

Abstract
Cell migration and invasion are highly regulated processes involved in both physiological and pathological conditions. Here we show that autophagy modulation regulates the migration and invasion capabilities of glioblastoma (GBM) cells. We observed that during autophagy occurrence, obtained by nutrient deprivation or by pharmacological inhibition of the mTOR complexes, GBM migration and chemokine‐mediated invasion were both impaired. We also observed that SNAIL and SLUG, two master regulators of the epithelial–mesenchymal transition (EMT process), were down‐regulated upon autophagy stimulation and, as a consequence, we found a transcriptional and translational up‐regulation of N‐ and R‐cadherins. Conversely, in BECLIN 1‐silenced GBM cells, an increased migration capability and an up‐regulation of SNAIL and SLUG was observed, with a resulting decrease in N‐ and R‐cadherin mRNAs. ATG5 and ATG7 down‐regulation also resulted in an increased migration and invasion of GBM cells combined to an up‐regulation of the two EMT regulators. Finally, experiments performed in primary GBM cells from patients largely confirmed the results obtained in established cell cultures.
Overall, our results indicate that autophagy modulation triggers a molecular switch from a mesenchymal phenotype to an epithelial‐like one in GBM cellular models. Since the aggressiveness and lethality of GBM is defined by local invasion and resistance to chemotherapy, we believe that our evidence provides a further rationale for including autophagy/mTOR‐based targets in the current therapeutical regimen of GBM patients.
Keywords: Autophagy, Cell migration, Glioma, EMT
Highlights
We were able to prove that autophagy modulates cell migration.
We show that autophagy modulation affects invasion of glioblastoma cells.
We provide evidence that some key EMT players are modulated by autophagy.
1. Introduction
Cell migration is a crucial process involved in physiological conditions during embryogenesis and post‐natal development, and in pathological contexts such as wound healing, inflammation and tumor metastatization. In order for cells to migrate, a highly regulated series of events occur within them. It has emerged that the epithelial‐to‐mesenchymal transition (EMT) and its reverse process (MET) are both essential for migration and invasion (De Craene and Berx, 2013; Nakaya and Sheng, 2013; Thompson and Williams, 2008; Acloque et al., 2009). In particular, EMT plays a critical role in tumor spreading and dissemination; once they activate EMT program, tumoral cells acquire an invasive phenotype that allows them to detach from the primary site and invade surrounding tissues and blood vessels. The reverse MET process is believed to be responsible for establishment and stabilization of metastases, following re‐acquisition by tumoral cells of epithelial‐like properties (Thompson and Williams, 2008). Differently from EMT, that has been widely investigated in recent years, only few studies exist on MET and its regulation during metastatization. A number of factors regulate the EMT/MET processes, among which are the SNAI family members SNAIL and SLUG that encode zinc finger‐containing transcriptional factors (Hemavathy et al., 2000; Peinado et al., 2007). SNAIL and SLUG are known to repress E‐cadherin expression in epithelial cells undergoing EMT, but no information exist on their role on other members of the cadherin family, neither additional roles on target genes. The Wnt/β‐catenin pathway has also been involved in EMT, by promoting transcription and stabilization of SNAIL (Zhou and Hung, 2005; Yook et al., 2005; Stemmer et al., 2008). Moreover, it has been demonstrated that SNAIL can interact with β‐catenin, thereby promoting Wnt‐dependent gene expression (Stemmer et al., 2008).
Autophagy is an evolutionary conserved process mediating degradation of cytoplasmic material, such as long‐lived proteins and old or damaged organelles (Mizushima and Komatsu, 2011; Di Bartolomeo et al., 2010a). During autophagy, double‐membrane vesicles form (the autophagosomes), surrounding and carrying the cytoplasmic material to lysosomes for degradation (Choi et al., 2013; Boya et al., 2013). In physiological conditions, autophagy contributes to the mantainance of a proper cellular homeostasis and is regulated by a signal transduction pathway involving the mTOR protein kinase. In the presence of nutrients, mTOR is activated and the autophagy initiation impaired, by inhibition of the Ulk1 complex (Nazio et al., 2013). In shortage of nutrients (mainly aminoacids) mTOR is inhibited and Ulk1 complexes can drive autophagosome formation (Boya et al., 2013; Di Bartolomeo et al., 2010b). The role of autophagy in tumor onset and progression is still a matter of debate; in fact, this process has been proposed to have a protective role for tumor initiation, by limiting genome instability and preventing accumulation of damaged organelles and proteins (Choi, 2012). Otherwise, established cancer cells probably use autophagy to promote their survival in a hypoxic and stressful microenvironment so as to escape both the physiological response to cancer and therapy (Kimmelman, 2011; Guo et al., 2013; White, 2012).
To date, only a few studies exist correlating autophagy to cell migration and invasion. Autophagy activation has been associated to the degradation of the EMT regulators SNAIL and TWIST in breast cancer models (Lv et al., 2012). Moreover, it has been demonstrated that there is high activity of mTOR protein kinase and low levels of autophagy during migration, and that pharmacological or genetic inhibition of mTOR signaling attenuated the migration of colon and hepatocellular cancer cells and that mTORC1/2 down‐regulation induced MET (Gulhati et al., 2011; Liao et al., 2015). mTOR inhibition induced cytoskeleton rearrangements, increased cell–cell contacts and decreased the activity of the small GTPases RhoA and Rac1 (Gulhati et al., 2011). By contrast, the knockdown of the autophagy regulators ATG7, or Atg5 or Atg3 stimulated cell migration of HeLa cells and MEFs respectively (Tuloup‐Minguez et al., 2013).
Autophagy impact on glioblastoma multiforme (GBM) remains poorly investigated, although it has been demonstrated that a number of autophagy regulators are highly expressed in GBM belonging to the mesenchymal subtype, and that the knockdown of some of these genes modifies the migration and invasion properties of GBM cells (Galavotti et al., 2013; Macintosh et al., 2012). Autophagy induction has been observed in GBM in response to radio‐ and temozolomide‐based therapy; even though a number of clinical trials aimed at inhibiting autophagy have been launched, others directed to inhibiting the mTOR pathway are ongoing (Amaravadi et al., 2011; Galanis et al., 2005; Nghiemphu et al., 2012; Dufour et al., 2011). Furthermore, recently, it has been shown that the combination of epidermal growth factor (EGFR) down‐regulation and autophagy stimulation impairs clonogenic and migration capability of T98G and U373MG glioma cells exposed to irradiation (Palumbo et al., 2014).
Here we show that autophagy modulation regulates the migration and the invasion capabilities of GBM cells by down‐regulating EMT master factors. GBM, the most common and lethal adult brain tumor, is characterized by an highly invasive behavior. Overall, our data suggest that therapeutical strategies targeting autophagy should take into account any possible effects on GBM invasiveness.
2. Results
2.1. Autophagy is induced in glioma cell lines upon aminoacid deprivation and mTOR inhibitors treatment
To determine the response of GBM cell lines to autophagic stimuli, the human GL15 and U87MG cells were incubated in an aminoacid‐free and serum‐free medium Earle's Balanced Salt Solution (EBSS), and then analyzed, at different time points, for autophagy occurrence by means of LC3I to LC3II conversion and by checking p62 down‐regulation. As shown in Figure 1a, both markers indicate a time‐dependent induction of autophagy. Upon 18 h of EBSS incubation, a further degradation and disappearance of LC3II can be observed in the absence of Chloroquine (CQ), a drug known to inhibit autophagosome to lysosome fusion and widely used to monitor the autophagic flux.
Figure 1.
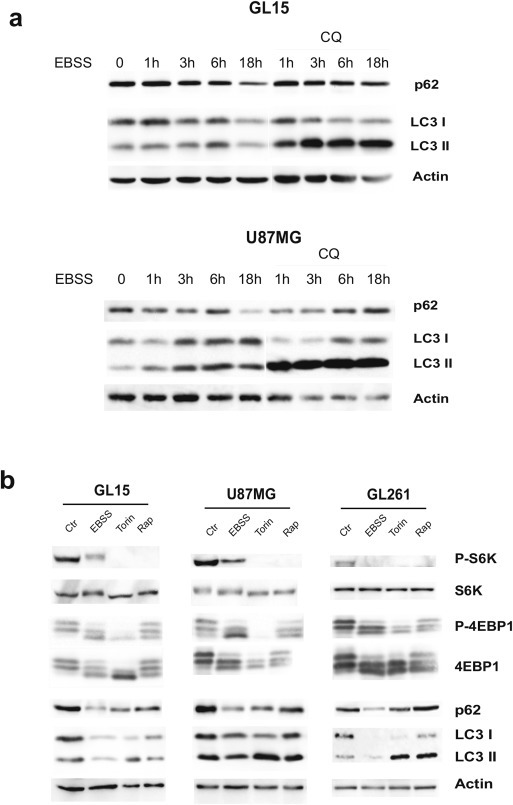
Induction of autophagy in glioma cells lines. (a) Human GL15 and U87MG cells were incubated in aminoacid‐ and serum‐free medium (EBSS) for the indicated time points in presence or absence of 20 μM Chloroquine (CQ) that impairs autophagosome‐to‐lysosome fusion and autophagy completion. After cell lysis, protein extracts were subjected to western blotting analysis by using specific antibodies for the autophagy markers LC3I/II and p62. Actin was used as loading control. The blots are representative of three independent experiments. (b) Human GL15 and U87MG and murine GL261 cell lines were incubated for 18 h in aminoacid‐ and serum‐free medium (EBSS) or in a rich medium containing 250 nM Torin 1 or 100 nM Rapamycin. After cell lysis, protein extracts were subjected to western blotting analysis by using specific antibodies for the mTOR substrates S6Kinase (S6K) and 4EBP1. Specific antibodies were used for the phosphorylated forms of both (phospho‐S6KThr389 and phospho‐4EBP1Thr37/46). The same protein extracts were subjected to western blotting analysis for the autophagy markers LC3I/II and p62. Actin was used as loading control. This blot is representative of three independent experiments.
In addition to starvation in EBSS medium, two potent pro‐autophagic drugs, the mTOR inhibitors Rapamycin and Torin 1 (Torin), were used and their capability to impair the mTOR pathway and to induce autophagy were tested. As shown in Figure 1b, in human GL15 and U87MG and in the mouse GL261 cell lines autophagy was fully activated following starvation and, at a lesser extent, upon mTOR inhibition at 18 h, as demonstrated by autophagy markers LC3 and p62. Dephosphorylation of the substrates p70S6 kinase and 4EBP1 demonstrated the effective mTOR signaling inhibition in the GBM cell lines treated with EBSS, Torin or Rapamycin (Figure 1b). None of the treatments affected cell viability for up to 48 h, as demonstrated by citofluorimetric analysis of propidium iodide‐stained cells and by morphological analysis (not shown).
2.2. Starvation and pharmacological mTOR inhibition negatively regulate cell migration and invasion in glioma cell lines
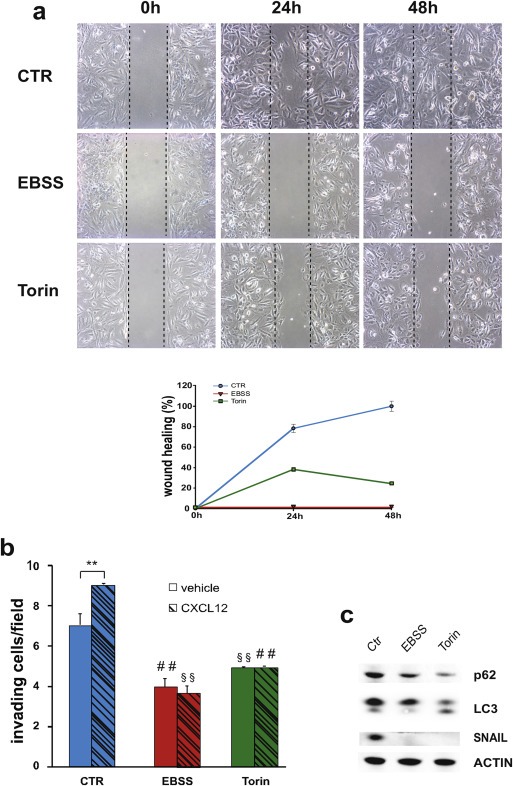
Next, we analyzed the migration capability of GBM cell lines in autophagy‐stimulating conditions (starvation and mTOR inhibition) by means of a wound healing assay. We observed a strong inhibition of chemokinesis in EBSS grown‐cells and, at a lesser extent, in Torin and Rapamycin‐treated cells in comparison with control cells (Figure 2a, Suppl. Figure S1 and Suppl. Video 1). The wound healing assay was performed in presence of 5 mM hydroxyurea to exclude the contribution of cell proliferation to the observed phenomena. Prompted by these results, we decided to test the effect, if any, of autophagy modulation on CXCL12‐induced invasion. GBM cells are known to express high levels of CXCR4 receptor that allow them to migrate both in vitro and in vivo models in response to the chemokine CXCL12 (Rubin et al., 2003). By means of a transwell‐based assay, in the presence of matrigel, we observed an evident reduction of spontaneous cell invasion in all the three cell lines analyzed when incubated in EBSS and in Torin‐containing medium (Figure 2b and Suppl. Figure S1, clear columns); moreover, an almost complete impairment of the capability to respond to CXCL12 was observed in these conditions (Figure 2b and Suppl. Figure S1, dark columns). We also analyzed the cellular invasion in Rapamycin‐containing medium and we found a slight but significant reduction of both spontaneous and CXCL12‐mediated invasion of GL15 and GL261, but not of U87MG cells (Figure 2b and Suppl. Figure S1). This result is in line with the evidence of a lesser capability of Rapamycin to induce autophagy in our model systems. To rule out the possibility that the invasion impairment observed in autophagic conditions was due to an autophagy‐mediated degradation of the CXCL12 receptor CXCR4, a western blotting analysis was performed with a specific antibody. In GL15 and U87MG cells grown for 24 h in EBSS or in the presence of Torin or Rapamycin, no differences were observed in CXCR4 expression levels (Suppl. Figure S2a), even if in the presence of the protein synthesis inhibitor cycloeximide (CHX) (Suppl. Figure S2b).
Figure 2.
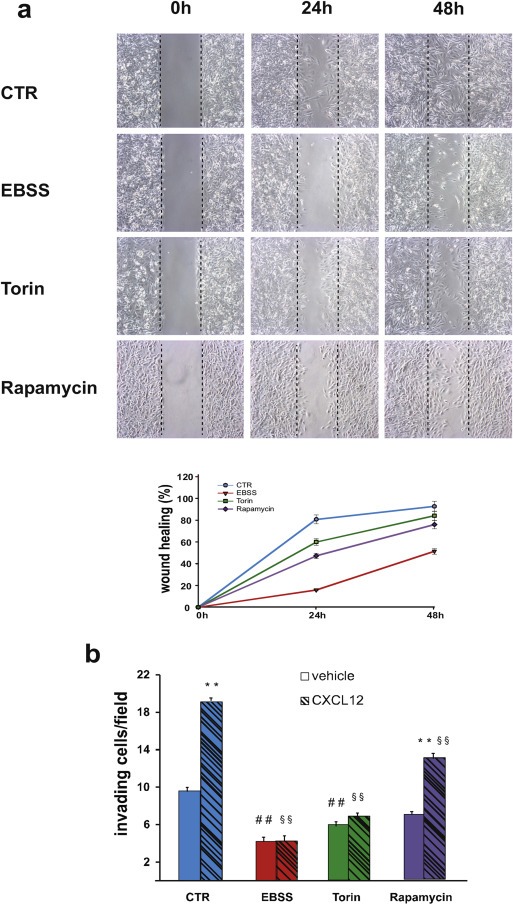
Impact of starvation and mTOR inhibition on glioma cell migration and invasion. (a) Wound healing assays were performed on GL15 cells in control rich medium (CTR) or in aminoacid‐ and serum‐free medium (EBSS) or in a rich medium containing 250 nM Torin 1 or 100 nM Rapamycin. 5 mM hydroxyurea was added to the culture media to impair cell proliferation. Phase‐contrast images (10× objective) were acquired at 0, 24 and 48 h after scratching and representative images of four independent experiments are shown. The wound healing area was analyzed by using ImageJ software (NIH) and the corresponding data, relative to 0 h, expressed in the graph. Statistical significance: P < 0.001, one‐way ANOVA. (b) Invasion assays were performed on GL15, in presence of vehicle (DMSO, light gray columns) or 100 nM CXCL12 (dark gray columns) in DMEM (CTR) or in EBSS or in DMEM containing 250 nM Torin 1 (Torin) or in DMEM containing 1 μM Rapamycin. The graphs shown represent the mean ± SE of at least three different experiments. Statistical significance of CXCL12 versus vehicle in control condition and in Rapamycin‐treated cells is indicated (**). Statistical significance of EBSS or Torin or Rapamycin vs CTR is indicated in the absence (##) and presence (§§) of CXCL12. Statistical significance: **P < 0.001, ##P < 0.001; §§P < 0.001, one‐way ANOVA.
Supplementary videos related to this article can be found online at http://dx.doi.org/10.1016/j.molonc.2015.04.016.
The following are the Supplementary videos related to this article:
2.3. Autophagy inhibition stimulates cell migration and invasion in GL15 glioma cells
To confirm the inhibitory role of autophagy on cell chemokinesis, we decided to analyze the migration capability of BECLIN 1‐depleted GBM cells. BECLIN 1, in fact, is one of the main autophagy regulators driving the autophagosome formation in basal conditions and upon canonical autophagy stimulation. GL15 cells were infected with a lentivirus carrying a BECLIN 1‐specific short hairpin RNA (shRNA) or an unrelated shRNA, and the effect on autophagy was evaluated by western blotting for p62 and LC3I/II. BECLIN 1 down‐regulation resulted in p62 accumulation and LC3I/II increase in GL15 cells, showing an impairment of basal autophagy (Figure 3a, left panel). When subjected to wound healing assay in complete medium (DMEM), BECLIN 1‐depleted GL15 cells were able to repair the wound much faster than control cells (Figure 3a, right panel and Suppl. Video 2), thus confirming that autophagy is inversely correlated to chemokinesis in this GBM cell line. A significant reduction of cell migration was also obtained upon ATG5 and ATG7 down‐regulation in GL15 cells (Figure 3b and Suppl. Figure S3).
Figure 3.
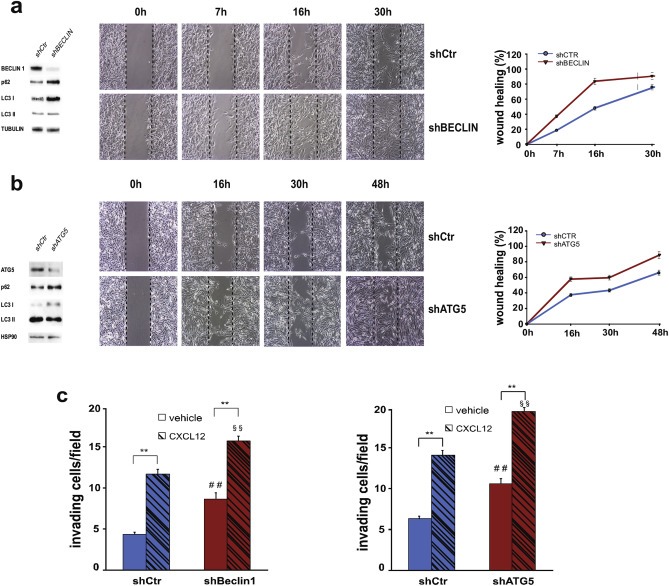
Impact of autophagy inhibition on glioma cell migration and invasion. (a–b) GL15 were transduced with a scramble RNA (shCtr) or with a BECN 1‐directed shRNA (shBECLIN) (a) or with an ATG5‐directed shRNA (b). Protein extracts were analyzed by western blotting in order to check the infection efficiency by using antibodies specific for BECLIN 1 and for ATG5 (a and b, respectively, left panels) and the effects of the interference on autophagy impairment by means of p62 and LC3I/II modulation. β‐tubulin and HSP‐90 were used as loading controls. Wound healing assays were performed on BECLIN 1‐ and ATG5‐depleted GL15 cells (shBECLIN and shATG5, respectively) and on control cells (shCtr) (a and b, right panels). Phase‐contrast images were acquired at the indicated time points following scratching. The images are representative of four independent experiments. The wound healing areas were analyzed by using ImageJ software (NIH) and the corresponding data, relative to 0 h, expressed in the graph. Statistical significance: P < 0.001, one‐way ANOVA. (c) Invasion assay on shCtr and shBECLIN 1‐silenced GL15 cells (left graph) and on shCtr and shATG5 (right graph) in presence of vehicle (DMSO, light gray columns) or 100 nM CXCL12 (dark gray columns). The graphs represent the mean ± SE of four different experiments. **P < 0.001, one‐way ANOVA. Statistical significance of shBeclin or shATG5 vs shCtr is indicated in the absence (##) and presence (§§) of CXCL12. ##P < 0.001; §§P < 0.001; one‐way ANOVA.
Supplementary videos related to this article can be found online at http://dx.doi.org/10.1016/j.molonc.2015.04.016.
The following are the Supplementary videos related to this article:
We also analyzed the chemotactic ability of BECLIN 1‐depleted GL15 cells and we found an increase of basal cell invasion, although we did not observe an increased response to CXCL12 in the absence of BECLIN 1 in comparison to control cells (Figure 3c, left panel). In order to corroborate the results obtained, we also analyze the invasion capability of ATG5‐ and ATG7‐silenced cells that indeed resulted increased (Figure 3c, right panel and Suppl. Figure S3).
Taken together, these data demonstrate that autophagy induction negatively correlates not only with chemokinesis, but also with cell invasion. Conversely, turning off autophagy results in a greater capability of GL15 cells to move across an extracellular‐like matrix.
2.4. The EMT regulators SNAI are down‐regulated during autophagy induction
In order to gain an insight into the molecular mechanisms involved in the autophagy‐mediated modulation of cell migration and invasion, an analysis of the main EMT/MET players was performed in the human GBM cell lines. As shown in Figure 4, both GL15 and U87MG express high levels of SNAI family members SNAIL and SLUG in control conditions, as expected for GBM and other highly invasive cancer cells (Jin‐Ku et al., 2014). Upon autophagy induction, obtained by culturing cells in EBSS for 24 h, a meaningful decrease in SNAIL and SLUG proteins was observed (Figure 4a). A significant reduction of SNAIL was also evident after incubation of the cells with mTOR inhibitors even though to lesser extent; notably, SLUG levels did not decrease in U87MG cells (Figure 4a). It was recently shown that SNAIL and TWIST are degraded by the autophagy‐lysosome degradation system in metastatic breast cancer cells upon PI3K or Beclin 1 overexpression and viceversa (Lv et al., 2012). To test whether the SNAIL decrease observed upon autophagy induction in GBM model systems was due to an autophagy‐mediated protein degradation, we induced autophagy in GL15, by EBSS or Torin administration, in the presence of MG132 or Chloroquine (CQ), which inhibit the ubiquitin–proteasome and the autophagy–lysosome degradation systems, respectively. The strong increase observed in control cells in the presence of MG132 (Figure 4b) confirms that SNAIL is mainly a physiological substrate of the proteasome machinery, as described in the literature (Viñas‐Castells et al., 2010; Lander et al., 2011); However, when autophagy is induced, an evident reduction of SNAIL expression was still observed, similar to that observed in the absence of inhibitors (Figure 4b). Moreover, in the presence of CQ, which inhibits the autophagosome fusion to lysosomes with a concomitant accumulation of LC3II protein, a slight accumulation of SNAIL was observed in control cells, this suggesting that autophagy contributes to SNAIL recycling in basal conditions (Figure 4b–c); in the presence of CQ, anyway, autophagy stimulation still leads to SNAIL down‐regulation in the long time range. Prompted by these results, we decided to better analyze the kinetics of SNAIL degradation and found that, 3 h exposure to EBSS or six hours of Torin treatment are sufficient to trigger an evident reduction of the protein (Figure 4d). In order to evaluate SNAIL half‐life in our model systems, we treated GL15 and U87MG cells with the protein synthesis inhibitor cycloeximide (CHX) and observed that the protein is completely depleted in U87MG and strongly down‐regulated in GL15 cells in a few hours in normal conditions (Figure 4e). These findings suggest that the SNAI decrease observed upon sustained autophagy induction, could be mainly due to a general impairment of the protein synthesis rather to an increase of protein degradation. We also decided to analyze SNAI expression levels in GL15 cells devoid of BECLIN 1 and ATG5; we found an increase of both SNAIL and SLUG proteins (Figure 4f) thus confirming our hypothesis about an involvement of the autophagic machinery in SNAI turnover in basal conditions.
Figure 4.
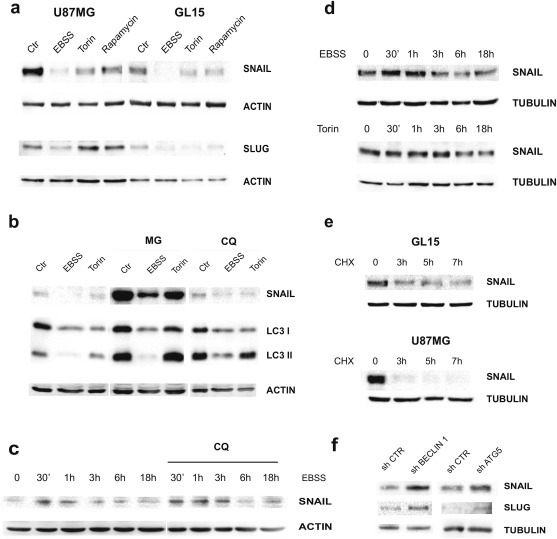
Effect of autophagy induction on EMT regulators. (a) Western blotting of protein extract from U87MG and GL15 cells cultured in DMEM (Ctr) or EBSS or DMEM containing 250 nM Torin 1 or 100 nM Rapamycin for 18 h was performed by using specific antibodies for SNAIL and SLUG. ACTIN was used as loading control. (b) GL15 were grown in DMEM (Ctr) or EBSS or incubated, for 18 h, with Torin 1, in presence of 3 μM MG132 (MG) or 20 μM Chloroquine (CQ). Western blotting analysis of SNAIL was then performed. ACTIN was used as loading control. Accumulation of LC3II band was used to check CQ efficiency. (c) GL15 were grown in EBSS or in Torin 1 (Torin) for the indicated time points and the corresponding protein extracts analyzed by Western blotting for SNAIL. TUBULIN was used as loading control. (d) GL15 and U87MG cells were incubated with 50 μg/ml cycloheximide (CHX) for the indicated times and Western blotting of SNAIL performed. TUBULIN was used as loading control. (e) GL15 were grown in EBSS for the indicated time points, in the presence or absence of 20 μM Chloroquine (CQ) and the corresponding protein extracts subjected to western blotting analysis by means of a specific antibody for SNAIL. ACTIN was used as loading control. (f) Western blotting analysis of SNAIL and SLUG proteins in shCtr, shBECLIN 1‐ and shATG5‐GL15 cells is shown. All the blot shown are representative of at least three different experiments.
2.5. N‐ and R‐Cadherin expression is affected by autophagy
SNAI factors are known to negatively regulate cadherin gene (CDH) transcription. The CDH superfamily encodes several types of cadherin differently expressed in tissues. The inhibitory role of SNAIL on E‐cadherin expression via repression of CDH1 promoter is well established. In order to investigate the effect of SNAI down‐regulation on cadherin expression in GBM cell lines undergoing autophagy, we performed a Real Time PCR analysis of selected cadherin mRNAs. In particular, we decided to analyze N‐ and R‐cadherin that are known to be expressed in the neural tissue. As shown in Figure 5a, we observed a remarkable increase in N‐CAD and R‐CAD mRNAs in both GL15 and U87MG cells grown in EBSS medium or in a nutrient‐rich medium containing Torin. We also analyzed E‐CAD and P‐CAD mRNAs that are not expressed in neural tissues in normal conditions; indeed, we found a very low expression level that did not further increase in autophagic conditions (data not shown). On the contrary, R‐CAD and N‐CAD mRNAs were significantly down‐regulated in BECLIN 1‐depleted GL15 cells, in which we know that SNAI proteins are up‐regulated (Figure 5b). Then we decided to analyze the effect of autophagy on cadherin protein levels by using an antibody against several members of cadherin superfamily and we observed an increase of expression in EBSS‐ and in Torin 1‐treated cells (Figure 5c). As expected, the specific analysis of E‐cadherin protein did not result in any signals by western blotting analysis (not shown).
Figure 5.
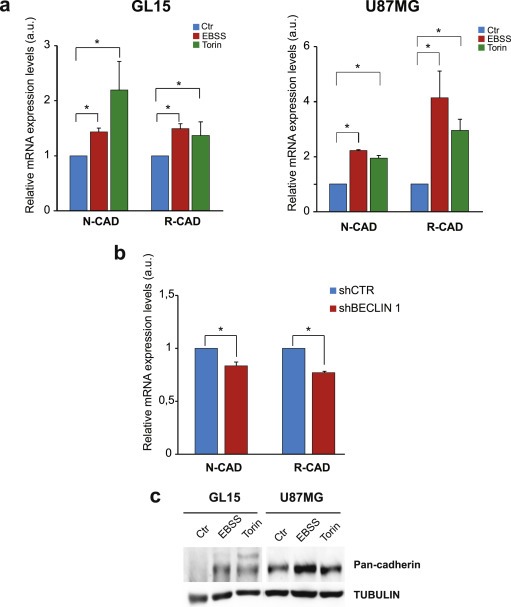
Cadherin expression upon autophagy modulation. (a) mRNA expression levels of N‐CADHERIN (N‐CAD) and R‐CADHERIN (R‐CAD) obtained by real‐time PCR on GL15 (left graph) and U87MG (right panel) cultured in DMEM, EBSS and 250 nM Torin 1‐containing medium, as indicated in the graph legend. The graphs represent the mean ± SE of three different experiments. Statistical significance: *P < 0.05; one‐way ANOVA. (b) mRNA expression levels of N‐CAD and R‐CAD obtained by real‐time PCR on GL15 cells after retroviral infection with scramble RNA (shCtr) or BECN‐1‐directed RNA (shBECLIN) grown in control conditions. Statistical significance: *P < 0.05; one‐way ANOVA. (c) Protein extracts from GL15 and U87MG cells grown in DMEM (Ctr), in EBSS and in Torin 1 (Torin) were subjected to Western blotting analysis of cadherins by using anti‐Pan‐Cadherin antibody. TUBULIN was used as loading control.
Overall, the results obtained on cadherin mRNAs and proteins demonstrate that the SNAI down‐regulation results in cadherin up‐regulation during autophagy induction and viceversa in GBM cell lines.
2.6. Autophagy induction regulates migration and invasion in GBM primary cells
To confirm the results obtained in established cell lines, we decided to extend our analysis to GBM primary cells obtained from patients after surgical resection (Sciaccaluga et al., 2013). Similarly to what we observed in cell lines, both primary cultures almost completely lost their migration capability when grown in EBSS medium (Figure 6a and Suppl. Figure S4a). Migration capability was also strongly reduced in cells grown in Torin‐containing medium (Figure 6a and Suppl. Figure S4a). Cellular invasion following CXCL12 exposure was also evaluated, demonstrating that autophagy induction drastically affects the invasion capability of primary GBM cells (Figure 6b and Suppl. Figure S4b). Accordingly, SNAIL expression strongly decreases in EBSS and in presence of Torin (Figure 6c and Suppl. Figure S4c). Overall, we were able to reproduce in primary cells the main results obtained in cell lines, thus confirming the physio‐pathological relevance of our study.
Figure 6.
Analysis of migration and invasion in primary glioblastoma cells upon autophagy induction. (a) Wound healing assay of a human primary glioblastoma culture, named GBM47, grown in DMEM (CTR), in EBSS or in Torin‐containing DMEM. Phase‐contrast microphotographs (10× objective) were acquired at 0, 24 and 48 h after scratching and representative images of three independent experiments are shown. The wound healing area was analyzed by using ImageJ software (NIH) and the corresponding data, relative to 0 h, expressed in the graph. Statistical significance: P < 0.001, one‐way ANOVA. (b) Graph showing the results of invasion assays performed on GBM47 grown in DMEM (CTR), in EBSS or in Torin‐containing DMEM, in presence of CXCL12 (dark gray columns) or vehicle (light gray columns). The graph represents the mean ± SE of three different experiments. Statistical significance of CXCL12 versus vehicle in control condition cells is indicated (**). Statistical significance of EBSS or Torin vs CTR is indicated in the absence (##) and presence (§§) of CXCL12. Statistical significance: **P < 0.001, ##P < 0.001; §§P < 0.001, one‐way ANOVA. (c) Western blotting analysis of the indicated proteins in cellular extracts from GBM47 grown in DMEM (CTR), in EBSS or in Torin‐containing DMEM for 18 h. ACTIN was used as loading control. This blot is representative of two different experiments.
3. Discussion
Here we demonstrate the existence of a direct effect of nutrient starvation and mTOR inhibition on the capability of GBM cells to move, both in the absence or presence of a chemoattractant agent. We clearly observed that autophagy activation impairs cell migration and invasion and that, conversely, autophagy inhibition stimulates the same process, similarly to what has been observed in highly metastatic breast, colon and hepatocellular cancer cells (Lv et al., 2012; Gulhati et al., 2011; Liao et al., 2015). Of note, other literature data show that the autophagic process is requested by glioma cells to migrate and that the down‐regulation of some autophagy genes limits the cell capability to migrate and invade (Galavotti et al., 2013; Macintosh et al., 2012). However, the different model systems (mouse or different human cell lines) used there as well as completely different time‐courses or most proteins analyzed could explain the apparent variation with our results. Moreover, we correlate the migration capabilities with a molecular shift from a mesenchymal phenotype to an “epithelial‐like” one (see Figure 7), similarly to what has been observed in other cancer cells (Lv et al., 2012; Gulhati et al., 2011). We found, in fact, a down‐regulation of the EMT player SNAIL upon prolonged starvation and Torin treatment in our GBM models; this seems to be due to a general impairment of the protein synthesis, as expected from the evidence that the mTORC1 complex regulates mRNA translation (Thoreen et al., 2012). Differently from starvation, that equally affects both SNAIL and SLUG expression levels, Torin treatment triggers SLUG down‐regulation in GL15 but not in U87MG cells, indicating that SLUG is differently regulated or that it is more stable than SNAIL in U87MG cells. Our results are in line with the previous evidence that SNAIL down‐regulation, by RNA interference, inhibited migration and invasion of U87MG and other glioma cell lines (Han et al., 2011). Notably, although both classified as IV grade GBMs, GL15 and U87MG show a very different phenotype and invasive behavior, with mouse GL261 partially possessing some U87MG‐like features (Guillamo et al., 2001; Candolfi et al., 2007; Szatmári et al., 2006). It is not surprising, therefore, to observe differences between the two cell lines both in protein pattern profiling and in response to stimuli; it is relevant that the final migration outcome is similar upon autophagy induction.
Figure 7.
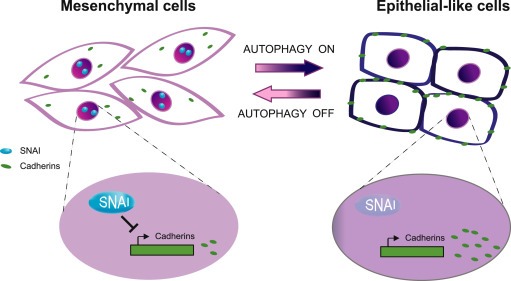
Proposed model illustrating the effect of autophagy on glioma phenotype. When autophagy is off, glioma cells show a typical mesenchymal phenotype with absence of cell–cell contacts, expression of SNAI proteins that repress cadherin transcription (left part of the cartoon). When autophagy is induced (right part of the cartoon), glioma cells loss SNAI proteins, with a consequent increase of cadherin expression so acquiring an epithelial‐like phenotype.
The decrease of SNAI proteins expression levels results in a transcriptional and post‐transcriptional increase in N‐ and R‐cadherin expression. Intriguingly, the increase of N‐ and R‐CAD mRNAs, upon Torin treatment, is less evident in U87MG than in GL15 cells, where both SNAI levels are reduced, thus supporting an important role for SLUG in CDH repression. Whilst it is well known that loss of epithelial E‐cadherin and cell polarity promotes EMT in carcinomas, the contribution of cadherins to the development of non‐epithelial tumors is less well documented (Onder et al., 2008; Gheldof and Berx, 2013). Classical cadherins are Ca2+‐dependent transmembrane proteins, which mediate homophilic interaction between adjacent cells and display a tissue‐specific expression (Wheelock and Johnson, 2003). Among others, E‐cadherin and N‐cadherin are the most studied and are mainly expressed in epithelial and in neural tissues, respectively. E‐cadherin expression in neural tissues is limited to stem cells and specific areas and it is rare in GBM, although it has been observed an abnormal E‐cadherin expression in a subset of highly aggressive GBM cells (Lewis‐Tuffin et al., 2010). N‐cadherin contributes to the polarity and migration of primary astrocytes (Camand et al., 2012). It has been observed that a decreased N‐cadherin expression is involved in the abnormal migratory behavior of GBM cells, which ‐differently from astrocytes, are unable to polarize and show a faster and less‐directed migration; re‐expression of N‐cadherin in gliomas, intriguingly, strongly reduces cell migration and restores polarity (Camand et al., 2012). The much less studied R‐cadherin is 74% identical to N‐cadherin and can form heterodimers with it in neural tissues. R‐cadherin role in GBM tumorigenesis has not yet been investigated, but its inappropriate expression in some epithelial cancer cells has been associated with an increased cell motility, dependent on higher activation of the GTPases Rac1 and Cdc42 (Johnson et al., 2004).
In our model, upon autophagy induction, we observe SNAI degradation which may be responsible for the impairment of cell movement as previously demonstrated in GBM cells (Han et al., 2011). Despite promising preclinical studies, targeting mTOR in cancer therapy has shown limited clinical benefits so far, probably due to the complexity of mTOR signaling; mTORC1 inhibition, in fact, can stimulate proliferative and pro‐survival pathway (Dufour et al., 2011).
Further, Ambra1 and Beclin 1, both upstream regulators of autophagy, have been recently shown to regulate cell cycle progression besides their direct pro‐autophagic activities (Qu et al., 2003; Cianfanelli et al., 2014), this being a key factor in tumorigenesis. However, approaches other than mTOR‐inhibiting drugs and autophagy blockers (such as chloroquine or its derivatives), aimed at targeting and modulating the autophagic process and its cross‐talk with other cellular pathways are currently lacking in therapy. Therefore, we believe that the findings presented here add important clues for the definition of future therapeutic strategies targeting the autophagic process as means to reduce the invasive properties of GBM, certainly one of the most serious obstacles to complete tumor eradication with the current multimodal therapy.
4. Materials and methods
4.1. Cell cultures
Human GL‐15 were kindly provided by Dr. E. Castigli, Perugia University, Italy. Mouse GL261 cells were kindly provided by Dr. S. Pellegatta, “C.Besta” Neurological Institute, Italy. All cell lines were cultured in DMEM (Invitrogen) supplemented with 10% heat‐inactivated FBS (Invitrogen), 100 IU/ml penicillin G, 100 μg/ml streptomycin, 2.5 μg/ml amphotericin B, 2 mM glutamine, and 1 mM sodium pyruvate. Cells were grown at 37 °C in a 5% CO2 humidified atmosphere.
Tumor specimens were obtained from the Department of Neurosurgery, Neuromed, from GBM patients who gave a written informed consent to the research proposals. The study was approved by the Institutional Ethics Committee of Neuromed and by Ministry of Health. Histopathological typing and tumor grading were done according to the WHO criteria resulting as grade IV (Sciaccaluga et al., 2013).
For autophagy induction, cells were treated with 250 nM Torin 1 (Sigma–Aldrich), 100 nM and 1 μM Rapamycin (Sigma–Aldrich) or cultured in Earle's Balanced Salt Solution (Sigma–Aldrich).
Where indicated, 3 μM MG132, 20 μM Chloroquine (CQ) and 50 μg/ml Cycloheximide (CHX) were added to the media.
4.2. Lentiviral preparation and infection
15 μg retroviral vectors was cotransfected with 5 μg expression plasmid for the vesicular stomatitis virus G protein into 293 gp/bsr cell line by using the calcium phosphate method. 48 h later, the supernatant containing the retroviral particles was recovered and supplemented with 4 μg/ml poly‐brene. GL15 cells were infected by incubation with lentivirus‐containing supernatant for 6–8 h.
4.3. Wound healing assay
A wound‐healing assay was used to compare the migratory ability of GBM cells in control and autophagic conditions. The same number of cells (4–9 × 104 cells/ml) were seeded and cultured into the inner wells of cell culture inserts (Ibidi, Germany) placed in a Petri dish. Once attached to the substratum, the inserts were removed from the surface letting a 500 μm cell‐free gap in which cells could migrate. To discriminate the contributions of cell proliferation and migration to wound closure, cell cycle blocker hydroxyurea (5 mM, Sigma–Aldrich) was added for the time of the experiment. To analyze the cell migration, the wounded areas were photographed at the indicated time points with CoolSNAP camera (Photometrics) coupled to an ECLIPSE Ti–S phase contrast microscope (Nikon) and processed using ImageJ analysis software (NIH). Percentage of wound healing was measured as following: [1 − (empty area X h/empty area 0 h)] × 100.
4.4. Invasion assay
Sub‐confluent cells were trypsinized, pre‐incubated with or without 250 nM Torin in invasion medium (DMEM without glutamine supplemented with 100 IU/ml penicillin G and 100 μg/ml streptomycin or EBSS with 0.1% BSA and 25 mM HEPES, pH 7.4) for 2 h, and plated at a density of 7 × 103 cells/cm2 on matrigel‐coated transwells (8 μm pore size, Costar). 100 nM CXCL12 (Peprotech) or vehicle were added to the lower chamber of the transwell system. After 24 h (for U87MG, GL‐15 and GBM primary cells) or 48 h (for GL261) of incubation at 37 °C, cells were treated with ice‐cold 10% trichloroacetic acid for 10 min. Cells adhering to the upper side of the filter were scraped off, whereas cells that have invaded through the insert were stained with a solution containing 50% isopropanol, 1% formic acid and 0.5% (wt/vol) brilliant blue R 250 (Sigma–Aldrich) and counted in more than 20 fields with a 20× objective. Data are the mean of cells that have invaded through the insert ± SE of at least 3 experiments.
4.5. Western blotting and antibodies
Proteins were separated on SDS‐PAGE electrophoresis gel or on NuPAGE Bis‐Tris gel (Invitrogen) and electroblotted onto nitrocellulose (Protran; Schleicher & Schuell). Blots were incubated with primary antibodies in 5% nonfat dry milk in TBS plus 0.1% Tween‐20 overnight at 4 °C. Detection was achieved using horseradish peroxidase‐conjugated secondary antibody (Bio‐Rad Laboratories) and visualized with ECL plus (GE Healthcare). The following primary antibodies were used in this study: anti‐p70S6K and anti‐phospho‐p70S6K (Thr389), anti‐4EBP1 and anti‐phospho‐4EBP1 (Thr37/46), anti LC3, anti‐ SNAIL, anti‐SLUG (Cell Signaling), polyconal anti‐p62, anti‐Beclin1 (Santa Cruz), anti‐Actin and anti β‐tubulin (Sigma).
4.6. RNA isolation and quantitative RT‐PCR
After treatments, total RNA was extracted with the RNeasy mini kit (Qiagen), following the producer's instructions. 1–2 μg of total RNA was then retrotranscribed with oligodT and M‐MLV (Promega) and used for the quantitative PCR (SYBR GREEN masetr mix, Roche) with the following primers: for N‐cadherin, 5′‐TATGCCCAAGACAAAGAGACC‐3′ (Fw) and 5′‐CAACTTCTGCTGACTCCTTCA‐3′ (Rev); for R‐cadherin, 5′‐CGAGTTCTGAGCTGTGCTATG‐3′ (Fw) and 5′‐GAGATTTCCAGTCCTTCCACTAC‐3′ (Rev); for P‐cadherin, 5′‐ATGACTTCACTGTGCGGAAT‐3′ (Fw) and 5′‐CCAATCTCTCTTGTGTCTTCGT‐3′ (Rev); for E‐cadherin, 5′‐CTCCCTTCACAGCAGAACTAAC‐3′ (Fw) and 5′‐CCACCTCTAAGGCCATCTTTG‐3′ (Rev). Relative expression levels were calculated after normalization over β‐Actin.
4.7. Statistical analysis
All experiments were performed at least three times. SigmaPlot software was used for statistical analysis. Statistical significance was determined by using one‐way analysis of variance (ANOVA) followed by Student–Newman–Keuls or Student–Holm–Sidak Method. *P < 0.05 was considered significant.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
We thank S. Pellegatta, E. Castigli, G. Schiavo and G. Velasco for kindly providing us with GL261 cells, GL15 and U87MG, respectively. We also thank G.M. Fimia for shBECN and shCtr viruses, F. Loreni for helpful discussion and F. Faienza and F. Pagani for research assistance. We also thank M. Acuña Villa and M.W. Bennett for editorial and secretarial work. This work was supported by grants from AIRC (IG 2010 and IG 2012 to FC and IG12774 to CL), from Telethon Foundation (GGP10225), the Italian Ministry of University and Research (PRIN 2009: 20095932WN and FIRB Accordi di Programma 2011: RBAP11LP2W‐003) and the Italian Ministry of Health (RF 2009).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.04.016.
Catalano Myriam, D'Alessandro Giuseppina, Lepore Francesca, Corazzari Marco, Caldarola Sara, Valacca Cristina, Faienza Fiorella, Esposito Vincenzo, Limatola Cristina, Cecconi Francesco, Di Bartolomeo Sabrina, (2015), Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.04.016.
References
- Acloque, H. , Adams, M.S. , Fishwick, K. , Bronner-fraser, M. , Nieto, M.A. , 2009. Review series epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. 119, (6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi, R.K. , Lippincott-Schwartz, J. , Yin, X.-M. , Weiss, W.A. , Takebe, N. , Timmer, W. , 2011. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 17, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya, P. , Reggiori, F. , Codogno, P. , 2013. Emerging regulation and functions of autophagy. [Internet] Nat. Cell Biol. 15, 713–720. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23817233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camand, E. , Peglion, F. , Osmani, N. , Sanson, M. , Etienne-Manneville, S. , 2012 Feb 15. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. [Internet] J. Cell Sci. 125, (Pt 4) 844–857. [cited 2014 May 1]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22275437 [DOI] [PubMed] [Google Scholar]
- Candolfi, M. , Curtin, J.F. , Nichols, W.S. , Muhammad, A.G. , King, G.D. , Pluhar, G.E. , 2007. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J. Neurooncol. 85, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.S. , 2012. Autophagy and cancer. Exp. Mol. Med. 109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, A.M.K. , Ryter, S.W. , Levine, B. , 2013 Mar 14. Autophagy in human health and disease. [Internet] N. Engl. J. Med. 368, (7) 651–662. [cited 2014 Apr 29]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23406030 [DOI] [PubMed] [Google Scholar]
- Cianfanelli, V. , Fuoco, C. , Lorente, M. , Salazar, M. , Quondamatteo, F. , Gherardini, P.F. , 2014. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 17, (1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene, B. , Berx, G. , 2013. Regulatory networks defining EMT during cancer initiation and progression. [Internet] Nat. Rev. Cancer. 13, 97–110. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23344542 [DOI] [PubMed] [Google Scholar]
- Di Bartolomeo, S. , Nazio, F. , Cecconi, F. , Oct 2010a. The role of autophagy during development in higher eukaryotes. [Internet] Traffic. 11, (10) 1280–1289. [cited 2014 Apr 30]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20633243 [DOI] [PubMed] [Google Scholar]
- Di Bartolomeo, S. , Corazzari, M. , Nazio, F. , Oliverio, S. , Lisi, G. , Antonioli, M. , 4 Oct 2010b. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. [Internet] J. Cell Biol. 191, (1) 155–168. [cited 2014 May 2]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2953445&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour, M. , Dormond-Meuwly, A. , Demartines, N. , Dormond, O. , 2011 Jan. Targeting the mammalian target of rapamycin (mTOR) in cancer therapy: lessons from past and future perspectives. [Internet] Cancers (Basel). 3, (2) 2478–2500. [cited 2014 Apr 28]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3757428&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis, E. , Buckner, J.C. , Maurer, M.J. , Kreisberg, J.I. , Ballman, K. , Boni, J. , 2005 Aug 10. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. [Internet] J. Clin. Oncol. 23, (23) 5294–5304. [cited 2014 May 15]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15998902 [DOI] [PubMed] [Google Scholar]
- Galavotti, S. , Bartesaghi, S. , Faccenda, D. , Shaked-Rabi, M. , Sanzone, S. , McEvoy, A. , 2013 Feb 7. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. [Internet]. Nature Publishing Group Oncogene. 32, (6) 699–712. [cited 2014 May 2]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22525272 [DOI] [PubMed] [Google Scholar]
- Gheldof, A. , Berx, G. , 2013. Cadherins and epithelial-to-mesenchymal transition. Prog. Mol. Biol. Transl. Sci. 116, 317–336. [DOI] [PubMed] [Google Scholar]
- Guillamo, J.S. , Doz, F. , Delattre, J.Y. , 2001. Brain stem gliomas. Curr. Opin. Neurol. 14, 711–715. [DOI] [PubMed] [Google Scholar]
- Gulhati, P. , Bowen, K.A. , Liu, J. , Stevens, P.D. , Rychahou, P.G. , Chen, M. , 2011 May 1. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. [Internet] Cancer Res. 71, (9) 3246–3256. [cited 2014 May 5]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3085654&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J.Y. , Xia, B. , White, E. , 2013. Autophagy-mediated tumor promotion. Cell. 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, S.P. , Kim, J.H. , Han, M.E. , Sim, H.E. , Kim, K.S. , Yoon, S. , 2011. SNAI1 is involved in the proliferation and migration of glioblastoma cells. Cell. Mol. Neurobiol. 31, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemavathy, K. , Ashraf, S.I. , Ip, Y.T. , 2000. Snail/Slug family of repressors: slowly going into the fast lane of development and cancer. Gene. 1–12. [DOI] [PubMed] [Google Scholar]
- Johnson, E. , Theisen, C.S. , Johnson, K.R. , Wheelock, M.J. , 2004 Jul 23. R-cadherin influences cell motility via Rho family GTPases. [Internet] J. Biol. Chem. 279, (30) 31041–31049. [cited 2014 May 15]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15143071 [DOI] [PubMed] [Google Scholar]
- Kimmelman, A.C. , 2011. The dynamic nature of autophagy in cancer. Genes Dev. 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, R. , Nordin, K. , LaBonne, C. , 2011 Jul 11. The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. [Internet] J. Cell Biol. 194, (1) 17–25. [cited 2014 Apr 30]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3135407&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.K. , Joo, K.M. , Lee, J. , Yoon, Y. , Nam, D.H. , 2014. Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling. Onco Targets Ther. 7, 1933–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis-Tuffin, L.J. , Rodriguez, F. , Giannini, C. , Scheithauer, B. , Necela, B.M. , Sarkaria, J.N. , 2010 Jan. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. [Internet] PLoS One. 5, (10) e13665 [cited 2014 May 15]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2965143&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, H. , Huang, Y. , Guo, B. , Liang, B. , Liu, X. , Ou, H. , 2015. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. 5, (1) 125–139. [PMC free article] [PubMed] [Google Scholar]
- Lv, Q. , Wang, W. , Xue, J. , Hua, F. , Mu, R. , Lin, H. , 2012 Jul 1. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. [Internet] Cancer Res. 72, (13) 3238–3250. [cited 2014 May 15]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22719072 [DOI] [PubMed] [Google Scholar]
- Macintosh, R.L. , Timpson, P. , Thorburn, J. , Anderson, K.I. , Thorburn, A. , Ryan, K.M. , 2012. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle. 11, 2022–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N. , Komatsu, M. , 2011. Autophagy: renovation of cells and tissues. Cell. 728–741. [DOI] [PubMed] [Google Scholar]
- Nakaya, Y. , Sheng, G. , 2013. EMT in developmental morphogenesis. Cancer Lett. 9–15. [DOI] [PubMed] [Google Scholar]
- Nazio, F. , Strappazzon, F. , Antonioli, M. , Bielli, P. , Cianfanelli, V. , Bordi, M. , 2013. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. [Internet] Nat. Cell Biol. 15, 406–416. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23524951 [DOI] [PubMed] [Google Scholar]
- Nghiemphu, P.L. , Lai, A. , Green, R.M. , Reardon, D.A. , Cloughesy, T. , 2012 Nov. A dose escalation trial for the combination of erlotinib and sirolimus for recurrent malignant gliomas. [Internet] J. Neurooncol. 110, (2) 245–250. [cited 2014 May 15]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3472078&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder, T.T. , Gupta, P.B. , Mani, S.A. , Yang, J. , Lander, E.S. , Weinberg, R.A. , 2008. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 68, 3645–3654. [DOI] [PubMed] [Google Scholar]
- Palumbo, S. , Tini, P. , Toscano, M. , Allavena, G. , Angeletti, F. , Manai, F. , 2014. Combined EGFR and autophagy modulation impairs cell migration and enhances radiosensitivity in human glioblastoma cells. [Internet] J. Cell. Physiol. 229, 1863–1873. http://www.ncbi.nlm.nih.gov/pubmed/24691646 [DOI] [PubMed] [Google Scholar]
- Peinado, H. , Olmeda, D. , Cano, A. , 2007. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?. Nat. Rev. Cancer. 7, 415–428. [DOI] [PubMed] [Google Scholar]
- Qu, X. , Yu, J. , Bhagat, G. , Furuya, N. , Hibshoosh, H. , Troxel, A. , 2003. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 112, (12) 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin, J.B. , Kung, A.L. , Klein, R.S. , Chan, J.A. , Sun, Y. , Schmidt, K. , 2003 Nov 11. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. [Internet] Proc. Natl. Acad. Sci. U. S. A. 100, (23) 13513–13518. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=263845&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaccaluga, M. , D'Alessandro, G. , Pagani, F. , Ferrara, G. , Lopez, N. , Warr, T. , 2013 Jan. Functional cross talk between CXCR4 and PDGFR on glioblastoma cells is essential for migration. [Internet] PLoS One. 8, (9) e73426 [cited 2014 May 13]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3759384&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer, V. , de Craene, B. , Berx, G. , Behrens, J. , 2008 Aug 28. Snail promotes Wnt target gene expression and interacts with beta-catenin. [Internet] Oncogene. 27, (37) 5075–5080. [cited 2014 Apr 30]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18469861 [DOI] [PubMed] [Google Scholar]
- Szatmári, T. , Lumniczky, K. , Désaknai, S. , Trajcevski, S. , Hídvégi, E.J. , Hamada, H. , 2006. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 97, 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, E.W. , Williams, E.D. , 2008. EMT and MET in carcinoma – clinical observations, regulatory pathways and new models. Clin. Exp. Metastasis. 25, 591–592. [DOI] [PubMed] [Google Scholar]
- Thoreen, C.C. , Chantranupong, L. , Keys, H.R. , Wang, T. , Gray, N.S. , Sabatini, D.M. , 2012 May 3. A unifying model for mTORC1-mediated regulation of mRNA translation. [Internet]. Nature Publishing Group Nature. 485, (7396) 109–113. [cited 2014 Apr 30]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3347774&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuloup-Minguez, V. , Hamaï, A. , Greffard, A. , Nicolas, V. , Codogno, P. , Botti, J. , 2013 Oct 15. Autophagy modulates cell migration and β1 integrin membrane recycling. [Internet] Cell Cycle. 12, (20) 3317–3328. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24036548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viñas-Castells, R. , Beltran, M. , Valls, G. , Gómez, I. , García, J.M. , Montserrat-Sentís, B. , 2010 Mar 5. The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. [Internet] J. Biol. Chem. 285, (6) 3794–3805. [cited 2014 Apr 30]. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2823521&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock, M.J. , Johnson, K.R. , 2003. Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol. 19, 207–235. [DOI] [PubMed] [Google Scholar]
- White, E. , 2012. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer. 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yook, J.I. , Li, X.-Y. , Ota, I. , Fearon, E.R. , Weiss, S.J. , 2005. Wnt-dependent regulation of the E-cadherin repressor snail. J. Biol. Chem. 280, 11740–11748. [DOI] [PubMed] [Google Scholar]
- Zhou, B.P. , Hung, M.-C. , 2005. Wnt, hedgehog and snail: sister pathways that control by GSK-3beta and beta-Trcp in the regulation of metastasis. Cell Cycle. 4, 772–776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data