

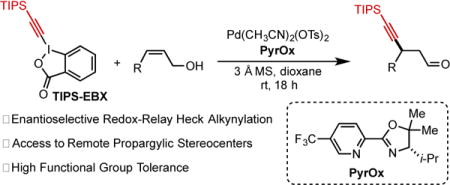
Abstract
An enantioselective redox-relay Heck alkynylation of di- and trisubstituted alkenols to construct propargylic stereocenters is disclosed using a new pyridine oxazoline ligand. This strategy allows direct access to chiral β-alkynyl carbonyl compounds employing allylic alcohol substrates in contrast to more traditional conjugate addition methods.
Keywords: Heck reaction, alkenes, propargylic stereocenter, alkynylation
Graphical abstract
A convenient redox-relay Heck strategy to synthesize enantiomerically enriched β-alkynyl carbonyl compounds from allylic alcohol substrates is described. Trisubstituted allylic alcohols are also promising substrates allowing for the formation of propargylic quaternary stereocenters.
Intermolecular Heck reactions generally feature the coupling of an sp2-hybridized reaction partner to an alkene followed by β-hydride elimination towards the site of initial migratory insertion. This formally yields a sp2-sp2 carbon-carbon connection.[1] Recently, this reaction has been expanded through both substrate and catalyst design to preferentially undergo β-hydride elimination away from the site of initial migratory insertion to both set a stereocenter and allow the formation of a sp2-sp3 C–C bond.[2] Specifically, our group has reported a suite of such enantioselective redox-relay Heck reactions of acyclic alkenyl alcohols using aryldiazonium salts,[3] arylboronic acids,[4] and alkenyl triflates,[5] which provides direct preparation of carbonyl compounds that contain remote alkenyl/aryl stereocenters (Scheme 1A). However, this emerging strategy has thus far been limited to sp2-hybridized nucleophiles/electrophiles as coupling partners.
Scheme 1.
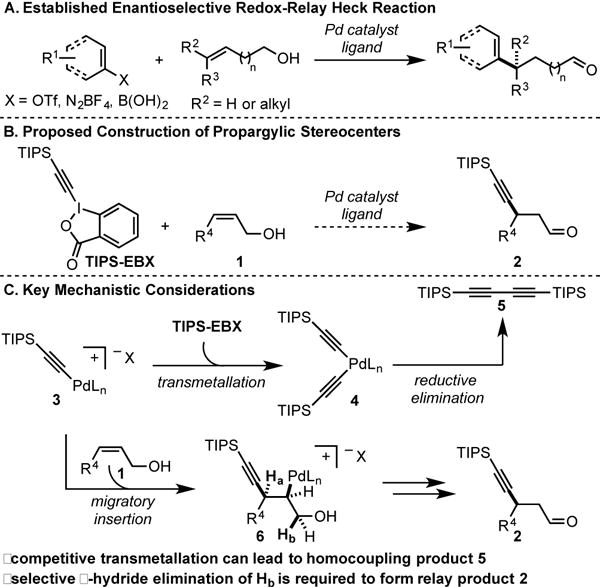
Proposed redox-relay Heck alkynylation to access propargylic stereocenters.
In an effort to expand the breadth of products one can access with this approach, we selected to investigate the enantioselective Heck alkynylation of alkenols to construct propargylic stereocenters and forge sp-sp3 C–C bonds (Scheme 1B). The successful development of an alkynyl Heck reaction would allow direct access to chiral β-alkynyl carbonyl compounds, which are versatile intermediates that have extensive applications in organic synthesis.[6] Traditionally, these types of compounds have been synthesized using enantioselective conjugate addition technologies, pioneered by Carreira,[7] Hayashi,[8] and others, through organometallic acetylide addition to α,β-unsaturated carbonyl substrates.[9] However, we envisioned that a redox-relay Heck approach that utilized allylic alcohol substrates would provide an attractive alternative to this field due to the ease of preparation, handling and improved stability of such alkenols. In addition, it was deemed possible, on the basis of our previous reports, that trisubstituted alkenols may be viable substrates. Using a traditional conjugate addition approach, only one report of conjugate alkynylation of β,β-disubstituted carbonyl compounds to establish propargylic quaternary stereocenters has appeared to date and is limited to β-aryl-β-trifluoromethyl enones.[10]
To achieve a successful alkynylative redox-relay Heck transformation three main challenges were carefully considered: (1) the identification of a suitable sp-hybridized carbon reagent (such as benziodoxole derived triisopropylsilyl variant (TIPS- EBX) initially developed by Waser), (2) avoiding competitive transmetallation with Pd-alkynyl species 3 (Scheme 1C), which would ultimately lead to Pd-bis(alkynyl) intermediate 4 followed by reductive elimination to yield homocoupling product 5, and (3) selective β-hydride elimination of Hb (6) since β-hydride elimination of Ha would produce the traditional Heck-type product (not shown) and remove the newly established propargylic stereocenter. Herein, we disclose the development of an enantioselective redox-relay Heck alkynylation of allylic alkenols as a complementary approach to access enantiomerically enriched β-alkynyl carbonyl compounds. This method utilizes easily accessible alkenol substrates, a simple chiral ligand, and a benziodoxole derived reagent as the alkyne source.
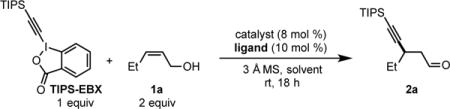
To initiate our studies, cis-2-penten-1-ol (1a, Table 1) and TIPS-EBX[11] were selected as model coupling partners with Pd(CH3CN)2(OTs)2 as a precatalyst, a chiral pyridine-oxazoline ligand (PyrOx, ligand 1), and CH2Cl2 as solvent (see SI for additional ligands and alkyne sources explored). Promisingly, the desired product (2a) was produced in 40% yield and 98:2 er (entry 1). To improve the reaction yield, a solvent screen was performed (entries 2–4). As a result, dioxane was found to increase the product yield to 60%. Replacing the t-Bu group (ligand 1) on the PyrOx ligand with an i-Pr group and adding a gem-dimethyl moiety on the oxazoline portion (ligand 2) gave a slight boost in yield to 65% while maintaining high enantioselectivity (entry 5). The CF3 group on the pyridine moiety of the ligand produces a more electrophilic catalyst, which has been reported to deliver higher product yields and enantioselectivities.[3] In addition, no significant change was observed when the reaction temperature was decreased to 15 °C (entry 6). Surprisingly, switching to a Pd(0) precatalyst (Pd2(dba)3) resulted in <5% product formation (entry 7). In contrast, Buchwald’s precatalyst, known to form Pd(0) in situ upon heating or adding base,[12] furnished the desired product (2a) in 65% yield and 96:4 er. These results suggest that the alkyne source (TIPS-EBX) can react with both Pd(0) and Pd(II), presumably through oxidative addition and transmetallation mechanisms, respectively. It remains unclear why Pd2(dba)3 is unable to catalyze this transformation.
Table 1.
Reaction Optimization[a]
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | catalyst | ligand | solvent | % yield | er |
| 1 | Pd(CH3CN)2(OTs)2 | ligand 1 | CH2Cl2 | 40 | 98:2 |
| 2 | Pd(CH3CN)2(OTs)2 | ligand 1 | THF | 48 | 92:8 |
| 3 | Pd(CH3CN)2(OTs)2 | ligand 1 | EtOAc | 42 | 91:9 |
| 4 | Pd(CH3CN)2(OTs)2 | ligand 1 | dioxane | 60 | 97:3 |
| 5 | Pd(CH3CN)2(OTs)2 | ligand 2 | dioxane | 65 | 96.5:3.5 |
| 6[b] | Pd(CH3CN)2(OTs)2 | ligand 2 | dioxane | 63 | 96.5:3.5 |
| 7[c] | Pd2(dba)3 | ligand 2 | dioxane | <5 | – |
| 8[d] | Buchwald precatalyst | ligand 2 | dioxane | 65 | 96:4 |
Each entry represents the isolated yield on 0.20 mmol scale. er values were determined by SFC.
The reaction was performed at 15 °C.
4 mol % catalyst was used. The Buchwald precatalyst (4 mol %) was heated to 80 °C for 10 min prior use to generate Pd(0) and TsOH (25 mol %) was added.
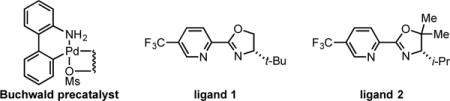
In should be noted that in addition to the TIPS protecting group, a range of other groups were also investigated on the benziodoxole derived alkyne reagent. The tert-butyldiphenylsilyl variant (TBDPS-EBX) resulted in 61% yield and 95:5 er using the conditions shown in entry 5. Unfortunately, smaller groups such as TBS and phenyl gave significantly lower yields and enantioselectivites presumably due to other competitive processes including homocoupling (see SI for complete details). While this is a limitation, the TIPS protecting group can be easily removed and the resulting terminal alkyne is amenable to many well vetted transforms.
Under the optimized conditions (entry 5), the scope of allylic alkenols was explored (Table 2). In general, the desired alkynyl carbonyl compounds were obtained with moderate to good yields and excellent enantiomeric ratios. The mass balance in all examples was overall excellent with mainly consumption of the TIPS-EBX reagent to the homocoupling byproduct (5, Scheme 1C). Compared with alkenol 1a, increasing the alkyl chain length at R1 did not have a significant effect (2b, 2c). Enhancing the size of the substituent at R1 to a benzyl or cyclohexylmethyl group led to slightly decreased yields (2d, 2e). It should be noted that when trans-1d was used instead of cis-1d the yield decreased to 38% with 24:76 er (ent-2d). Alkenol 1f, a substrate containing both di- and trisubstituted alkenes, reacted selectively at the less-hindered disubstituted alkene to afford the desired product (2f) in 62% yield and 97:3 er. The reaction also tolerates an ester (1g), an alcohol (1h) and a benzyl ether (1i) delivering the corresponding products in good yields and high enantioselectivites. Substrates containing a primary tosylate (1j) or primary chloride (1k) could also be subjected to the reaction conditions to yield products 2j and 2k in 55% yield and 95.5:4.5 er and 60% yield and 97:3 er, respectively. A nitrile group was also well suited under the reaction conditions providing product 2l in 62% yield and 96.5:3.5 er. Alkenols containing a phthalimide (1m) or a sulfide (1n) were also viable substrates resulting in a modest reduction in yield of products 2m and 2n. Finally, a substrate containing a secondary alcohol (1o) furnished ketone product 2o in 41% yield and 83:17 er. The er of product 2o could be improved to 94:6 when ligand 1 was employed. The absolute configuration for product 2d was determined to be (R) through [α]D comparison with the previously reported (S)-compound. All other compounds were assigned by analogy to product 2d.[8b]
Table 2.
Evaluation of Disubstituted Alkenol Substrates[a]
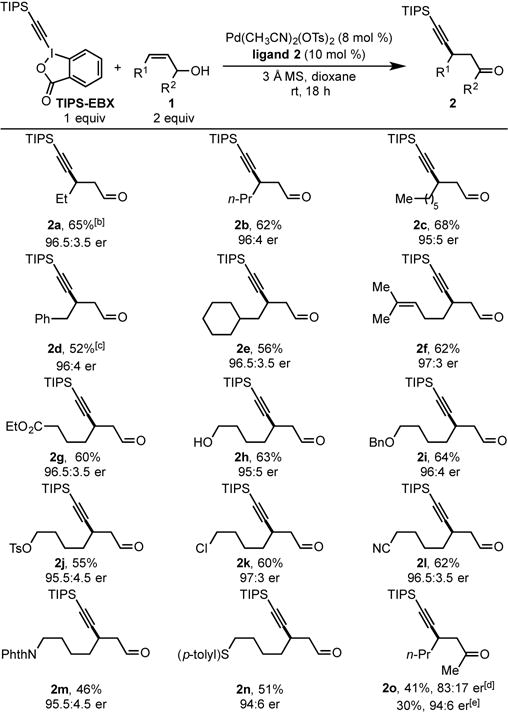
|
Each entry represents the isolated yield on 0.20 mmol scale. er values were determined by SFC.
64% yield and 96:4 er on 2.0 mmol scale.
38% yield and 24:76 er when trans-1d was used.
4.0 equiv of alkenol 1o was used. [e] Ligand 1 was used.
As homoallylic substrates (and longer chain alkenols) have previously been excellent substrates in sp2 coupling processes, we evaluated these substrates under the reaction conditions. Unfortunately, the reactions delivered low amounts of the desired redox-relay product (<20% yield). In these cases, the major byproduct was the traditional Heck product. This indicates that β-hydride elimination is not selective when an additional methylene unit is added between the alkene and the alcohol moiety suggestive that the biasing of β-hydride elimination is significantly reduced with the sp-center installed. Likely, a substantial redesign of the system will be required to overcome this limitation.
Given the difficulty associated with the enantioselective formation of propargylic quaternary stereocenters using metal-catalyzed conjugate addition approaches, we sought to extend this methodology to trisubstituted alkenol substrates. Due to the sluggish migratory insertion associated with trisubstituted alkenes,[13] we were cognizant that competitive transmetallation with Pd-alkynyl species 3 (Scheme 1c) would lead to homocoupling byproduct 5. To increase the relative rate of migratory insertion, four equivalents of alkenol were used. As a result, when a simple alkyl group was positioned at R (1p, Table 3), the desired product containing a proparygylic quaternary stereocenter was obtained in 36% yield and 94:6 er (2p). The incorporation of a benzyl group at R delivered product 2q in 25% yield and 95:5 er. Lastly, the presence of an additional trisubstituted alkene was also tolerated furnishing product 2r in 31% yield and 94:6 er. The lower yields for trisubstituted alkene substrates are attributed to competitive alkyne homocoupling. Efforts to improve the product yield by increasing the relative rate of migratory insertion or slowing the rate of transmetallation have been unsuccessful thus far.
Table 3.
Evaluation of Trisubstituted Alkenol Substrates[a]
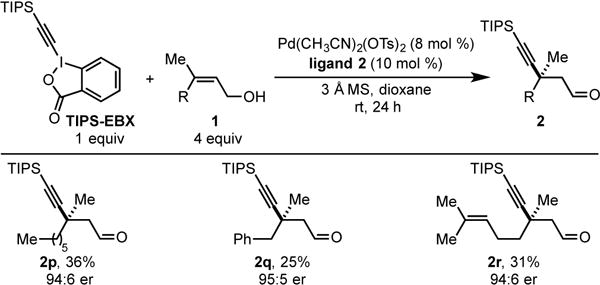
|
Each entry represents the isolated yield on 0.20 mmol scale. er values were determined by SFC.
In order to interrogate the reaction mechanism to determine if Pd migrates toward the alcohol functional group in a similar fashion to our previous reports, deuterated alkenol 1a–d2 was subjected to the optimized Heck alkynylation reaction conditions. As a result, one deuterium atom was transposed to the β carbon of the newly formed aldehyde delivering 2a–d2, in accordance with our previous mechanistic reports. This implies that 1a–d2 undergoes a migratory insertion with the Pd-alkynyl species resulting in Pd-alkyl intermediate 7. Through selective β-deuteride elimination (8) and reinsertion, intermediate 9 is formed resulting in the transposition of one deuterium atom. Furthermore, this suggests if intermediate 9 is formed the reaction terminates through alcohol oxidation and establishment of Pd(0), presumably through β-hydride elimination or an E2-type elimination. Moreover, since Pd(0) is formed with each catalytic turnover, TIPS-EBX (or TsO-EBX, a possible byproduct of transmetallation) must oxidize Pd(0) back to Pd(II) in this case.
In summary, we have developed an enantioselective redox-relay Heck alkynylation of disubstituted alkenols in good yields and high enantioselectivity. The β-alkynyl carbonyl compounds obtained using this methodology contain a vast array of functionality. The ability to use allylic alcohol substrates provides a complementary approach to access such products to the more traditional conjugate addition strategies. Finally, promising results using trisubstituted allylic alkenol substrates to access propargylic quaternary stereocenters are provided. Future efforts are aimed at developing ligands and systems that prevent homocoupling of the alkyne to overcome this limitation.
Supplementary Material
Scheme 2.
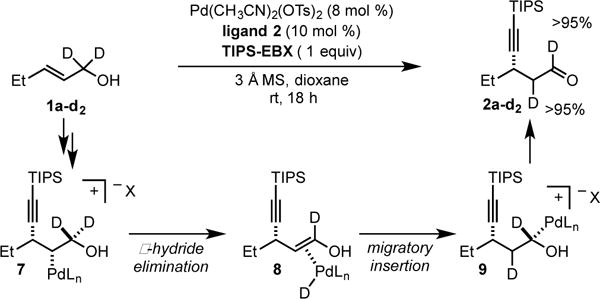
Deuterium Labeling Study.
Acknowledgments
The work was supported by National Institute of Health (NIGMS R01GM063540). C.S.N acknowledges the NSF for a graduate research fellowship. Z.-M.C. would like to thank Shanghai Jiao Tong University for a postdoctoral fellowship. We acknowledge Dr. Nicholas Race for preparation of the deuterated alkenol substrate.
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/
Conflict of interest
The authors declare no conflict of interest.
References
- 1.For selected reviews, see:; a) Heck RF. Acc Chem Res. 1979;12:146–151. [Google Scholar]; b) Crisp GT. Chem Soc Rev. 1998;27:427–436. [Google Scholar]; c) Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]; d) Shibasaki M, Vogl EM, Ohshima T. Adv Synth Catal. 2004;346:1533–1552. [Google Scholar]; e) Zeni G, Larock RC. Chem Rev. 2006;106:4644–4680. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]; f) Le Bras J, Muzart J. Chem Rev. 2011;111:1170–1214. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]; g) McCartney D, Guiry PJ. Chem Soc Rev. 2011;40:5122–5150. doi: 10.1039/c1cs15101k. [DOI] [PubMed] [Google Scholar]; h) Seechurn CCCJ, Kitching MO, Colacot TJ, Snieckus V. Angew Chem. 2012;124:5150–5174. [Google Scholar]; Angew Chem Int Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 2.For selected works, see:; a) Larock RC, Leung WY, Stolz-Dunn S. Tetrahedron Lett. 1989;30:6629–6632. [Google Scholar]; b) Bouquillon S, Ganchegui B, Estrine B, Hénin F, Muzart J. J Organomet Chem. 2001;634:153–156. [Google Scholar]; c) Berthiol F, Doucet H, Santelli M. Tetrahedron. 2006;62:4372–4383. [Google Scholar]; d) Oliveira CC, Pfaltz A, Correia CRD. Angew Chem. 2015;127:14242–14245. [Google Scholar]; Angew Chem Int Ed. 2015;54:14036–14039. doi: 10.1002/anie.201507927. [DOI] [PubMed] [Google Scholar]; e) Singh S, Bruffaerts J, Vasseur A, Marek I. Nat Commun. 2017;8 doi: 10.1038/ncomms14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werner EW, Mei TS, Burckle AJ, Sigman MS. Science. 2012;338:1455–1458. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Mei TS, Werner EW, Burckle AJ, Sigman MS. J Am Chem Soc. 2013;135:6830–6833. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mei TS, Patel HH, Sigman MS. Nature. 2014;508:340–344. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang C, Santiago CB, Kou L, Sigman MS. J Am Chem Soc. 2015;137:7290–7293. doi: 10.1021/jacs.5b04289. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chen ZM, Hilton MJ, Sigman MS. J Am Chem Soc. 2016;138:11461–11464. doi: 10.1021/jacs.6b06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Patel HH, Sigman MS. J Am Chem Soc. 2015;137:3462–3465. doi: 10.1021/ja5130836. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Patel HH, Sigman MS. J Am Chem Soc. 2016;138:14226–14229. doi: 10.1021/jacs.6b09649. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang C, Tutkowski B, DeLuca RJ, Joyce LA, Wiest O, Sigman MS. Chem Sci. 2017;8:2277–2282. doi: 10.1039/c6sc04585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected works, see:; a) Evans DA, Ripin DHB, Halstead DP, Campos KR. J Am Chem Soc. 1999;121:6816–6826. [Google Scholar]; b) Fürstner A, De Souza D, Parra-Rapado L, Jensen JT. Angew Chem. 2003;115:5516–5518. doi: 10.1002/anie.200352413. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:5358–5360. doi: 10.1002/anie.200352413. [DOI] [PubMed] [Google Scholar]; c) Yuan Y, Men H, Lee C. J Am Chem Soc. 2004;126:14720–14721. doi: 10.1021/ja0447154. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nakajima R, Ogino T, Yokoshima S, Fukuyama T. J Am Chem Soc. 2010;132:1236–1237. doi: 10.1021/ja9103233. [DOI] [PubMed] [Google Scholar]; e) Sugimoto K, Toyoshima K, Nonaka S, Kotaki K, Ueda H, Tokuyama H. Angew Chem. 2013;125:7309–7312. doi: 10.1002/anie.201303067. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:7168–7171. doi: 10.1002/anie.201303067. [DOI] [PubMed] [Google Scholar]; f) Trost BM, Ehmke V. Org Lett. 2014;16:2708–2711. doi: 10.1021/ol5009872. [DOI] [PubMed] [Google Scholar]
- 7.For selected works, see:; a) Knöpfel TF, Zarotti P, Ichikawa T, Carreira EM. J Am Chem Soc. 2005;127:9682–9683. doi: 10.1021/ja052411r. [DOI] [PubMed] [Google Scholar]; b) Fujimori S, Knöpfel TF, Zarotti P, Ichikawa T, Boyall D, Carreira EM. Bull Chem Soc Jpn. 2007;80:1635–1657. [Google Scholar]; c) Zarotti P, Knöpfel TF, Aschwanden P, Carreira EM. ACS Catal. 2012;2:1232–1234. [Google Scholar]
- 8.For selected works, see:; a) Nishimura T, Guo XX, Uchiyama N, Katoh T, Hayashi T. J Am Chem Soc. 2008;130:1576–1577. doi: 10.1021/ja710540s. [DOI] [PubMed] [Google Scholar]; b) Nishimura T, Sawano T, Hayashi T. Angew Chem. 2009;121:8201–8203. doi: 10.1002/anie.200904486. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:8057–8059. doi: 10.1002/anie.200904486. [DOI] [PubMed] [Google Scholar]; c) Dou X, Huang Y, Hayashi T. Angew Chem. 2016;128:1145–1149. [Google Scholar]; Angew Chem Int Ed. 2016;55:1133–1137. doi: 10.1002/anie.201509778. [DOI] [PubMed] [Google Scholar]
- 9.For selected works, see:; a) Fillion E, Zorzitto AK. J Am Chem Soc. 2009;131:14608–14609. doi: 10.1021/ja905336p. [DOI] [PubMed] [Google Scholar]; b) Cui S, Walker SD, Woo JCS, Borths CJ, Mukherjee H, Chen MJ, Faul MM. J Am Chem Soc. 2010;132:436–437. doi: 10.1021/ja909105s. [DOI] [PubMed] [Google Scholar]; c) Yazaki R, Kumagai N, Shibasaki M. J Am Chem Soc. 2010;132:10275–10277. doi: 10.1021/ja105141x. [DOI] [PubMed] [Google Scholar]; d) Larionov OV, Corey EJ. Org Lett. 2010;12:300–302. doi: 10.1021/ol902643w. [DOI] [PubMed] [Google Scholar]; e) Trost BM, Taft BR, Masters JT, Lumb JP. J Am Chem Soc. 2011;133:8502–8505. doi: 10.1021/ja203171x. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Blay G, Cardona L, Pedro JR, Sanz-Marco A. Chem Eur J. 2012;18:12966–12969. doi: 10.1002/chem.201201765. [DOI] [PubMed] [Google Scholar]; g) Sanz-Marco A, García-Ortiz A, Blay G, Fernández I, Pedro JR. Chem Eur J. 2014;20:668–672. doi: 10.1002/chem.201303920. [DOI] [PubMed] [Google Scholar]; h) Sanz-Marco A, García-Ortiz A, Blay G, Pedro JR. Chem Commun. 2014;50:2275–2278. doi: 10.1039/c3cc48508k. [DOI] [PubMed] [Google Scholar]
- 10.Sanz-Marco A, Blay G, Vila C, Pedro JR. Org Lett. 2016;18:3538–3541. doi: 10.1021/acs.orglett.6b01494. [DOI] [PubMed] [Google Scholar]
- 11.For selected reviews, see:; a) Li Y, Hari DP, Vita MV, Waser J. Angew Chem. 2016;128:4512–4531. doi: 10.1002/anie.201509073. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2016;55:4436–4454. doi: 10.1002/anie.201509073. [DOI] [PubMed] [Google Scholar]; b) Zhdankin VV, Stang PJ. Chem Rev. 2008;108:5299–5358. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; For selected works, see:; c) Le Vaillant F, Courant T, Waser J. Angew Chem. 2015;127:11352–11356. doi: 10.1002/anie.201505111. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2015;54:11200–11204. doi: 10.1002/anie.201505111. [DOI] [PubMed] [Google Scholar]; d) Nicolai S, Piemontesi C, Waser J. Angew Chem. 2011;123:4776–4779. doi: 10.1002/anie.201100718. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2011;50:4680–4683. doi: 10.1002/anie.201100718. [DOI] [PubMed] [Google Scholar]
- 12.For selected works, see:; a) Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bruno NC, Buchwald SL. Org Lett. 2013;15:2876–2879. doi: 10.1021/ol401208t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Senecal TD, Shu W, Buchwald SL. Angew Chem. 2013;125:10219–10223. doi: 10.1002/anie.201304188. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:10035–10039. doi: 10.1002/anie.201304188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilton MJ, Cheng B, Buckley BR, Xu L, Wiest O, Sigman MS. Tetrahedron. 2015;71:6513–6518. doi: 10.1016/j.tet.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.