

Abstract
Characterization of genetic alterations in tumor biopsies serves as useful biomarkers in prognosis and treatment management. Circulating tumor cells (CTCs) obtained non‐invasively from peripheral blood could serve as a tumor proxy. Using a label‐free CTC enrichment strategy that we have established, we aimed to develop sensitive assays for qualitative assessment of tumor genotype in patients. Blood consecutively obtained from 44 patients with local and advanced colorectal cancer and 18 healthy donors were enriched for CTCs using a size‐based microsieve technology. To screen for CTC mutations, we established high‐resolution melt (HRM) and allele‐specific PCR (ASPCR) KRAS‐codon 12/13‐ and BRAF‐codon 600‐ specific assays, and compared the performance with pyrosequencing and Sanger sequencing. For each patient, the resulting CTC genotypes were compared with matched tumor and normal tissues. Both HRM and ASPCR could detect as low as 1.25% KRAS‐ or BRAF‐mutant alleles. HRM detected 14/44 (31.8%) patients with KRAS mutation in CTCs and 5/44 (11.3%) patients having BRAF mutation in CTCs. ASPCR detected KRAS and BRAF mutations in CTCs of 10/44 (22.7%) and 1/44 (2.3%) patients respectively. There was an increased detection of mutation in blood using these two methods. Comparing tumor tissues and CTCs mutation status using HRM, we observed 84.1% concordance in KRAS genotype (p = 0.000129, Fishers' exact test; OR = 38.7, 95% CI = 4.05–369) and 90.9% (p = 0.174) concordance in BRAF genotype. Our results demonstrate that CTC enrichment, coupled with sensitive mutation detection methods, may allow rapid, sensitive and non‐invasive assessment of tumor genotype.
Keywords: Non-invasive, Circulating tumor cells, Targeted phenotyping, Colorectal cancer
Highlights
KRAS and BRAF mutation analysis are current clinical standard for patients receiving chemotherapy.
These markers predict outcomes of anti‐EGFR therapy in colorectal patients.
Circulating tumor cells (CTCs) from blood enables non‐invasive evaluation of tumor status.
Our optimized PCR‐based assays could detect concordant mutation in CTCs and tissues.
This approach enables a rapid, qualitative and non‐invasive assessment of tumor genotype.
Abbreviations
- CTC
circulating tumor cell
- CRC
colorectal cancer
- HRM
high-resolution melt
- ASPCR
allele-specific PCR
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- BRAF
proto-oncogene B-Raf
- EGFR
epidermal growth factor receptor
1. Introduction
Tumor genotyping is instrumental in prognosis and treatment management of cancer patients as characterization of genetic alterations serve as useful biomarkers of disease. Typically, mutations in genes coding for signaling proteins such as KRAS (Kirsten rat sarcoma viral oncogene), BRAF (proto‐oncogene B‐Raf) and EGFR (epidermal growth factor receptor) are routinely detected in tissue biopsies by PCR‐based methods (Bihl et al., 2012; Sgambato et al., 2012). In colorectal cancer (CRC), KRAS and BRAF mutations are useful markers for patients receiving systemic chemotherapy; besides predicting the therapeutic efficiency of anti‐EGFR therapy, patients with poor prognosis may be identified (Richman et al., 2009; Yokota et al., 2011). There has been significant attention on these mutations as potential prognostic and predictive biomarkers in patients with metastatic disease treated with anti‐EGFR therapies, such as panitumumab and cetuximab (Karapetis et al., 2008; Laurent‐Puig et al., 2009). Prospective studies have shown that patients with KRAS mutations do not benefit from cetuximab treatment. Since almost 40% of CRC patients harbor the KRAS mutation, identifying this subset of patients would be valuable in making informed clinical decisions (Asghar et al., 2010; Modest et al., 2011; Price et al., 2011). In addition, retrospective analysis has shown that wild‐type BRAF is necessary for a successful response to panitumumab or cetuximab therapies in metastatic CRC (De Roock et al., 2010; Laurent‐Puig et al., 2009). Together, KRAS and BRAF serve as useful predictive markers for anti‐EGFR therapies.
Genotyping tumor may be challenging due to invasiveness and limited availability of tissue biopsies. Circulating tumor cells (CTCs) are seen as an alternative source of tumor cells circulating in the peripheral blood of cancer patients (Hüsemann et al., 2008; Klein, 2009). The concept of hematogenous dissemination of cancer dates back to mid‐19th century, supported directly by pioneering postmortem observations of the presence of cancer cells in the blood via microscopy (Langenbeck, 1841; Paget, 1853), potentially providing a dynamic pool of genetic biomarkers that can be assessed repeatedly. The ability to capture and profile CTCs presents an attractive platform to genotype tumors non‐invasively.
Numerous approaches have been employed to capture CTCs for diagnostics and discovery applications. To date, the CellSearch® system (Veridex) is the only FDA‐approved and leading automated immunomagnetic separation platform for routine clinical detection and analysis of CTCs in patients with metastatic breast cancer, prostate cancer and CRC. The platform utilizes ferrofluid nanoparticles coated with anti‐EpCAM antibodies to capture CTCs passing through a magnetic field (Allard et al., 2004). However, such targeted approach introduces a bias on the profile of captured CTCs since CTCs are largely heterogeneous; thus, some subpopulations of cells might not be properly captured. (Spizzo et al., 2011). Other non‐targeted approaches such as size‐based microfiltration and density centrifugation have shown the ability to enrich CTCs for downstream analysis. Regardless of the CTCs enrichment approaches (Cima et al., 2013), both methodologies still demonstrate sub‐optimal performance in isolation of pure CTCs.
CTC isolates typically contain rare tumor cells mixed white blood cells. As such, sensitive platforms are required to detect low fractions of mutant alleles in the background of normal cells. Although Sanger sequencing is the gold standard for mutant identification, it is only reliable in detecting 20% mutant copies at the allelic level (Tsiatis et al., 2010). Lower level of mutations, which are typically masked by high background of wild‐type DNA, cannot be detected by conventional sequencing. Other PCR‐based approaches for detecting genetic alterations have also been demonstrated, with advantages and disadvantages in terms of cost, sensitivity and specificity. These reported techniques include pyrosequencing (Tsiatis et al., 2010), co‐amplification at lower denaturation temperature‐PCR (cold‐PCR) (Zuo et al., 2009), amplification refractory mutation system using a bifunctional self‐probing primer (Franklin et al., 2010), massively‐parallel sequencing (Peeters et al., 2013), and high‐resolution melting (HRM) analysis (Simi et al., 2008).
In this study, we report the use of in‐house silicon microsieve technology (Lim et al., 2012) to capture and enrich CTCs from peripheral blood of CRC patients. Using the microfiltered cells, we demonstrate the ability to perform non‐invasive mutation analysis using HRM and allele‐specific PCR (ASPCR) assays. HRM is a rapid, simple in‐tube scanning methodology that detects changes in nucleotide sequences by monitoring the melting profile of PCR amplicons and is useful in scanning for unknown mutations. Coupled with the use of ASPCR to ascertain the mutations, we were able to identify CRC patients harboring KRAS and BRAF hotspot mutations in the CTCs. Most importantly, we observed a reasonable concordance in mutation status between the tumor tissues and CTCs.
2. Materials and methods
2.1. Patient samples
All study subjects gave informed written consent to participate and biological samples were obtained according to protocols approved by the Institutional Review Board (IRB), National University of Singapore and Fortis. Blood samples from 44 cancer patients were provided by Fortis Surgical Hospital, while blood samples from 18 subjects without cancer were provided by the National University Hospital Singapore. Blood was collected in EDTA vacutainer tubes (Becton–Dickinson), and was processed within 6 h. Fresh frozen tumors obtained from surgical resections at Fortis Surgical Hospital were stored at −80 °C until use. Clinicopathological data for the subjects are described in Table 1 and Supplementary Table 1.
Table 1.
Clinicopathologic characteristics of study subjects.
| Healthy controls | Colorectal cancer patients | |
|---|---|---|
| n = 18 | n = 44 | |
| Age (median, range) | 50 (34–70) | 58.5 (26–74) |
| Sex | ||
| Female | 12 (66.7%) | 20 (45.5%) |
| Male | 6 (33.3%) | 24 (54.5%) |
| Race | ||
| Chinese | 12 (66.7%) | 37 (84.1%) |
| Indian | 5 (27.7%) | 1 (2.3%) |
| Malay | 1 (2.3%) | |
| Others | 1 (5.6%) | 5 (11.3%) |
| Site of primary tumor | ||
| R (Ascending colon) | 2 (4.5%) | |
| R (Caecum) | 4 (9.1%) | |
| L (Descending colon) | 3 (6.8%) | |
| L (Rectum) | 20 (45.5%) | |
| L (Rectosigmoid) | 3 (6.8%) | |
| L (Sigmoid) | 10 (22.7%) | |
| L (Splenic flexure) | 2 (4.5%) | |
| Dukes tumor stage | ||
| Adenoma | 4 (9.1%) | |
| A | 8 (18.2%) | |
| B | 14 (31.8%) | |
| C | 15 (34.1%) | |
| D | 3 (6.8%) | |
| Metastasis | ||
| Yes | 4 (9%) | |
| No | 40 (91%) | |
| Neo‐adjuvant chemotherapy at point of primary tumor | ||
| Yes | 8 (18.2%) | |
| No | 35 (79.5%) | |
| Incomplete data | 1 (2.3%) | |
| Neo‐adjuvant radiotherapy at point of primary tumor | ||
| Yes | 5 (11.4%) | |
| No | 38 (86.4%) | |
| Incomplete data | 1 (2.3%) | |
| Adjuvant chemotherapy at point of primary tumor | ||
| Yes | 20 (45.5%) | |
| No | 22 (50%) | |
| Incomplete data | 2 (4.5%) | |
| Adjuvant radiotherapy at point of primary tumor | ||
| Yes | 4 (9%) | |
| No | 39 (88.6%) | |
| Incomplete data | 1 (2.3%) | |
2.2. Microsieve technology for capturing CTCs
CTCs were isolated using a microfabricated silicon microsieve serving as a size‐based filtration unit (IBN microsieve) (Figure 1A). The IBN microsieve is integrated in a customized filtration cartridge as described previously (Lim et al., 2012). The fluidic flow of whole blood was controlled by a peristaltic pump (IPC 4, Ismatec) connected to the IBN microsieve through peristaltic tubes, with a column at an inlet to contain whole blood and a bottle at the outlet to collect waste.
Figure 1.
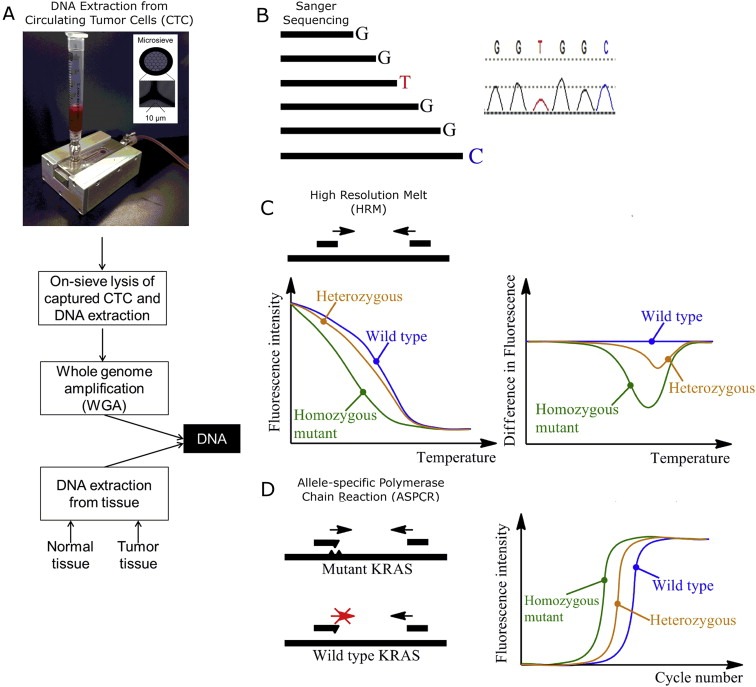
Targeted mutation analysis of CTCs: an overview. (A) Matched normal, tumor and CTC DNA were extracted from patient. (B) Patients were sequenced for KRAS and BRAF mutations using Sanger sequencing. (C) Mutations were identified using HRM, which rapidly detects sequence variation based on melting profile. (D) Mutations were validated using ASPCR followed by gel electrophoresis.
Briefly, 3 ml of blood from CRC patients or healthy subjects was passed through the IBN microsieve followed by 3–4 washes of 1 ml of 1X PBS/0.5% BSA/2 mM EDTA wash buffer at a flow rate of 0.5 ml/min. The sieve was removed from the cartridge, placed in a nuclease‐free 2‐ml ultracentrifuge tube, and immediately stored at −80 °C until further use. The presence of CTCs (as defined by cytokeratin‐positive and CD45 negative cells) in all 44 patient samples was documented in a separate run using 1 mL of blood. After filtering, the captured cells were fixed with 4% paraformaldehyde and permeabilized using 0.1% Triton X‐100 containing buffer solution. Cells were immunostained with antibody cocktail consisting of DAPI (BUF061, AbD Serotec), pan‐keratin (C11) mouse mAb (Alexa fluor® 488 conjugate) (4523S, Cell Signaling Technology), vimentin antibody (V9) TRITC (sc‐6260 TRITC, Santa Cruz Biotechnology) and mouse anti human CD45:alexa fluor® 647 (MCA87A647, AbD Serotec) for 1 h. After that, the stained cells were imaged using BX61 microscope (Olympus). Serial enumeration studies are ongoing.
2.3. DNA extraction and whole‐genome amplification of CTCs
The QIAamp DNA Mini Kit (Qiagen) was used according to the manufacturer's protocol. Briefly, 200 μl of PBS containing 20 μl of proteinase K was added to the tube containing IBN microsieve, followed by 200 μl of lysis buffer and incubation at 56 °C for 1 h. The solution containing DNA from lysed cells was placed and washed in the spin column. DNA was eluted and subsequently amplified using the RepliG Ultra‐Fast Mini Kit (Qiagen). Whole‐genome amplified DNA was cleaned using ethanol precipitation. DNA quality and concentration were measured with NanoDrop 1000 spectrophotometer (Thermo Scientific).
2.4. Conventional PCR and sequencing
PCR was performed as previously described, with primers for KRAS exon 2 and BRAF exon 15 generating 280 bp and 224 bp products, respectively (Feng et al., 2005; Lee et al., 2011). PCR products were purified using FastAP Thermosensitive Alkaline Phosphatase and Exonuclease I (Thermo Scientific) prior to DNA sequencing.
2.5. High resolution melt (HRM) assay and sensitivity testing
Samples were assayed using the Viia7 real‐time PCR machine (Applied Biosystems). Each 20 μl‐triplicate reaction contained 20 ng of DNA diluted in Melt Doctor HRM Master Mix and 200 nM HRM primers (KRAS exon 2 forward primer 5′‐GGCCTGCTGAAAATGACTGA‐3′ and KRAS exon 3 reverse primer 5′‐CCTCTATTGTTGGATCATATTCG‐3′; or BRAF exon 15 forward primer 5′‐TTCATGAAGACCTCACAGTAAAAA and BRAF exon 15 reverse primer 5′‐AACTCAGCAGCATCTCAGGG‐3′), with the following PCR program and melting conditions used for all amplicons: 95 °C for 10 min; 50 cycles of 95 °C for 15 s and 60 °C for 1 min; denaturation at 90 °C for 10 s, annealing at 60 °C for 1 min, followed by a high‐resolution melt of 60–95 °C (0.01 °C/s, 45 acquisitions/°C). Data were acquired and analyzed using the accompanying High Resolution Melt software.
For sensitivity testing, cell lines‐derived DNA carrying a heterozygous KRAS G13D mutation (DLD‐1, ATCC® CCL‐221™) or a heterozygous BRAF V600E mutation (HT29, ATCC® HTB‐38™) was mixed with wild‐type DNA of healthy donor (IBN‐A01) with various percentages of mutant alleles. The resulting mixtures of mutant alleles serve as templates for sensitivity testing.
2.6. Allele‐specific PCR (ASPCR)
Samples were assayed using CFX96 C1000 real‐time thermal cycler (Bio‐rad). Each 10 μl‐quadruplicate reaction comprised of 20 ng of template DNA in QuantiFast SYBR Green PCR Master Mix (Qiagen) and 250 nM allele‐specific primers (nucleotide changes of c.34G > T, c.34G > A, c.34G > C, c.35G > T, c.35G > A, c.35G > C, and c.38G > A, corresponding to amino acid changes of G12C, G12S, G12R, G12V, G12D, G12A, and G13D for KRAS and c.1799T > A corresponding to amino acid change of V600E for BRAF) (Lang et al., 2011). The following cycling conditions were used: 95 °C for 3 min, 40 cycles of 95 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s. For CTC, reactions containing 10 ng of template DNA were subjected to 50 cycles of amplification. PCR reactions were separated on a 2% agarose gel for visual confirmation of products.
2.7. Pyrosequencing
10 ng input DNA was amplified using PyroMark® PCR kit (Qiagen) and biotinylated primers (BRAF forward 5′‐TGAAGACCTCACAGTAAAAATAGGTG‐3′ and BRAF reverse 5′‐biotinCCACAAAATGGATCCAGACA‐3′; or KRAS forward 5′‐AGGCCTGCTGAAAATGACTG‐3′ and KRAS reverse 5′‐biotinCAAGATTTACCTCTATTGTTG‐3′) on the following PCR program: 95 °C for 15 min; 45 cycles of 94 °C for 30 s, 56 °C for 30 s, 72 °C for 30 s; and 72 °C for 5 min. The PCR product was incubated with Streptavidin Sepharose beads (GE Healthcare) and PyroMark® Binding Buffer (Qiagen) at room temperature for 30 min followed by hyridization to the sequencing primers. The pyrosequencing reaction was performed on the PyroMark® Q24 (Qiagen) platform using PyroMark® Gold Q24 reagents. The pyrosequencing result was analyzed using PyroMark® Q24 software. Samples with more than 5% mutated alleles were scored as positive.
2.8. Statistical analysis and calculation of sensitivity and specificity
Concordance in mutational status between tumor tissue and CTCs using the various approaches were evaluated in 2 × 2 contingency tables. Fisher's exact test of probability, odds ratio (OR) and 95% confidence interval (CI) were determined for each approach. Sensitivity was calculated as the number of positive samples as defined by Sanger sequencing divided by a sum of true‐positive and false‐negative samples, multiplied by 100. Specificity was calculated as number of true‐negative samples as defined by Sanger sequencing divided by the sum of true‐negative and false‐positive samples, multiplied by 100.
3. Results
DNA from matched normal tissues, tumor tissues and CTCs was extracted from each patient, and subjected to Sanger sequencing, HRM and ASPCR as illustrated in Figure 1.
3.1. Analytical sensitivity of the HRM and ASPCR assay
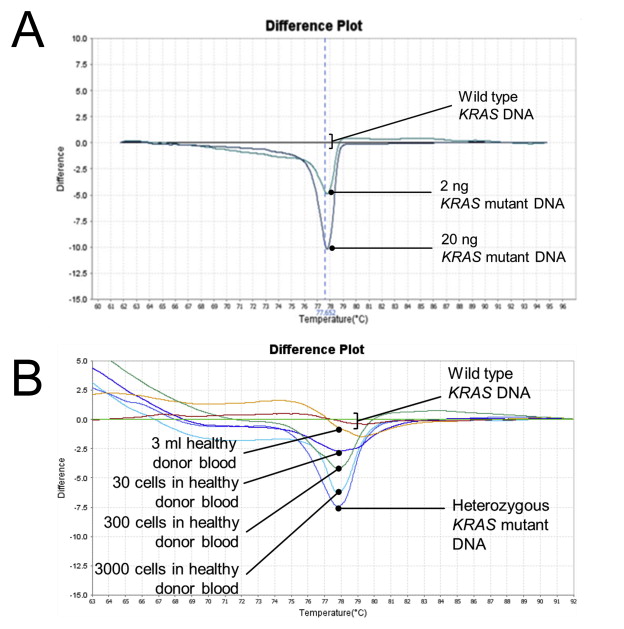
Genomic DNA of the heterozygous KRAS or BRAF mutant samples from DLD‐1 and HT29 cells were mixed in wild‐type DNA of a healthy donor (labeled as IBN‐A01) in various dilutions of mutant alleles. A non‐linear difference in fluorescence was observed with decreasing percentage of mutant alleles (Figure 2A). Both HRM and ASPCR could detect as low as 1.25% KRAS and BRAF mutant alleles (Figure 2B–E), which corresponded to 2.5% tumor cells carrying a heterozygous mutation. To investigate the lowest concentration of mutant DNA that could be reliably identified as being qualitatively present in the sample, the HRM assay was performed with 20 ng–0.2 ng of KRAS G13D mutant DNA template. KRAS mutation in non‐degraded samples could be detected using only 2 ng of mutant DNA (Supplementary Figure 1A). Finally, to determine the minimal number of mutant cells that can be reliably detected in healthy blood, known numbers of mutant cells were spiked in healthy blood and processed through the microsieve. Our data indicates that a minimal of 100 mutant cells needs to be captured on the microsieve for confident detection of mutation (Supplementary Figure 1B).
Figure 2.
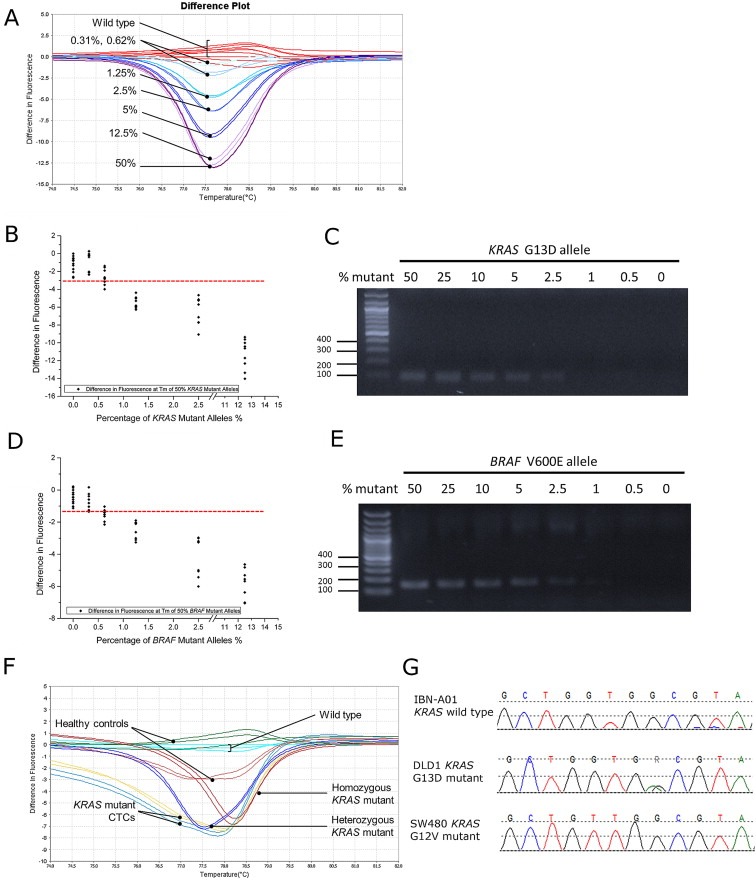
Sensitivity and specificity of HRM and ASPCR. (A) HRM sensitivity using varying percentages of mutant alleles. (B, C) HRM and ASPCR could detect as low as 1.25% mutant alleles for KRAS and (D, E) BRAF. (F) DNA extracted from blood of subjects without cancer was passed through the IBN microsieve and served as negative controls. (G) No KRAS mutation detected in subjects without cancer.
3.2. Specificity of KRAS and BRAF HRM assay
An accurate diagnostic assay that can correctly identify samples with mutations while excluding wild‐type samples without mutation is critical in clinical diagnosis. To establish specificity of the assays, 3 ml of blood from 18 subjects without known history of cancer was processed through the IBN microsieve. DNA extracted from the captured cells on the sieve was subjected to whole‐genome amplification and were used as negative control. Neither HRM nor ASPCR detected KRAS and BRAF mutations in the blood of these healthy subjects (Figure 2F and G), indicating 100% specificity for the tested samples. These results were confirmed by Sanger sequencing.
3.3. KRAS mutation detected in tissues and CTCs of CRC patients
44 patients with early‐ and advanced‐stage CRC were screened for mutations. DNA obtained from the matched normal and tumor tissues of each patient was profiled for mutations using Sanger sequencing, HRM and ASPCR whereas the CTCs were profiled using the latter two techniques. We first profiled KRAS mutations in tumor tissues; mutations were detected in 9 (20.4%) out of 44 patients by Sanger sequencing, in 12 (27.3%) patients by HRM analysis, and in 11 (25%) cases by ASPCR. Five different substitutions were detected at KRAS codon 12/13, with 82% mutations occurring at codon 12 as determined by ASPCR. The agreement between these 3 approaches can be found in Supplementary Table 2. HRM could identify patients with and without mutation, as validated by Sanger sequencing (Figure 3A–D). Using HRM and ASPCR, we were able to profile the mutation status of CTCs (Figure 3E and F), and observed 70% concordance in the mutation status of tumor tissues and CTCs from patients. Notably, the HRM and ASPCR approaches detected KRAS mutations in 2 patients who were classified as KRAS wild‐type by Sanger sequencing (Figure 3G–I). These results provide evidence that our technique is able to detect mutant KRAS in CTCs that mirror the primary tumor.
Figure 3.
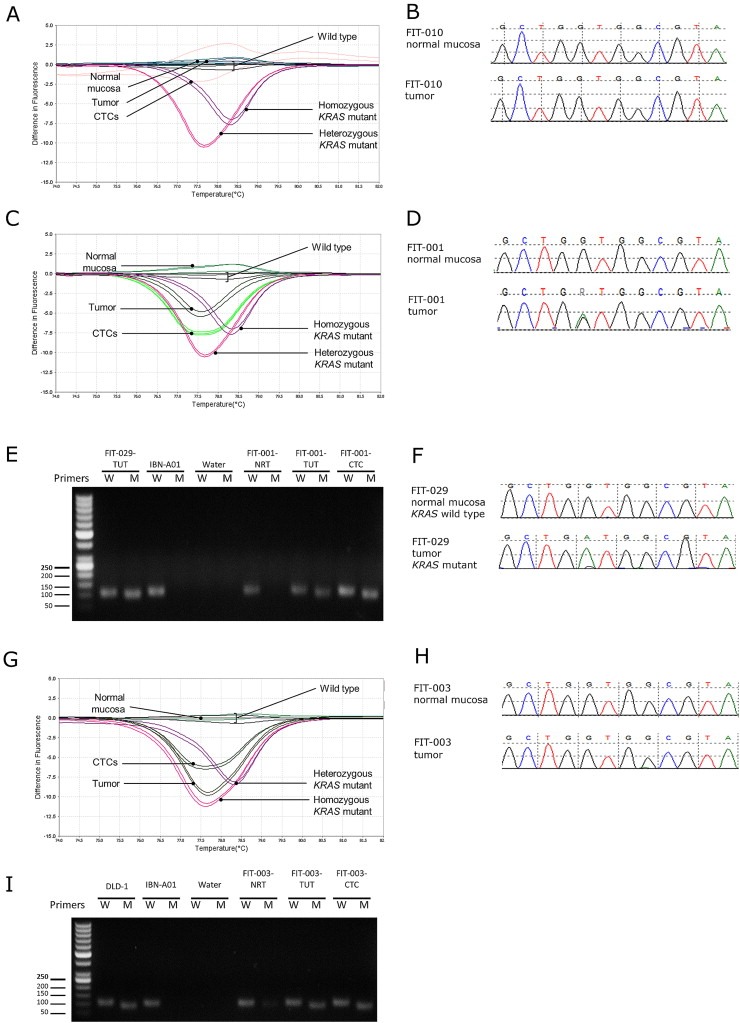
Detection of KRAS mutations in CTCs of CRC patients. Illustration of a patient with wild‐type KRAS using (A) HRM and (B) Sanger. KRAS mutation was detected in patient FIT‐001's tumor and CTCs by (C) HRM and (D) Sanger. (E) ASPCR identified the mutations in FIT‐001 and FIT‐029 as KRAS G12D, as verified by (F) Sanger. (G) KRAS mutation detected by HRM in a (H) Sanger sequencing‐negative patient FIT‐003. (I) ASPCR identified the mutation in FIT‐003 as KRAS G13D mutation.
3.4. BRAF mutation detected in tissues and CTCs of CRC patients
Mutation at codon 600 accounted for most of the BRAF mutations found in CRC (Yokota et al., 2011). In our study, BRAF mutation was detected in 1 (2.5%) out of 44 patients by conventional sequencing method, in 3 (7%) patients by HRM analysis, and in 2 (4.5%) cases by ASPCR. HRM and ASPCR were able to detect the mutant sample identified by Sanger sequencing (Figure 4A–C). Furthermore, both HRM and ASPCR could detect the mutation in the CTCs of this patient. However, we did not detect a BRAF mutation in the CTCs of 2 patients with BRAF‐mutant tumor tissues (Figure 4D–F).
Figure 4.
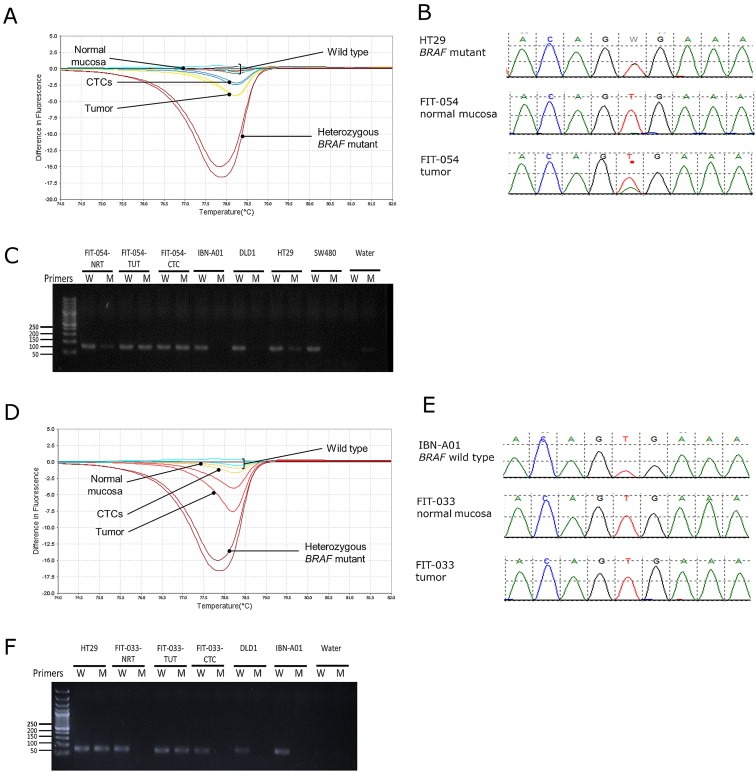
Detection of BRAF mutations in CTCs of CRC patients. (A) BRAF mutation detected by HRM in patient FIT‐054's tumor tissue and CTCs. (B) Sanger sequencing and (C) ASPCR identified the mutation in FIT‐054 as BRAF V600E. (D) HRM detected BRAF mutation in tumor tissues, but not in CTCs in a (E) sequencing‐negative clinical patient FIT‐033. (F) ASPCR identified the mutation in FIT‐033's tumor tissue as BRAF V600E, but no mutation was detected in CTCs.
3.5. Concordance in tumor genotyping between tumor tissues and CTCs using Sanger sequencing, HRM, ASPCR and pyrosequencing
DNA obtained from the normal tissues, tumor tissues and CTCs of each of the 44 patients was profiled for KRAS and BRAF mutations (See Supplementary Tables 2 and 3, respectively). For HRM, we obtained 84.1% concordance (p = 0.000129, Fishers' exact probability test) in KRAS genotype between tumor tissues and CTCs (OR = 38.7, 95% CI = 4.05–369) (Table 2). Similarly for the ASPCR approach, we obtained 70.5% concordance (p = 0.659) in KRAS genotype (OR = 2, 95% CI = 0.398–10.1). Despite having insufficient positive BRAF‐mutant cases, we observed high concordance of 90.9% (p = 0.175) and 100% (p = 0.0227) in BRAF genotype between tumor and CTCs using HRM and ASPCR approaches, respectively.
Table 2.
2 × 2 table of mutation status between tumor and CTCs.
| Tumor tissue (Sanger sequencing) | Concordance | 95% confidence interval | Odds ratio | 95% confidence interval | Fisher's exact probability test | |||
|---|---|---|---|---|---|---|---|---|
| Mutant | Wild‐type | |||||||
| CTCs (KRAS HRM assay) | Mutant | 8 | 1 | 84.1% | [73.3%–94.9%] | 38.7 | [4.05–369] | 0.000129 |
| Wild‐type | 6 | 29 | ||||||
| CTCs (KRAS ASPCR assay) | Mutant | 3 | 6 | 70.5% | [57.1%–83.4%] | 2 | [0.398–10.1] | 0.659 |
| Wild‐type | 7 | 28 | ||||||
| CTCs (BRAF HRM assay) | Mutant | 1 | 0 | 90.9% | [82.4%–99.4%] | NA | NA | 0.114 |
| Wild‐type | 4 | 39 | ||||||
| CTCs (BRAF ASPCR assay) | Mutant | 1 | 0 | 100.0% | NA | NA | NA | 0.0227 |
| Wild‐type | 0 | 43 | ||||||
Nonetheless, we observed that there were discrepancies in mutation status of several patients using the 3 methods described above. Using pyrosequencing, we were able to determine the presence of mutation with a minimal requirement of 5% mutant alleles that needs to be present in the tumor or CTCs before a positive call for mutation was made (See Supplementary Figure 2).
3.6. Sensitivity, specificity and predictive value of assays
The apparent sensitivity, specificity and predictive values of the HRM‐ and ASPCR‐ KRAS and BRAF assays were analyzed. Regardless of the approach used for detection of KRAS and BRAF mutations, we obtained 100% apparent sensitivity, with variable specificity of 91.4%–97.6%, depending on the approach and assay that was utilized (See Supplementary Tables 4 and 5).
4. Discussion
In the classical cancer paradigm, cell detachment and metastasis are the final steps in cancer progression, whereby tumor cells finally acquire sufficient mutations to endow them with the ability to escape from the primary tumor site and circulate freely in the bloodstream, before seeding at secondary sites (Fidler, 2003). Emerging data, however, suggest that metastasis initiation occurs as a relatively early event in tumor development (Mego et al., 2010); early dissemination of tumor cells has been observed before primary tumor became morphologically invasive (Hüsemann et al., 2008; Klein, 2009). The early dissemination of tumor cells has significant clinical and biological implications – it provides a potential source of tumor cells that can be obtained non‐invasively and analyzed at the molecular level. CTCs are thus regarded as “virtual” biopsy, providing a dynamic pool of genetic biomarkers during cancer progression.
Biomarkers are crucial to guide treatment decisions. In CRC, information on KRAS and BRAF genotype is extremely valuable in systemic chemotherapy; besides predicting the therapeutic efficiency of anti‐EGFR therapy, patients with poor prognosis can be identified. Herein we present an effective way to screen for KRAS and BRAF mutations in CRC patients using CTCs isolated from blood through microsieve filtration.
A recently published study aimed at molecular profiling of CTCs enriched using the CellSearch® system have demonstrated variable degree of success (Mostert et al., 2013). Mutations were assessed by cold‐PCR, real‐time PCR (EntroGen™), and nested allele‐specific blocker (ASB)‐PCR. Despite being unable to correlate tissue‐based mutational status with CTCs, the authors were able to detect KRAS and BRAF mutations in CTCs of 6 (14%) out of 43 CRC patients, with ASB‐PCR proving to be the most sensitive method for mutation detection. Interestingly, 5 out of 6 patients with mutations had CTC counts above 3 in 30 ml of blood. Thus, they concluded that improvements in both CTC enrichment and mutational analysis approaches were necessary to extract mutational data from the majority of the patients who has less than 3 CTCs. In contrast, the IsoFlux System isolated more than 4 CTCs in 87% of their 15‐patient CRC cohort (Harb et al., 2013). By using the competitive allele‐specific Taqman® PCR method (Life Technologies), they obtained a KRAS mutant rate of 50%, with 46% of patients displaying a CTC KRAS mutational status that differed from their previously acquired tissue biopsy data. Together, these studies indicate that a combination of good CTC enrichment step and sensitive molecular profiling approaches is key to successful non‐invasive molecular profiling.
To the best of our knowledge, both the HRM and ASPCR assays that we have presented here offers a novel non‐invasive genetic assay for tumor profiling, providing us the means to verify the presence of CTCs through molecular approach. In our study, HRM clearly has an edge over sequencing methods in terms of ease of use, speed and sensitivity in scanning for mutations in amplicon of interest, while ASPCR is sensitive and could be quantitative. The performance of our size‐based CTCs enrichment platform is critical in the successful detection of mutation in CTCs from only 3 ml of blood using the assays that we have developed. Remarkably, we able to observe a high concordance in mutation status between the tumor tissues and CTCs, which might have several major implications: (i) the HRM and ASPCR approaches have adequate sensitivity to detect low abundance mutation in CTCs amidst the high background of wild‐type normal blood cells, (ii) CTCs originate from the primary tumor since they have the same tumor genotype, and (iii) CTCs are potentially present in non‐metastatic CRC patients. Detection of mutations in CTCs provides the opportunity to profile this rare population of cells as a mean to genotype tumor non‐invasively.
Interestingly, we observed an increased detection of mutation in the blood which was not readily detected in the tumor. On the other hand, we were unable to detect mutation in some of the CTCs, despite the presence of mutation in the tumor. To resolve this disparity between tumor and CTCs, we proposed that microdissection of different areas of the same primary tumor could be taken and compared to the respective CTCs in the same patients. Since clinical biopsy samples often contain populations of tumor cells that present intra‐ and inter‐tumor heterogeneity at the genetic level, mutation could be present at periphery of tumor but not in the core of the tumor, and vice versa (Kosmidou et al., 2014). Therefore, it is conceivable that we would expect similar discordance in genotypes, especially between the tumor and CTCs, which has been described by several published studies (Harb et al., 2013; Mostert et al., 2013; Powell et al., 2012; Raimondi et al., 2014). Furthermore, a recent study had found significant heterogeneity within single CTCs from the same patient (Gasch et al., 2013), and it is conceivable that the pooled CTCs that we have captured would contain a mixed population of CTCs along with the contaminating leukocytes.
We also acknowledged that there may be limitations in our microsieve technology, resulting in variable performance during the enrichment of CTCs, which could influence the number and purity of CTCs captured. Likewise, we also recognize that there could be some technical limitations in our HRM and ASPCR approaches. While it is clear that HRM is useful in detecting a change in nucleotide and detecting unknown changes in amplicon of interest, single nucleotide polymorphisms could not be distinguished from mutations, invariably resulting in false‐positives calls. Also, unlike ASPCR, HRM is non‐quantitative; thus, quantification of the percentage of mutant alleles in the sample is not possible. Alternative methods such as pyrosequencing, which allows detection and quantification of mutant alleles, could also be used. We have employed the pyrosequencing platform in an attempt to verify the source of discrepancy in mutation status in several of our patients. Representative examples of pyrograms, along with PCR, HRM and ASPCR results indicate that the sensitivity of these methods are important factors in deciding a positive call for mutation. While having a high‐sensitivity enables detection of rare mutant alleles, this may increase the false‐positive rates.
Despite these limitations that have been discussed, it is apparent that our CTC enrichment platform, coupled with the HRM and ASPCR approaches, exhibit high sensitivity and specificity in mutation detection in CRC patients. The ease of scanning for mutations using our approaches offers the added advantage of speed and simplicity in assessment of mutation in CTCs with minimal blood sample requirement. Ideally, CTCs would be useful as a liquid biopsy to select a specific personalized therapy on the basis of the molecular features of a patient's tumor. CTCs could present as complementary biomarkers for identification of predictive targets and real‐time monitoring of disease in clinical practice. The assays that we have described could serve as a valuable tool to identify mutations in CRC patients non‐invasively.
5. Conclusion
Mutation analysis of clinical samples involve the use of legacy material, and testing of circulating tumor cells (CTCs) obtained from peripheral blood may allow non‐invasive evaluation of a patient's tumor status. Herein we have systematically tested this approach using a label‐free enrichment platform to capture CTCs from blood in a consecutive series of patients undergoing surgery. Coupled with the application of our optimized PCR‐based assays to detect KRAS or BRAF mutation, we have shown that CTC genotyping may be achieved with high sensitivity and specificity. This approach enables a rapid, qualitative and non‐invasive assessment of tumor genotype in the clinical setting, allowing better treatment decisions.
Funding
This work is supported by the Institute of Bioengineering and Nanotechnology, Biomedical Research Council (Diagnostics Grant and Strategic Positioning Fund), and Agency for Science, Technology and Research, Singapore.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
We thank Fortis Surgical Hospital and the Fortis‐IBN Tissue Bank for their support in provision of samples.
1.
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.molonc.2014.12.011.
Mohamed Suhaimi Nur-Afidah, Foong Yu Miin, Lee Daniel Yoke San, Phyo Wai Min, Cima Igor, Lee Esther Xing Wei, Goh Wei Lin, Lim Wei-Yen, Chia Kee Seng, Kong Say Li, Gong Min, Lim Bing, Hillmer Axel M., Koh Poh Koon, Ying Jackie Y., Tan Min-Han, (2015), Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients, Molecular Oncology 9, doi: 10.1016/j.molonc.2014.12.011.
References
- Allard, W.J. , Matera, J. , Miller, M.C. , Repollet, M. , Connelly, M.C. , Rao, C. , Tibbe, A.G. , Uhr, J.W. , Terstappen, L.W. , 2004. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 10, 6897–6904. [DOI] [PubMed] [Google Scholar]
- Asghar, U. , Hawkes, E. , Cunningham, D. , 2010. Predictive and prognostic biomarkers for targeted therapy in metastatic colorectal cancer. Clin. Colorectal Cancer 9, 274–281. [DOI] [PubMed] [Google Scholar]
- Bihl, M.P. , Hoeller, S. , Andreozzi, M.C. , Foerster, A. , Rufle, A. , Tornillo, L. , Terracciano, L. , 2012. KRAS mutation testing in colorectal cancer: comparison of the results obtained using 3 different methods for the analysis of codons G12 and G13. Diagn. Mol. Pathol. 21, 14–23. [DOI] [PubMed] [Google Scholar]
- Cima, I. , Chay, W.Y. , Iliescu, F.S. , Phyo, W.M. , Lim, K.H. , Iliescu, C. , Tan, M.H. , 2013. Label-free isolation of circulating tumor cells in microfluidic devices: current research and perspectives. Biomicrofluidics 7, 11810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roock, W. , Claes, B. , Bernasconi, D. , De Schutter, J. , Biesmans, B. , Fountzilas, G. , Kalogeras, K.T. , Kotoula, V. , Papamichael, D. , Laurent-Puig, P. , Penault-Llorca, F. , Rougier, P. , Vincenzi, B. , Santini, D. , Tonini, G. , Cappuzzo, F. , Frattini, M. , Molinari, F. , Saletti, P. , De Dosso, S. , Martini, M. , Bardelli, A. , Siena, S. , Sartore-Bianchi, A. , Tabernero, J. , Macarulla, T. , Di Fiore, F. , Gangloff, A.O. , Ciardiello, F. , Pfeiffer, P. , Qvortrup, C. , Hansen, T.P. , Van Cutsem, E. , Piessevaux, H. , Lambrechts, D. , Delorenzi, M. , Tejpar, S. , 2010. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 11, 753–762. [DOI] [PubMed] [Google Scholar]
- Feng, Y.-Z. , Shiozawa, T. , Miyamoto, T. , Kashima, H. , Kurai, M. , Suzuki, A. , Konishi, I. , 2005. BRAF mutation in endometrial carcinoma and hyperplasia: correlation with KRAS and p53 mutations and mismatch repair protein expression. Clin. Cancer Res. 11, 6133–6138. [DOI] [PubMed] [Google Scholar]
- Fidler, I.J. , 2003. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat. Rev. Cancer 3, 453–458. [DOI] [PubMed] [Google Scholar]
- Franklin, W.A. , Haney, J. , Sugita, M. , Bemis, L. , Jimeno, A. , Messersmith, W.A. , 2010. KRAS mutation: comparison of testing methods and tissue sampling techniques in colon cancer. J. Mol. Diagn.: JMD 12, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch, C. , Bauernhofer, T. , Pichler, M. , Langer-Freitag, S. , Reeh, M. , Seifert, A.M. , Mauermann, O. , Izbicki, J.R. , Pantel, K. , Riethdorf, S. , 2013. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal Cancer. Clin. Chem. 59, 252–260. [DOI] [PubMed] [Google Scholar]
- Harb, W. , Fan, A. , Tran, T. , Danila, D.C. , Keys, D. , Schwartz, M. , Ionescu-Zanetti, C. , 2013. Mutational analysis of circulating tumor cells using a novel microfluidic collection device and qPCR assay. Transl. Oncol. 6, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüsemann, Y. , Geigl, J.B. , Schubert, F. , Musiani, P. , Meyer, M. , Burghart, E. , Forni, G. , Eils, R. , Fehm, T. , Riethmüller, G. , Klein, C.A. , 2008. Systemic spread is an early step in breast cancer. Cancer Cell 13, 58–68. [DOI] [PubMed] [Google Scholar]
- Karapetis, C.S. , Khambata-Ford, S. , Jonker, D.J. , O'Callaghan, C.J. , Tu, D. , Tebbutt, N.C. , Simes, R.J. , Chalchal, H. , Shapiro, J.D. , Robitaille, S. , Price, T.J. , Shepherd, L. , Au, H.-J. , Langer, C. , Moore, M.J. , Zalcberg, J.R. , 2008. K-ras mutations and benefit from cetuximab in advanced colorectal Cancer. New Engl. J. Med. 359, 1757–1765. [DOI] [PubMed] [Google Scholar]
- Klein, C.A. , 2009. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 9, 302–312. [DOI] [PubMed] [Google Scholar]
- Kosmidou, V. , Oikonomou, E. , Vlassi, M. , Avlonitis, S. , Katseli, A. , Tsipras, I. , Mourtzoukou, D. , Kontogeorgos, G. , Zografos, G. , Pintzas, A. , 2014. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA Pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum. Mutat. 35, 329–340. [DOI] [PubMed] [Google Scholar]
- Lang, A.H. , Drexel, H. , Geller-Rhomberg, S. , Stark, N. , Winder, T. , Geiger, K. , Muendlein, A. , 2011. Optimized allele-specific real-time PCR assays for the detection of common mutations in KRAS and BRAF. J. Mol. Diagn.: JMD 13, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbeck, 1841. On the development of cancer in the veins, and the transmission of cancer from man to the lower animals. Edinb. Med. Surg. J. 55, 251–253. [Google Scholar]
- Laurent-Puig, P. , Cayre, A. , Manceau, G. , Buc, E. , Bachet, J.-B. , Lecomte, T. , Rougier, P. , Lievre, A. , Landi, B. , Boige, V. , Ducreux, M. , Ychou, M. , Bibeau, F. , Bouché, O. , Reid, J. , Stone, S. , Penault-Llorca, F. , 2009. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 27, 5924–5930. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Lee, I. , Han, B. , Park, J.O. , Jang, J. , Park, C. , Kang, W.K. , 2011. Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations. J. Natl. Cancer Inst. 103, 674–688. [DOI] [PubMed] [Google Scholar]
- Lim, L.S. , Hu, M. , Huang, M.C. , Cheong, W.C. , Gan, A.T.L. , Looi, X.L. , Leong, S.M. , Koay, E.S.-C. , Li, M.-H. , 2012. Microsieve lab-chip device for rapid enumeration and fluorescence in situ hybridization of circulating tumor cells. Lab Chip 12, 4388–4396. [DOI] [PubMed] [Google Scholar]
- Mego, M. , Mani, S.A. , Cristofanilli, M. , 2010. Molecular mechanisms of metastasis in breast cancer–clinical applications. Nat. Rev. Clin. Oncol. 7, 693–701. [DOI] [PubMed] [Google Scholar]
- Modest, D.P. , Stintzing, S. , Laubender, R.P. , Neumann, J. , Jung, A. , Giessen, C. , Haas, M. , Aubele, P. , Schulz, C. , Boeck, S. , Stemmler, H.J. , Kirchner, T. , Heinemann, V. , 2011. Clinical characterization of patients with metastatic colorectal cancer depending on the KRAS status. Anticancer Drugs 22, 913–918. [DOI] [PubMed] [Google Scholar]
- Mostert, B. , Jiang, Y. , Sieuwerts, A.M. , Wang, H. , Bolt-de Vries, J. , Biermann, K. , Kraan, J. , Lalmahomed, Z. , van Galen, A. , de Weerd, V. , van der Spoel, P. , Ramírez-Moreno, R. , Verhoef, C. , Ijzermans, J.N.M. , Wang, Y. , Gratama, J.-W. , Foekens, J.A. , Sleijfer, S. , Martens, J.W.M. , 2013. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int. J. Cancer 133, 130–141. [DOI] [PubMed] [Google Scholar]
- Paget, J. , 1853. Lectures on Surgical Pathology second ed. Longrnan, Brown,Green, & Longman; London: 536–581. [Google Scholar]
- Peeters, M. , Oliner, K.S. , Parker, A. , Siena, S. , Van Cutsem, E. , Huang, J. , Humblet, Y. , Van Laethem, J.-L. , André, T. , Wiezorek, J. , Reese, D. , Patterson, S.D. , 2013. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal Cancer. Clin. Cancer Res. 19, 1902–1912. [DOI] [PubMed] [Google Scholar]
- Powell, A.A. , Talasaz, A.H. , Zhang, H. , Coram, M.A. , Reddy, A. , Deng, G. , Telli, M.L. , Advani, R.H. , Carlson, R.W. , Mollick, J.A. , Sheth, S. , Kurian, A.W. , Ford, J.M. , Stockdale, F.E. , Quake, S.R. , Pease, R.F. , Mindrinos, M.N. , Bhanot, G. , Dairkee, S.H. , Davis, R.W. , Jeffrey, S.S. , 2012. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One 7, e33788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, T.J. , Hardingham, J.E. , Lee, C.K. , Weickhardt, A. , Townsend, A.R. , Wrin, J.W. , Chua, A. , Shivasami, A. , Cummins, M.M. , Murone, C. , Tebbutt, N.C. , 2011. Impact of KRAS and BRAF gene gutation status on outcomes from the phase III AGITG MAX trial of capecitabine alone or in combination with bevacizumab and mitomycin in advanced colorectal cancer. J. Clin. Oncol. 29, 2675–2682. [DOI] [PubMed] [Google Scholar]
- Raimondi, C. , Nicolazzo, C. , Gradilone, A. , Giannini, G. , De Falco, E. , Chimenti, I. , Varriale, E. , Hauch, S. , Plappert, L. , Cortesi, E. , Gazzaniga, P. , 2014. Circulating tumor cells: exploring intratumor heterogeneity of colorectal cancer. Cancer Biol. Ther. 15, 0–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman, S.D. , Seymour, M.T. , Chambers, P. , Elliott, F. , Daly, C.L. , Meade, A.M. , Taylor, G. , Barrett, J.H. , Quirke, P. , 2009. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J. Clin. Oncol. 27, 5931–5937. [DOI] [PubMed] [Google Scholar]
- Sgambato, A. , Casaluce, F. , Maione, P. , Rossi, A. , Rossi, E. , Napolitano, A. , Palazzolo, G. , Bareschino, M. , Schettino, C. , Sacco, P.C. , Ciardiello, F. , Gridelli, C. , 2012. The role of EGFR tyrosine kinase inhibitors in the first-line treatment of advanced non small cell lung cancer patients harboring EGFR mutation. Curr. Med. Chem. 19, 3337–3352. [DOI] [PubMed] [Google Scholar]
- Simi, L. , Pratesi, N. , Vignoli, M. , Sestini, R. , Cianchi, F. , Valanzano, R. , Nobili, S. , Mini, E. , Pazzagli, M. , Orlando, C. , 2008. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am. J. Clin. Pathol. 130, 247–253. [DOI] [PubMed] [Google Scholar]
- Spizzo, G. , Fong, D. , Wurm, M. , Ensinger, C. , Obrist, P. , Hofer, C. , Mazzoleni, G. , Gastl, G. , Went, P. , 2011. EpCAM expression in primary tumour tissues and metastases: an immunohistochemical analysis. J. Clin. Pathol. 64, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiatis, A.C. , Norris-Kirby, A. , Rich, R.G. , Hafez, M.J. , Gocke, C.D. , Eshleman, J.R. , Murphy, K.M. , 2010. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J. Mol. Diagn.: JMD 12, 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota, T. , Ura, T. , Shibata, N. , Takahari, D. , Shitara, K. , Nomura, M. , Kondo, C. , Mizota, A. , Utsunomiya, S. , Muro, K. , Yatabe, Y. , 2011. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br. J. Cancer 104, 856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, Z. , Chen, S.S. , Chandra, P.K. , Galbincea, J.M. , Soape, M. , Doan, S. , Barkoh, B.A. , Koeppen, H. , Medeiros, L.J. , Luthra, R. , 2009. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod. Pathol. 22, 1023–1031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data