

Abstract
Hepatocellular carcinoma (HCC) is a complex and heterogeneous tumor most commonly associated with underlying chronic liver disease, especially hepatitis. It is a growing problem in the United States and worldwide. There are two potential ways to prevent HCC. Primary prevention which is based on vaccination or secondary prevention involving agents that slow down carcinogenesis. Several pathways have been thought to play a role in the development of HCC; specifically, those involving vascular endothelial growth factor (VEGF)‐mediated angiogenesis, WNT, phosphatidylinositol 3‐kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), AMP‐activated protein kinase (AMPK), and c‐MET. Currently, there are only a limited number of drugs which have been proven as effective treatment options for HCC and several clinical trials are testing drugs which target aberrations in the pathways mentioned above. In this review, we discuss currently approved therapies, monotherapies and combination therapy for the treatment of HCC.
Keywords: Hepatocellular carcinoma, PI3K, AKT, mTOR, Molecular targets
Highlights
Limited drugs of proven efficacy are available for treatment of patients with HCC.
Cross‐talk between signaling pathways hampers the efficacy of targeted agents.
Pre‐ and post‐treatment biopsy may shed light on pathways of resistance in HCC.
Development of combination therapies may help to overcome resistance in HCC.
1. Introduction
Hepatocellular carcinoma (HCC) is a complex and heterogeneous tumor most commonly associated with underlying chronic liver disease, especially hepatitis. It is a growing problem in the United States and worldwide. Globally, HCC is the third leading cause of cancer death and it is the most common cause of death among patients with cirrhosis in western countries (Villanueva et al., 2008; Goyal et al., 2013). The prevalence of HCC is increasing by 1.75% in the US per year (Buitrago‐Molina and Vogel, 2012), and advanced HCC has a poor prognosis, with a 5‐year overall survival rate of less than 10% (Goyal et al., 2013). Typically, hepatitis B and C infections, exposure to alphatoxins, and alcoholic and non‐alcoholic steatohepatitis (NASH) are risk factors for HCC (Buitrago‐Molina and Vogel, 2012). It is estimated that about 20–30% of patients diagnosed with HCC are eligible for curative treatments such as resection, ablation, or liver transplantation (Llovet and Bruix, 2008). The prognosis for patients with advanced disease is rather bleak (Buitrago‐Molina and Vogel, 2012). No standard systemic therapy was available for patients with advanced disease until 2007, when the molecular therapy sorafenib was approved for unresectable HCC (Llovet and Bruix, 2008).
There are two potential ways to prevent HCC. Primary prevention is based on vaccination, for example, the hepatitis B vaccine (Buitrago‐Molina and Vogel, 2012). Secondary prevention involves the use of agents that slow down carcinogenesis in patients with potential cancer precursors (Buitrago‐Molina and Vogel, 2012). For both prevention and treatment, in recent years, the practice of oncology has undergone a paradigm shift from broader disease‐based treatments to therapies that target specific aberrant cellular pathways and subcellular structures. In keeping with this shift, an explosive number of “targeted agents” have been developed to address a broad range of tumor types. In this article, we describe the molecular pathways that have been identified in HCC and efforts to target them, both established and investigational.
2. Molecular pathways in HCC
Several molecular alterations have been observed in HCC (Figure 1). Accumulation of these genetic alterations and aberrant activation of several signaling pathways have been thought to play a role in the development of HCC (Llovet and Bruix, 2008); specifically, those involving vascular endothelial growth factor (VEGF)‐mediated angiogenesis, WNT, phosphatidylinositol 3‐kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), AMP‐activated protein kinase (AMPK), and c‐MET.
Figure 1.
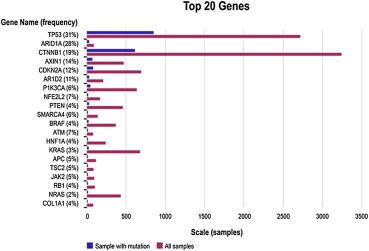
Key molecular alterations in HCC as documented in the Catalogue of Somatic Mutations in Cancer (COSMIC) database. This is a diagram illustrating the top 20 genes with highest prevalence of molecular alterations in patients with HCC. The most common among them are mutations in the TP53, ARID1A, CTNNB1, CDKN2A and ARID2 gene (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) at the time of this manuscript (March 24, 2014).
2.1. VEGF‐mediated angiogenesis pathway
Genetically unstable cancer cells and mutations that have accumulated within these cells are therapeutic targets of most novel agents. However, anti‐angiogenic therapy targets endothelial cells because they do not have any genetic mutations. The genetic stability of endothelial cells may render them less susceptible to acquired drug resistance. Thus, angiogenesis inhibitors are emerging as single agent therapies as well as in novel combinations.
Angiogenesis role in the initial progression from pre‐malignant tumor to cancer prompted investigators to study the role of VEGF in the natural history of HCC (Hanahan and Folkman, 1996). In 1999, a group of researchers suggested that the degree of tissue VEGF expression increased in accord with the stepwise development of HCC (Park et al., 2000). In addition, studies showed that VEGF was frequently expressed in HCC. A quantitative study reported VEGF tissue expression in 89% (32/36) of HCCs (Huang et al., 2005). Notably, studies have suggested a correlation between the degree of tissue VEGF expression and the intensity of both the magnetic resonance signal and the computed tomographic enhancement of the hepatic artery, which represent radiological vascular signals (Kanematsu et al., 2004, 2005, 2005). Hence, the hypervascular nature of HCC has led to increasing interest in exploring the potential of anti‐angiogenic therapy in this disease.
In HCC, hypoxia is believed to increase the expression of VEGF through the expression of hypoxia inducible factor‐1α (Torimura et al., 2004). VEGF is also known as the vascular permeability factor and stimulates proliferation of endothelial cells specifically through tyrosine kinase receptors (Ferrara and Davis‐Smyth, 1997). Angiopoietin 1 and 2 are ligands for the tyrosine kinase receptor Tie2. VEGF and angiopoietin‐2 (Ang‐2) are expressed on cancer cells, whereas angiopoietin‐1 (Ang‐1) is predominantly in supportive cells of large blood cells, stromal cells, endothelial cells, and tumor cells. Ang‐2 is a partial agonist and antagonist of Ang‐1 and is expressed for vascular remodeling preventing vascular stability thus allowing VEGF to stimulate endothelial cells (Moon et al., 2003).
2.2. WNT pathway
The WNT pathway has a crucial role in the regulation of diverse disease processes, which include cell proliferation, survival, migration, and cell fate. This pathway is involved in multiple normal physiological processes and embryonic development. In this pathway, when normally active, WNT ligand binds to a receptor leading to cytosolic accumulation of β‐catenin; thereafter, β‐catenin can translocate to the nucleus to initiate transcription (Klaus and Birchmeier, 2008). WNT can be activated if there is a mutation in the β‐catenin gene (CTNNB1), which is the third most frequent mutation seen in HCC after p53 mutation (Boyault et al., 2007).
2.3. PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway plays an important role in cell regulation. This pathway is involved in many cellular processes, such as cell division, cell growth, and programmed cell death (Yothaisong et al., 2013), and can be activated by many different stimuli, such as activated tyrosine kinase growth factor receptors, G‐protein‐coupled receptors, and oncogenes such as RAS (Hassan et al., 2013). Furthermore, the mTOR complex is an important therapeutic target, as it is a key intracellular node for a number of cellular signaling pathways. Inhibiting mTOR can prevent abnormal cell proliferation, tumor angiogenesis, and abnormal cellular metabolism, thus providing the rationale for mTOR inhibitors as potential targetable agents (Naing et al., 2012). Several studies have shown that selective mTOR inhibition can lead to AKT activation, which is mediated by the insulin growth factor 1 receptor pathway (Naing, 2013). Activation of AKT can limit anti‐tumor activity of mTOR inhibitors thus leading to resistance. This pathway is activated in a subset of patients with HCC, and the use of rapalogs has been shown to inhibit growth in HCC cell lines and experimental models (Llovet and Bruix, 2008).
2.4. AMPK pathway
AMPK is a conserved heterotrimeric serine/threonine kinase that has a central role in linking metabolism and cancer development. AMPK is activated when there is increased activity of AMP/ATP under a stressful condition such as low glucose level, hypoxia, ischemia, or heat shock. Once activated, AMPK suppresses cell proliferation in tumor and non‐tumor cells, through regulation of the cell cycle, apoptosis, autophagy, and inhibition of protein synthesis. There is growing evidence to suggest that the levels of AMPK are altered in various cancers, specifically HCC (Zheng et al., 2013).
A study by Zheng et al. (2013) showed that AMPK was down‐regulated in a majority of HCC patients, which correlated with a worse prognosis. Another study, by Lee et al. (2012), showed that AMPK is a tumor suppressor in HCC and that its inactivation can lead to hepatocarcinogenesis by destabilizing p53. These studies indicate that AMPK plays a role in HCC and its loss can contribute to the progression of HCC.
Many agents, such as metformin, a commonly used drug for diabetes mellitus, act by stimulating the AMPK pathway (Hajjar et al., 2013). Downstream of AMPK, the tumor suppressor proteins tuberous sclerosis 1 and 2 are activated, which leads to apoptosis. Activation of this pathway has been shown to inhibit or delay the onset of various tumors (Zheng et al., 2013).
2.5. c‐MET pathway
Hepatocyte growth factor (HGF)/mesenchymal–epithelial transition factor (MET), which is a receptor tyrosine kinase, is frequently dysregulated in many malignancies and has a pivotal role in HCC (Santoro et al., 2013a). When c‐MET is activated, it promotes tumor cell growth, survival, migration, invasion, angiogenesis, and metastasis (Trojan and Zeuzem, 2013). Therefore, activation of this pathway can lead to a more aggressive form of HCC and a poor outcome (Santoro et al., 2013a). Aberrant c‐MET signaling is seen in HCC and in patients who have received sorafenib, and overexpression of MET has been associated with a poor prognosis (Trojan and Zeuzem, 2013).
The c‐MET proto‐oncogene encodes the receptor for the ligand HGF. Binding of a ligand to the HGF receptor leads to homodimerization, subsequent autophosphorylation of downstream residues, and downstream activation of the MAPK and PI3K pathways. c‐MET phosphorylation can occur in the absence of HGF through interaction with the epidermal growth factor receptor (EGFR), cell attachment, or binding of the alternative ligand des‐gamma carboxyprothrombin. Irrespective of how HGF is activated, it leads to c‐MET dimerization, autophosphorylation, and kinase activity, which are necessary for malignant transformation (Goyal et al., 2013).
In addition to the pathways which were discussed above, other pathways such as NRAS, are involved in HCC. Furthermore, the figure illustrates many other pathways which are involved such as RAS/RAF/MEK. Similar to other targeted agents, inhibitors of BRAF/KRAS/NRAS have been shown to modulate growth factors and chemokine receptors in HCC cells but that has not been noticed in healthy liver tissues (Breunig et al., 2014).
3. Current targeted agents in HCC
Currently only a limited number of drugs have been proven effective for treatment of HCC. Several early‐phase clinical trials in patients are testing drugs that target aberrations in the pathways described above.
3.1. Sorafenib
Currently, sorafenib is the only FDA‐approved treatment for unresectable HCC. Sorafenib is an oral multityrosine kinase and angiogenesis inhibitor that has activity against the VEGF, platelet‐derived growth factor receptor (PDGFR), c‐KIT receptor, BRAF, and p38 signaling pathways (Trojan and Zeuzem, 2013). The median time to disease progression for patients with advanced HCC taking sorafenib is 5.5 months, and the median overall survival (OS) time is 10.7 months (Llovet et al., 2008). In a phase II study (Abou‐Alfa et al., 2006), 137 patients with inoperable HCC in Belgium, France, Italy, Israel, and the United States received sorafenib 400 mg twice daily. Disease control was reported in 42% of the patients. The median time to progression (TTP) was 5.5 months and the median OS was 9.2 months (Abou‐Alfa et al., 2006). In two large phase III randomized trials done by Llovet et al. (2008) and Cheng et al. (2012), sorafenib significantly improved overall survival compared to placebo. Study done by Cheng et al. (2012), was conducted in the Asian population, which showed improvement in median OS (5.9 vs 4.1 months) and median TTP (2.7 vs 1.4 months). In the SHARP study by Llovet et al. (2008), majority of the patients had hepatitis C and alcohol compared to few patients with hepatitis B. In contrast, in the study done by Cheng et al. (2012), 71% and 78% of the patients in the sorafenib and placebo group were positive for hepatitis B. Though both trials had similar design, the higher rate of hepatitis C in the SHARP compared to hepatitis B in Asian population indicates that these two populations are distinct from each other (Llovet et al., 2008; Cheng et al., 2012). Furthermore, sorafenib has a category 1 recommendation for patients with a Child‐Pugh class A and category 2A for those with Child‐Pugh class B with a special caution in the latter group specifically the bilirubin level. Presently there is no additional therapy for individuals whose disease progresses on sorafenib or who have poor tolerance for this drug (NCCN Clinical Practice Guidelines in Oncology Hepatobiliary Cancers Version 2.2015). Some common side effects seen with the use of sorafenib are skin rash, hand–foot syndrome, diarrhea, and hypertension (Alexandrescu et al., 2008).
3.2. Tivantinib
Initially tivantinib was considered a selective oral MET receptor tyrosine kinase inhibitor that interrupts downstream MET‐dependent signaling. Early pre‐clinical data show that tivantinib inhibits MET autophosphorylation, thereby leading to inhibition of downstream MET effectors (Munshi et al., 2010). However recent data from Katayama et al. (2013) suggests that tivantinib causes microtubule disruption, instability similar to vincristine, in addition to inhibiting c‐MET. In a phase I study done by Rosen et al. (2011), 79 patients, all with advanced solid tumors, were treated with tivantinib. The most common drug‐related dose‐limiting toxicities of grade 3 or more were leucopenia, neutropenia, thrombocytopenia, vomiting and dehydration. These side effects were observed in two patients being treated at 360 mg two times daily. There was no dose‐limiting toxicity observed at any dose less than 360 mg taken orally twice a day. In this study, three patients (3.8%) had achieved a partial response (PR) and 40 patients (50.6%) had stable disease (SD) for 19.9 weeks. Based on this dose, the phase II recommended dose was 360 mg twice daily (Rosen et al., 2011). In this phase I study, none of the patients had HCC. In a phase Ib multicenter study conducted by Santoro et al. (2013a), 21 patients who did not respond to or had relapses after prior treatment were treated with tivantinib 360 mg taken orally twice daily. The median age of patients was 69 years; ECOG performance status was 0–1, patients had preserved liver function at the start of the study, and they had received at least one prior systemic therapy. The most common side effects were neutropenia (52%), anemia (48%), asthenia (48%), leucopenia (38%), anorexia (38%), diarrhea (29%) and fatigue (29%). The most common serious drug‐related side effects (grades 3–4) observed were neutropenia (52%), anemia (24%), and leucopenia (19%). In this study, the mean time to progression was 3.3 months.
The above‐described data led to a phase I study with tivantinib in combination with sorafenib (Puzanov et al., 2015). In this study, there were a total of 87 patients who were enrolled. At the maximum tolerated dose (MTD) there was an expansion cohort in five tumor types that included 20 patients with HCC. The most common adverse effects were rash (40%), diarrhea (38%), and anorexia (33%). The overall response rate in all patients was 12% and 10% in patients with HCC. The median progression free survival (PFS) in all patients was 3.6 months and 3.5 months in patients with HCC. Furthermore, patients with Child‐Pugh A status had similar PFS compared to those with Child‐Pugh B, and those with prior anti‐VEGF (n = 8) had a longer median PFS (15.9 months) compared to those without any prior anti‐VEGF (n = 12, 3.5 months). The combination treatment could not be administered at MTD for patient with HCC due to toxicity, thus tivantinib had to be dose reduced (Puzanov et al., 2015).
In a phase II randomized multicenter study, tivantinib was used as monotherapy in patients with locally advanced or metastatic HCC after failure of prior therapy (Santoro et al., 2013b). Patients were randomized to receive either tivantinib or placebo and continued on the treatment until there was suspicion of disease progression. Patients being treated on the placebo arm were allowed to cross over to the treatment arm in this study. Overall survival in the tivantinib group was 6.6 months, and median time to progression was 1.6 months, versus 6.2 and 1.4 months, respectively, in the placebo group (p = 0.04 and p = 0.63 respectively). However, dose reductions were done in 24% of patients in the tivantinib group due to toxicity. In this study, 77 patients had adequate tissue to be tested by immunohistochemistry MET expression, of which 37 (48%) patient had MET‐high tumors. High MET expression by IHC was noted to be a poor prognostic factor; overall survival was 9 months in patients with low MET expression and 3.8 months in those with high MET expression (p = 0.02). For patients with high MET expression median time to progression was longer with tivantinib compared to placebo (2.7 months vs 1.4 months respectively p = 0.03). The median overall survival was 7.2 months for patients with MET high tumors who received tivantinib compared to 3.8 months in patients who received placebo (p = 0.01). In contrast, there was no difference noted in the efficacy between tivantinib and placebo in patients with low MET expression. This study demonstrated that tivantinib has promising anti‐tumor activity in MET‐amplified tumors (Santoro et al., 2013b).
3.3. Other investigational therapies
3.3.1. Cancer stem cells
Patients often respond initially to targeted therapies. However, a significant percentage of these patients relapse with metastatic disease. An increasing body of evidence supports the ‘cancer stem cell hypothesis’ which suggests that the potential for tumor growth and metastatic spread is dependent on a small subset of cells within the tumor – cancer stem cells, which have the ability to self‐renew and proliferate extensively (Lobo et al., 2007). Novel therapies targeting stem cells are being investigated in different tumor types including HCC. WNT signaling pathway has been implicated in the development of HCC and regulation of cancer stem cells (Pez et al., 2013). A phase I trial with OMP‐54F28 targeting the WNT pathway is ongoing (Table 1).
Table 1.
Summay of clinical trials in HCC that were ongoing at the time of this writing (March 2, 2015) (ClinicalTrials.gov, 2013).
| Agent | Molecular targets | Company/Sponsor | Phase |
|---|---|---|---|
| Sorafenib + BIIB022 | N/A | Biogen | I |
| Cetuximab | EGFR inhibitor | Massachusetts General Hospital | II |
| Lenvatiniba | VEGFR, FGFR, PDGFR, RET, SCFR | Eisai Limited | III |
| Regorafeniba | VEGFR, FGFR, PDGFR, KIT, BRAF, RET | Bayer | III |
| Cabozantiniba | c‐MET, VEGFR, RET | Exelixis | III |
| Tremelimumab with Chemoembolization or Ablation | anti CTLA‐4 | National Cancer Institute | I |
| Nivolumaba (BMS‐936559) | anti PD‐1 | Bristol‐Myers Squibb | I |
| Fluorouracil implants (regional chemotherapy) | N/A | Simcere Pharmaceutical Group | II |
| AZD9150 | ISIS‐STAT3 | AstraZeneca | I |
| Axitiniba | VEGFR, PDGFR, c‐KIT | University Health Network, Toronto | II |
| SOM230 | Somatostatin peptidomimetic | University of Miami | II |
| ADI‐PEG 20 vs placebo | N/A | Polaris Group | III |
| BYL719 | PIK3CA | Novartis | I |
| AZD5363 | AstraZeneca | I | |
| BGJ398/BYL719 | Novartis | I | |
| MEK162 + BYL719 | Novartis | I/II | |
| MK2206 | AKT | NCI | II |
| Vaccine therapy +/− sirolimus with NY‐ESO‐1 | mTOR | Roswell Park Cancer Institute | I |
| Temsirolimus + sorafeniba | Philipps University Marburg Medical Center | I/II | |
| CC‐223 | Dual mTOR | Celgene | I/II |
| MSC2156119J | cMET | Germany | I |
| OMP‐54F28 | WNT | Norris Comprehensive Cancer CenterUniversity of ColoradoIU Health University HospitalMassachusetts General HospitalMount Sinai Medical CenterFox Chase Cancer Center | I |
| TRC105 | Endoglin | NIH | I/II |
Indicates clinical trials which are being conducted internationally.
3.3.2. Immunotherapies
Spontaneous regression of HCC has been reported in about 70 cases in the literature (Arora and Madhusudhana, 2011). Besides ischemia, anti‐tumor immunity has been proposed as one of the possible mechanisms by which this phenomenon occurs. Moreover, chemotherapeutic agents may compromise the liver function as most of them are hepatotoxic. In this context, immunotherapy may represent an attractive alternative to traditional therapies (Pardee and Butterfield, 2012). However, the development of effective immune‐based therapies is challenging due to the inherent tolerogenic nature of the liver favoring immunosuppression (Pardee and Butterfield, 2012). Consequent to promising pre‐clinical activity, 35 patients with advanced HCC were administered intravenous vaccination with mature autologous dendritic cells (DCs) pulsed ex vivo with a liver tumor cell line lysate (HepG2). Disease control rate was 28% and serum alpha‐feto protein (AFP) dropped to less than 10% of baseline levels in 4 of the 17 patients with baseline AFP ≥1000 ng/mL (Palmer et al., 2009). In another phase II clinical trial, tremelimumab was administered to 20 patients with advanced HCC (Sangro et al., 2013). Tremelimumab (a fully human IgG2 monoclonal antibody) enhances T cell activation and proliferation by cytotoxic T‐lymphocyte‐associated antigen 4 (CTLA‐4) blockade. Disease control rate was 76.4%. Three patients (17.6%) had confirmed PR and 10 patients (58.8%) achieved SD. AFP dropped by >50% of baseline levels in 36% of the 11 patients with elevated AFP at baseline. The median TTP was 6.48 months (95% CI 3.95–9.14) and median OS was 8.2 months (95% CI 4.64–21.34), which is comparable to the anti‐tumor activity seen with sorafenib (Sangro et al., 2013). The results of immune‐based therapies in HCC are promising and warrant further investigation. A phase I trial of tremelimumab with chemoembolization or ablation and a phase I trial of nivolumab (BMS‐936559; anti‐programmed death‐1) in HCC is ongoing (Table 1) (ClinicalTrials.gov, 2013).
4. Combination therapeutic strategies for overcoming resistant disease
As described above, HCC has limited treatment options and there are many pathways by which it can progress. Although many investigational targeted agents have held early promise, the reality is that the success of single‐agent targeted therapy is often disappointing owing to the rapid development of resistance or alternative signaling. Each of the cellular pathways described above interact with each other; thus, blocking one pathway can lead to signaling via another. For example, when using the current rapalogs to inhibit mTOR1, signaling can still occur via mTOR and the negative feedback loop of S6K, thus leading to tumor progression. Furthermore, activation of a pathway leads to many cellular activities and not just cell proliferation. Combination of drugs affecting different pathways is one way to overcome resistance.
4.1. VEGF and Ang‐2 inhibition
Elevated expression levels of angiopoietin‐2 (Ang‐2) have been shown to be associated with resistance to anti‐angiogenesis and poor overall survival in HCC (Llovet et al., 2012; Reig et al., 2010). Ang‐2 is a ligand of the Tie2 tyrosine‐kinase receptor, which was reported to promote angiogenesis and tumor growth (Maisonpierre et al., 1997; Yu and Stamenkovic, 2001). Recent studies showed elevated plasma expression of Ang‐2 (Scholz et al., 2007) and upregulation of Ang‐2 mRNA (Torimura et al., 2004; Moon et al., 2003) in tissues from HCC patients. Overexpression of Ang‐2 in HCC tissues was reported to be associated with advanced histopathologic features and poor outcome in HCC. Additionally, recent studies of HCC patients treated with sorafenib found that patients with elevated baseline plasma Ang‐2 levels had shorter overall survival and time to progression (Llovet et al., 2012; Reig et al., 2010; Kaseb et al., 2012). Finally, pre‐clinical studies have shown that Ang‐2 synergistically augments VEGF‐mediated angiogenesis in HCC (Yoshiji et al., 2005; Lobov et al., 2002). Thus, combined inhibition of VEGF and Ang‐2 may have complementary effects on inhibiting HCC angiogenesis (Lobov et al., 2002; Coxon and Sun, 2008).
4.2. VEGF and EGF inhibition
In HCC, the vascular endothelial growth factor receptor (VEGFR) pathway has been recognized as the driving force for HCC initiation and progression, as outlined in a recent review (Pang et al., 2008). Furthermore, EGFR is frequently expressed in human hepatoma cells, and EGF may be one of the mitogens needed for the growth of hepatoma cells (Fausto, 1991; Hisaka et al., 1999). Erlotinib is an orally active and selective inhibitor of the EGFR/HER1 (human epidermal receptor)‐related tyrosine kinase enzyme. In two phase II studies of erlotinib in HCC (Philip et al., 2005; Thomas et al., 2007), the response rates were 0% and 9%, but the disease control rates were 43% and 50%, and median survival times were 10.75 and 13 months, respectively. EGFR/HER1 expression was detected in 71% and 88% of evaluable patients in the two studies, respectively, but there was no significant difference in terms of overall survival between the high‐EGFR and low‐EGFR groups. It should be noted that this assay detects simply the presence of EGFR and does not determine the receptors' functional status (e.g., phosphorylation status).
Notably, the VEGF and EGF pathways share common downstream signals, and several pre‐clinical studies have shown indications of either direct or indirect pro‐angiogenic effects of EGFR signaling (Hirata et al., 2002; Tamesa et al., 2009; Giannelli et al., 2008). Furthermore, up‐regulation of VEGF has been implicated in poor prognosis, high recurrence rate, and resistance to EGFR inhibition (Moller and Becker, 1992). Therefore, our group initiated combination systemic therapy trials targeting VEGF and EGFR in untreated HCC; the results were published recently (Kaseb et al., 2012; Thomas et al., 2009). Interestingly, our study (Kaseb et al., 2012) showed that elevated plasma expression levels of EGFR were marginally associated with shorter progression‐free survival times. However, this observation has not been reported to date in HCC patients treated with erlotinib, which is a selective inhibitor of the EGFR/HER1‐related tyrosine kinase enzyme. Although two phase II studies of erlotinib found elevated EGFR/HER1 expression in HCC specimens (Philip et al., 2005; Thomas et al., 2007), neither of these studies showed that overexpression of EGFR was related to efficacy of the treatment. Thus, the potential utility of circulating EGFR as a surrogate marker of resistance to anti‐EGFR systemic therapy in HCC patients is inconclusive and needs further study.
Notably, our exploratory biomarker study (Kaseb et al., 2012) showed that patients with elevated (above the median) levels of endothelin‐1 (ET‐1) had shorter progression‐free survival times than did patients whose ET‐1 levels were below the median. If validated, this finding, which has not been reported previously, may have implications for the design of future HCC clinical trials. ET‐1 receptors have been found on liver endothelial cells and are thought to mediate vasoregulation, and elevated circulating ET‐1 has been found to be associated with cirrhosis and HCC (Jorgensen et al., 1993, 1994, 1993). Because elevated ET‐1 levels have been shown to play a role in HCC progression (Moller et al., 1993) and to be associated with advanced tumors (Moller et al., 1991, 1993), strategies to target this pathway in the treatment of HCC patients are worth investigating.
4.3. VEGF and FGF inhibition
Aberrant FGF signaling has been associated with tumorigenesis and development of resistance to anti‐VEGF inhibitors in HCC (Siegel et al., 2010). Brivanib, a dual inhibitor of FGF and VEGF receptor tyrosine kinases, demonstrated strong pre‐clinical activity and 45.7% disease control rate in a phase II trial in patients with advanced HCC who had failed prior anti‐angiogenic therapy (Finn et al., 2012). A phase III double‐blinded, randomized, placebo‐controlled trial was conducted in 395 patients with advanced HCC who had progressed on or after receiving sorafenib (Llovet et al., 2013). The ORR was 10% for brivanib versus 2% for placebo (p = 0.003). The median TTP was longer for patients on brivanib (4.2 months) compared to those on placebo (2.7 months; p < 0.001). However, the primary endpoint of statistically significant improvement in OS with brivanib was not met. The median OS with brivanib was 9.4 months compared to 8.2 months on placebo (p = 0.3307). Despite the promising pre‐clinical and phase II data, brivanib failed to demonstrate improved OS in the large phase III trial (Llovet et al., 2013).
4.4. Metformin and mTOR inhibition
Both mTOR inhibitor and metformin individually have potential effect in HCC. The tolerability and efficacy of the combination thus merit investigation. mTOR inhibitor and metformin inhibit the mTOR pathway by different mechanisms. Upregulation of AKT through a positive feedback loop due to mTOR inhibition is frequently noticed. Metformin inhibits the mTOR pathway through several mechanisms, involving AMPK, REED1, and IRS 1, thus enhancing mTOR‐targeted anti‐cancer therapy (Hajjar et al., 2013; Zakikhani et al., 2010).
4.5. Sorafenib and other agents
Typically, oncologists stop sorafenib when disease progresses. However, when sorafenib is stopped, HCC patients develop accelerated portal hypertension. It is possible that HCC tissues contain various clone types and that in response to sorafenib controlling the growth of a sensitive clone, the growth of another preexisting clone or newly acquired clone may be facilitated (Naing and Kurzrock, 2010, 2013). If sorafenib is stopped because of these clones' growth, growth of the sorafenib‐sensitive clones may resume. One alternative to future HCC second‐line clinical trials would be to continue treating with sorafenib and to add additional targeted agents to block the resistance pathways. A phase I trial combining sorafenib and everolimus was conducted in 30 patients with advanced HCC of Child‐Pugh class A liver function who were naïve to systemic therapy (Finn et al., 2013). Majority of these patients were Asian. Sixteen of the 30 patients were administered 2.5 mg once daily dose of everolimus with standard dose of sorafenib, and 14 of them received 5 mg once daily dose of everolimus with standard dose of sorafenib. Adverse events on this trial were consistent with known toxicities seen with everolimus and sorafenib in patients with cancer. Twenty‐five of the 30 were evaluable for MTD determination. The MTD for everolimus was 2.5 mg once daily in combination with standard dose of sorafenib. Disease stabilization was reported in 62.5%, the median TTP was 4.5 months, and the median OS was 7.4 months at this dose level. However, phase II study of this combination was not done because of the failure to achieve a biologically effective everolimus concentration at the MTD (Finn et al., 2013).
5. Conclusion
Treatment of HCC remains challenging, and the efficacy of current therapies such as sorafenib has been dismal. Better understanding of molecular pathways involved in pathogenesis of HCC is needed. In the era of personalized therapy, baseline biopsy and blood samples, in addition to serial blood samples and biopsy at the time of progression, are becoming essential to understanding resistance mechanisms in individual patients and hence tailoring treatment on an individual basis. Additionally, advances in HCC molecular profiling and noninvasive tools to assess the status of concomitant underlying liver disease are critical to progress in this field given the independent effect of chronic liver disease on patients' tolerance of therapies and on overall survival.
Conflict of interest
None for all authors.
Bupathi Manojkumar, Kaseb Ahmed, Meric-Bernstam Funda, Naing Aung, (2015), Hepatocellular carcinoma: Where there is unmet need, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.06.005.
References
- Abou-Alfa, G.K. , Schwartz, L. , Ricci, S. , 2006. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 24, (26) 4293–4300. [DOI] [PubMed] [Google Scholar]
- Alexandrescu, D.T. , McClure, R. , Farzanmehr, H. , Dasanu, C.A. , 2008. Secondary erythrocytosis produced by the tyrosine kinase inhibitors sunitinib and sorafenib. J. Clin. Oncol. 26, (24) 4047–4048. [DOI] [PubMed] [Google Scholar]
- Arora, N. , Madhusudhana, S. , 2011. Spontaneous regression of hepatocellular cancer: case report and review of literature. Gastrointest. Cancer Res. – GCR. 4, (4) 141–143. [PMC free article] [PubMed] [Google Scholar]
- Boyault, S. , Rickman, D.S. , de Reynies, A. , 2007. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 45, (1) 42–52. [DOI] [PubMed] [Google Scholar]
- Breunig, C. , Mueller, B.J. , Umansky, L. , 2014. B-Raf and MEK inhibitors differentially regulate cell fate and microenvironment in human hepatocellular carcinoma. Clin. Cancer Res. 20, (9) 2410–2423. [DOI] [PubMed] [Google Scholar]
- Buitrago-Molina, L.E. , Vogel, A. , 2012. mTor as a potential target for the prevention and treatment of hepatocellular carcinoma. Curr. Cancer Drug Targets. 12, (9) 1045–1061. [DOI] [PubMed] [Google Scholar]
- Cheng, A.L. , Guan, Z. , Chen, Z. , 2012. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: subset analyses of the phase III sorafenib Asia-Pacific trial. Eur. J. Cancer. 48, (10) 1452–1465. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov., 2013. http://clinicaltrials.gov/.
- Coxon, A.R.K. , Sun, J. , 2008. Combined treatment of angiopoietin and VEGF pathway antagonists enhances antitumor activity in preclinical models of colon carcinoma [abstract 1113]. Proceedings of the 99th Annual Meeting of the American Association for Cancer Research; 2008 Apr 12–16; San Diego, CA. AACR; Philadelphia (PA) [Google Scholar]
- Fausto, N. , 1991. Growth factors in liver development, regeneration and carcinogenesis. Prog. Growth Factor Res. 3, (3) 219–234. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. , Davis-Smyth, T. , 1997. The biology of vascular endothelial growth factor. Endocr. Rev. 18, (1) 4–25. [DOI] [PubMed] [Google Scholar]
- Finn, R.S. , Kang, Y.K. , Mulcahy, M. , 2012. Phase II, open-label study of brivanib as second-line therapy in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 18, (7) 2090–2098. [DOI] [PubMed] [Google Scholar]
- Finn, R.S. , Poon, R.T. , Yau, T. , 2013. Phase I study investigating everolimus combined with sorafenib in patients with advanced hepatocellular carcinoma. J. Hepatol. 59, (6) 1271–1277. [DOI] [PubMed] [Google Scholar]
- Giannelli, G. , Sgarra, C. , Porcelli, L. , Azzariti, A. , Antonaci, S. , Paradiso, A. , 2008. EGFR and VEGFR as potential target for biological therapies in HCC cells. Cancer Lett. 262, (2) 257–264. [DOI] [PubMed] [Google Scholar]
- Goyal, L. , Muzumdar, M.D. , Zhu, A.X. , 2013. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin. Cancer Res. 19, (9) 2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar, J. , Habra, M.A. , Naing, A. , 2013. Metformin: an old drug with new potential. Expert Opin. Investig. Drugs. 22, (12) 1511–1517. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Folkman, J. , 1996. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 86, (3) 353–364. [DOI] [PubMed] [Google Scholar]
- Hassan, B. , Akcakanat, A. , Holder, A.M. , Meric-Bernstam, F. , 2013. Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg. Oncol. Clin. N. Am. 22, (4) 641–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata, A. , Ogawa, S. , Kometani, T. , 2002. ZD1839 (Iressa) induces antiangiogenic effects through inhibition of epidermal growth factor receptor tyrosine kinase. Cancer Res. 62, (9) 2554–2560. [PubMed] [Google Scholar]
- Hisaka, T. , Yano, H. , Haramaki, M. , Utsunomiya, I. , Kojiro, M. , 1999. Expressions of epidermal growth factor family and its receptor in hepatocellular carcinoma cell lines: relationship to cell proliferation. Int. J. Oncol. 14, (3) 453–460. [DOI] [PubMed] [Google Scholar]
- Huang, G.W. , Yang, L.Y. , Lu, W.Q. , 2005. Expression of hypoxia-inducible factor 1alpha and vascular endothelial growth factor in hepatocellular carcinoma: impact on neovascularization and survival. World J. Gastroenterol. 11, (11) 1705–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, J.O. , Blum, W.F. , Horn, N. , 1993. Insulin-like growth factors (IGF) I and II and IGF binding proteins 1, 2 and 3 during low-dose growth hormone (GH) infusion and sequential euglycemic and hypoglycemic glucose clamps: studies in GH-deficient patients. Acta Endocrinol. (Copenh.). 128, (6) 513–520. [DOI] [PubMed] [Google Scholar]
- Jorgensen, J.O. , Moller, N. , Moller, J. , Weeke, J. , Blum, W.F. , 1994. Insulin-like growth factors (IGF)-I and -II and IGF binding protein-1, -2, and -3 in patients with acromegaly before and after adenomectomy. Metabolism. 43, (5) 579–583. [DOI] [PubMed] [Google Scholar]
- Kanematsu, M. , Osada, S. , Amaoka, N. , 2004. Expression of vascular endothelial growth factor in hepatocellular carcinoma and the surrounding liver: correlation with angiographically assisted CT. AJR – Am. J. Roentgenol. 183, (6) 1585–1593. [DOI] [PubMed] [Google Scholar]
- Kanematsu, M. , Semelka, R.C. , Osada, S. , Amaoka, N. , 2005. Magnetic resonance imaging and expression of vascular endothelial growth factor in hepatocellular nodules in cirrhosis and hepatocellular carcinomas. Top. Magn. Reson. Imaging. 16, (1) 67–75. [DOI] [PubMed] [Google Scholar]
- Kaseb, A.O. , Garrett-Mayer, E. , Morris, J.S. , 2012. Efficacy of bevacizumab plus erlotinib for advanced hepatocellular carcinoma and predictors of outcome: final results of a phase II trial. Oncology. 82, (2) 67–74. [DOI] [PubMed] [Google Scholar]
- Katayama, R. , Aoyama, A. , Yamori, T. , 2013. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 73, (10) 3087–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus, A. , Birchmeier, W. , 2008. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer. 8, (5) 387–398. [DOI] [PubMed] [Google Scholar]
- Lee, C.W. , Wong, L.L. , Tse, E.Y. , 2012. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 72, (17) 4394–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet, J.M. , Bruix, J. , 2008. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 48, (4) 1312–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet, J.M. , Ricci, S. , Mazzaferro, V. , 2008. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, (4) 378–390. [DOI] [PubMed] [Google Scholar]
- Llovet, J.M. , Pena, C.E.A. , Lathia, C.D. , 2012. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 18, (8) 2290–2300. [DOI] [PubMed] [Google Scholar]
- Llovet, J.M. , Decaens, T. , Raoul, J.L. , 2013. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. J. Clin. Oncol. 31, (28) 3509–3516. [DOI] [PubMed] [Google Scholar]
- Lobo, N.A. , Shimono, Y. , Qian, D. , Clarke, M.F. , 2007. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 23, 675–699. [DOI] [PubMed] [Google Scholar]
- Lobov, I.B. , Brooks, P.C. , Lang, R.A. , 2002. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. U. S. A. 99, (17) 11205–11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre, P.C. , Suri, C. , Jones, P.F. , 1997. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 277, (5322) 55–60. [DOI] [PubMed] [Google Scholar]
- Moller, S. , Becker, U. , 1992. Insulin-like growth factor 1 and growth hormone in chronic liver disease. Dig. Dis. 10, (4) 239–248. [DOI] [PubMed] [Google Scholar]
- Moller, S. , Jensen, M. , Svensson, P. , Skakkebaek, N.E. , 1991. Insulin-like growth factor 1 (IGF-1) in burn patients. Burns. 17, (4) 279–281. [DOI] [PubMed] [Google Scholar]
- Moller, S. , Gronbaek, M. , Main, K. , Becker, U. , Skakkebaek, N.E. , 1993. Urinary growth hormone (U-GH) excretion and serum insulin-like growth factor 1 (IGF-1) in patients with alcoholic cirrhosis. J. Hepatol. 17, (3) 315–320. [DOI] [PubMed] [Google Scholar]
- Moon, W.S. , Rhyu, K.H. , Kang, M.J. , 2003. Overexpression of VEGF and angiopoietin 2: a key to high vascularity of hepatocellular carcinoma?. Mod. Pathol. 16, (6) 552–557. [DOI] [PubMed] [Google Scholar]
- Munshi, N. , Jeay, S. , Li, Y. , 2010. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol. Cancer Ther. 9, (6) 1544–1553. [DOI] [PubMed] [Google Scholar]
- Naing, A. , 2013. Overcoming resistance to mTOR inhibition for enhanced strategies in clinical trials. Expert Opin. Investig. Drugs. 22, (6) 679–685. [DOI] [PubMed] [Google Scholar]
- Naing, A. , Kurzrock, R. , 2010. Chemotherapy resistance and retreatment: a dogma revisited. Clin. Colorectal Cancer. 9, (2) E1–E4. [DOI] [PubMed] [Google Scholar]
- Naing, A. , Kurzrock, R. , 2013. Dodging a dogma: is treating beyond progression beneficial?. Cancer Chemother. Pharmacol. 71, (5) 1385–1386. [DOI] [PubMed] [Google Scholar]
- Naing, A. , Aghajanian, C. , Raymond, E. , 2012. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br. J. Cancer. 107, (7) 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCCN Clinical Practice Guidelines in Oncology Hepatobiliary Cancers Version 2.2015. http://www.nccn.org/professionals/physician_gls/pdf/hepatobiliary.pdf (accessed 17.04.15.) [Google Scholar]
- Palmer, D.H. , Midgley, R.S. , Mirza, N. , 2009. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 49, (1) 124–132. [DOI] [PubMed] [Google Scholar]
- Pang, R.W. , Joh, J.W. , Johnson, P.J. , Monden, M. , Pawlik, T.M. , Poon, R.T. , 2008. Biology of hepatocellular carcinoma. Ann. Surg. Oncol. 15, (4) 962–971. [DOI] [PubMed] [Google Scholar]
- Pardee, A.D. , Butterfield, L.H. , 2012. Immunotherapy of hepatocellular carcinoma: unique challenges and clinical opportunities. Oncoimmunology. 1, (1) 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, Y.N. , Kim, Y.B. , Yang, K.M. , Park, C. , 2000. Increased expression of vascular endothelial growth factor and angiogenesis in the early stage of multistep hepatocarcinogenesis. Arch. Pathol. Lab. Med. 124, (7) 1061–1065. [DOI] [PubMed] [Google Scholar]
- Pez, F. , Lopez, A. , Kim, M. , Wands, J.R. , Caron de Fromentel, C. , Merle, P. , 2013. Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. J. Hepatol. 59, (5) 1107–1117. [DOI] [PubMed] [Google Scholar]
- Philip, P.A. , Mahoney, M.R. , Allmer, C. , 2005. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J. Clin. Oncol. 23, (27) 6657–6663. [DOI] [PubMed] [Google Scholar]
- Puzanov, I. , Sosman, J. , Santoro, A. , 2015. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Invest. New Drugs. 33, (1) 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reig, M.E. , Rodriguez de Lope, C. , Forner, A. , 2010. Impact of early treatment interruption and biomarker profiling in patients with HCC treated with sorafenib. The Liver Meeting 2010 (AASLD), Abstract No 1748. [Google Scholar]
- Rosen, L.S. , Senzer, N. , Mekhail, T. , 2011. A phase I dose-escalation study of Tivantinib (ARQ 197) in adult patients with metastatic solid tumors. Clin. Cancer Res. 17, (24) 7754–7764. [DOI] [PubMed] [Google Scholar]
- Sangro, B. , Gomez-Martin, C. , de la Mata, M. , 2013. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 59, (1) 81–88. [DOI] [PubMed] [Google Scholar]
- Santoro, A. , Simonelli, M. , Rodriguez-Lope, C. , 2013. A phase-1b study of tivantinib (ARQ 197) in adult patients with hepatocellular carcinoma and cirrhosis. Br. J. Cancer. 108, (1) 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, A. , Rimassa, L. , Borbath, I. , 2013. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 14, (1) 55–63. [DOI] [PubMed] [Google Scholar]
- Scholz, A. , Rehm, V.A. , Rieke, S. , 2007. Angiopoietin-2 serum levels are elevated in patients with liver cirrhosis and hepatocellular carcinoma. Am. J. Gastroenterol. 102, (11) 2471–2481. [DOI] [PubMed] [Google Scholar]
- Siegel, A.B. , Olsen, S.K. , Magun, A. , Brown, R.S. , 2010. Sorafenib: where do we go from here?. Hepatology. 52, (1) 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamesa, T. , Iizuka, N. , Mori, N. , 2009. High serum levels of vascular endothelial growth factor after hepatectomy are associated with poor prognosis in hepatocellular carcinoma. Hepatogastroenterology. 56, (93) 1122–1126. [PubMed] [Google Scholar]
- Thomas, M.B. , Chadha, R. , Glover, K. , 2007. Phase 2 study of erlotinib in patients with unresectable hepatocellular carcinoma. Cancer. 110, (5) 1059–1067. [DOI] [PubMed] [Google Scholar]
- Thomas, M.B. , Morris, J.S. , Chadha, R. , 2009. Phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced hepatocellular carcinoma. J. Clin. Oncol. 27, (6) 843–850. [DOI] [PubMed] [Google Scholar]
- Torimura, T. , Ueno, T. , Kin, M. , 2004. Overexpression of angiopoietin-1 and angiopoietin-2 in hepatocellular carcinoma. J. Hepatol. 40, (5) 799–807. [DOI] [PubMed] [Google Scholar]
- Trojan, J. , Zeuzem, S. , 2013. Tivantinib in hepatocellular carcinoma. Expert Opin. Investig. Drugs. 22, (1) 141–147. [DOI] [PubMed] [Google Scholar]
- Villanueva, A. , Chiang, D.Y. , Newell, P. , 2008. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 135, (6) 1972–1983. (1983 e1971–1911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Gao, Z.Q. , Yan, X. , 2005. Correlative study of angiogenesis and dynamic contrast-enhanced magnetic resonance imaging features of hepatocellular carcinoma. Acta Radiol. 46, (4) 353–358. [DOI] [PubMed] [Google Scholar]
- Yoshiji, H. , Kuriyama, S. , Noguchi, R. , 2005. Angiopoietin 2 displays a vascular endothelial growth factor dependent synergistic effect in hepatocellular carcinoma development in mice. Gut. 54, (12) 1768–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yothaisong, S. , Dokduang, H. , Techasen, A. , 2013. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumour Biol. 34, (6) 3637–3648. [DOI] [PubMed] [Google Scholar]
- Yu, Q. , Stamenkovic, I. , 2001. Angiopoietin-2 is implicated in the regulation of tumor angiogenesis. Am. J. Pathol. 158, (2) 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakikhani, M. , Blouin, M.J. , Piura, E. , Pollak, M.N. , 2010. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res. Treat. 123, (1) 271–279. [DOI] [PubMed] [Google Scholar]
- Zheng, L. , Yang, W. , Wu, F. , 2013. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 19, (19) 5372–5380. [DOI] [PubMed] [Google Scholar]