Scholar]
- Moll, R. , Lowe, A. , Laufer, J. , Franke, W.W. , 1992. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am. J. Pathol. 140, 427–447. [PMC free article] [PubMed] [Google Scholar]
- Mori, Y. , Yin, J. , Sato, F. , Sterian, A. , Simms, L.A. , Selaru, F.M. , Schulmann, K. , Xu, Y. , Olaru, A. , Wang, S. , Deacu, E. , Abraham, J.M. , Young, J. , Leggett, B.A. , Meltzer, S.J. , 2004. Identification of genes uniquely involved in frequent microsatellite instability colon carcinogenesis by expression profiling combined with epigenetic scanning. Cancer Res. 64, 2434–2438. [DOI] [PubMed] [Google Scholar]
- Mostert, B. , Kraan, J. , Bolt-de Vries, J. , van der Spoel, P. , Sieuwerts, A.M. , Schutte, M. , Timmermans, A.M. , Foekens, R. , Martens, J.W. , Gratama, J.W. , Foekens, J.A. , Sleijfer, S. , 2011. Detection of circulating tumor cells in breast cancer may improve through enrichment with anti-CD146. Breast Cancer Res. Treat. 127, 33–41. [DOI] [PubMed] [Google Scholar]
- Mostert, B. , Kraan, J. , Sieuwerts, A.M. , van der Spoel, P. , Bolt-de Vries, J. , Prager-van der Smissen, W.J.C. , Smid, M. , Timmermans, A.M. , Martens, J.W.M. , Gratama, J.W. , Foekens, J.A. , Sleijfer, S. , 2012. CD49f-based selection of circulating tumor cells (CTCs) improves detection across breast cancer subtypes. Cancer Lett. 319, (1) 49–55. [DOI] [PubMed] [Google Scholar]
- Mouradov, D. , Domingo, E. , Gibbs, P. , Jorissen, R.N. , Li, S. , Soo, P.Y. , Lipton, L. , Desai, J. , Danielsen, H.E. , Oukrif, D. , Novelli, M. , Yau, C. , Holmes, C.C. , Jones, I.T. , McLaughlin, S. , Molloy, P. , Hawkins, N.J. , Ward, R. , Midgely, R. , Kerr, D. , Tomlinson, I.P. , Sieber, O.M. , 2013. Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am. J. Gastroenterol. 108, 1785–1793. [DOI] [PubMed] [Google Scholar]
- Phipps, A.I. , Buchanan, D.D. , Makar, K.W. , Win, A.K. , Baron, J.A. , Lindor, N.M. , Potter, J.D. , Newcomb, P.A. , 2013. KRAS-mutation status in relation to colorectal cancer survival: the joint impact of correlated tumour markers. Br. J. Cancer 108, 1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethdorf, S. , Muller, V. , Zhang, L. , Rau, T. , Loibl, S. , Komor, M. , Roller, M. , Huober, J. , Fehm, T. , Schrader, I. , Hilfrich, J. , Holms, F. , Tesch, H. , Eidtmann, H. , Untch, M. , von Minckwitz, G. , Pantel, K. , 2010. Detection and HER2 expression of circulating tumor cells: prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin. Cancer Res. 16, 2634–2645. [DOI] [PubMed] [Google Scholar]
- Russo, A.L. , Borger, D.R. , Szymonifka, J. , Ryan, D.P. , Wo, J.Y. , Blaszkowsky, L.S. , Kwak, E.L. , Allen, J.N. , Wadlow, R.C. , Zhu, A.X. , Murphy, J.E. , Faris, J.E. , Dias-Santagata, D. , Haigis, K.M. , Ellisen, L.W. , Iafrate, A.J. , Hong, T.S. , 2014. Mutational analysis and clinical correlation of metastatic colorectal cancer. Cancer 120, 1482–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Pamplona, R. , Berenguer, A. , Cordero, D. , Riccadonna, S. , Sole, X. , Crous-Bou, M. , Guino, E. , Sanjuan, X. , Biondo, S. , Soriano, A. , Jurman, G. , Capella, G. , Furlanello, C. , Moreno, V. , 2012. Clinical value of prognosis gene expression signatures in colorectal cancer: a systematic review. PLoS One 7, e48877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieuwerts, A.M. , Kraan, J. , Bolt-de Vries, J. , van der Spoel, P. , Mostert, B. , Martens, J.W. , Gratama, J.W. , Sleijfer, S. , Foekens, J.A. , 2009. Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex real-time PCR. Breast Cancer Res. Treat. 118, 455–468. [DOI] [PubMed] [Google Scholar]
- Sieuwerts, A.M. , Kraan, J. , Bolt-de Vries, J. , van der Spoel, P. , Mostert, B. , Martens, J.W.M. , Gratama, J.W. , Sleijfer, S. , Foekens, J.A. , 2009. Molecular characterization of circulating tumor cells in large quantities of contaminating leukocytes by a multiplex real-time PCR. Breast Cancer Res. Treat. 118, 455–468. [DOI] [PubMed] [Google Scholar]
- Sieuwerts, A.M. , Mostert, B. , Bolt-de Vries, J. , Peeters, D. , de Jongh, F.E. , Stouthard, J.M. , Dirix, L.Y. , van Dam, P.A. , Van Galen, A. , de Weerd, V. , Kraan, J. , van der Spoel, P. , Ramirez-Moreno, R. , van Deurzen, C.H. , Smid, M. , Yu, J.X. , Jiang, J. , Wang, Y. , Gratama, J.W. , Sleijfer, S. , Foekens, J.A. , Martens, J.W. , 2011. mRNA and microRNA expression profiles in circulating tumor cells and primary tumors of metastatic breast Cancer patients. Clin. Cancer Res. 17, 3600–3618. [DOI] [PubMed] [Google Scholar]
- Smirnov, D.A. , Zweitzig, D.R. , Foulk, B.W. , Miller, M.C. , Doyle, G.V. , Pienta, K.J. , Meropol, N.J. , Weiner, L.M. , Cohen, S.J. , Moreno, J.G. , Connelly, M.C. , Terstappen, L.W. , O'Hara, S.M. , 2005. Global gene expression profiling of circulating tumor cells. Cancer Res. 65, 4993–4997. [DOI] [PubMed] [Google Scholar]
- Su, M.C. , Yuan, R.H. , Lin, C.Y. , Jeng, Y.M. , 2008. Cadherin-17 is a useful diagnostic marker for adenocarcinomas of the digestive system. Mod. Pathol. 21, 1379–1386. [DOI] [PubMed] [Google Scholar]
- Taylor, I. , 1996. Liver metastases from colorectal cancer: lessons from past and present clinical studies. Br. J. Surg. 83, 456–460. [DOI] [PubMed] [Google Scholar]
- Terrin, L. , Rampazzo, E. , Pucciarelli, S. , Agostini, M. , Bertorelle, R. , Esposito, G. , DelBianco, P. , Nitti, D. , De Rossi, A. , 2008. Relationship between tumor and plasma levels of hTERT mRNA in patients with colorectal cancer: implications for monitoring of neoplastic disease. Clin. Cancer Res. 14, 7444–7451. [DOI] [PubMed] [Google Scholar]
- van de Stolpe, A. , Pantel, K. , Sleijfer, S. , Terstappen, L.W. , den Toonder, J.M. , 2011. Circulating tumor cell isolation and diagnostics: toward routine clinical use. Cancer Res. 71, 5955–5960. [DOI] [PubMed] [Google Scholar]
- van der Pool, A.E. , de Wilt, J.H. , Lalmahomed, Z.S. , Eggermont, A.M. , Ijzermans, J.N. , Verhoef, C. , 2010. Optimizing the outcome of surgery in patients with rectal cancer and synchronous liver metastases. Br. J. Surg. 97, 383–390. [DOI] [PubMed] [Google Scholar]
- Verhoef, C. , van der Pool, A.E. , Nuyttens, J.J. , Planting, A.S. , Eggermont, A.M. , de Wilt, J.H. , 2009. The “liver-first approach” for patients with locally advanced rectal cancer and synchronous liver metastases. Dis. Colon Rectum 52, 23–30. [DOI] [PubMed] [Google Scholar]
- Vlems, F.A. , Diepstra, J.H.S. , Punt, C.J.A. , Ligtenberg, M.J.L. , Cornelissen, I.M.H.A. , van Krieken, J.H.J.M. , Wobbes, T. , van Muijen, G.N.P. , Ruers, T.J.M. , 2003. Detection of disseminated tumour cells in blood and bone marrow samples of patients undergoing hepatic resection for metastasis of colorectal cancer. Br. J. Surg. 90, 989–995. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Jatkoe, T. , Zhang, Y. , Mutch, M.G. , Talantov, D. , Jiang, J. , McLeod, H.L. , Atkins, D. , 2004. Gene expression profiles and molecular markers to predict recurrence of Dukes' B colon cancer. J. Clin. Oncol. 22, 1564–1571. [DOI] [PubMed] [Google Scholar]
- Xi, L. , Nicastri, D.G. , El-Hefnawy, T. , Hughes, S.J. , Luketich, J.D. , Godfrey, T.E. , 2007. Optimal markers for real-time quantitative reverse transcription PCR detection of circulating tumor cells from melanoma, breast, colon, esophageal, head and neck, and lung cancers. Clin. Chem. 53, 1206–1215. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T. , Kanda, T. , Sakai, Y. , Hatakeyama, K. , 1999. Liver fatty acid-binding protein is a new prognostic factor for hepatic resection of colorectal cancer metastases. J. Surg. Oncol. 72, 83–87. [DOI] [PubMed] [Google Scholar]
- Yie, S.M. , Lou, B. , Ye, S.R. , Cao, M. , He, X. , Li, P. , Hu, K. , Rao, L. , Wu, S.M. , Xiao, H.B. , Gao, E. , 2008. Detection of survivin-expressing circulating cancer cells (CCCs) in peripheral blood of patients with gastric and colorectal cancer reveals high risks of relapse. Ann. Surg. Oncol. 15, 3073–3082. [DOI] [PubMed] [Google Scholar]
- Zakaria, S. , Donohue, J.H. , Que, F.G. , Farnell, M.B. , Schleck, C.D. , Ilstrup, D.M. , Nagorney, D.M. , 2007. Hepatic resection for colorectal metastases: value for risk scoring systems?. Ann. Surg. 246, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary Table 1 Specifics of the Taqman assays used for detection of mRNA transcripts. Official gene symbol, reason for inclusion in our gene expression panel, Assay ID and Gene description of 95 mRNAs included in initial selection for measurement in CTCs. CTCM, well‐known CTC marker; DT, drug target; EM, epithelial marker (as discovered in our previous breast cancer studies (Sieuwerts et al., 2011)); ENM, endothelial marker; LM, leukocyte marker; REF, reference gene marker; SCM, stem cell marker.
Supplementary Table 2 Top 75% variably expressed between HDs and patients without detectable CTCs (a) and between HDs and patients with ≥3 CTCs (b). Panel a: The 34 mRNAs, which were at a Permutation P‐value <0.05 (two‐sample t‐test) differentially expressed between HDs and patients with ≥3 CTCs are depicted. All mRNAs were higher expressed in patients than in HDs. For reference purposes, the parametric P‐value and false discovery rate (FDR) are also given. Panel b: The 69 mRNAs, which were the top 75% variably, expressed genes between 30 HDs and 33 patients without detectable CTCs are depicted.
Supplementary Figure 2 Stability and detectability of RNA in CellSave preservative versus EDTA blood at different time points. Ct‐values of reference genes and percentages of 93 target genes that were detectable of MCF7 breast cancer cell line cells by RTq‐PCR. Both spiked and unspiked samples were analyzed, at 0, 24, 48, 72 and 96 hours after blood draw, and compared between EDTA versus CellSave preservative tubes.
Supplementary Figure 1 Clustering of patients (pat) from the primary set with 0–1 enumerated CTC and healthy donors (HDs). Unsupervised hierarchical clustering analysis comparing mRNA gene expression profiles in mCRC CTC‐enriched fractions from 66 patients with 0–1 detectable CTC and QC mRNA available and 30 HDs. Data shown have been subjected to median normalization of each individual sample across all genes followed by median normalization of each individual gene across all samples. Columns represent patient samples, rows represent genes. Red color indicates a transcript level above the median level, white color indicates a median transcript level, and blue color indicates a transcript level below the median level of the particular mRNA in all samples. Depicted gene clusters were identified at an average linkage correlation greater than 0.2.