

Abstract
Resistance to paclitaxel chemotherapy frequently develops in ovarian cancer. Oncolytic adenoviruses are a novel therapy for human malignancies that are being evaluated in early phase trials. However, there are no reliable predictive biomarkers for oncolytic adenovirus activity in ovarian cancer. We investigated the link between paclitaxel resistance and oncolytic adenovirus activity using established ovarian cancer cell line models, xenografts with de novo paclitaxel resistance and tumour samples from two separate trials. The activity of multiple Ad5 vectors, including dl922‐947 (E1A CR2‐deleted), dl1520 (E1B‐55K deleted) and Ad5 WT, was significantly increased in paclitaxel resistant ovarian cancer in vitro and in vivo. This was associated with greater infectivity resulting from increased expression of the primary receptor for Ad5, CAR (coxsackie adenovirus receptor). This, in turn, resulted from increased CAR transcription secondary to histone modification in resistant cells. There was increased CAR expression in intraperitoneal tumours with de novo paclitaxel resistance and in tumours from patients with clinical resistance to paclitaxel. Increased CAR expression did not cause paclitaxel resistance, but did increase inflammatory cytokine expression. Finally, we identified dysregulated cell cycle control as a second mechanism of increased adenovirus efficacy in paclitaxel‐resistant ovarian cancer. Ad11 and Ad35, both group B adenoviruses that utilise non‐CAR receptors to infect cells, are also significantly more effective in paclitaxel‐resistant ovarian cell models. Inhibition of CDK4/6 using PD‐0332991 was able both to reverse paclitaxel resistance and reduce adenovirus efficacy. Thus, paclitaxel resistance increases oncolytic adenovirus efficacy via at least two separate mechanisms – if validated further, this information could have future clinical utility to aid patient selection for clinical trials.
Keywords: Ovarian cancer, Paclitaxel resistance, Adenovirus, Coxsackie adenovirus receptor, Cell cycle control
Highlights
Oncolytic adenoviruses are more effective in paclitaxel‐resistant ovarian cancer, via multiple mechanisms.
Paclitaxel increases expression of coxsackie adenovirus receptor (CAR) through altered histone modification.
Increased CAR expression can increase inflammatory cytokine expression.
Paclitaxel resistance also increases adenovirus activity via dysregulated cell cycle control.
1. Introduction
The microtubule stabilising agent paclitaxel is widely used in ovarian cancer management, both in combination with platinum (du Bois et al., 2003) and as a single agent (Karlan et al., 2011). Multiple mechanisms underpin paclitaxel resistance, including overexpression of ATP‐dependent efflux pumps (Brooks et al., 2003), alterations of tubulin isoforms and microtubule associated proteins (MAPs) (Mozzetti et al., 2005), aberrant expression of apoptosis regulating proteins (Youle and Strasser, 2008), epigenetic modulation of genes such as polo‐like kinase 2 (Syed et al., 2011), overexpression of chromosome instability genes (Swanton et al., 2009), loss of the extracellular matrix protein TGFBI (Ahmed et al., 2007) and expression of focal adhesion kinase (FAK) (Kang et al., 2013).
Oncolytic viruses are promising new cancer treatments. E1A CR2‐deleted adenovirus type 5 vectors, such as dl922‐947 (Heise et al., 2000) and Δ24 (Fueyo et al., 2000), selectively replicate within and kill cells with defective pRb/G1‐S cell cycle checkpoint, a near‐universal abnormality in human malignancies, including ovarian cancers (Sherr and McCormick, 2002; TCGA, 2011). We have previously shown that dl922‐947 has activity in ovarian cancer (Lockley et al., 2006). It can abrogate multiple cell cycle checkpoints (Connell et al., 2008) and its efficacy is partially determined by genomic DNA damage and ATR‐Chk1 signalling (Connell et al., 2011). Clinical trials of E1A CR2‐deleted Ad5 vectors have taken place (Kimball et al., 2010), and we recently opened a phase I trial of the group B virus ColoAd1 (Kuhn et al., 2008) (ClinicalTrials.gov reference NCT02028117).
The main route for cell entry for most adenoviruses (including Ad5) is by primary binding to the coxsackie adenovirus receptor (CAR),1 followed by a secondary interaction with αvβ3‐or αvβ5‐integrins, leading to viral internalisation. For group B adenovirus, the primary receptors are CD46 (Ad16, 11, 21, 35) and/or desmoglein‐2 (Ad3, 7 and 14). Internalisation is followed by viral nuclear translocation in a microtubule‐dependent manner (Suomalainen et al., 1999). Paclitaxel can synergise with oncolytic adenoviruses (Cheong et al., 2008) and we recently showed that mitotic slippage may also play a role in the synergy seen between dl922‐947 and paclitaxel in ovarian cancer (Ingemarsdotter et al., 2010).
Here, we observed that oncolytic adenovirus activity is markedly increased in two separate paclitaxel‐resistant ovarian cancer models. Using tumour samples from two ovarian cancer clinical trials as well as in vitro and in vivo models, we demonstrate that there is increased expression of CAR in paclitaxel‐resistant ovarian cancer, allowing greater virus infectivity. In addition, cell cycle abnormalities in paclitaxel‐resistant ovarian cancers contribute to greater efficacy of multiple adenovirus serotypes, including Ad5, Ad11 and Ad35. Our data suggest paclitaxel resistance in ovarian cancer increases responsiveness to oncolytic adenovirus therapy through multiple mechanisms.
2. Methods
2.1. Cell lines, viruses and chemicals
A2780 cells were originally obtained from Dr A. Eliopoulos (University of Birmingham, UK) in 1998 (Eliopoulos et al., 1995). A2780tx1000 cells were generated by pulse‐treating A2780 with increasing concentrations of paclitaxel. SKOV3 and SKOV3‐TR (Duan et al., 1999) cells were obtained from Dr James Brenton (CRUK Cambridge Institute, Cambridge, UK) in 2010. Cell lines underwent 16 locus STR validation (LGC Standards, Middlesex UK) in 2011 and were most recently revalidated by 10 locus STR validation in June 2014 (CRUK Beatson Institute, Glasgow, UK; Supplementary Table S1). A2780 and A2780tx1000 cells were cultured in DMEM plus penicillin/streptomycin and 10% FCS, (PAA Laboratories, Austria). SKOV3 and SKOV3‐TR cells were cultured in RPMI plus penicillin/streptomycin and 10% FCS. A2780tx1000 and SKOV3‐TR were grown in medium supplemented with 1 μM and 300 nM paclitaxel, respectively. In all experiments, cells were released from paclitaxel at least four days prior to cell plating.
dl922‐947 is an Ad5 vector deleted in amino acids 122–129 of E1A‐CR2 as well as E3B (Heise et al., 2000). dlCR2‐pIX‐dsRed is an E1A‐CR2‐deleted Ad5 vector in which the minor capsid protein pIX is fused to dsRed as previously described (Ingemarsdotter et al., 2010). Wildtype Ad5, Ad11, Ad35 and dl1520 were all obtained from Dr Yaohe Wang, Barts Cancer Institute. ON‐TARGETplus Smartpool siRNA oligos were purchased from ThermoScientific (Loughborough, UK) with ON‐TARGETplus Control Pool as scrambled control. Cell survival was assessed by MTT assay (Lockley et al., 2006). Ad LM‐X is an E1‐deleted non‐replicating Ad5 vector containing no transgene as previously described (McNeish et al., 2001).
2.2. GFP transduction assays and flow cytometry
Cells were infected with E1‐deleted Ad5‐CMV‐GFP virus, and analysed for GFP expression 24 h post‐infection by flow cytometry (BD FACSCalibur, BectonDickinson, Oxford, UK). GFP fluorescence intensity was analysed on a Victor3 multilabel plate reader (Perkin Elmer, Cambridge, UK). Primary antibodies for flow cytometry are listed in Supplementary Methods. CAR expression was assessed in non‐permeabilised cells.
2.3. Immunofluorescence
Cells were grown on poly‐l‐lysine coated coverslips and fixed in 95% ethanol/5% acetic acid for 10 min at room temperature followed by four washes in PBS. Cells were permeabilised in 0.5% Triton‐X100 in PBS for 5 min except for Figure 2E, where the permeabilisation step was abolished. blocking was performed in 3% BSA in PBS for 30 min at room temperature. Cells were washed once in PBS followed by incubation with primary antibodies for 1 h at room‐temperature in 3% BSA in PBS‐Tween (0.2% Tween‐20 in PBS), washed four times in PBS‐Tween and incubated with secondary antibodies for 1 h at room‐temperature. Coverslips were washed followed by staining with DAPI (Invitrogen) for 1 min. Cells were then washed twice in PBS and mounted onto glass slides.
Figure 2.
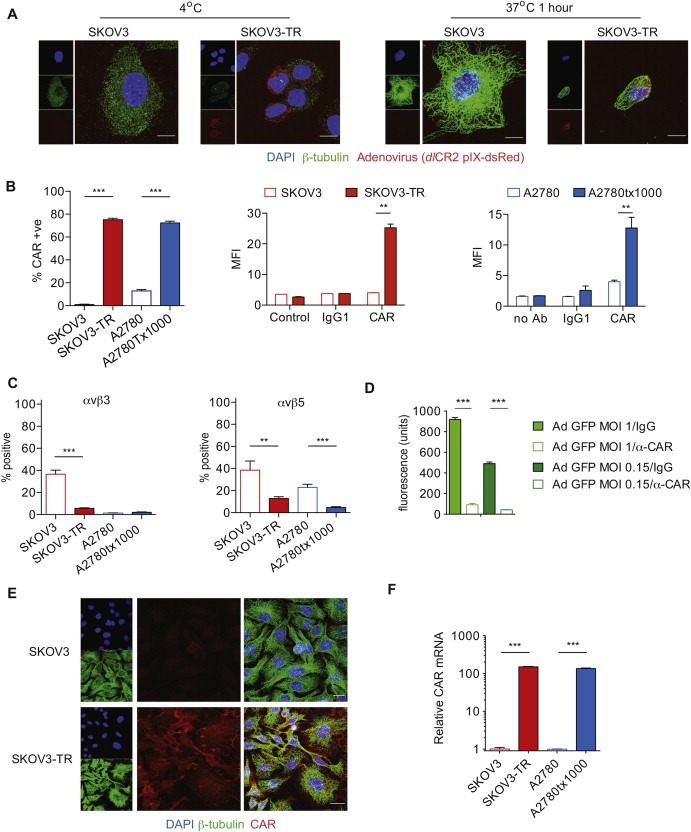
Coxsackie adenovirus receptor expression. A. SKOV3 and SKOV3‐TR cells were infected with dlCR2‐pIX‐dsRed on ice. Cells were warmed to 37 °C for 1 h, stained with β‐tubulin, DAPI and subjected to confocal analysis. Scale bar = 10 μm. B. CAR expression was assessed by flow cytometry; percentage positive cells (left) and mean fluorescence intensity (right). ***; p < 0.001, **; p < 0.01. C. Expression of αvβ3 and αvβ5 integrin. **; p < 0.01. ***; p < 0.001. D. SKOV3‐TR cells were infected with Ad CMV‐GFP (MOI 1 and 0.15) in the presence of anti‐CAR blocking Ab (1:25) or control IgG (1:25). GFP fluorescence was assessed 24 h later using a multilabel plate reader. ***; p < 0.0001. E. Localisation of CAR was assessed by confocal microscopy. F. CXADR transcription in both cell pairs, relative to 18S. ***; p < 0.001.
2.4. Viral internalisation assay
5 × 105 cells were seeded overnight in 6 cm dishes, washed in cold 1% BSA in PBS, and then infected with dl922‐947 (5000 virus particles/cell) in 1% BSA in PBS. Unbound viral particles were removed by washing in cold 1% BSA in PBS. Infection then continued for 1 h at 37 °C. DNA was extracted with the QIAamp DNA Blood Mini kit (Qiagen, Manchester, UK). The number of internalised viral genomes was determined by qPCR (Applied Biosystems 7500 system). Primers and probes and conditions are listed in Supplementary Methods. Absolute genome number was quantified using a standard curve of viral DNA equivalent to 104–109 genomes.
2.5. RNA extraction, reverse transcription and quantitative PCR
Cells were lysed with Trizol (Invitrogen, Paisley, UK) as per manufacturer's instructions. Samples were mixed, incubated for 5 min at room temperature and centrifuged for 15 min, 14000 rpm at 4 °C. The RNA was precipitated with 0.7× volumes of isopropanol at −20 °C overnight. The RNA was pelleted by centrifugation as above and resuspended in 70% ethanol in DEPC‐H20 followed by another centrifugation step. The supernatant was discarded and the pellet dried and dissolved in RNase‐free H2O. The isolated RNA was cleaned up with the RNeasy Mini Kit (Qiagen), according to the manufacturer's instructions. 1 μg of RNA was reverse transcribed using the First Strand cDNA synthesis kit for RT‐PCR (Roche). Expression was normalised to 18S (Applied Biosciences, UK).
2.6. Microscopy
Confocal analysis was performed with an inverted Zeiss LSM 510 META laser‐scanning microscope with a Plan‐Apochromat 63×/1.4 Oil objective. Images were acquired in the x,y,z direction, with a line average of 4. Z‐sections were acquired at optimum interval levels with sections of 0.43 μm. Maximal intensity Z‐projections were assembled with the LSM5 Image browser software.
2.7. Viral binding and trafficking assay
1 × 105 cells were seeded overnight on poly‐l‐lysine coated 24 well plates, washed in cold phenol red‐free medium, and infected with dlCR2‐pIX‐dsRed (10000vp/cell) on ice for 1 h. Unbound viral particles were removed by washing. Cells were fixed for immunofluorescence (0 h timepoint), or the infection was continued for 1 h at 37 °C. Cells were then washed, fixed and stained.
2.8. Virion production and viral exit assay
Following infection, adherent cells were washed twice in 0.1 M Tris pH8, scraped and resuspended in 0.1 M Tris pH8 followed by three rounds of freeze thawing (liquid N2/37 °C). Virus was titered on JH293 cells. For the viral exit assay, cells were washed twice in PBS and refed 24 h post‐infection. Medium was collected 48 h post‐infection, centrifuged for 5 min at 4000 rpm and titered on JH293 cells.
2.9. Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed with a LowCell#ChIP kit (Diagenode, Liège, Belgium) as detailed in Supplementary Methods.
2.10. vivo models
All experiments complied with the National Cancer Research Institute guidelines for the welfare and use of animals in cancer research (Workman et al., 2010) and were conducted after specific UK government Home Office personal (reference 70/19481) and project (reference 70/7263) licence approval. Experiments were approved locally by the ethics review committee of Queen Mary University of London Biological Services Unit (reference PAC60‐3). 5 × 106 cells were inoculated IP in 6‐week old ICRF nu/nu female mice. For virus efficacy, 5 × 109 particles dl922‐947 or control virus Ad LM‐X (McNeish et al., 2001) were injected daily for 5 days in 400 μl PBS. For generation of paclitaxel‐resistant tumours, mice were treated with intraperitoneal paclitaxel (10 mg/kg) or PBS weekly for up to 14 weeks.
2.11. Clinical trial samples
The SaPPrOC trial (Clinicaltrials.gov reference NCT01196741) was a randomised phase II study of weekly paclitaxel ± oral saracatinib in women with relapsed, platinum‐resistant ovarian cancer (McNeish et al., 2014). The study was approved by the UK National Research Ethics Committee – West Midlands (Coventry and Warwickshire) reference 10/H1211/26. A tissue microarray was created with duplicate 5 mm cores from formalin‐fixed paraffin‐embedded tumour material collected at the time of diagnosis. 5 μm sections were stained for CAR, and scored for intensity of staining (scores 0, 1, 2, 3) in tumour cells by two readers (IAMcN, DE) who were blind to the clinical response data. Final score for each patient was the average of scores of the two readers. Best clinical responses were determined by combined RECIST/GCIG CA125 criteria (Rustin et al., 2004). Details of the Cambridge Translational Cancer Research Ovary 01 study (UKCRN reference 1206 http://public.ukcrn.org.uk/search/StudyDetail.aspx?StudyID=1206) have been described previously (Ahmed et al., 2007).
2.12. Gene expression and bioinformatics analyses
Total RNA from SKOV3 and SKOV3‐TR cells was extracted using RNeasy kit (Qiagen) in combination with the QIAshredder kit (Qiagen) for cell homogenization. Total RNA quality was assessed using an Agilent 2100 Bioanalyser prior to sample preparation, starting with 100 ng of total RNA and 16 h IVT as per 3′ IVT Express protocol (Affymetrix). Samples were hybridized to GeneChip Human Genome U133 Plus 2.0 Arrays. Bioinformatics analyses are described in Supplementary Methods. Raw SKOV3/SKOV3‐TR gene expression data are deposited at GEO, accession number GSE54772. CTCR OV01 gene expression data were previously deposited at GEO, accession numbers GSE9455 and GSE15622. Previous SKOV3/SKOV3‐TR gene expression data (acquired using a DMF Human 6.5K array) were also previously deposited at GEO, accession number GSE2627.
2.13. Statistics
All other statistical analyses were generated with Prism 6.0, (GraphPad, San Diego, CA). Unless otherwise stated, statistical analyses are unpaired, two‐tailed, student's t‐test.
3. Results
3.1. Increased adenovirus efficacy in paclitaxel‐resistant ovarian cancer cells
The paclitaxel‐resistant ovarian cancer cell line, SKOV3‐TR, which is at least 100‐fold more resistant to paclitaxel than parental SKOV3 cells, is highly sensitive to the E1A CR2‐deleted oncolytic adenovirus type 5 vector dl922‐947 (Figure 1A). We also evaluated a second resistant ovarian cancer cell line generated in our lab, A2780tx1000; again, the paclitaxel‐resistant cells showed a marked increase in dl922‐947‐induced cytotoxicity (Figure 1B). Both resistant lines were also highly sensitive to other Ad5‐based vectors, including Ad5 WT and dl1520 (Figure S1). This in vitro sensitivity was mirrored in vivo: mice bearing SKOV3‐TR intraperitoneal xenografts survived long term following intraperitoneal (IP) dl922‐947 treatment (Figure 1C). Of note, there was no difference in growth rate between parental and paclitaxel‐resistant cells in vitro (Figure S2) or following control treatment in vivo (Figure 1C).
Figure 1.
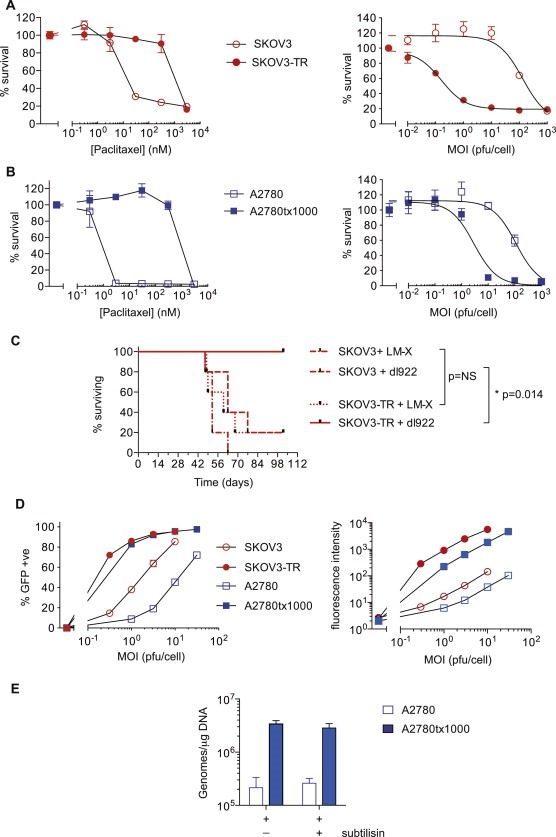
Paclitaxel‐resistant cells are more sensitive to dl922‐947‐induced cytotoxicity with greater infectivity. A. SKOV3 vs SKOV3‐TR and B. A2780 vs A2780tx1000 cells were treated with paclitaxel for 96 h (left) or infected with E1A CR2‐deleted Ad5 vector dl922‐947 for 144 h (right). Cell survival was analysed by MTT assay. C. 5 × 106 SKOV3 or SKOV3‐TR cells were injected IP into female ICRF nu/nu mice in groups of 5. On days 4–8 inclusive, dl922‐947 or control vector Ad LM‐X was injected IP (3 × 109 particles daily). D. Cells were infected in triplicate with increasing concentrations of Ad5 CMV‐GFP and infectivity (left) was analysed by flow cytometry. Mean fluorescence intensity (right) was analysed on a multilabel plate reader. E. Viral internalisation was assessed 1 h following infection with dl922‐947 (5000vp/cell). Cells were also treated with subtilisin (2 mg/ml). Viral genome copy number per μg DNA is shown. (**p < 0.01 and ***p < 0.001).
3.2. Increased Ad5 infectivity in paclitaxel‐resistant ovarian cancer cells
GFP assays indicated a marked increase in transduction of both paclitaxel‐resistant cell lines, with marked increases in both number of infected cells and mean fluorescence intensity (Figure 1D). This was confirmed on quantitative internalisation assay, with at least a one‐log increase in genome copy number/μg DNA (Figure 1E; data not shown for SKOV3‐TR). Subtilisin treatment indicated that virus was truly internalised rather than simply adhering to the cell surface.
Using the fluorescent capsid‐labelled adenovirus dlCR2‐pIX‐dsRed (Ingemarsdotter et al., 2010), many more viral particles were detected at the cell surface in SKOV3‐TR cells after infection at 4 °C compared with SKOV3 cells. After 1 h at 37 °C, dlCR2‐pIX‐dsRed could be detected in the perinuclear area in SKOV3‐TR cells, with only single focus in SKOV3 cells (Figure 2A). These data confirm that more virus is bound to the cell surface in SKOV3‐TR cells than SKOV3, allowing greater internalisation and nuclear translocation.
3.3. Paclitaxel‐resistant cells express high levels of coxsackie adenovirus receptor (CAR)
By flow cytometry (Figure 2B, Figure S3), both paclitaxel‐resistant lines expressed significantly more CAR than parental cells, and expression remained stable even after 80 days growth in paclitaxel‐free medium (Figure S4). By contrast, adenovirus secondary receptor αvβ3 integrin was expressed at lower levels on SKOV3‐TR cells compared with SKOV3 cells and was undetectable in both A2780 lines (Figure 2C), whilst αvβ5 expression was reduced on both resistant lines. Treatment with a CAR blocking Ab significantly reduced infection (Figure 2D). CAR was distributed in the periphery of SKOV3‐TR cells with areas of intense expression at cell:cell junctions and some co‐localisation with β‐tubulin (Figure 2E). Quantitative RT‐PCR (Figure 2F) revealed that CAR mRNA levels increased over 100 fold in both resistant lines compared with their parental counterparts. Thus, CAR is transcriptionally upregulated in paclitaxel‐resistant ovarian cancer cells, and expressed on the cell surface.
To understand the mechanisms of CAR transcriptional upregulation, we undertook methylation‐specific PCR and bisulphite sequencing of CpG islands around the CXADR promoter: these showed no consistent change (data not shown). However, chromatin immunoprecipitation using antibodies specific for trimethylated Lysine 4 (H3K4me3) and acetylated Lysine 27 (H3K27ac) on histone H3, both of which are associated with increased transcription, indicated significant changes around the CXADR promoter (Figure 3A). This finding was reinforced by treatment with the histone deacetylase inhibitor TSA, which significantly increased CAR transcript levels in both parental cell lines (Figure 3B). CAR expression varies throughout the cell cycle, peaking in M phase (Seidman et al., 2001). However, short‐term exposure of both parental cell lines to nocodazole and paclitaxel at concentrations sufficient to induce G2/M arrest (Figure S5) had a minimal effect on CAR expression cells (Figure 3C). Overall, these data suggest that CAR transcript levels are increased in paclitaxel‐resistant ovarian cancer, likely secondary to epigenetic modification, specifically changes in histone acetylation and methylation.
Figure 3.
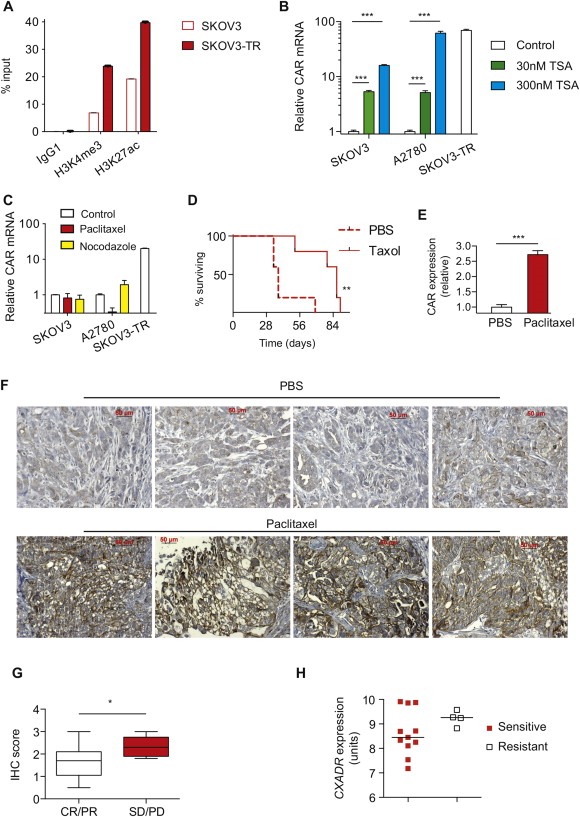
Increased CXADR transcript levels results from histone modification; increased expression in paclitaxel‐resistant ovarian cancer in mice and humans. A. ChIP was performed with sheared chromatin with antibodies against H3K4me3 or H3K27ac. Percentage input was calculated as per Supplementary Methods. B. SKOV3 and A2780 cells were treated with 30–300 nM TSA for 24 h. CXADR transcript number was assessed by qRT‐PCR. ***; p < 0.001. C. SKOV3 and A2780 cells were also treated with 30 nM paclitaxel or 300 nM nocodazole for 24 h to induce G2/M arrest. CXADR transcription was assessed by qRT‐PCR. D. Female ICRF nu/nu mice bearing intraperitoneal SKOV3 xenografts were treated with weekly intraperitoneal PBS or paclitaxel (10 mg/kg). **; p < 0.01. E. CXADR transcript level was quantified in tumours excised from PBS‐ and paclitaxel‐treated mice, relative to 18S. ***; p < 0.001. F. Expression of CAR was assessed by IHC in 5 μm sections. Bars represent 50 μm. G. Expression of CAR in duplicate cores from archival tumour samples from patients in the SaPPrOC trial of weekly paclitaxel chemotherapy was assessed and correlated against best clinical response. *; p < 0.05. H. CXADR transcription in tumours samples from CTCR OV01 trial was correlated with best clinical outcome.
3.4. CAR expression increases in paclitaxel‐resistant ovarian xenografts and human tumour samples
To assess whether the changes seen in cell lines were reproduced in vivo, we firstly generated de novo paclitaxel‐resistant tumours by treating mice bearing intraperitoneal parental SKOV3 xenografts with weekly doses of paclitaxel (10 mg/kg) or PBS. Despite the therapeutic effect of this regime (Figure 3D), all mice eventually developed solid intra‐abdominal tumour nodules. There was no ascites in these mice, so assessment of CAR expression by flow cytometry in ascites cells was not possible. However, we found a 2.7‐fold increase in CAR transcript levels in excised paclitaxel‐resistant tumours (Figure 3E; p < 0.0001), with a demonstrable increase in expression by IHC (Figure 3F).
To assess the clinical relevance of our findings, we evaluated CAR expression by IHC in tumours from women enrolled in the SaPPrOC trial of weekly paclitaxel chemotherapy (McNeish et al., 2014) – even though samples were taken at the time of diagnosis, there was a significantly higher IHC score in resistant tumours (best response of Progressive Disease or Stable Disease) compared to sensitive (best response of Partial or Complete response; Figure 3G and Figure S6). We also analysed the publicly available tumour gene expression data from the cohort of patients with advanced ovarian cancer who received first‐line single agent paclitaxel chemotherapy in CTCR OV01 trial (Ahmed et al., 2007). Data from 15 tumour samples passed initial quality control: the median level of CXADR transcript in those who were resistant to paclitaxel (n = 4) was higher than in those who were sensitive (n = 11) (Figure 3H), although the trend did not reach statistical significance (t test: p = 0.10).
3.5. CAR overexpression does not cause paclitaxel resistance but increases inflammatory cytokine production
One intriguing question was whether the increased expression of CAR seen in cell lines, xenografts and tumour samples was a direct cause of resistance to paclitaxel or simply an epiphenomenon. Expression of full‐length or truncation CAR constructs lacking various parts of the CAR intracytoplasmic tail (Farmer et al., 2009) increased adenovirus infectivity and efficacy as expected (Figure 4A and data not shown) but had no effect on paclitaxel sensitivity in either SKOV3 or A2780 cells (Figure 4A and B). Knockdown of CAR had only modest effects on paclitaxel resistance in SKOV3‐TR cells and none in A2780tx1000 cells (Figure 4B). In two other ovarian cancer cell lines that express CAR at high levels, CAR siRNA also generated only minor alterations in paclitaxel sensitivity (Figure 4C and D). When resistant cells are cultured out of paclitaxel for over 80 days, there was some loss of drug resistance (Figure 4E), but CAR expression remained stable (Figure S4). These data together suggest that CAR expression does not directly cause paclitaxel resistance in our cells.
Figure 4.
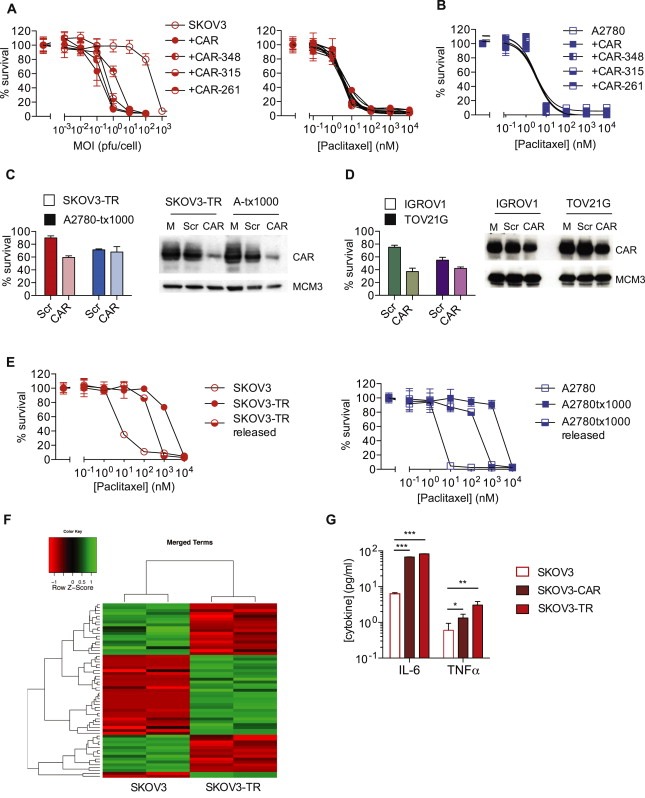
CAR expression increases inflammatory cytokine production but does not induce paclitaxel resistance. A. SKOV3 cells expressing full expressing full length CAR and CAR truncation mutants were infected with dl922‐947 (left) or treated with paclitaxel (right). Cell survival was assessed after 120 h (dl922‐947) and 96 h (paclitaxel). B. A2780 cells expressing full length CAR and CAR truncation mutants were treated with paclitaxel. Cell survival was assessed after 96 h. C. Twenty‐four hours following siRNA‐mediated CAR knockdown A2780tx1000, SKOV3‐TR, and D. IGROV1 and TOV21G cells were treated with paclitaxel (100 nM for A2780tx1000 and SKOV3‐TR, 1 nM for IGROV1 and TOV21G) for 96 h. Cell survival was assessed by MTT assay. E. SKOV3‐TR and A2780‐tx1000 were grown for 80 days in paclitaxel‐free medium. 96 h paclitaxel dose response curves were performed as previously. F. Gene expression in SKOV3 and SKOV3‐TR cells was assessed using GeneChip Human Genome U133 Plus 2.0 Arrays. Differential gene expression analysis was carried out using the O‐miner web tool and genes differentially expressed between the two cell lines were identified using limma. Using the GOStats package in Bioconductor, differential expression was assessed according to the GO terms ‘Inflammatory Response’, ‘Cytokine Activity’ and ‘Chemokine Activity’. G. Release of IL‐6 and TNF‐α from SKOV3, SKOV3‐CAR and SKOV3‐TR cells was assessed using Mesoscale MSD Discovery platform. *; p < 0.05. **; p < 0.01. ***; p < 0.001.
However, we hypothesised that CAR upregulation may contribute to the expression of inflammatory cytokines seen in paclitaxel‐resistance (Duan et al., 2002; Wang et al., 2010). CAR expression can induce an inflammatory cardiomyopathy in mice, with expression of cytokines that include interleukin‐6 (IL‐6) and TNF‐α (Yuen et al., 2011). We undertook gene expression profiling of SKOV3 and SKOV3‐TR cells, which confirmed increased CXADR (CAR) transcript levels (Log2FC 3.349, p = 0.001) as well as alterations in other genes previously implicated in paclitaxel resistance, including TGFBI (Log2FC −9.509, p = 2.610 × 10−5), ABCB1 (Log2FC 10.669, p = 1.233 × 10−6) and β‐tubulin (TUBB2B; Log2FC 3.708, p = 0.002). In addition, there was highly significant upregulation of both cytokine and chemokine activity (Figure 4F and Supplementary Table S2). Confirming a potential role for CAR in this cytokine activity, expression of full length CAR alone in SKOV3 cells significantly increased both IL‐6 and TNF‐α production (Figure 4G).
3.6. Aberrant cell cycle control in paclitaxel‐resistant cells also improves adenovirus efficacy
To test if increased infectivity via increased CAR expression was the only explanation for increased adenovirus efficacy in paclitaxel‐resistant cells, we iso‐infected cells with dl922‐947 ‐ that is, we altered the MOI to ensure that all cells were infected equally (Figure 5A). Iso‐infection was defined by the number of GFP positive cells, using consistent thresholds, rather than mean GFP fluorescence in order to compensate for any potential differences in GFP intensity between cells. There was significantly more cell death in both paclitaxel‐resistant lines (Figure 5B). Furthermore, both resistant cells showed increased sensitivity to Ad11 and Ad35, which do not use CAR as their primary receptor (Figure 5C). The increased sensitivity to Ad11 and Ad35 did not result from greater infectivity; there was no significant difference in Ad11 or Ad35 internalisation in resistant cells compared to parental (Figure S7) and no increase in expression of group B adenovirus receptors CD46 or desmoglein‐2 (Figure S8). Thus, paclitaxel resistance was able to increase viral efficacy through mechanisms beyond simple infectivity.
Figure 5.
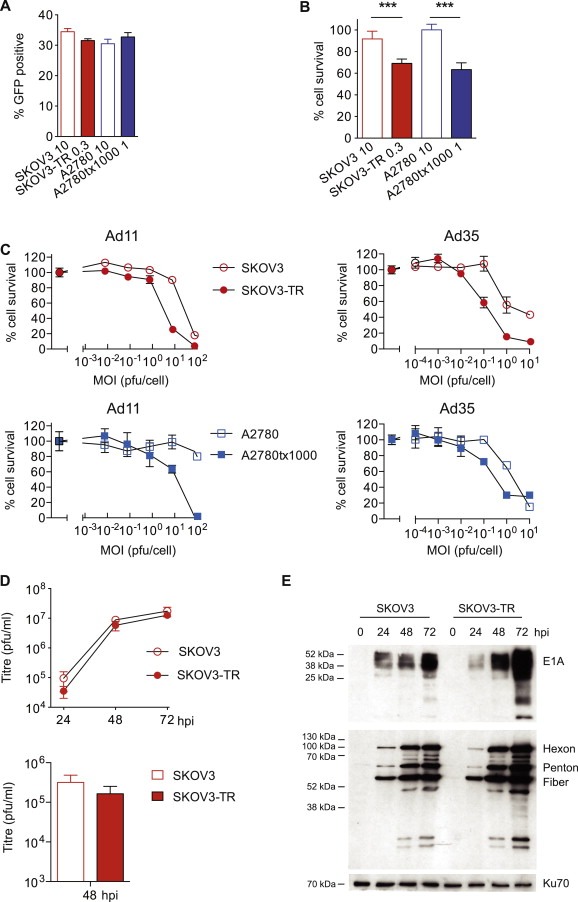
Paclitaxel‐resistant ovarian cancer cells show increased sensitivity independent of CAR‐mediated infection. A. SKOV3, SKOV3‐TR, A2780 and A2780tx1000 cells were infected with AdCMV‐GFP at MOI 10, 0.3, 10 and 1 respectively. The percentage of GFP positive cells was determined by flow cytometry. B. SKOV3, SKOV3‐TR, A2780 and A2780tx1000 cells were infected with dl922‐947 at MOI 10, 0.3, 10 and 1 respectively. Cell survival was determined 120 h later. ***; p < 0.001. C. Both cell pairs were infected with Ad11 and Ad35. Cell survival was determined 120 h later. D. SKOV3 and SKOV3‐TR cells were isoinfected with dl922‐947. Intracellular (above) and released (below) infectious virion production was quantified up to 72 h post‐infection by TCID50 assay. E. Protein was extracted from SKOV3 and SKOV3‐TR cells isoinfected with dl922‐947 up to 72 h post‐infection. Expression of E1A and adenoviral structural proteins was assessed by immunoblot.
The increased killing following iso‐infection with dl922‐947 did not result from increased virion production or exit (Figure 5D), nor was there convincing increase in adenovirus structural protein expression (Figure 5E). We did, however, observe increased E1A expression in the resistant cells 48 and 72 h post‐infection, in keeping with our previous data, which indicated that levels of E1A expression in ovarian cancer cells correlated closely with cytotoxicity (Connell et al., 2011; Flak et al., 2010).
We investigated cell cycle changes in paclitaxel‐resistant cells because our previous results have indicated that cell cycle status at the time of infection influences virus activity (Flak et al., 2010), and that adenovirus infection abrogates multiple cell cycle checkpoints (Connell et al., 2008) and induces profound over‐replication of genomic DNA in highly sensitive cells (Connell et al., 2011). In asynchronous populations, there were profound alterations in cell cycle profiles by flow cytometry, especially the amount of 4N and >4N DNA (Figure 6A), which persisted following iso‐infection with dl922‐947 (Figure 6B). Over‐expression of CAR alone did not alter cell cycle profiles (Figure S9). Gene expression confirmed highly significant changes in multiple cell cycle‐related pathways and processes in SKOV3‐TR cells (Figure 6C and Supplementary Table S3), and Ingenuity Pathway Analysis of our previously published data (Syed et al., 2011) indicated that the biological function ‘Cell cycle’ was significantly deregulated in A2780tx1000 compared to parental A2780 (p = 0.00015–0.0486; Supplementary Table S4).
Figure 6.
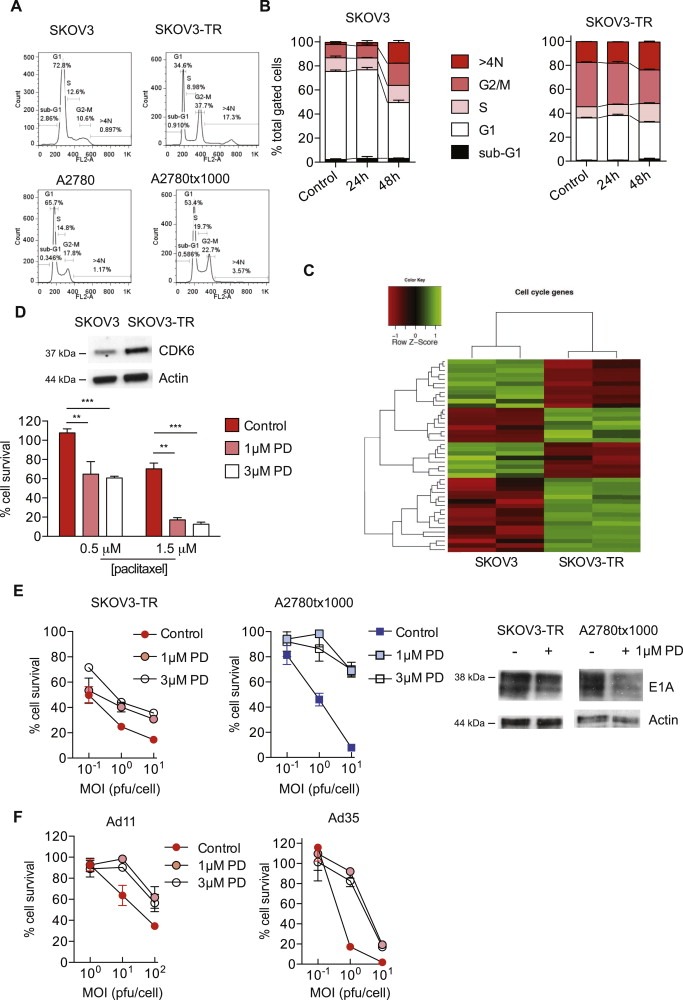
Cell cycle abnormalities in paclitaxel‐resistant cells mediate increased adenovirus sensitivity. A. Cell cycle state was assessed in asynchronous cells by flow cytometry following propidium iodide staining. Paclitaxel‐resistant cells were released from paclitaxel for 96 h prior to analysis. B. SKOV3 and SKOV3‐TR cells were iso‐infected with dl922‐947. Cell cycle state was assessed up to 48 h post‐infection by flow cytometry following propidium iodide staining. C. Differential expression in SKOV3 and SKOV3‐TR cells was assessed according to the GO terms ‘M phase’, ‘cell cycle process’, ‘mitosis’, ‘cell division’ and ‘mitotic cell cycle’. D. Expression of CDK6 was assessed in SKOV3 and SKOV3‐TR cells by immunoblot (upper). One hour following treatment with paclitaxel, SKOV3‐TR cells were exposed to CDK4/6 inhibitor PD‐0332991 (0, 1 and 3 μM). Cell survival was assessed 72 h later (lower). **; p < 0.01. ***; p < 0.001. E. SKOV3‐TR and A2780tx1000 cells were infected with dl922‐947 and re‐fed with CDK4/6 inhibitor PD‐0332991 (0, 1, 3 μM) 2 h later. Medium was replaced 72 h later. Cell survival was assessed at 120 h post‐infection. Expression of E1A was assessed following infection with dl922‐947 (MOI 1) for 72h in the presence and absence of 1 μM PD‐0332991 (right). F. SKOV3‐TR cells were infected with Ad 11 and Ad 35 and re‐fed with CDK4/6 inhibitor PD‐0332991 (0, 1, 3 μM) 2 h later. Medium was replaced 72 h thereafter. Cell survival was assessed at 120 h post‐infection.
Using matched Mad2+/+ and Mad2+/− Hct116 cells, we found that chromosomal instability (Swanton et al., 2009) did not influence adenovirus sensitivity (data not shown). CDK6 was one of the upregulated genes in resistant cells (Supplementary Table S3, Figure 6D). siRNA‐mediated CDK6 knockdown could not be achieved (Figure S10), but pharmacological inhibition using the combined CDK4/6 inhibitor PD‐0332991 (Toogood et al., 2005) at concentrations sufficient to alter cell cycle progression (Figure S11) significantly increased paclitaxel sensitivity (Figure 6D, Figure S12) and reduced both the activity of dl922‐947 and expression of E1A in both resistant lines (Figure 6E). Moreover, PD‐0332991 treatment was also able to reduce activity of Ad11 and Ad35 in the paclitaxel‐resistant lines (Figure 6F, Figure S13). CDK4/6 inhibition alone had a very minor effect on survival of both resistant cells (Figure S14) and did not alter CAR expression (data not shown).
4. Discussion and conclusion
4.1. Discussion
Previously, the clinical potential of adenovirus‐based therapies in ovarian cancer has been questioned due to low expression of the primary adenovirus receptor CAR in primary human tumours (Zeimet et al., 2002). However, using ovarian cancer cell lines and xenograft, we demonstrate that paclitaxel resistance in ovarian cancer is associated with increased sensitivity to adenoviruses of multiple serotypes that results both from increased expression of CAR and aberrant cell cycle control. These data are largely supported by analysis of clinical samples from the SaPPrOC and CTCR OV01 trials. However, further prospective clinical validation will be necessary.
Although previous work has demonstrated an increase in CAR and markers of stem‐like behaviour in paclitaxel resistant cell non‐small cell lung carcinoma cell lines (Zhang et al., 2012), the mechanism of this upregulation has not been explored nor has there been evaluation of xenografts or primary patient tumours. We found that the increase in CAR expression was associated with increased transcript levels with accompanying histone modification. These changes were paralleled by transient exposure of parental cells to the histone deacetylase inhibitor (HDACi) trichostatin A, which also increased CAR transcript levels. Previous investigators have noted that CAR expression increases through the cell cycle, peaking at M phase (Seidman et al., 2001), suggesting that paclitaxel‐induced G2/M arrest could cause the increased expression that we noted. However, in the parental cells, exposure to both nocodazole and paclitaxel at doses sufficient to cause profound G2/M arrest did not increase CAR expression to levels seen in paclitaxel‐resistant cells. It is possible that some of the increase in CAR expression in the resistant cells did results from prolonged relative G2/M arrest. However, this could not be substantiated as it was not possible sustain arrest in the parental cells beyond 48 h due to cytotoxicity. Widespread epigenetic changes have been described in chemotherapy‐resistant malignancies (Balch et al., 2010). We have previously identified that altered methylation contributes to the paclitaxel resistance of A2780tx1000 cells, via dysregulated expression of the polo‐like kinase 2 (Syed et al., 2011), whilst the current expression data analysis also showed marked alterations in HDAC4 and 9 expression in SKOV3‐TR cells (Supplementary Table 3). Our finding here that increased CAR expression did not result from altered promoter methylation concurs with studies in urothelial cancer (Pong et al., 2003). The region upstream of the CXADR transcriptional start site contains potential binding sites for several transcription factors, including E2F and Sp1; treatment with hypomethylating agents had no effect on transcript levels, in contrast to exposure to HDACi (Pong et al., 2003).
In keeping with others, our gene expression data here and previously (Ahmed et al., 2007; Syed et al., 2011) identified that MDR‐1 (ABCB1) was highly upregulated in both SKOV3‐TR and A2780tx1000. One question is whether increased CAR expression is observed in other chemotherapy resistance models associated with upregulated MDR‐1. In vincristine‐resistant SKOV3 cells (Buys et al., 2007), there was dramatic upregulation of ABCB1 (Log2FC 10.708). However, CXADR was downregulated (Log2FC −2.486 – GEO accession number GSE7556). Similarly, in an analysis of gene expression data (GEO accession number GSE3001) from ten ovarian cancer cell lines (Komatsu et al., 2006), we found no significant correlation between ABCB1 and CXADR transcript levels. Thus, we believe that the increase in CAR expression seen in paclitaxel resistance occurs independently of increases in MDR‐1.
We also demonstrate clearly that the secondary receptors for most adenoviruses, αvβ3 and αvβ5, are downregulated in paclitaxel resistant cells – this concurs with previous work by one of us (JDB), which demonstrated that loss of the extracellular matrix protein TGFBI (transforming growth factor beta induced) was able to induce resistance to paclitaxel and that restoration of sensitivity upon exposure to TGFBI was integrin dependent. Thus, downregulation of integrin and integrin‐mediated signalling appears a common feature of paclitaxel resistance (Ahmed et al., 2007). Reassuringly, we found also marked downregulation of TGFBI transcript levels in the SKOV3‐TR cells here. It is also interesting to note here that both our paclitaxel resistant cells show dramatically greater infectivity despite loss of integrin expression, suggesting that CAR is the dominant determinant of overall infection.
Productive adenovirus replication depends upon deregulation of host cell cycle checkpoints. E1A expression is critical. The E1A CR2 region, which binds with high affinity to pRb, dissociates pRb from E2F, permitting transactivation of genes necessary for viral DNA replication. E1A is the first viral gene to be expressed and its expression is entirely dependent upon host cell factors. Thus, the environment within cells at the time of infection is critical to expression of E1A and hence the entire virus life cycle. We have previously shown that expression of E1A correlates with overall cytotoxicity and that increased ovarian cancer cell sensitivity to the Ad5‐mediated killing is associated with abnormal G1/S cell cycle control (Flak et al., 2010). Thus, it was highly relevant that we identified increased E1A expression in resistant cells compared to parental following iso‐infection (Figure 5E). We have also shown that the E1A CR2‐deleted Ad5 vector dl922‐947 is capable of over‐riding multiple cell cycle checkpoints (Connell et al., 2008). Indeed, the synergy between low‐dose paclitaxel and adenovirus activity in ovarian cancer partially depends upon the induction of mitotic slippage (Ingemarsdotter et al., 2010). Thus, there is a close link between aberrant cell cycle control, E1A expression and oncolytic adenovirus activity.
The recent analysis by the Cancer Genome Atlas consortium suggested that Rb pathway signalling is abnormal in at least 66% all high grade serous ovarian cancers (TCGA, 2011), whilst amplification of CCNE1 (cyclin E1) at 19q12 is a potentially important mechanism for platinum resistance (Etemadmoghadam et al., 2009). Chromosomal instability and regulators of mitotic arrest have also been shown previously to regulate taxane resistance (Swanton et al., 2007, 2009). We show here that paclitaxel‐resistant ovarian cancer cell lines have profound deregulation of cell cycle control genes and that inhibition of CDK4/6 can partially reverse both paclitaxel resistance and sensitivity to adenoviruses of multiple serotypes, suggesting again a close link between virus function and paclitaxel activity. Of note, we also showed that inhibition of CDK4/6 reduced expression of E1A, the critical gene in overall adenovirus activity. Interestingly, Konecny et al. have previously shown parental A2780 and SKOV3 cells to be sensitive to the CDK4/6 inhibitor PD‐0332991 (IC50 0.06 and 0.24 μM respectively) (Konecny et al., 2011), whereas we found that A2780tx1000 and SKOV3‐TR cells were able to tolerate concentrations of 3 μM for 120 h with only minor loss of cell viability (Figure S10). Like us, Konecny et al. also found that CDK4/6 inhibition using PD‐0332991 sensitised ovarian cancer cells to paclitaxel, whilst Zhang et al. found that a different CDK4/6 inhibitor sensitised non‐small cell lung carcinoma cells to paclitaxel (Zhang et al., 2013). However, our attempts to validate these findings with siRNA‐mediated knockdown of CDK6 were unsuccessful in both paclitaxel‐resistant lines, despite multiple attempts – only minimal knockdown of CDK6 protein expression was achieved (Figure S6). siRNA knockdown of CAR was successful (Figure 4B), so SKOV3‐TR and A2780tx1000 cells are not intrinsically resistant to siRNA transfection. However, we believe that the present findings utilising PD‐0332991, which are consistent across two different cells lines, multiple doses of paclitaxel and multiple viruses, validate the concept that inhibition of CDK4/6 can alter both virus activity and cell sensitivity to paclitaxel.
4.2. Conclusions
Resistance to taxane chemotherapy is clearly multi‐factorial (reviewed in Murray et al. (2012)). Our investigations were initiated following data generated in cell lines in which resistance had been generated by in vitro culture of established tumour cells in increasing concentrations of chemotherapy. Such cell lines, although useful lab tools, may not be truly representative of clinical chemotherapy resistance, a fact attested to by clinical trials of chemotherapy resistance modifiers that do not meet their primary endpoints (McNeish et al., 2014). However, although our data suggest that increased CAR expression is not a cause of paclitaxel resistance – rather it appears to be a by‐product of altered transcriptional activity induced by altered histone modification – we believe that the results seen in xenografts and patient samples strongly suggest that CAR expression is genuinely upregulated in paclitaxel‐resistant ovarian cancers. Elucidating more clearly the link between dysregulated cell cycle control and adenovirus activity will be the focus of future work. However, taken together, our data suggest that adenoviruses, especially E1A CR‐2‐deleted vectors such as dl922‐947 and others that utilise CAR as their primary receptor, may have particular utility in paclitaxel resistance ovarian cancers.
Funding source
This work was supported by the Medical Research Council (grant reference G0601891) and Cancer Research UK (grant reference A11569).
Conflict of interest
The authors have no conflict of interest to declare.
Supporting information
The following are the supplementary data related to this article:
S1. Paclitaxel‐resistant cells show increased sensitivity to Ad5 wild‐type and dl1520 induced cytotoxicity. SKOV3 and SKOV3‐TR, and A2780 and A2780tx1000 cells were infected with wild‐type Ad5 or the E1B55K‐deleted Ad5 vector dl1520 for 120 h. Cell survival was analysed by MTT assay.
S2. In vitro growth of paclitaxel resistant ovarian cancer cells. SKOV3 and SKOV3‐TR, and A2780 and A2780tx1000 cells were plated in 24 well plates in triplicate. MTT assays were performed every 24 h until 72 h.
S3. CAR flow cytometry (Figure 2B). CAR expression was assessed by flow cytometry in SKOV3, SKOV3‐TR, A2780 and A2780tx1000.
S4. CAR flow cytometry in cells released from paclitaxel for 80 days. CAR expression was assessed by flow cytometry in SKOV3‐TR and A2780tx1000 that were maintained in paclitaxel (Tx) or released (R) for 80 days. Percentage cells positive (left) and flow histograms (right).
S5. Growth arrest following treatment with nocodazole or paclitaxel. Parental SKOV3 and A2780 cells were treated for 24 h with paclitaxel (30 nM) or nocodazole (300 nM). Cell cycle state was assessed by flow cytometry following propidium iodide staining.
S6. CAR staining by IHC on SaPPrOC trial tumour samples. Representative tumour samples showing histoscores of 0, 1, 2 and 3. Magnification is 10×. Intensity of CAR staining in tumour cells was assessed by two independent observers, who were blind to clinical response data.
S7. Ad11 and Ad35 internalisation in paclitaxel‐resistant cell lines. Viral internalisation was assessed 1 h following infection with Ad11 and Ad35 (5000vp/cell). Viral genome copy number per μg DNA is shown. NS = non‐significant (unpaired t‐test).
S8. Expression of group B adenovirus receptors on ovarian cancer cell pairs. Cell pairs were assayed for cell surface protein expression of CD46 and desmoglein 2 by flow cytometry. IgG1 stained cells served as a negative control.
S9. Expression of CAR alone does not alter cell cycle profile. Cell cycle state was assessed in asynchronous parental SKOV3 and SKOV3 cells expressing full length CAR and CAR truncation mutants by flow cytometry following propidium iodide staining.
S10. Failure to achieve CDK6 knockdown using siRNA. SKOV3‐TR cells were transfected with CDK6 siRNA oligonucleotides at different concentrations. 24 h later, CDK6 expression was assessed by immunoblot.
S11. CDK4/6 inhibition alters cell cycle dynamics in paclitaxel‐resistant ovarian cancer cells. SKOV3‐TR and A2780tx1000 cells were treated for 72 h with PD‐0332991. Cell cycle status was assessed by flow cytometry following propidium iodide staining.
S12. CDK4/6 inhibition increases paclitaxel sensitivity in A2780tx1000 cells. One hour following treatment with paclitaxel, A2780tx1000 cells were exposed to CDK4/6 inhibitor PD‐0332991 (0, 1 and 3 μM). Cell survival was assessed 72 h later by MTT assay (lower). *; p < 0.05.
S13. CDK4/6 inhibition reduces Ad 11 and Ad35 efficacy in A2780tx1000 cells. A2780tx1000 cells were infected with Ad 11 (MOI 0.1–10) and Ad35 (MOI 1–100). They were re‐fed with medium containing CDK4/6 inhibitor PD‐0332991 (0, 1, 3 μM) 2 h later. Medium was replaced 72 h later. Cell survival was assessed at 120 h post‐infection by MTT assay.
S14. CDK4/6 inhibition alone has minor effect on survival in paclitaxel‐resistant cells. Survival of SKOV3‐TR and A2780tx1000 cells treated with PD‐0332991 alone from experiments depicted in Figure 6E and F. Bars represent mean ± s.d., n = 6.
Table S1. Results of most recent 10 locus STR validation of SKOV3, SKOV3‐TR, A2780 and A2780tx1000 cells.
Table S2. Genes significantly up‐ and downregulated in SKOV3‐TR cells compared to SKOV3 according to the GO terms ‘Inflammatory Response’, ‘Cytokine Activity’ and ‘Chemokine Activity’.
Table S3. Genes significantly up‐ and downregulated in SKOV3‐TR cells compared to SKOV3 according to the GO terms ‘M phase’, ‘Cell Cycle Process’, ‘Mitosis’, ‘Cell Division and ‘Mitotic Cell Cycle’.
Table S4. Differential biological functions in A2780tx1000 compared to A2780 cells, as analysed by Ingenuity Pathway Analysis.
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
{kind=link}
Acknowledgements
We would like to thank Dr Maddy Parsons for the GFP‐tagged CAR constructs. We also thank Penny Morton, Nikolay Popov, Verity Mynors, Linda Hammond and Tracy Chaplin for technical assistance and advice.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.molonc.2014.12.007.
Ingemarsdotter Carin K., Tookman Laura A., Browne Ashley, Pirlo Katrina, Cutts Rosalind, Chelela Claude, Khurrum Karisma F., Leung Elaine Y.L., Dowson Suzanne, Webber Lee, Khan Iftekhar, Ennis Darren, Syed Nelofer, Crook Tim R., Brenton James D., Lockley Michelle, McNeish Iain A., (2015), Paclitaxel resistance increases oncolytic adenovirus efficacy via upregulated CAR expression and dysfunctional cell cycle control, Molecular Oncology 9, doi: 10.1016/j.molonc.2014.12.007.
Footnotes
Non‐standard abbreviations: CAR – coxsackie adenovirus receptor.
References
- Ahmed, A.A. , Mills, A.D. , Ibrahim, A.E. , Temple, J. , Blenkiron, C. , Vias, M. , Massie, C.E. , Iyer, N.G. , McGeoch, A. , Crawford, R. , Nicke, B. , Downward, J. , Swanton, C. , Bell, S.D. , Earl, H.M. , Laskey, R.A. , Caldas, C. , Brenton, J.D. , 2007. The extracellular matrix protein TGFBI induces microtubule stabilization and sensitizes ovarian cancers to paclitaxel. Cancer Cell 12, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch, C. , Matei, D.E. , Huang, T.H. , Nephew, K.P. , 2010. Role of epigenomics in ovarian and endometrial cancers. Epigenomics 2, 419–447. [DOI] [PubMed] [Google Scholar]
- Brooks, T.A. , Minderman, H. , O'Loughlin, K.L. , Pera, P. , Ojima, I. , Baer, M.R. , Bernacki, R.J. , 2003. Taxane-based reversal agents modulate drug resistance mediated by P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Mol. Cancer Ther. 2, 1195–1205. [PubMed] [Google Scholar]
- Buys, T.P. , Chari, R. , Lee, E.H. , Zhang, M. , MacAulay, C. , Lam, S. , Lam, W.L. , Ling, V. , 2007. Genetic changes in the evolution of multidrug resistance for cultured human ovarian cancer cells. Genes Chromosomes Cancer 46, 1069–1079. [DOI] [PubMed] [Google Scholar]
- Cheong, S.C. , Wang, Y. , Meng, J.H. , Hill, R. , Sweeney, K. , Kirn, D. , Lemoine, N.R. , Hallden, G. , 2008. E1A-expressing adenoviral E3B mutants act synergistically with chemotherapeutics in immunocompetent tumor models. Cancer Gene Ther. 15, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell, C.M. , Shibata, A. , Tookman, L.A. , Archibald, K.M. , Flak, M.B. , Pirlo, K.J. , Lockley, M. , Wheatley, S.P. , McNeish, I.A. , 2011. Genomic DNA damage and ATR-Chk1 signaling determine oncolytic adenoviral efficacy in human ovarian cancer cells. J. Clin. Invest. 121, 1283–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell, C.M. , Wheatley, S.P. , McNeish, I.A. , 2008. Nuclear survivin abrogates multiple cell cycle checkpoints and enhances viral oncolysis. Cancer Res. 69, 7923–7931. [DOI] [PubMed] [Google Scholar]
- du Bois, A. , Luck, H.J. , Meier, W. , Adams, H.P. , Mobus, V. , Costa, S. , Bauknecht, T. , Richter, B. , Warm, M. , Schroder, W. , Olbricht, S. , Nitz, U. , Jackisch, C. , Emons, G. , Wagner, U. , Kuhn, W. , Pfisterer, J. , 2003. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Natl. Cancer Inst. 95, 1320–1329. [DOI] [PubMed] [Google Scholar]
- Duan, Z. , Feller, A.J. , Penson, R.T. , Chabner, B.A. , Seiden, M.V. , 1999. Discovery of differentially expressed genes associated with paclitaxel resistance using cDNA array technology: analysis of interleukin (IL) 6, IL-8, and monocyte chemotactic protein 1 in the paclitaxel-resistant phenotype. Clin. Cancer Res. 5, 3445–3453. [PubMed] [Google Scholar]
- Duan, Z. , Lamendola, D.E. , Penson, R.T. , Kronish, K.M. , Seiden, M.V. , 2002. Overexpression of IL-6 but not IL-8 increases paclitaxel resistance of U-2OS human osteosarcoma cells. Cytokine 17, 234–242. [DOI] [PubMed] [Google Scholar]
- Eliopoulos, A. , Kerr, D. , Herod, J. , Hodgkins, E. , Krajewski, S. , Reed, J. , Young, L. , 1995. The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and Bcl-2. Oncogene 11, 1217–1228. [PubMed] [Google Scholar]
- Etemadmoghadam, D. , deFazio, A. , Beroukhim, R. , Mermel, C. , George, J. , Getz, G. , Tothill, R. , Okamoto, A. , Raeder, M.B. , Harnett, P. , Lade, S. , Akslen, L.A. , Tinker, A.V. , Locandro, B. , Alsop, K. , Chiew, Y.E. , Traficante, N. , Fereday, S. , Johnson, D. , Fox, S. , Sellers, W. , Urashima, M. , Salvesen, H.B. , Meyerson, M. , Bowtell, D. , Group, A.S. , 2009. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin. Cancer Res. 15, 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, C. , Morton, P.E. , Snippe, M. , Santis, G. , Parsons, M. , 2009. Coxsackie adenovirus receptor (CAR) regulates integrin function through activation of p44/42 MAPK. Exp. Cell Res. 315, 2637–2647. [DOI] [PubMed] [Google Scholar]
- Flak, M.B. , Connell, C.M. , Chelala, C. , Archibald, K. , Salako, M.A. , Pirlo, K.J. , Lockley, M. , Wheatley, S.P. , Balkwill, F.R. , McNeish, I.A. , 2010. p21 promotes oncolytic adenoviral activity in ovarian cancer and is a potential biomarker. Mol. Cancer 9, 175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fueyo, J. , Gomez-Manzano, C. , Alemany, R. , Lee, P. , McDonnell, T. , Mitlianga, P. , Shi, Y.-X. , VA, L. , Yung, W. , Kyritsis, A. , 2000. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo . Oncogene 19, 2–12. [DOI] [PubMed] [Google Scholar]
- Heise, C. , Hermiston, T. , Johnson, L. , Brooks, G. , Sampson-Johannes, A. , Williams, A. , Hawkins, L. , Kirn, D. , 2000. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 6, 1134–1139. [DOI] [PubMed] [Google Scholar]
- Ingemarsdotter, C.K. , Baird, S.K. , Connell, C.M. , Oberg, D. , Hallden, G. , McNeish, I.A. , 2010. Low-dose paclitaxel synergizes with oncolytic adenoviruses via mitotic slippage and apoptosis in ovarian cancer. Oncogene 29, 6051–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, Y. , Hu, W. , Ivan, C. , Dalton, H.J. , Miyake, T. , Pecot, C.V. , Zand, B. , Liu, T. , Huang, J. , Jennings, N.B. , Rupaimoole, R. , Taylor, M. , Pradeep, S. , Wu, S.Y. , Lu, C. , Wen, Y. , Huang, J. , Liu, J. , Sood, A.K. , 2013. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J. Natl. Cancer Inst. 105, 1485–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlan, B.Y. , Oza, A.M. , Richardson, G.E. , Provencher, D.M. , Hansen, V.L. , Buck, M. , Chambers, S.K. , Ghatage, P. , Pippitt, C.H. , Brown, J.V. , Covens, A. , Nagarkar, R.V. , Davy, M. , Leath, C.A. , Nguyen, H. , Stepan, D.E. , Weinreich, D.M. , Tassoudji, M. , Sun, Y.N. , Vergote, I.B. , 2012. Randomized, Double-blind, placebo-controlled phase II study of AMG 386 combined with weekly paclitaxel in patients with recurrent ovarian Cancer. J. Clin. Oncol. 30, 362–371. [DOI] [PubMed] [Google Scholar]
- Kimball, K.J. , Preuss, M.A. , Barnes, M.N. , Wang, M. , Siegal, G.P. , Wan, W. , Kuo, H. , Saddekni, S. , Stockard, C.R. , Grizzle, W.E. , Harris, R.D. , Aurigemma, R. , Curiel, D.T. , Alvarez, R.D. , 2010. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin. Cancer Res. 16, 5277–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu, M. , Hiyama, K. , Tanimoto, K. , Yunokawa, M. , Otani, K. , Ohtaki, M. , Hiyama, E. , Kigawa, J. , Ohwada, M. , Suzuki, M. , Nagai, N. , Kudo, Y. , Nishiyama, M. , 2006. Prediction of individual response to platinum/paclitaxel combination using novel marker genes in ovarian cancers. Mol. Cancer Ther. 5, 767–775. [DOI] [PubMed] [Google Scholar]
- Konecny, G.E. , Winterhoff, B. , Kolarova, T. , Qi, J. , Manivong, K. , Dering, J. , Yang, G. , Chalukya, M. , Wang, H.J. , Anderson, L. , Kalli, K.R. , Finn, R.S. , Ginther, C. , Jones, S. , Velculescu, V.E. , Riehle, D. , Cliby, W.A. , Randolph, S. , Koehler, M. , Hartmann, L.C. , Slamon, D.J. , 2011. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin. Cancer Res. 17, 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, I. , Harden, P. , Bauzon, M. , Chartier, C. , Nye, J. , Thorne, S. , Reid, T. , Ni, S. , Lieber, A. , Fisher, K. , Seymour, L. , Rubanyi, G.M. , Harkins, R.N. , Hermiston, T.W. , 2008. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One 3, e2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockley, M. , Fernandez, M. , Wang, Y. , Li, N.F. , Conroy, S.E. , Lemoine, N.R. , McNeish, I.A. , 2006. Activity of the adenoviral E1A deletion mutant dl922-947 in ovarian cancer: comparison with adenovirus wild-type, bioluminescence monitoring and intraperitoneal delivery in icodextrin. Cancer Res. 66, 989–998. [DOI] [PubMed] [Google Scholar]
- McNeish, I.A. , Ledermann, J.A. , Webber, L. , James, L. , Kaye, S.B. , Hall, M. , Hall, G. , Clamp, A. , Earl, H. , Banerjee, S. , Kristeleit, R. , Raja, F. , Feeney, A. , Lawrence, C. , Dawson-Athey, L. , Persic, M. , Khan, I. , 2014. A randomised placebo-controlled trial of weekly paclitaxel and saracatinib (AZD0530) in platinum-resistant ovarian, fallopian tube or primary peritoneal cancer. Ann. Oncol. 25, 1988–1995. [DOI] [PubMed] [Google Scholar]
- McNeish, I.A. , Tenev, T. , Bell, S. , Marani, M. , Vassaux, G. , Lemoine, N. , 2001. Herpes simplex virus thymidine kinase/ganciclovir-induced cell death is enhanced by co-expression of caspase-3 in ovarian carcinoma cells. Cancer Gene Ther. 8, 308–319. [DOI] [PubMed] [Google Scholar]
- Mozzetti, S. , Ferlini, C. , Concolino, P. , Filippetti, F. , Raspaglio, G. , Prislei, S. , Gallo, D. , Martinelli, E. , Ranelletti, F.O. , Ferrandina, G. , Scambia, G. , 2005. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 11, 298–305. [PubMed] [Google Scholar]
- Murray, S. , Briasoulis, E. , Linardou, H. , Bafaloukos, D. , Papadimitriou, C. , 2012. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev. 38, 890–903. [DOI] [PubMed] [Google Scholar]
- Pong, R.C. , Lai, Y.J. , Chen, H. , Okegawa, T. , Frenkel, E. , Sagalowsky, A. , Hsieh, J.T. , 2003. Epigenetic regulation of coxsackie and adenovirus receptor (CAR) gene promoter in urogenital cancer cells. Cancer Res. 63, 8680–8686. [PubMed] [Google Scholar]
- Rustin, G.J. , Quinn, M. , Thigpen, T. , du Bois, A. , Pujade-Lauraine, E. , Jakobsen, A. , Eisenhauer, E. , Sagae, S. , Greven, K. , Vergote, I. , Cervantes, A. , Vermorken, J. , 2004. Re: new guidelines to evaluate the response to treatment in solid tumors (ovarian cancer). J. Natl. Cancer Inst. 96, 487–488. [DOI] [PubMed] [Google Scholar]
- Seidman, M.A. , Hogan, S.M. , Wendland, R.L. , Worgall, S. , Crystal, R.G. , Leopold, P.L. , 2001. Variation in adenovirus receptor expression and adenovirus vector-mediated transgene expression at defined stages of the cell cycle. Mol. Ther. 4, 13–21. [DOI] [PubMed] [Google Scholar]
- Sherr, C.J. , McCormick, F. , 2002. The RB and p53 pathways in cancer. Cancer Cell 2, 103–112. [DOI] [PubMed] [Google Scholar]
- Suomalainen, M. , Nakano, M.Y. , Keller, S. , Boucke, K. , Stidwill, R.P. , Greber, U.F. , 1999. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144, 657–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton, C. , Marani, M. , Pardo, O. , Warne, P.H. , Kelly, G. , Sahai, E. , Elustondo, F. , Chang, J. , Temple, J. , Ahmed, A.A. , Brenton, J.D. , Downward, J. , Nicke, B. , 2007. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 11, 498–512. [DOI] [PubMed] [Google Scholar]
- Swanton, C. , Nicke, B. , Schuett, M. , Eklund, A.C. , Ng, C. , Li, Q. , Hardcastle, T. , Lee, A. , Roy, R. , East, P. , Kschischo, M. , Endesfelder, D. , Wylie, P. , Kim, S.N. , Chen, J.G. , Howell, M. , Ried, T. , Habermann, J.K. , Auer, G. , Brenton, J.D. , Szallasi, Z. , Downward, J. , 2009. Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. U. S. A. 106, 8671–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed, N. , Coley, H.M. , Sehouli, J. , Koensgen, D. , Mustea, A. , Szlosarek, P. , McNeish, I. , Blagden, S.P. , Schmid, P. , Lovell, D.P. , Hatzimichael, E. , Crook, T. , 2011. Polo-like kinase Plk2 is an epigenetic determinant of chemosensitivity and clinical outcomes in ovarian Cancer. Cancer Res. 71, 3317–3327. [DOI] [PubMed] [Google Scholar]
- TCGA, 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood, P.L. , Harvey, P.J. , Repine, J.T. , Sheehan, D.J. , VanderWel, S.N. , Zhou, H. , Keller, P.R. , McNamara, D.J. , Sherry, D. , Zhu, T. , Brodfuehrer, J. , Choi, C. , Barvian, M.R. , Fry, D.W. , 2005. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 48, 2388–2406. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Niu, X.L. , Qu, Y. , Wu, J. , Zhu, Y.Q. , Sun, W.J. , Li, L.Z. , 2010. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 295, 110–123. [DOI] [PubMed] [Google Scholar]
- Workman, P. , Aboagye, E.O. , Balkwill, F. , Balmain, A. , Bruder, G. , Chaplin, D.J. , Double, J.A. , Everitt, J. , Farningham, D.A. , Glennie, M.J. , Kelland, L.R. , Robinson, V. , Stratford, I.J. , Tozer, G.M. , Watson, S. , Wedge, S.R. , Eccles, S.A. , Committee of the National Cancer Research, I.2010. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 102, 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle, R.J. , Strasser, A. , 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. [DOI] [PubMed] [Google Scholar]
- Yuen, S. , Smith, J. , Caruso, L. , Balan, M. , Opavsky, M.A. , 2011. The coxsackie-adenovirus receptor induces an inflammatory cardiomyopathy independent of viral infection. J. Mol. Cell. Cardiol. 50, 826–840. [DOI] [PubMed] [Google Scholar]
- Zeimet, A.G. , Muller-Holzner, E. , Schuler, A. , Hartung, G. , Berger, J. , Hermann, M. , Widschwendter, M. , Bergelson, J.M. , Marth, C. , 2002. Determination of molecules regulating gene delivery using adenoviral vectors in ovarian carcinomas. Gene Ther. 9, 1093–1100. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Fang, B. , Mohan, R. , Chang, J.Y. , 2012. Coxsackie-adenovirus receptor as a novel marker of stem cells in treatment-resistant non-small cell lung cancer. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 105, 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.H. , Cheng, Y. , Shin, J.Y. , Kim, J.O. , Oh, J.E. , Kang, J.H. , 2013. A CDK4/6 inhibitor enhances cytotoxicity of paclitaxel in lung adenocarcinoma cells harboring mutant KRAS as well as wild-type KRAS. Cancer Biol. Ther. 14, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
S1. Paclitaxel‐resistant cells show increased sensitivity to Ad5 wild‐type and dl1520 induced cytotoxicity. SKOV3 and SKOV3‐TR, and A2780 and A2780tx1000 cells were infected with wild‐type Ad5 or the E1B55K‐deleted Ad5 vector dl1520 for 120 h. Cell survival was analysed by MTT assay.
S2. In vitro growth of paclitaxel resistant ovarian cancer cells. SKOV3 and SKOV3‐TR, and A2780 and A2780tx1000 cells were plated in 24 well plates in triplicate. MTT assays were performed every 24 h until 72 h.
S3. CAR flow cytometry (Figure 2B). CAR expression was assessed by flow cytometry in SKOV3, SKOV3‐TR, A2780 and A2780tx1000.
S4. CAR flow cytometry in cells released from paclitaxel for 80 days. CAR expression was assessed by flow cytometry in SKOV3‐TR and A2780tx1000 that were maintained in paclitaxel (Tx) or released (R) for 80 days. Percentage cells positive (left) and flow histograms (right).
S5. Growth arrest following treatment with nocodazole or paclitaxel. Parental SKOV3 and A2780 cells were treated for 24 h with paclitaxel (30 nM) or nocodazole (300 nM). Cell cycle state was assessed by flow cytometry following propidium iodide staining.
S6. CAR staining by IHC on SaPPrOC trial tumour samples. Representative tumour samples showing histoscores of 0, 1, 2 and 3. Magnification is 10×. Intensity of CAR staining in tumour cells was assessed by two independent observers, who were blind to clinical response data.
S7. Ad11 and Ad35 internalisation in paclitaxel‐resistant cell lines. Viral internalisation was assessed 1 h following infection with Ad11 and Ad35 (5000vp/cell). Viral genome copy number per μg DNA is shown. NS = non‐significant (unpaired t‐test).
S8. Expression of group B adenovirus receptors on ovarian cancer cell pairs. Cell pairs were assayed for cell surface protein expression of CD46 and desmoglein 2 by flow cytometry. IgG1 stained cells served as a negative control.
S9. Expression of CAR alone does not alter cell cycle profile. Cell cycle state was assessed in asynchronous parental SKOV3 and SKOV3 cells expressing full length CAR and CAR truncation mutants by flow cytometry following propidium iodide staining.
S10. Failure to achieve CDK6 knockdown using siRNA. SKOV3‐TR cells were transfected with CDK6 siRNA oligonucleotides at different concentrations. 24 h later, CDK6 expression was assessed by immunoblot.
S11. CDK4/6 inhibition alters cell cycle dynamics in paclitaxel‐resistant ovarian cancer cells. SKOV3‐TR and A2780tx1000 cells were treated for 72 h with PD‐0332991. Cell cycle status was assessed by flow cytometry following propidium iodide staining.
S12. CDK4/6 inhibition increases paclitaxel sensitivity in A2780tx1000 cells. One hour following treatment with paclitaxel, A2780tx1000 cells were exposed to CDK4/6 inhibitor PD‐0332991 (0, 1 and 3 μM). Cell survival was assessed 72 h later by MTT assay (lower). *; p < 0.05.
S13. CDK4/6 inhibition reduces Ad 11 and Ad35 efficacy in A2780tx1000 cells. A2780tx1000 cells were infected with Ad 11 (MOI 0.1–10) and Ad35 (MOI 1–100). They were re‐fed with medium containing CDK4/6 inhibitor PD‐0332991 (0, 1, 3 μM) 2 h later. Medium was replaced 72 h later. Cell survival was assessed at 120 h post‐infection by MTT assay.
S14. CDK4/6 inhibition alone has minor effect on survival in paclitaxel‐resistant cells. Survival of SKOV3‐TR and A2780tx1000 cells treated with PD‐0332991 alone from experiments depicted in Figure 6E and F. Bars represent mean ± s.d., n = 6.
Table S1. Results of most recent 10 locus STR validation of SKOV3, SKOV3‐TR, A2780 and A2780tx1000 cells.
Table S2. Genes significantly up‐ and downregulated in SKOV3‐TR cells compared to SKOV3 according to the GO terms ‘Inflammatory Response’, ‘Cytokine Activity’ and ‘Chemokine Activity’.
Table S3. Genes significantly up‐ and downregulated in SKOV3‐TR cells compared to SKOV3 according to the GO terms ‘M phase’, ‘Cell Cycle Process’, ‘Mitosis’, ‘Cell Division and ‘Mitotic Cell Cycle’.
Table S4. Differential biological functions in A2780tx1000 compared to A2780 cells, as analysed by Ingenuity Pathway Analysis.
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data