

Abstract
The intestinal epithelium forms an essential barrier between a host and its microbiota. Protozoa and helminths are members of the gut microbiota of mammals, including humans, yet the many ways that gut epithelial cells orchestrate responses to these eukaryotes remain unclear. Here we show that tuft cells, which are taste-chemosensory epithelial cells, accumulate during parasite colonization and infection. Disruption of chemosensory signaling through the loss of TRMP5 abrogates the expansion of tuft cells, goblet cells, eosinophils, and type 2 innate lymphoid cells during parasite colonization. Tuft cells are the primary source of the parasite-induced cytokine interleukin-25, which indirectly induces tuft cell expansion by promoting interleukin-13 production by innate lymphoid cells. Our results identify intestinal tuft cells as critical sentinels in the gut epithelium that promote type 2 immunity in response to intestinal parasites.
The mammalian gut microbiota is a collective of bacteria, archaea, viruses, fungi, and parasites that reside in the lumen and mucosal surface of the intestine. These microbes are sequestered from interior tissues by a single layer of epithelial cells lining the gut that acts as a barrier and sensor. Intestinal epithelial cells (IECs) express pattern recognition receptors that detect microbial components and thus are critical sensors for and orchestrators of mucosal immunity (1–4).
Beyond pattern recognition receptors, hosts monitor and respond to the microbiota via heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs). For example, microbially produced short-chain fatty acids are sensed via GPR41 and GPR43 (5,6), and sinonasal epithelial cells can detect the pathogen Pseudomonas aeruginosa via a taste-chemosensory GPCR (7–12). Many taste-chemosensory GPCRs require the taste-specific G protein subunit gustducin and the cation channel TRPM5 to transduce their signals (7,9). The disruption of either gustducin or TRPM5 can perturb physiological responses to P. aeruginosa (13–15). In the gut, TRPM5 and other canonical taste-chemosensory components are predominantly expressed by an intestinal epithelial subset called tuft cells (16). Tuft cells, which are identified by the expression of doublecortin-like kinase 1 (DCLK1), comprise a minor fraction of small intestinal epithelial cells (17–19) and are putative quiescent stem cells (20). Although tuft cells express taste-chemosensory machinery, it is unknown whether tuft cells sense the gut microbiota by means of taste chemosensation or transduce signals to the mucosal immune system (21).
We began by evaluating the frequency of DCLK1+ tuft cells in the distal small intestine of wild-type (WT) specific-pathogen-free mice that were bred in-house (BIH). We found markedly more intestinal DCLK1+ tuft cells (7.2%) (Fig. 1A) than previous reports (0.4%) (19,22) and confirmed this discrepancy with an alternative tuft cell marker, GFI1B (fig. S1) (23). As interinstitutional differences in microbiota can contribute to substantial variation among mucosal immune cell populations (24), we compared tuft cell abundance in mice obtained from The Jackson Laboratory (JAX) with BIH mice. Similar to previous reports (19,25), tuft cells constituted 1.0% of the total IEC population of JAX mice (Fig. 1A). Feeding the cecal contents from BIH mice to JAX mice was sufficient to increase tuft cell populations to BIH levels (fig. S2), suggesting that transmissible components of the BIH microbiota may drive tuft cell expansion when introduced to JAX mice. In support of this idea, intestinal histology revealed numerous single-celled protozoa in BIH but not in JAX mice (Fig. 1B). To identify these protozoa, we purified and imaged them by means of scanning electron microscopy (SEM); we identified them as tritrichomonads (Fig. 1C) (26–28). Quantitative polymerase chain reaction (qPCR) confirmed that they were Tritrichomonas muris (Tm), a common but understudied member of the rodent microbiota (Fig. 1D).
Fig. 1. Symbiotic protozoa or helminths increase intestinal tuft cell abundance.
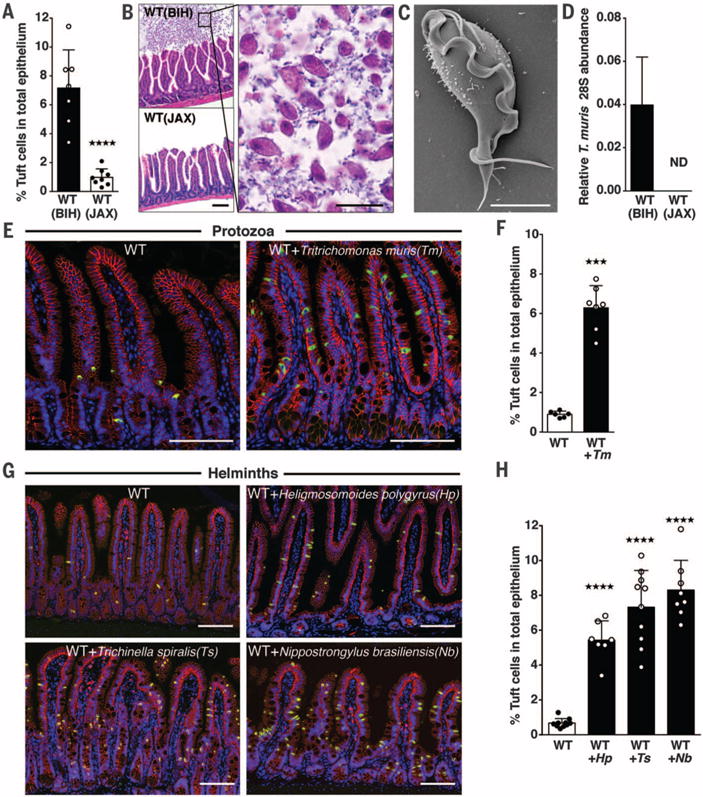
(A) DCLK1+ tuft cell frequency in the small intestine (SI) of WT BIH and WT JAX mice. (B) Hemtoxylin and eosin-stained SI sections from WT BIH and WT JAX mice (scale bar, 50 μm) (left). A higher magnification of the WT BIH section is shown on the right (scale bar, 20 μm). (C) SEM micrograph of protozoa isolated from WT BIH mice (scale bar, 4 μm). (D) Tm abundance in stool DNA (Tm 28S rRNA relative to Eubacteria 16S rRNA), determined by qPCR (ND, not detectable). (E) Representative SI images from uninfected and Tm-colonized mice and (F) tuft cell frequency. (G) Representative SI images from uninfected and helminth-colonized mice and (H) tuft cell frequency. DCLK1 is shown in green, E-cadherin in red, and DAPI (4′,6-diamidino-2-phenylindole) in blue [scale bars in (E) and (G), 100 μm]. Each symbol represents an individual mouse, and all data are representative of two [(D), (F), and (H)] or three (A) independent experiments. Tm infection was 17 days in (E) and (F). In (G) and (H), Hp infection was 21 days, Ts infection was 15 days, and Nb infection was 8 days. Data are plotted as means with SD. Four stars, P < 0.0001; three stars, P = 0.0001; one-way analysis of variance (ANOVA) or Mann-Whitney test.
To eradicate Tm from BIH mice, we added metronidazole (2.5 g/liter) to their drinking water for 1 week. This eliminated Tm and concomitantly reduced tuft cell abundance (fig. S3). Because this treatment does not exclude the possibility that other metronidazole-sensitive organisms may contribute to tuft cell expansion, we cultured Tm (28,29) and colonized unexposed mice. Tm colonization significantly elevated tuft cell numbers in conventional (Fig. 1, E and F) and germ-free mice (fig. S4), suggesting that this symbiotic protozoa is sufficient to increase tuft cell frequency.
Helminths are common eukaryotic inhabitants of the mammalian intestine, but they are evolutionarily distinct from protozoa. These parasites inflict a substantial global health burden, yet worms may also provide therapeutic benefits (8). To investigate the effect of helminth infection on tuft cell abundance, we infected mice with a diverse set of parasitic worms including Heligmosomoides polygyrus (Hp), Trichinella spiralis (Ts), and Nippostrongylus brasiliensis (Nb). Similar to our results with Tm, infections with all three helminths increased tuft cell abundance, indicating that expansion of tuft cells is a broadly conserved feature of parasite colonization (Fig. 1, G and H).
Because tuft cells are postulated to be chemosensory cells (30), we considered whether perturbations to tuft chemosensory pathways may affect their expansion in response to parasites and/or to the type 2 immune response typically initiated by parasites. Multiple taste-chemosensory GPCRs sense sweet, bitter, and umami compounds; engagement of these different receptors activates a common signal transduction pathway involving gustducin, PLCβ2, and TRPM5 (Fig. S5) (7, 9). We confirmed that GFI1B+ tuft cells are the primary IEC subset expressing the canonical taste-associated components gustducin, PLCβ2, and TRPM5 (Fig. 2A) (16, 23, 31).
Fig. 2. Tuft cells influence type 2 immunity through TRPM5.
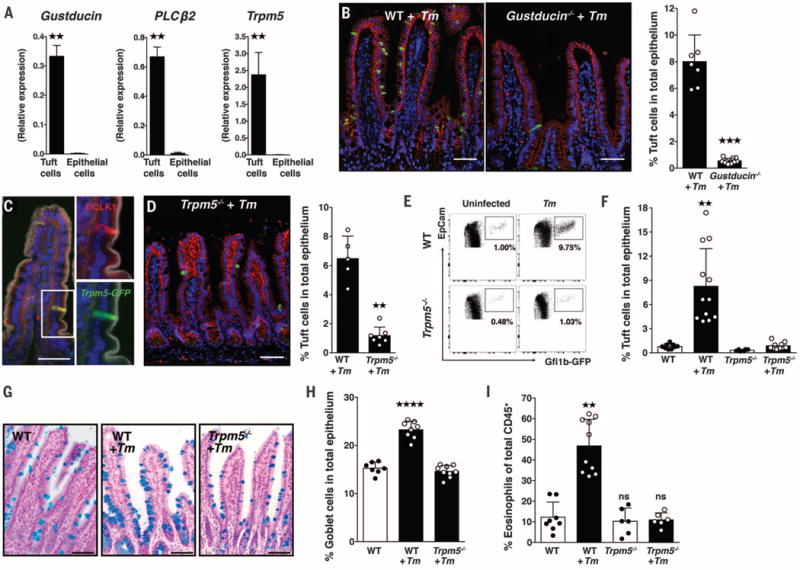
(A) Gustducin, PLCβ2, and TRPM5 expression in sorted tuft cells, compared with the non-tuft cell epithelium. (B) Representative images of Tm-colonized WT and gustducin−/− mice and tuft cell frequencies. (C) Representative image from Trpm5eGFP mice. GFP is shown in green), DCLK1 in red), DAPI in blue, and phalloidin in white. (D) Representative image of Tm-colonized Trpm5−/− mice and tuft cell frequencies. Scale bars in (B), (C), and (D), 50 μm. (E) Representative flow cytometry plots of IECs from uninfected (left) or Tm-colonized (right) WT (Gfi1beGFP/+, top) and Trpm5−/− (Gfi1beGFP/+ Trpm5−/−, bottom) mice and (F) tuft cell frequency. (G) Goblet cells in SI sections stained with Alcian blue and nuclear red in uninfected WT and Tm-colonized WT and Trpm5−/− mice and (H) goblet cell frequency. (I) Eosinophil frequency in the distal SI lamina propria (LP) of uninfected and Tm-colonized WT and Trpm5−/− mice. Scale bars, 50 μm. Each symbol represents an individual mouse, and all data are representative of at least three independent experiments. Data are plotted as means with SD. Four stars, P < 0.0001; three stars, P = 0.0001; two stars, P < 0.01; ns, not significant; one-way ANOVA, Kruskal-Wallis, or Mann-Whitney tests.
We compared tuft cell abundance in WT and gustducin-deficient (gustducin−/−) mice colonized with Tm and found significantly fewer tuft cells in gustducin−/− animals (Fig. 2B). Using Trpm5eGFP (eGFP, enhanced green fluorescent protein) reporter mice, we validated that TRPM5 is restricted to the epithelium and expressed by DCLK1+ tuft cells in the distal small intestine (Fig. 2C and fig. S6). Given the multiplicity of taste-chemosensory GPCRs, the established role of TRPM5 in taste chemosensation (7,32), and the predominant intestinal TRPM5 expression by tuft cells, we used TRPM5-deficient mice to evaluate whether these pathways affect tuft cell parasite responses. Similar to gustducin−/− mice, tuft cells failed to expand in Trpm5−/− mice during Tm colonization (Fig. 2, D to F). To determine whether the blunted response was due to reduced parasite colonization, we measured Tm in the distal small intestine (fig. S7A). We found slightly more parasites in both gustducin−/− and Trpm5T−/− mice than in WT mice (fig. S7B), indicating that the lack of tuft cell response was not due to decreased Tm colonization. Because Tm is a stable component of the microbiota, we tested how the loss of TRPM5 would affect clearance of a pathogenic helminth such as Hp. Thirty-six days after infection, we determined that Trpm5−/− mice had a significantly higher worm burden than WT mice (fig. S7C). Collectively, these data suggest that pathways initiated upstream of TRPM5 may mediate tuft cell responses to intestinal parasites.
If tuft cell responses represent an early step in parasite recognition, we hypothesized that other antiparasitic responses may be altered in parasite-burdened Trpm5−/− mice. Consistent with helminth infections (33), Tm colonization also induced goblet cell hyperplasia in WT (P < 0.0001) but not in Trpm5−/− mice (Fig. 2, G and H). Similarly, we observed eosinophilia in WT but not in Trpm5−/− mice colonized with Tm (Fig. 2I).
Because epithelial cells are a key source of the parasite-induced cytokines thymic stromal lymphopoietin (TSLP) and interleukin-33 (IL-33) and -25 (17), we isolated tuft cells and the remaining epithelial fraction to determine TSLP, IL-33, and IL-25 expression patterns. Consistent with recent reports, we found that tuft cells expressed less TSLP and IL-33 than other epithelial cells and are the main source of epithelial IL-25 (Fig. 3A and fig. S8A) (34,35). To determine whether TRPM5 affects parasite-induced IL-25 expression, we infected WT and Trpm5−/− mice with Tm and measured both parasite colonization and the corresponding epithelial IL-25 expression over time. Tm rapidly colonized both WT and Trpm5−/− mice, but Trpm5−/− mice had significantly reduced IL-25 expression 12 days after infection (P = 0.0006) (Fig. 3B).
Fig. 3. Tuft cells express IL-25 and elicit ILC2s in a TRPM5-dependent manner in response to symbiotic protozoa.
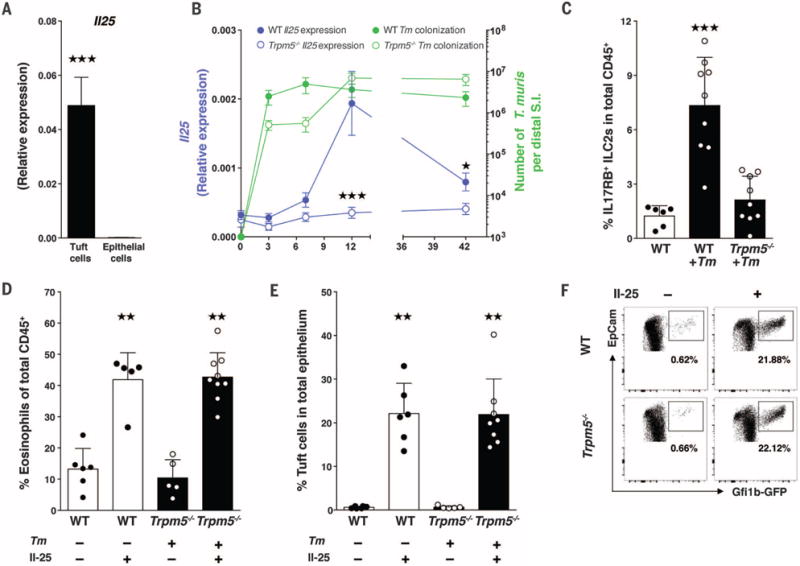
(A) IL-25 expression from sorted tuft cells. (B) WT (solid circles) and Trpm5−/− (open circles) mice were colonized with Tm for 3, 7, 12, and 42 days. At each time point, epithelial cell IL-25 expression was measured (purple line) and Tm colonization was quantified (green line). (C) Frequency of IL17RB+ (IL-25R) ILC2s in the distal SI LP of uninfected WT mice and WT and Trpm5−/− mice colonized with Tm for 12 days. (D) Eosinophil frequency in the distal SI LP of uninfected WT or Tm-colonized Trpm5−/− mice intraperitoneally injected with IL-25 or phosphate-buffered saline (PBS) control. (E) Tuft cell frequencies and (F) flow plots of epithelial cells isolated from Trpm5−/− mice intraperitoneally injected with IL-25 or PBS. Each symbol in (C), (D), and (E) represents an individual mouse, and all data are representative of three independent experiments. Data are plotted as means with SD.Three stars, P < 0.001; two stars, P < 0.01; Kruskal-Wallis or Mann-Whitney tests.
IL-25 promotes proliferation and activation of type 2 innate lymphoid cells (ILC2s) via the receptor subunit IL17RB (11, 36,37). Accordingly, the frequency of intestinal lamina propria IL17RB+ ILC2s significantly increased in WT but not Trpm5−/− mice after 12 days of Tm infection (Fig. 3C). To determine whether the parasite response in Trpm5−/− mice could be complemented by exogenous IL-25, we injected IL-25 intraperitoneally into Trpm5−/− mice; we observed restoration of distal small intestinal eosinophilia and tuft cell abundance (Fig. 3, D to F), suggesting that tuft cells may influence their own abundance.
Epithelial cells are not only a crucial source of IL-25 but also signal in an autocrine manner via IL17RB (22). Therefore, we examined tuft cell IL17RB expression and found that it was significantly higher (P = 0.0043) than for other epithelial cells (fig. S8B). This raised the question of whether IL-25 induces tuft cell expansion via autocrine signaling or indirectly through recruitment of ILC2s. To evaluate factors that affect tuft cell abundance independently of the microbiota or immune system, we used an in vitro primary intestinal organoid system (38,39). Small intestinal organoids reconstitute all the epithelial subsets from IEC stem cells. By generating organoids from Gfi1beGFP/+ mice, we detected GFP+ tuft cells (Fig. 4A and fig. S9A). Both WT and Trpm5−/− organoids contained ~0.3% tuft cells, but IL-25 did not increase tuft cell numbers (Fig. 4B and fig. S9A), suggesting that IL-25 does not act in an autocrine manner to expand tuft cell abundance. Because IL-25 promotes expansion of ILC2s, which are critical sources of IL-13 (11,36,40), a cytokine previously shown to increase goblet cell numbers (25), we considered that IL-13 may also increase tuft cell abundance. IL-13 significantly expanded tuft cells from 0.3% of total organoid cells to 11.9% and 10.9% (WT and Trpm5−/−, respectively) (Fig. 4B and fig. S9A). In agreement with these results, expression of DCLK1 and TRPM5 also increased in IL-13-treated organoids (fig. S9, B and C).
Fig. 4. Innate lymphoid cells and IL-13 increase tuft cells in organoids and the small intestine.
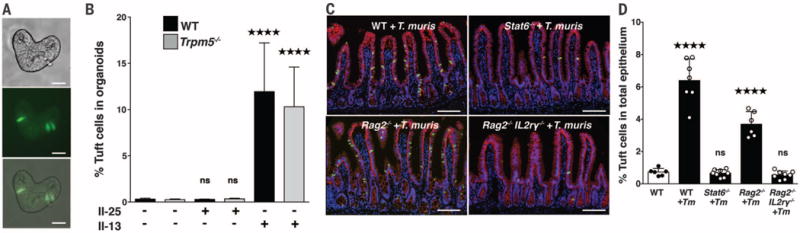
(A) Differential interference contrast, fluorescent, and merged images of small intestinal organoids generated from Gfi1beGFP/+ mice (scale bars, 25 μm). (B) GFP+ tuft cell abundance by flow cytometry of WT and Trpm5−/− organoids treated with recombinant IL-13 or IL-25. (C) Representative images of SI from WT, Stat6−/−, Rag2−/−, and Rag2−/−Il2rγ−/− mice colonized with Tm and (D) tuft cell frequency. DCLK1 is shown in green, E-cadherin in red, and DAPI in blue (scale bars, 100 μm). Each symbol in (D) represents an individual mouse, and all data are representative of (D) two or (B) three independent experiments. Data are plotted as means with SD. Four stars, P < 0.0001; one-way ANOVA or Mann-Whitney tests.
To determine whether type 2 cytokine production by ILC2s may contribute to tuft cell expansion in vivo, we colonized WT, Stat6−/−, Rag2−/−, and Rag2−/−Il2rγ−/− mice with Tm (fig. S10). STAT6 is activated by the type 2 cytokines IL-4 and IL-13 and is required for intestinal helminth expulsion (26). Consistent with our organoid data demonstrating that IL-13 potently induces tuft cell expansion, tuft cells did not expand when Tm colonized Stat6−/− mice (Fig. 4, C and D). Although both T helper 2 (TH2) and ILC2 cells can produce IL-13 in mucosal tissue (41), parasite-induced IL-25 potently activates IL-13 expression in ILCs (11,36,42). We compared tuft cell abundance in Rag2−/− mice that lack TH2 cells but contain ILC2s versus Rag2−/−Il2rγ−/− mice that lack both TH2 and ILC2s cells (8,11,12). Infected Rag2−/− mice had elevated tuft cell abundance compared with uninfected WT mice; however, similar to both Trpm5−/− and Stat6−/− mice, Rag2−/−Il2rγ−/− mice showed no tuft cell increase during Tm infection (Fig. 4, C and D). Collectively, these data suggest that tuft cells may detect Tm through TRPM5 taste chemosensation to elicit ILCs, which in turn produce IL-13 to expand tuft cell abundance (fig. S11).
IECs are positioned for direct contact with lumenal microbes and microbial products, and they function as sensory nodes to promote homeostasis with symbiotic microbes and initiate immunity against pathogens. Eukaryota, including helminths and protozoa, are common members of the gut microbiota (43, 44) that profoundly modulate the host immune system (45,46). Many of the pattern recognition receptor systems that recognize bacterial members of the microbiota do not contribute to recognition of parasites (47,48). Here we show that tuft cells orchestrate type 2 immunity, in agreement with two recent studies (34,35). Taste receptors respond to a panoply of ingested agonists (18), and we speculate that tuft cells and taste chemosensation within the gut provide similarly broad recognition of parasitic signals.
Supplementary Material
Acknowledgments
We thank members of the Garrett Lab for helpful discussion, T. Stappenbeck for supplying L-WRN cells, R. Montgomery for help with organoid imaging, and W. Fowle for help with SEM. The data from this study are tabulated in the main paper and in the supplementary materials. Trpm5eGFP, Trpm5−/−, and gustducin−/− mice are available from Monell Chemical Senses Center under a material transfer agreement. This work was supported by NIH National Research Service Award (NRSA) F32DK098826 to M.R.H.; NIH NRSA F31DK105653 to S.L.; and NIH grants R01 CA154426 and R01 GM099531, a Burroughs Wellcome Career in Medical Sciences Award, and a Searle Scholars Award to W.S.G.
Footnotes
The authors declare no competing financial interests.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/351/6279/1329/suppl/DC1
Materials and Methods
References (49–53)
REFERENCES AND NOTES
- 1.Kobayashi KS, et al. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 2.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Proc Natl Acad Sci USA. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaishnava S, et al. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith PM, et al. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Gastroenterology. 2013;145:396–406.e10. doi: 10.1053/j.gastro.2013.04.056. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, et al. Cell. 2003;112:293–301. doi: 10.1016/s0092-8674(03)00071-0. [DOI] [PubMed] [Google Scholar]
- 8.Mishra PK, Palma M, Bleich D, Loke P, Gause WC. Mucosal Immunol. 2014;7:753–762. doi: 10.1038/mi.2014.23. [DOI] [PubMed] [Google Scholar]
- 9.Wong GT, Gannon KS, Margolskee RF. Nature. 1996;381:796–800. doi: 10.1038/381796a0. [DOI] [PubMed] [Google Scholar]
- 10.Lee RJ, et al. J Clin Invest. 2012;122:4145–4159. doi: 10.1172/JCI64240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Y, et al. Nat Immunol. 2015;16:161–169. doi: 10.1038/ni.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurst SD, et al. J Immunol. 2002;169:443–453. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- 13.Tizzano M, et al. Proc Natl Acad Sci USA. 2010;107:3210–3215. doi: 10.1073/pnas.0911934107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee RJ, et al. J Clin Invest. 2014;124:1393–1405. doi: 10.1172/JCI72094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saunders CJ, Christensen M, Finger TE, Tizzano M. Proc Natl Acad Sci USA. 2014;111:6075–6080. doi: 10.1073/pnas.1402251111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bezenpon C, et al. J Comp Neurol. 2008;509:514–525. doi: 10.1002/cne.21768. [DOI] [PubMed] [Google Scholar]
- 17.Peterson LW, Artis D. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 18.Meyerhof W, et al. Chem Senses. 2010;35:157–170. doi: 10.1093/chemse/bjp092. [DOI] [PubMed] [Google Scholar]
- 19.Gerbe F, et al. J Cell Biol. 2011;192:767–780. doi: 10.1083/jcb.201010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westphalen CB, et al. J Clin Invest. 2014;124:1283–1295. doi: 10.1172/JCI73434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerbe F, Legraverend C, Jay P. Cell Mol Life Sci. 2012;69:2907–2917. doi: 10.1007/s00018-012-0984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang Z, et al. Immunity. 2012;36:821–833. doi: 10.1016/j.immuni.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bjerknes M, et al. Dev Biol. 2012;362:194–218. doi: 10.1016/j.ydbio.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivanov II, et al. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenzie GJ, Bancroft A, Grencis RK, McKenzie ANJ. Curr Biol. 1998;8:339–342. doi: 10.1016/s0960-9822(98)70134-4. [DOI] [PubMed] [Google Scholar]
- 26.Urban JF, Jr, et al. Immunity. 1998;8:255–264. doi: 10.1016/s1074-7613(00)80477-x. [DOI] [PubMed] [Google Scholar]
- 27.Baker DG. In: Flynn’s Parasites of Laboratory Animals. Baker DG, editor. Blackwell Publishing; 2008. pp. 303–397. [Google Scholar]
- 28.Materials and methods are available as supplementary materials on Science Online
- 29.Saeki H, Togo M, Imai S, Ishii T. Japan J Vet Sci. 1983;45:151–156. doi: 10.1292/jvms1939.45.151. [DOI] [PubMed] [Google Scholar]
- 30.Schütz B, et al. Front Physiol. 2015;6:87. doi: 10.3389/fphys.2015.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kusumakshi S, et al. Chem Senses. 2015;40:413–425. doi: 10.1093/chemse/bjv023. [DOI] [PubMed] [Google Scholar]
- 32.Pérez CA, et al. Nat Neurosci. 2002;5:1169–1176. doi: 10.1038/nn952. [DOI] [PubMed] [Google Scholar]
- 33.Khan WI, Blennerhasset P, Ma C, Matthaei KI, Collins SM. Parasite Immunol. 2001;23:39–42. doi: 10.1046/j.1365-3024.2001.00353.x. [DOI] [PubMed] [Google Scholar]
- 34.von Moltke J, Ji M, Liang HE, Locksley RM. Nature. 2016;529:221–225. doi: 10.1038/nature16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerbe F, et al. Nature. 2016;529:226–230. doi: 10.1038/nature16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neill DR, et al. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saenz SA, et al. Nature. 2010;464:1362–1366. doi: 10.1038/nature08901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyoshi H, Stappenbeck TS. Nat Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato T, et al. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 40.Fallon PG, et al. J Exp Med. 2006;203:1105–1116. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang HE, et al. Nat Immunol. 2012;13:58–66. doi: 10.1038/ni.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price AE, et al. Proc Natl Acad Sci USA. 2010;107:11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parfrey LW, et al. Front Microbiol. 2014;5:298. doi: 10.3389/fmicb.2014.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hotez PJ, et al. J Clin Invest. 2008;118:1311–1321. doi: 10.1172/JCI34261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osborne LC, et al. Science. 2014;345:578–582. doi: 10.1126/science.1256942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reese TA, et al. Science. 2014;345:573–577. doi: 10.1126/science.1254517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helmby H, Grencis RK. Eur J Immunol. 2003;33:2974–2979. doi: 10.1002/eji.200324264. [DOI] [PubMed] [Google Scholar]
- 48.Reynolds LA, et al. J Immunol. 2014;193:2984–2993. doi: 10.4049/jimmunol.1401056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.