

Abstract
Chemoresistance is the main obstacle to cancer cure. Contrasting studies focusing on single gene mutations, we hypothesize chemoresistance to be due to inactivation of key pathways affecting cellular mechanisms such as apoptosis, senescence, or DNA repair. In support of this hypothesis, we have previously shown inactivation of either TP53 or its key activators CHK2 and ATM to predict resistance to DNA damaging drugs in breast cancer better than TP53 mutations alone. Further, we hypothesized that redundant pathway(s) may compensate for loss of p53‐pathway signaling and that these are inactivated as well in resistant tumour cells. Here, we assessed genetic alterations of the retinoblastoma gene (RB1) and its key regulators: Cyclin D and E as well as their inhibitors p16 and p27. In an exploratory cohort of 69 patients selected from two prospective studies treated with either doxorubicin monotherapy or 5‐FU and mitomycin for locally advanced breast cancers, we found defects in the pRB‐pathway to be associated with therapy resistance (p‐values ranging from 0.001 to 0.094, depending on the cut‐off value applied to p27 expression levels). Although statistically weaker, we observed confirmatory associations in a validation cohort from another prospective study (n = 107 patients treated with neoadjuvant epirubicin monotherapy; p‐values ranging from 7.0 × 10−4 to 0.001 in the combined data sets). Importantly, inactivation of the p53‐and the pRB‐pathways in concert predicted resistance to therapy more strongly than each of the two pathways assessed individually (exploratory cohort: p‐values ranging from 3.9 × 10−6 to 7.5 × 10−3 depending on cut‐off values applied to ATM and p27 mRNA expression levels). Again, similar findings were confirmed in the validation cohort, with p‐values ranging from 6.0 × 10−7 to 6.5 × 10−5 in the combined data sets. Our findings strongly indicate that concomitant inactivation of the p53‐ and pRB‐ pathways predict resistance towards anthracyclines and mitomycin in breast cancer in vivo.
Keywords: p53, pRB, Resistance, Breast cancer
Highlights
Alterations of pRB's upstream regulators may substitute for RB1 mutations.
The pRB‐pathway may direct response to chemotherapy.
Inactivation of the p53‐and the pRB‐pathways predict resistance to chemotherapy.
Concomitant p53‐and pRB‐pathway inactivation is a strong resistance predictor.
Concomitant p53‐and pRB‐pathway inactivation predicts poor prognosis.
1. Introduction
Resistance towards chemotherapy remains the main reason for treatment failure and death among cancer patients. Considering breast cancers exposed to optimal adjuvant chemo‐ and/or endocrine therapy, on average, the death rate is reduced by about 30–50% (Albain et al., 2012). In metastatic disease, however, the effect of therapy is temporary, and resistance in general develops within less than one year for each individual regimen (Alba et al., 2004; Greenberg et al., 1996).
Despite extensive experimental research, the mechanisms underlying resistance to chemotherapy in vivo remains poorly understood. Regarding treatment with anthracyclines in breast cancer, topo‐II amplifications have been associated with a dose‐responsiveness different from what is observed in non‐amplified tumours (Knoop et al., 2005; Tanner et al., 2006), but no single factor predictive of drug resistance has reached clinical application. Breast cancers may be stratified into different biological subgroups based on their global gene expression profiles (Perou et al., 2000; Sorlie et al., 2001). These subgroups differ in their mutational profile and may respond differently to chemotherapy (Hugh et al., 2009; Rouzier et al., 2005; Sorlie et al., 2006). However, resistance to given therapy regimens may not be predicted based on gene expression signatures. Rather, the results, so far, are more consistent with the underlying mechanisms of drug resistance being unequally distributed between the subclasses. Similarly, other gene expression signatures have revealed correlations to outcome but to be modest with respect to predicting drug resistance (Albain et al., 2010; Paik et al., 2006; Straver et al., 2010).
To explore potential mechanisms of anthracycline resistance in breast cancer, we have undertaken a different approach, searching for defects in key functional pathways regulating vital cellular functions like growth arrest, DNA repair and apoptosis (Lonning et al., 2013). It is well established that p53 (the protein encoded by the TP53 gene) plays a key role in executing DNA damage induced apoptosis and growth arrest (Enoch et al., 1995). Previously, we reported mutations in the L2 and L3 domains of p53 to be associated with resistance to chemotherapy with anthracycline monotherapy (Chrisanthar et al., 2008; Geisler et al., 2001) or mitomycin plus 5‐fluoro‐uracil in combination (Geisler et al., 2003) and similar results have been reported by others (Kandioler‐Eckersberger et al., 2000). The finding that these TP53 mutations were highly associated with, but not fully predictive for, chemoresistance made us postulate that inactivation of other genes involved in the p53 pathway could substitute for TP53 mutations and generate a similar chemoresistant phenotype (Lonning, 2004). In support of our hypothesis, we found nonsense mutations in the CHEK2 gene and low expression levels of ATM (the two factors mainly responsible for activating p53 in response to genotoxic stress (Buscemi et al., 2004; Chehab et al., 1999; Hirao et al., 2000)) to substitute for TP53 mutations predicting resistance toward DNA damaging drugs in vivo (Chrisanthar et al., 2008; Knappskog et al., 2012).
The findings that inactivation of the p53‐pathway (ATM – CHK2 – p53) predicts resistance to chemotherapy while similar inactivation is also observed in some chemosensitive tumours made us hypothesize that response to therapy may be executed through redundant pathways (Lonning, 2004; Lonning et al., 2007). In this respect, several lines of evidence indicate a role for the retinoblastoma protein (pRB), a key regulator of cell cycle arrest upon DNA damage (Harrington et al., 1998). Concomitant inactivation of p53 and pRB leads to immortalization of keratinocytes (Smeets et al., 2011) as well as lymphoma development and chemoresistance in a transgenic mouse model (Schmitt et al., 2002). While mutations affecting the RB1 gene are rare in breast cancer, we have previously found such mutations associated with a poor response to anthracycline chemotherapy in both primary and metastatic breast cancer (Berge et al., 2010, 2011).
In the present study, we found inactivation of the pRB‐pathway to predict resistance to therapy with DNA damaging agents in patients suffering from locally advanced breast cancer (Figure 1). Importantly, we found concomitant inactivation of both the p53‐pathway and the pRB‐pathway to be more strongly associated with chemoresistance as compared to inactivation of each pathway alone. Based on these findings, we propose a model where in vivo resistance to DNA damaging drugs occurs as a consequence of concomitant inactivation of both the p53‐ and the pRB‐ pathways.
Figure 1.
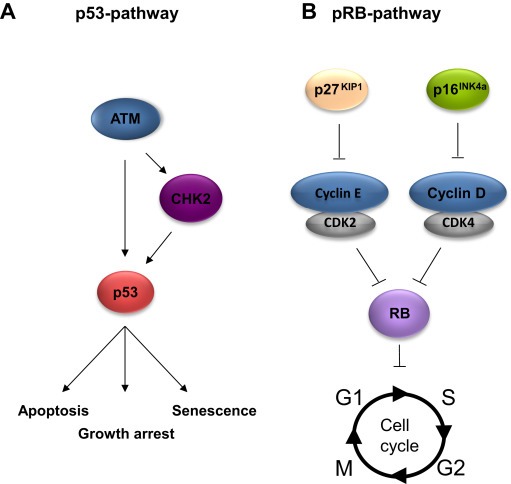
Schematic illustrations of the p53 (A) and the pRB (B) functional pathways.
2. Materials and methods
2.1. Patients and evaluation of response
This study included an exploratory cohort (n = 69) (Knappskog et al., 2012), containing patients from two single institution studies combined (Geisler et al., 2001, 2003), and a validation cohort (n = 107), based on patients from a multicenter study (Chrisanthar et al., 2008; Knappskog et al., 2012). All patients suffered from locally advanced breast cancers. The study protocols were approved by the regional ethical committee and all patients provided informed consent. Tumour samples were obtained prior to chemotherapy by an open biopsy, and all tissue samples were snap‐frozen in liquid nitrogen immediately upon excision in the operating theatre. The patients for this study were enrolled between 1991 and 2001, therefore, the responses were consistently graded by the UICC system (Hayward et al., 1977) and not the more recently implemented “RECIST” criteria (Therasse et al., 2000). For further details on response evaluation criteria and patient cohorts, see Supplementary information.
2.2. Nucleic acid purification
Genomic DNA was isolated by using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer's instructions. Total RNA was purified using Trizol (Life Technologies) extraction and cDNA was synthesized by reverse transcription using the Transcriptor reverse transcriptase system (Roche) according to the manufacturer's instructions, using both Oligo‐T and random hexamers as primers.
2.3. Mutation screening
Point mutations and indels were analyzed for RB1, CDKN2A (p16) and CDKN1B (p27) by PCR amplification and Sanger sequencing. Primers and reaction conditions for RB1 and CDKN2A were as previously described (Berge et al., 2010; Knappskog et al., 2006). The p27 coding region (CDKN1B) was amplified using the primer pairs listed in Supplementary Table S1. Amplifications were carried out using the DyNazyme DNA EXT polymerase (FINNZYMES) according to the manufacturer's instructions. For detailed protocol, see Supplementary information.
2.4. DNA sequencing
DNA sequencing was performed using Big Dye terminator mixture (Applied Biosystems) and capillary gel electrophoresis on an ABI 3700 DNA sequencer (Perkin–Elmer Biosystems). For detailed protocol, see Supplementary information.
2.5. Quantitative PCR
Levels of p27 mRNA were determined using hydrolysis probe‐assays on the LightCycler 480 system (Roche). All analyses were performed as duplex reactions amplifying the gene of interest in the same reaction well as the ribosomal protein P2 (rpP2), as internal reference. All obtained Cp values were converted to relative concentrations via internal standard curves in each run. Each analysis was performed in triplicate. For detailed protocol, see Supplementary information. ATM mRNA levels were extracted from previously published data (Knappskog et al., 2012).
2.6. Methylation specific PCRs (MSP)
For promoter methylation analyses, genomic DNA from tumour samples were subjected to methylated‐ and unmethylated‐specific PCRs, using the AmpliTaq Gold DNA Polymerase (Applied Biosystems) after bisulfite modification using the CpGenome DNA Modification Kit (Intergen). For detailed protocol, see Supplementary information.
2.7. Fluorescence in situ hybridization (FISH)
All samples in the exploratory cohort were analyzed for CCND1 and CCNE1 amplifications by fluorescence in situ hybridization on sections from formalin fixed and paraffin embedded tumour tissue. Samples were scored as amplified if the ratio between gene specific (CCND1 and CCNE1) and a centromeric reference was higher than 2. (For detailed protocol, see Supplementary information).
2.8. MLPA
Copy number alterations for RB1, CCND1 and CCNE1 (validation cohort) and CDKN2A were assessed by MLPA. Analyses were performed using the SALSA MLPA RB1, the P078‐Breast tumour and the CDKN2A/2B probemix‐kits respectively (MRC‐Holland, Amsterdam, The Netherlands), as previously described (Berge et al., 2010; Knappskog et al., 2006). In brief, in the patient samples, the peak areas of all MLPA products resulting from probes specific for the genes of interest were first normalized by the average of peak areas resulting from control probes (specific for locations other than the chromosome harbouring the gene of interest). A ratio was then calculated where this normalized value was divided by the corresponding value from a sample consisting of pooled DNA from >6 healthy individuals. A sample was scored as having a reduced copy number at a specific location if this ratio was below 0.75, and as having an increased copy number if the ratio was above 1.25.
2.9. Statistical analyses
Samples were assessed for potential factors predicting drug resistance by comparing tumours progressing on therapy (PD) to the combined group of tumours classified as SD/PR/CR (responders), as previously described (Chrisanthar et al., 2008, 2001, 2003, 2012). The theoretical rationale for this approach has been outlined in detail elsewhere (Lønning, 2003).
For TP53, mutations affecting the L2/L3 domains (including nonsense and frameshift mutations upstream of the sequence encoding these domains) were included in the model based on previous findings (Chrisanthar et al., 2008, 2001, 2003, 2012). For CHEK2, mutations previously found to maintain wild‐type kinase activity were excluded (Chrisanthar et al., 2008). Since the thresholds for what may be regarded as pathologically low levels of ATM and p27 are not known, we applied a set of different cut‐offs, ranging from the lower 50% to the lower 5% of the patients in each cohort (ranked by their expression levels).
Comparison of mutations found (binary variable comparisons) between the different groups of patients were performed using Fisher's Exact Test, while mRNA expression levels were compared using the Mann–Whitney rank test for independent samples.
Multivariate models were calculated using binary logistic regression analyses defining mutation status for the different genes as categorical variables and mRNA expression levels as continuous variables.
Comparisons between patient groups with respect to relapse‐free survival and disease specific survival were performed by Log‐rank tests. For all patients included (both in the exploratory and in the validation cohorts) follow‐up data for up to 10 years or time of death was available. As for these calculations, we used the median value of ATM and p27 expression within each cohort as cut‐off.
All p‐values are given as two‐sided, and the p‐values obtained by Fischer exact tests are given as cumulative. All statistical analyses were performed using the SPSS software version 19 (IBM).
3. Results
3.1. Patient cohorts and treatment
This study contains an exploratory cohort consisting of 69 patients selected from two prospective studies assessing factors predictive for response to weekly doxorubicin monotherapy (n = 36) (Geisler et al., 2001) or mitomycin in concert with 5‐fluoro‐uracil (n = 33) (Geisler et al., 2003), as outlined in materials and methods and Figure 2.
Figure 2.
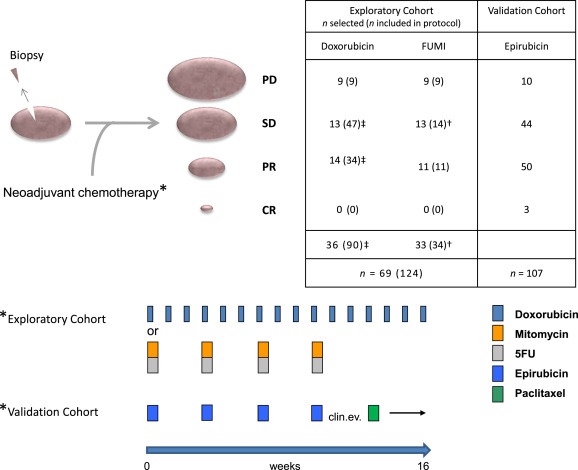
Study design and treatment regimens of included patients. All patients were diagnosed with locally advanced breast cancers and subject to biopsy for genetic analyses before commencement of neoadjuvant chemotherapy. Post therapy, all patients were evaluated for primary response to therapy before surgery (CR = Complete response, PR = Partial Response, SD = Stable Disease, PD = Progressive Disease, see Supplementary Information for details). In the exploratory cohort (n = 69), 36 of the patients were originally selected from the 90 patients enrolled in a prospective study assessing resistance to treatment with doxorubicin in locally advanced breast cancer (‡ selection of all patients with PD along with representative control groups of SD and PR). These patients received weekly doxorubicin for 16 weeks or until progressive disease was recorded. The remaining 33 patients were from a prospective study assessing resistance to mitomycin and 5‐fluorouracil (FUMI; † one patient was omitted due to lack of biological material). These patients received mitomycin and FUMI every third week (four cycles). Patients in the validation cohort received epirubicin every third week (four cycles) before clinical evaluation of response and change of therapy to paclitaxel every third week (four cycles) in case of an inferior response to the first line treatment. For further information regarding the cohorts, see Supplementary Information.
In addition, we studied a validation cohort, containing 107 patients treated with epirubicin monotherapy on a 3‐weekly basis for locally advanced breast cancer in a prospective study (Chrisanthar et al., 2008).
Detailed descriptions of these cohorts are given elsewhere (Chrisanthar et al., 2008, 2001, 2003, 2012) and in the Supplementary information.
3.2. The p53‐pathway and chemoresistance
All findings are summarized in Figure 3 and Supplementary Tables S3A and S3B. Among the patients included in the exploratory cohort, we previously reported mutations in TP53 (L2/L3 domains) or CHEK2 in 12 out of 18 patients with progressive disease (PD), contrasting 16 out of 51 revealing an objective response or stable disease upon therapy (p = 0.0123). This strong link between p53 inactivation and drug resistance was further corroborated by including ATM expression levels in the model (association between defects in the ATM – CHK2 – p53 pathway and PD; p‐values varying between 0.027 and 0.001 depending on the percentile used to define ATM levels as “low”; p = 0.010, binary logistic multivariate model; Figure 2A; Supplementary Table 4) (Knappskog et al., 2012).
Figure 3.
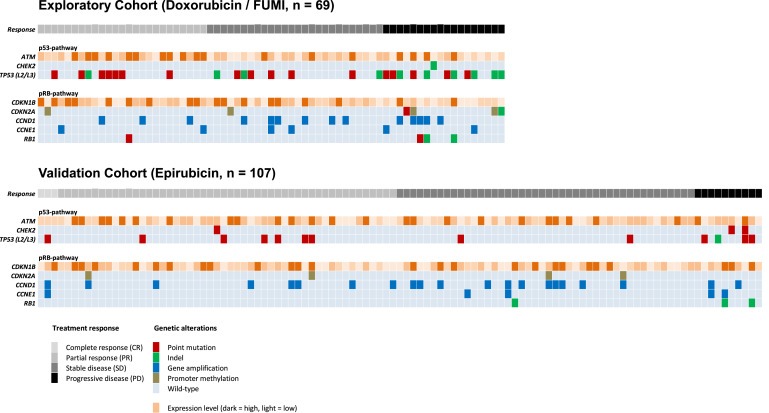
Graphical summary of the detected genetic alterations. Each column represent one of the 176 patients analyzed (Exploratory Cohort; n = 69 and Validation Cohort; n = 107) while each row represent one of the analyzed genes in the p53‐or the pRB‐pathway. Treatment response and genetic alterations for each individual patient is given as colored squares according to the color‐key (bottom panel). A detailed summary of these findings are listed in Supplementary Tables S3A and S3B.
These findings were confirmed in the validation cohort (Chrisanthar et al., 2008; Knappskog et al., 2012) where 5 out of 10 patients with progressive disease harboured mutations in TP53 (L2/L3 domains) or CHEK2, while corresponding numbers among therapy responders were 10 out of 97 patients (p = 0.005). Similar to what was recorded in the exploratory cohort, including ATM expression strengthened the observed association (association between defects in the ATM – CHK2 – p53‐pathway and PD; p‐values varying between 0.186 and 1.66 × 10−4 depending on the percentile used to define ATM levels as “low”; p = 0.007, binary logistic multivariate model; Figure 4A; Supplementary Table 4) (Knappskog et al., 2012).
Figure 4.
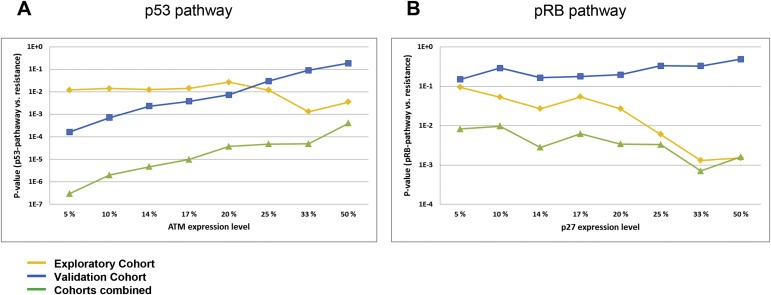
Correlations between p53‐/or pRB‐pathway defects and resistance to DNA damaging drugs. (A) Lines representing p‐values (Y‐axis) for the correlations between defects in the p53‐pathway (TP53 mutations, CHEK2 mutation or low levels of ATM mRNA) and resistance to DNA damaging drugs in vivo, plotted as a function of different cut‐offs applied to define “pathologically” low levels of ATM (X‐axis). The percentages refer to the lower percentile of patients in each cohort. (B) Lines representing p‐values (Y‐axis) for the correlations between defects in the pRb pathway (RB1 mutations, Cyclin D or E amplifications, CDKN2A defects or low levels of p27 mRNA) and resistance to DNA damaging drugs in vivo, plotted as a function of different cut‐offs applied to define “pathologically” low levels of p27 (X‐axis). Exploratory cohort (Cohort 1; treated with doxorubicin or 5‐FU/mitomycin; yellow line), validation cohort (Cohort 2; treated with epirubicin; blue line) and combined data from cohort 1 and 2 (green line).
Combining the data from the two cohorts, we here found the correlation between defects in the p53 pathway and resistance to therapy to be strong and robust across all expression cut‐off values applied for ATM (p < 0.001 for all calculations; Figure 4A).
3.3. RB1 mutations and chemoresistance
Previously, we reported a significant association between RB1 mutations and PD in our explorative cohort, revealing RB1 mutations to occur among 3 out of 17 patients with PD in response to anthracycline or mitomycin treatment, contrasting a single mutation among 51 individuals with response to the therapy (p = 0.046) (Berge et al., 2010).
In the present analysis, we were able to expand our material including one additional patient with PD (Berge et al., 2010). All findings in individual patients are listed in Supplementary Table S3. Analyzing this dataset, we found RB1 mutations in 3 out of 18 tumours with a PD as compared to 1 out of 51 responders (p = 0.052).
In the Validation Cohort, we found no point mutations in RB1 (among 30 patients screened for point mutations in the entire coding region; 10 PD patients and 20 responders). Analyzing the total data set for intragenetic deletions, we found defects in 3 out of 107 patients (deletion of exon 1, exon 2–10 and exon 10, respectively). Among these 3 patients, 2 belonged to the group of 10 patients displaying progressive disease upon treatment, while 1 was among the 97 therapy responders (p = 0.023).
Combining these data from the exploratory and the validation cohorts, we found RB1 defects to be strongly associated with lack of response to therapy (p = 0.001).
3.4. The pRB‐ pathway and chemoresistance
The pRB protein is inactivated by different cyclin/cdk‐complexes, the most important ones being Cyclin D/cdk4/6 and Cyclin E/cdk2. Both CCNE1 (Cyclin E) and CCND1 (Cyclin D) are known to be amplified in breast cancers (Sutherland et al., 2004). In order to assess the impact of alterations potentially inactivating the pRB‐pathway, we analyzed for amplifications of CCND1 and CCNE1, as well as alterations affecting the most relevant Cyclin‐cdk inhibitors:
While amplifications of CCND1 were observed in multiple tumours, no association to treatment response was recorded in the exploratory or in the validation cohort (Figure 3, Supplementary Table S3A; p > 0.5). As for CCNE1, we found this gene to be amplified in 6 tumours (2 of the 18 PD patients and 4 of the 51 responders) in the exploratory cohort (p > 0.5; Figure 5). As for the validation Cohort, we observed CCNE1 amplifications in 2 of the PD patients and 3 of the responders (p = 0.068; p = 0.077, both cohorts combined).
Figure 5.
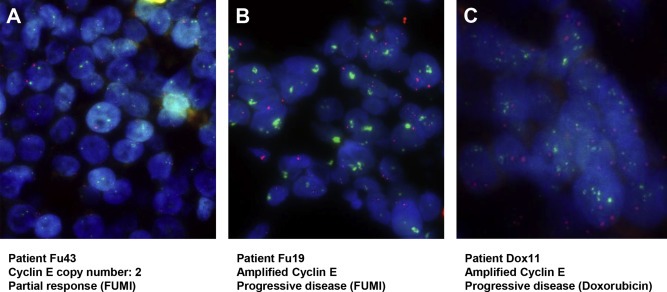
Cyclin E copy number analysis. Representative pictures of Cyclin E copy number analyses as performed by fluorescence in situ hybridization (FISH) using Cyclin E specific probe (green) and centromere 11 specific probe (red). (A) Normal copy number. (B) Highly amplified Cyclin E locus in patient with progressive disease upon 5‐FU and mitomycin treatment. (C) Highly amplified Cyclin E locus in patient with progressive disease upon doxorubicin monotherapy treatment. Magnification 630×.
Regarding the most important regulator of CyclinD/cdk4‐activity, CDKN2A (encoding p16), we found 6 patients in the exploratory cohort to harbour mutations or promoter methylation affecting this gene (Figure 3, Supplementary Table S3A), 4 of which were resistant to therapy (p = 0.036). In the validation cohort, in contrast, we observed alterations (promoter methylation) in 4 out of 97 responders but no methylation or mutations in the 10 PD‐patients (p = 0.055, both cohorts combined).
Next, we extended our analysis to alterations of CyclinE inhibitors. We have previously shown mutations or SNPs affecting CDKN1A (encoding p21 and p21B) not to be associated with chemoresistance (Knappskog et al., 2007; Staalesen et al., 2004) and therefore focused the present analyses on the CDKN1B (p27KIP1) locus. We found one patient in the exploratory cohort to harbour a point mutation affecting the coding region of p27KIP1, while no hypermethylation of the CDKN1B promoter was detected in any of the patients. However, analyzing p27KIP1 for mRNA expression levels, we found low levels to be significantly associated with lack of response to therapy in the exploratory cohort (p = 0.007; p = 0.005 when restricting the analysis to patients with no other alterations in the pRB‐pathway). In the validation cohort, p27 expression levels were not significantly associated with resistance (p > 0.2). However, 5 out of the 6 patients with PD and no RB1 alteration or CCND1/CCNE1 amplifications displayed p27 expression levels below the average of the cohort, with 2 of these patients displaying expression levels in the lower 12% of the cohort.
Regarding the data from analyses of factors in the pRB‐pathway in concert, according to the hypothesis that one “hit” would inactivate the biological function of the pathway in the tumour cells, we found a strong correlation between inactivation of the pRB‐pathway and resistance to therapy in our exploratory cohort (p‐values ranging from 0.001 to 0.094, depending on the cut‐off value for the definition of “low” levels of p27 expression; p = 0.003, binary logistic multivariate model; Figure 4B; Supplementary Table 4).
Similarly, we observed a clear, although not always significant, association between inactivation of the pRB‐pathway and resistance to therapy in the validation cohort (p < 0.3 for all calculations where a cut‐off of 20% or lower is used to define “low” p27 expression levels; p = 0.121, binary logistic multivariate model; Figure 4B; Supplementary Table 4). Combining the data from the exploratory and the validation cohorts, we found a strong and robust association across all cut‐offs applied for p27 expression levels (p < 0.01 for all calculations; Figure 4B).
3.5. Concomitant disturbances in the p53‐and pRB‐pathways and chemoresistance
We have previously hypothesized that concomitant inactivation of the p53‐and redundant‐pathways may lead to chemoresistance toward anthracyclines and mitomycin in breast cancer (Lønning, 2004). Analyzing our data with respect to the p53‐and pRB‐pathway in concert, we found concomitant inactivation of both pathways to be strongly associated with therapy resistance in the exploratory cohort (p‐values ranging from 0.008 to 3.9 × 10−6 depending on the cut‐off values used to dichotomize ATM and p27 mRNA into “low” versus “high”; p = 0.001, binary logistic multivariate model including all variables; Figure 6A; Supplementary Table 4). Most importantly, regardless of the cut‐offs applied, the two‐pathway‐hit model revealed a stronger association to chemoresistance as compared to either the p53‐or the pRB‐pathways individually.
Figure 6.
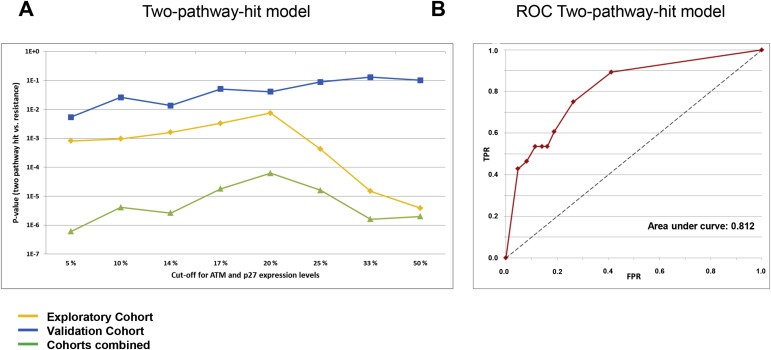
Correlations between the two‐pathway‐hit model and resistance to DNA damaging drugs. (A) Lines representing p‐values for the correlations between concomitant defects in the p53 and the pRB pathway and resistance to DNA damaging drugs in vivo, plotted as a function of different cut‐offs applied to define “pathologically” low levels of ATM and p27. Exploratory cohort (treated with doxorubicin or 5‐FU/mitomycin; yellow line), validation cohort (treated with epirubicin; blue line), and combined data from both cohorts (green line). (B) ROC‐curve for the twopathway‐ hit model, based on the combined data from the exploratory‐ and the validation cohorts (green line in Figure 6A). TPR: True positive rate; FPR: False positive rate. The eight points on the ROC‐curve corresponds to the eight cut offs on the x‐axis of panel A.
Considering our validation cohort, inevitably, the correlations were weaker due to fewer patients in the PD‐group (n = 10), and for some of the high cut‐offs applied for ATM expression, the model was non‐significant. However, in general the validation cohort strongly supported the findings from the exploratory cohort, with p‐values for the two‐pathway‐hit model ranging from 0.068 to 2.6 × 10−4 when applying low ATM cut‐off values (<14%) and any cut‐off for p27 levels (p = 0.008, binary logistic multivariate model including all variables; Figure 6A; Supplementary Table 4).
Finally, analyzing the data from both cohorts combined with respect to the two‐pathway‐hit model, we found strong correlations between concomitant inactivation of the p53‐and pRB‐pathways and resistance to therapy (p‐values ranging from 2.4 × 10−4 to 1.0 × 10−8 depending on the cut‐off values used to dichotomize ATM and p27 mRNA into “low” versus “high”; Figure 6A; Supplementary Table 4). Calculating the ROC‐curve for this dataset, we found the area under the curve to be 0.809 (Figure 6B).
3.6. Concomitant disturbances in the p53‐and pRB‐pathways and long‐term outcome
In addition to the impact of concomitant inactivation of the p53‐and the pRB‐pathway on primary resistance to therapy, we further assessed the impact of the two‐pathway‐hit model on long term outcome. While the number of patients within each cohort is limited for such analyses, there was a clear trend indicating an inferior relapse‐free as well as disease‐specific survival for patients with concomitant inactivation of both the p53‐and the RB‐pathway, in both cohorts (Figure 7). Combining data from both cohorts, we found a significantly lower relapse free survival (p = 0.004) as well as disease specific survival (p = 0.013; Figure 7).
Figure 7.
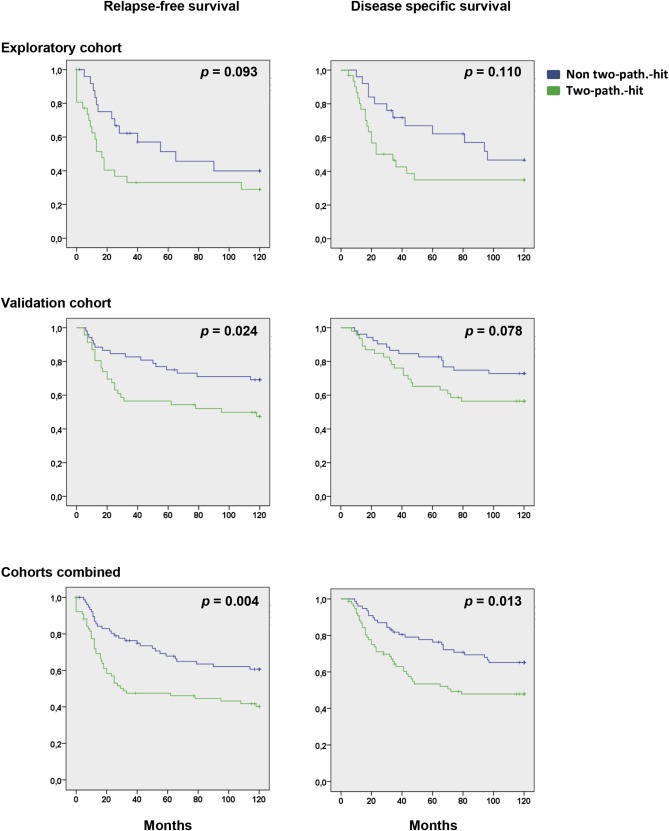
Kaplan–Meier plots illustrating the impact of concomitant inactivation of the p53‐and pRB‐functional pathways on 10 years relapse‐free survival and disease specific survival. Green lines represent patients with concomitant inactivation (two‐pathway‐hit). Blue lines represent patients without concomitant inactivation.
4. Discussion
Despite much effort, neither single parameters nor gene expression profiles have been able to identify the mechanisms underlying resistance to chemotherapy in patients (Lonning, 2004; Pollack et al., 2002). In this study, we evaluated the hypothesis postulating that concomitant inactivation of redundant gene cascades (focusing on p53 and pRB) may cause resistance towards anthracyclines and related compounds in breast cancer (Lonning et al., 2013).
Previous studies by us (Chrisanthar et al., 2008, 2001, 2003) and others (Kandioler‐Eckersberger et al., 2000) have revealed an association between TP53 mutations and lack of response to anthracyclines. This association was strengthened by including mutations in the CHEK2 gene (Chrisanthar et al., 2008) and low ATM expression levels (Knappskog et al., 2012) to the model. While de Thé and colleagues previously have reported TP53 mutations to be associated with an improved chance of having a complete response (Bertheau et al., 2007), this was limited to patients receiving cyclophosphamide at high doses in concert with anthracyclines. Among patients receiving anthracyclines without cyclophosphamide or cyclophosphamide at standard doses, the chance of having a complete response was lower among tumours harbouring mutated versus wild‐type TP53 (Lehmann‐Che et al., 2010).
The fact that defects in the p53 pathway alone were not fully predictive for anthracycline/mitomycin resistance made us postulate that redundant pathways act in concert (Lonning, 2004). While conflicting evidence has linked RB1 mutations to enhanced chemosensitivity in experimental models (Bosco et al., 2004), the role of RB1 mutations in vivo, in particular when acting in concert with p53‐defects, may be different. Based on previous findings revealing RB1 mutations to be associated with anthracycline resistance in primary (Berge et al., 2010) as well as metastatic (Berge et al., 2011) breast cancer, the aim of this study was i) to explore alterations in the pRB‐pathway with respect to drug resistance, and ii) most importantly, to explore whether inactivation of the p53‐and pRB‐ pathways in concert may be a stronger predictor for anthracycline/mitomycin resistance as compared to alterations in each individual pathway alone, a “nature's corollary” to the pharmacological concept of synthetic lethality (Farmer et al., 2005).
Although RB1 mutations were rare in our study, we observed a strong association between such mutations and resistance to DNA damaging drugs in both cohorts analyzed. This association was strengthened when defects in key regulators of pRB function were included in the model. While each individual factor revealed a trend for an association with chemoresistance, taking these factors in concert into a functional pathway analysis significantly improved the association. Similar findings have been reported by others with respect to other tumour characteristics (Hortobagyi 2013; Oricchio et al., 2014). The only individual parameter for which no association was recorded was Cyclin D. While the reason for this difference between Cyclin D and E may be difficult to pinpoint, previous studies reported elevated Cyclin D expression to be associated with an improved prognosis in breast cancer (Loden et al., 2002; Xu et al., 2013).
Notably, some tumours responded to chemotherapy despite alterations inactivating both pathways. A likely explanation for this is that mutations may be present in subclones in the tumour, and that these subclones are large enough to allow detection in the laboratory, but not large enough to dominate the changes in tumour size. Another potential scenario could be that the alterations affecting the two pathways are present in different subclones within the tumour.
We could not identify defects in both pathways across all tumours with a PD. Considering that our analyses were restricted to DNA and the RNA and the multiple mechanisms by which p53 and pRB can be modified and inactivated post‐translationally (Munro et al., 2012; Toledo et al., 2006), most likely, some patients may have the analyzed pathways inactivated through mechanisms not yet identified. Interestingly, two tumours in the validation cohort revealing PD upon therapy harboured non‐sense mutations in CHEK2 but no detectable defects in the pRB‐pathway. Chk2 phosphorylation of the pRB protein at Ser612 has been reported to activate its pro‐apoptotic function (Inoue et al., 2007) thus, the possibility exists that CHEK2 non‐sense mutations may inactivate pRB function in addition to inactivating the p53 pathway.
A major advantage of this study is that the doxorubicin treated patients (∼50% of the exploratory cohort) and all the epirubicin treated patients (validation cohort) were treated with anthracycline monotherapy. While the chemotherapy regimens applied to patients in the exploratory cohort (using doxorubicin monotherapy for weekly treatment, or mitomycin and 5‐fluoro‐uracil combined) may be considered archaic, patients in the validation cohort received epirubicin monotherapy at conventional doses at 3‐weekly intervals. Importantly, our findings, that the same predictive factors are identified across the different patient cohorts, indicate these factors may predict primary resistance to anthracyclines administered at both low and regular doses to a similar extent.
Although the main focus of the present work was to assess the impact of concomitant inactivation of the p53‐and the pRB‐pathway on primary response to therapy, the dismal biological effects of this two‐pathway‐hit model was further supported by our findings of a significantly reduced relapse‐free as well as disease‐specific survival among patients with both pathways inactivated.
In conclusion, we show that genetic alterations inactivating the p53 and pRB functional pathways in concert are associated with primary resistance to DNA‐damaging drugs as well as long term outcome in patients with locally advanced breast cancer. Based on these findings, we propose a model where in vivo resistance to DNA damaging drugs is dependent on a “two‐pathway‐hit”, inactivating both pathways. While our findings do not provide a complete identification of all factors potentially associated with anthracycline resistance, we believe they outline a general model explaining primary resistance to DNA damaging drugs in breast cancer in vivo. Further studies, using genome‐/exome‐wide sequencing and epigenetics assessments, should be able to sort out the details missing, making this a complete model predicting anthracycline resistance. Further, our findings outline a general conceptual approach that may be applicable when exploring the mechanisms of resistance against other drugs across different cancer forms in vivo.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgements
Most of this work was performed in the Mohn Cancer Research Laboratory. The project was supported by grants from the Bergen Medical Research Foundation, the Norwegian Cancer Society, and the Norwegian Health Region West. We thank Johan R. Lillehaug for scientific and technical advice and Dagfinn Ekse, Hildegunn Helle, Linda Ramsevik, Sandra H. Haugen and Nhat K. Duong for technical assistance.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.04.008.
Knappskog Stian, Berge Elisabet O., Chrisanthar Ranjan, Geisler Stephanie, Staalesen Vidar, Leirvaag Beryl, Yndestad Synnøve, de Faveri Elise, Karlsen Bård O., Wedge David C., Akslen Lars A., Lilleng Peer K., Løkkevik Erik, Lundgren Steinar, Østenstad Bjørn, Risberg Terje, Mjaaland Ingvild, Aas Turid, Lønning Per E, (2015), Concomitant inactivation of the p53‐ and pRB‐ functional pathways predicts resistance to DNA damaging drugs in breast cancer in vivo, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.09.008.
References
- Alba, E. , Martin, M. , Ramos, M. , Adrover, E. , Balil, A. , Jara, C. , Barnadas, A. , Fernandez-Aramburo, A. , Sanchez-Rovira, P. , Amenedo, M. , Casado, A. , 2004. Multicenter randomized trial comparing sequential with concomitant administration of doxorubicin and docetaxel as first-line treatment of metastatic breast cancer: a Spanish Breast Cancer Research Group (GEICAM-9903) phase III study. J. Clin. Oncol. 22, (13) 2587–2593. [DOI] [PubMed] [Google Scholar]
- Albain, K.S. , Barlow, W.E. , Shak, S. , Hortobagyi, G.N. , Livingston, R.B. , Yeh, I.T. , Ravdin, P. , Bugarini, R. , Baehner, F.L. , Davidson, N.E. , Sledge, G.W. , Winer, E.P. , Hudis, C. , Ingle, J.N. , Perez, E.A. , Pritchard, K.I. , Shepherd, L. , Gralow, J.R. , Yoshizawa, C. , Allred, D.C. , Osborne, C.K. , Hayes, D.F. , Breast Cancer Intergroup of North A, 2010. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol. 11, (1) 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albain, K. , Anderson, S. , Arriagada, R. , Barlow, W. , Bergh, J. , Bliss, J. , Buyse, M. , Cameron, D. , Carrasco, E. , Clarke, M. , Correa, C. , Coates, A. , Collins, R. , Costantino, J. , Cutter, D. , 2012. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100000 women in 123 randomised trials. Lancet. 379, 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge, E.O. , Knappskog, S. , Geisler, S. , Staalesen, V. , Pacal, M. , Borresen-Dale, A.L. , Puntervoll, P. , Lillehaug, J.R. , Lonning, P.E. , 2010. Identification and characterization of retinoblastoma gene mutations disturbing apoptosis in human breast cancers. Mol. Cancer. 9, 173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge, E.O. , Knappskog, S. , Lillehaug, J.R. , Lonning, P.E. , 2011. Alterations of the retinoblastoma gene in metastatic breast cancer. Clin. Exp. Metastasis. 28, (3) 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertheau, P. , Turpin, E. , Rickman, D.S. , Espie, M. , de Reynies, A. , Feugeas, J.P. , Plassa, L.F. , Soliman, H. , Varna, M. , de Roquancourt, A. , Lehmann-Che, J. , Beuzard, Y. , Marty, M. , Misset, J.L. , Janin, A. , de The, H. , 2007. Exquisite sensitivity of TP53 mutant and basal breast cancers to a dose-dense epirubicin-cyclophosphamide regimen. PLoS Med. 4, (3) e90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco, E.E. , Mayhew, C.N. , Hennigan, R.F. , Sage, J. , Jacks, T. , Knudsen, E.S. , 2004. RB signaling prevents replication-dependent DNA double-strand breaks following genotoxic insult. Nucleic Acids Res. 32, (1) 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscemi, G. , Perego, P. , Carenini, N. , Nakanishi, M. , Chessa, L. , Chen, J. , Khanna, K. , Delia, D. , 2004. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene. 23, (46) 7691–7700. [DOI] [PubMed] [Google Scholar]
- Chehab, N.H. , Malikzay, A. , Stavridi, E.S. , Halazonetis, T.D. , 1999. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. U S A. 96, (24) 13777–13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisanthar, R. , Knappskog, S. , Lokkevik, E. , Anker, G. , Ostenstad, B. , Lundgren, S. , Berge, E.O. , Risberg, T. , Mjaaland, I. , Maehle, L. , Engebretsen, L.F. , Lillehaug, J.R. , Lonning, P.E. , 2008. CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS ONE. 3, (8) e3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortobagyi, G.N. , Piccart-Gebhart, M.J. , Rugo, H.S. , 2013. Correlation of molecular alterations with efficacy of everolimus in hormone receptor-positive, HER-2 Negative Advanced Breast cancer. Results from Bolero-2 ASCO; Chicago, IL, USA: [Google Scholar]
- Enoch, T. , Norbury, C. , 1995. Cellular responses to DNA damage: cell-cycle checkpoints, apoptosis and the roles of p53 and ATM. Trends Biochem. Sci. 20, (10) 426–430. [DOI] [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C.J. , Tutt, A.N. , Johnson, D.A. , Richardson, T.B. , Santarosa, M. , Dillon, K.J. , Hickson, I. , Knights, C. , Martin, N.M. , Jackson, S.P. , Smith, G.C. , Ashworth, A. , 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434, (7035) 917–921. [DOI] [PubMed] [Google Scholar]
- Geisler, S. , Lonning, P.E. , Aas, T. , Johnsen, H. , Fluge, O. , Haugen, D.F. , Lillehaug, J.R. , Akslen, L.A. , Borresen-Dale, A.L. , 2001. Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res. 61, (6) 2505–2512. [PubMed] [Google Scholar]
- Geisler, S. , Borresen-Dale, A.L. , Johnsen, H. , Aas, T. , Geisler, J. , Akslen, L.A. , Anker, G. , Lonning, P.E. , 2003. TP53 gene mutations predict the response to neoadjuvant treatment with 5-fluorouracil and mitomycin in locally advanced breast cancer. Clin. Cancer Res. 9, (15) 5582–5588. [PubMed] [Google Scholar]
- Greenberg, P.A. , Hortobagyi, G.N. , Smith, T.L. , Ziegler, L.D. , Frye, D.K. , Buzdar, A.U. , 1996. Long-term follow-up of patients with complete remission following combination chemotherapy for metastatic breast cancer. J. Clin. Oncol. 14, (8) 2197–2205. [DOI] [PubMed] [Google Scholar]
- Harrington, E.A. , Bruce, J.L. , Harlow, E. , Dyson, N. , 1998. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc. Natl. Acad. Sci. U S A. 95, (20) 11945–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, J.L. , Carbone, P.P. , Heusen, J.C. , Kumaoka, S. , Segaloff, A. , Rubens, R.D. , 1977. Assessment of response to therapy in advanced breast cancer. Br. J. Cancer. 35, (3) 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao, A. , Kong, Y.Y. , Matsuoka, S. , Wakeham, A. , Ruland, J. , Yoshida, H. , Liu, D. , Elledge, S.J. , Mak, T.W. , 2000. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 287, (5459) 1824–1827. [DOI] [PubMed] [Google Scholar]
- Hugh, J. , Hanson, J. , Cheang, M.C. , Nielsen, T.O. , Perou, C.M. , Dumontet, C. , Reed, J. , Krajewska, M. , Treilleux, I. , Rupin, M. , Magherini, E. , Mackey, J. , Martin, M. , Vogel, C. , 2009. Breast cancer subtypes and response to docetaxel in node-positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. J. Clin. Oncol. 27, (8) 1168–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, Y. , Kitagawa, M. , Taya, Y. , 2007. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 26, (8) 2083–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandioler-Eckersberger, D. , Ludwig, C. , Rudas, M. , Kappel, S. , Janschek, E. , Wenzel, C. , Schlagbauer-Wadl, H. , Mittlbock, M. , Gnant, M. , Steger, G. , Jakesz, R. , 2000. TP53 mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin. Cancer Res. 6, (1) 50–56. [PubMed] [Google Scholar]
- Knappskog, S. , Geisler, J. , Arnesen, T. , Lillehaug, J.R. , Lonning, P.E. , 2006. A novel type of deletion in the CDKN2A gene identified in a melanoma-prone family. Genes Chromosomes Cancer. 45, (12) 1155–1163. [DOI] [PubMed] [Google Scholar]
- Knappskog, S. , Chrisanthar, R. , Staalesen, V. , Borresen-Dale, A.L. , Gram, I.T. , Lillehaug, J.R. , Lonning, P.E. , 2007. Mutations and polymorphisms of the p21B transcript in breast cancer. Int. J. Cancer. 121, (4) 908–910. [DOI] [PubMed] [Google Scholar]
- Knappskog, S. , Chrisanthar, R. , Lokkevik, E. , Anker, G. , Ostenstad, B. , Lundgren, S. , Risberg, T. , Mjaaland, I. , Leirvaag, B. , Miletic, H. , Lonning, P.E. , 2012. Low expression levels of ATM may substitute for CHEK2/TP53 mutations predicting resistance towards anthracycline and mitomycin chemotherapy in breast cancer. Breast Cancer Res.: BCR. 14, (2) R47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoop, A.S. , Knudsen, H. , Balslev, E. , Rasmussen, B.B. , Overgaard, J. , Nielsen, K.V. , Schonau, A. , Gunnarsdottir, K. , Olsen, K.E. , Mouridsen, H. , Ejlertsen, B. , 2005. retrospective analysis of topoisomerase IIa amplifications and deletions as predictive markers in primary breast cancer patients randomly assigned to cyclophosphamide, methotrexate, and fluorouracil or cyclophosphamide, epirubicin, and fluorouracil: Danish Breast Cancer Cooperative Group. J. Clin. Oncol. 23, (30) 7483–7490. [DOI] [PubMed] [Google Scholar]
- Lehmann-Che, J. , Andre, F. , Desmedt, C. , Mazouni, C. , Giacchetti, S. , Turpin, E. , Espie, M. , Plassa, L.F. , Marty, M. , Bertheau, P. , Sotiriou, C. , Piccart, M. , Symmans, W.F. , Pusztai, L. , de The, H. , 2010. Cyclophosphamide dose intensification may circumvent anthracycline resistance of p53 mutant breast cancers. The Oncologist. 15, (3) 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loden, M. , Stighall, M. , Nielsen, N.H. , Roos, G. , Emdin, S.O. , Ostlund, H. , Landberg, G. , 2002. The cyclin D1 high and cyclin E high subgroups of breast cancer: separate pathways in tumorogenesis based on pattern of genetic aberrations and inactivation of the pRb node. Oncogene. 21, (30) 4680–4690. [DOI] [PubMed] [Google Scholar]
- Lønning, P.E. , 2003. Study of suboptimum treatment response: lessons from breast cancer. Lancet Oncol. 4, (3) 177–185. [DOI] [PubMed] [Google Scholar]
- Lonning, P.E. , Knappskog, S. , 2013. Mapping genetic alterations causing chemoresistance in cancer: identifying the roads by tracking the drivers. Oncogene. 32, 5315–5330. [DOI] [PubMed] [Google Scholar]
- Lonning, P.E. , Knappskog, S. , Staalesen, V. , Chrisanthar, R. , Lillehaug, J.R. , 2007. Breast cancer prognostication and prediction in the postgenomic era. Ann. Oncol. 18, (8) 1293–1306. [DOI] [PubMed] [Google Scholar]
- Lonning, P.E. , 2004. Genes causing inherited cancer as beacons to identify the mechanisms of chemoresistance. Trends Mol. Med. 10, (3) 113–118. [DOI] [PubMed] [Google Scholar]
- Munro, S. , Carr, S.M. , La Thangue, N.B. , 2012. Diversity within the pRb pathway: is there a code of conduct?. Oncogene. 31, (40) 4343–4352. [DOI] [PubMed] [Google Scholar]
- Oricchio, E. , Ciriello, G. , Jiang, M. , Boice, M.H. , Schatz, J.H. , Heguy, A. , Viale, A. , de Stanchina, E. , Teruya-Feldstein, J. , Bouska, A. , McKeithan, T. , Sander, C. , Tam, W. , Seshan, V.E. , Chan, W.C. , Chaganti, R.S. , Wendel, H.G. , 2014. Frequent disruption of the RB pathway in indolent follicular lymphoma suggests a new combination therapy. J. Exp. Med. 211, (7) 1379–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik, S. , Tang, G. , Shak, S. , Kim, C. , Baker, J. , Kim, W. , Cronin, M. , Baehner, F.L. , Watson, D. , Bryant, J. , Costantino, J.P. , Geyer, C.E. , Wickerham, D.L. , Wolmark, N. , 2006. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J. Clin. Oncol. 24, (23) 3726–3734. [DOI] [PubMed] [Google Scholar]
- Perou, C.M. , Sørlie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Rees, C.A. , Pollack, J.R. , Ross, D.T. , Johnsen, H. , Akslen, L.A. , Fluge, Ø. , Pargamenschikov, A. , Williams, C. , Zhu, S.X. , Lønning, P.E. , Børresen-Dale, A.-L. , Brown, P.O. , Botstein, D. , 2000. Molecular portraits of human breast tumours. Nature. 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Pollack, J.R. , Sorlie, T. , Perou, C.M. , Rees, C.A. , Jeffrey, S.S. , Lonning, P.E. , Tibshirani, R. , Botstein, D. , Borresen-Dale, A.L. , Brown, P.O. , 2002. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. U S A. 99, (20) 12963–12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzier, R. , Pusztai, L. , Delaloge, S. , Gonzalez-Angulo, A.M. , Andre, F. , Hess, K.R. , Buzdar, A.U. , Garbay, J.R. , Spielmann, M. , Mathieu, M.C. , Symmans, W.F. , Wagner, P. , Atallah, D. , Valero, V. , Berry, D.A. , Hortobagyi, G.N. , 2005. Nomograms to predict pathologic complete response and metastasis-free survival after preoperative chemotherapy for breast cancer. J. Clin. Oncol. 23, (33) 8331–8339. [DOI] [PubMed] [Google Scholar]
- Schmitt, C.A. , Fridman, J.S. , Yang, M. , Lee, S. , Baranov, E. , Hoffman, R.M. , Lowe, S.W. , 2002. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 109, (3) 335–346. [DOI] [PubMed] [Google Scholar]
- Smeets, S.J. , van der Plas, M. , Schaaij-Visser, T.B. , van Veen, E.A. , van Meerloo, J. , Braakhuis, B.J. , Steenbergen, R.D. , Brakenhoff, R.H. , 2011. Immortalization of oral keratinocytes by functional inactivation of the p53 and pRb pathways. Int. J. Cancer. 128, (7) 1596–1605. [DOI] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , Hastie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Thorsen, T. , Quist, H. , Matese, J.C. , Brown, P.O. , Botstein, D. , Eystein Lonning, P. , Borresen-Dale, A.L. , 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U S A. 98, (19) 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Fan, C. , Geisler, S. , Aas, T. , Nobel, A. , Anker, G. , Akslen, L.A. , Botstein, D. , Borresen-Dale, A.L. , Lonning, P.E. , 2006. Gene expression profiles do not consistently predict the clinical treatment response in locally advanced breast cancer. Mol. Cancer Ther. 5, (11) 2914–2918. [DOI] [PubMed] [Google Scholar]
- Staalesen, V. , Leirvaag, B. , Lillehaug, J.R. , Lonning, P.E. , 2004. Genetic and epigenetic changes in p21 and p21B do not correlate with resistance to doxorubicin or mitomycin and 5-fluorouracil in locally advanced breast cancer. Clin. Cancer Res. 10, (10) 3438–3443. [DOI] [PubMed] [Google Scholar]
- Straver, M.E. , Glas, A.M. , Hannemann, J. , Wesseling, J. , van de Vijver, M.J. , Rutgers, E.J. , Vrancken Peeters, M.J. , van Tinteren, H. , Van't Veer, L.J. , Rodenhuis, S. , 2010. The 70-gene signature as a response predictor for neoadjuvant chemotherapy in breast cancer. Breast Cancer Res. Treat. 119, (3) 551–558. [DOI] [PubMed] [Google Scholar]
- Sutherland, R.L. , Musgrove, E.A. , 2004. Cyclins and breast cancer. J. Mammary Gland Biol. Neopl. 9, (1) 95–104. [DOI] [PubMed] [Google Scholar]
- Tanner, M. , Isola, J. , Wiklund, T. , Erikstein, B. , Kellokumpu-Lehtinen, P. , Malmstrom, P. , Wilking, N. , Nilsson, J. , Bergh, J. , 2006. Topoisomerase IIalpha gene amplification predicts favorable treatment response to tailored and dose-escalated anthracycline-based adjuvant chemotherapy in HER-2/neu-amplified breast cancer: Scandinavian Breast Group Trial 9401. J. Clin. Oncol. 24, (16) 2428–2436. [DOI] [PubMed] [Google Scholar]
- Therasse, P. , Arbuck, S.G. , Eisenhauer, E.A. , Wanders, J. , Kaplan, R.S. , Rubinstein, L. , Verweij, J. , Van Glabbeke, M. , van Oosterom, A.T. , Christian, M.C. , Gwyther, S.G. , 2000. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 92, (3) 205–216. [DOI] [PubMed] [Google Scholar]
- Toledo, F. , Wahl, G.M. , 2006. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer. 6, (12) 909–923. [DOI] [PubMed] [Google Scholar]
- Xu, X.L. , Chen, S.Z. , Chen, W. , Zheng, W.H. , Xia, X.H. , Yang, H.J. , Li, B. , Mao, W.M. , 2013. The impact of cyclin D1 overexpression on the prognosis of ER-positive breast cancers: a meta-analysis. Breast Cancer Res. Treat. 139, (2) 329–339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data