

Abstract
Estrogen receptor (ER) positive tumors represent the majority of breast malignancies, and are effectively treated with hormonal therapies, such as tamoxifen. However, in the recurrent disease resistance to tamoxifen therapy is common and a major cause of death. In recent years, in‐depth proteome analyses have enabled identification of clinically useful biomarkers, particularly, when heterogeneity in complex tumor tissue was reduced using laser capture microdissection (LCM). In the current study, we performed high resolution proteomic analysis on two cohorts of ER positive breast tumors derived from patients who either manifested good or poor outcome to tamoxifen treatment upon recurrence. A total of 112 fresh frozen tumors were collected from multiple medical centers and divided into two sets: an in‐house training and a multi‐center test set. Epithelial tumor cells were enriched with LCM and analyzed by nano‐LC Orbitrap mass spectrometry (MS), which yielded >3000 and >4000 quantified proteins in the training and test sets, respectively. Raw data are available via ProteomeXchange with identifiers PXD000484 and PXD000485. Statistical analysis showed differential abundance of 99 proteins, of which a subset of 4 proteins was selected through a multivariate step‐down to develop a predictor for tamoxifen treatment outcome. The 4‐protein signature significantly predicted poor outcome patients in the test set, independent of predictive histopathological characteristics (hazard ratio [HR] = 2.17; 95% confidence interval [CI] = 1.15 to 4.17; multivariate Cox regression p value = 0.017). Immunohistochemical (IHC) staining of PDCD4, one of the signature proteins, on an independent set of formalin‐fixed paraffin‐embedded tumor tissues provided and independent technical validation (HR = 0.72; 95% CI = 0.57 to 0.92; multivariate Cox regression p value = 0.009). We hereby report the first validated protein predictor for tamoxifen treatment outcome in recurrent ER‐positive breast cancer. IHC further showed that PDCD4 is an independent marker.
Keywords: Breast cancer, Proteomics, Tamoxifen resistance, Biomarker, Mass spectrometry
Highlights
Analysis of hormonal naïve cohorts of patients with recurrent breast cancer.
Laser microdissection based enrichment of epithelial breast tumors.
High resolution mass spectrometric analysis of epithelial breast tumors.
Development and validation of a protein signature predicting tamoxifen outcome.
Validation by immunohistochemistry in independent cohort.
Abbreviations
- MS
mass spectrometry
- IHC
immunohistochemistry
- LCM
laser capture microdissection
- TMA
tissue micro-array
- LFQ
label-free quantification
- ER
estrogen receptor
- SRM/MRM MS
selected reaction monitoring/multiple reaction monitoring mass spectrometry
- SD
standard deviation
- WTL
whole tissue lysate
- PgR
progesterone receptor
- TTP
time to progression
- DFI
disease free interval
- PEP
posterior error probability score
- EMC
Erasmus MC, University Medical Center
- NKI-AVL
Netherlands Cancer Institute – Antoni van Leeuwenhoek Hospital
- RadboudUMC
Radboud University Medical Center
- HR
hazard ratio
- CI
confidence interval
- PPV
positive predicted value
- NPV
negative predicted value
- ROC
receiver operating characteristic
- CR
complete remission
- PR
partial remission
- NC
no change
- PD
progressive disease
- SD
stable disease
- PEN
polyethylene naphthalate
- IQR
interquartile range
- AUC
area under the curve
- LC-MS
liquid chromatography mass spectrometry
- IPA
ingenuity pathway analysis
1. Introduction
ER positive tumors constitute the majority of all breast malignancies. Tamoxifen therapy has been shown to significantly improve survival and cure of patients with primary ER positive breast tumors, but upon recurrence about half of the patients show intrinsic resistance, while those initially responding will ultimately develop acquired resistance (Cardoso et al., 2012; Milani, 2014). The need for biomarkers capable of determining mechanisms of resistance has led to the development of several predictive signatures, though none has been introduced in the clinic so far (Beelen et al., 2012). With the recent advancements in MS techniques, in‐depth quantification of the human proteome has become possible and the ability of measuring protein abundance over a broad dynamic range has established proteomics as a robust tool for biomarker discovery (Drabovich et al., 2014; Kim et al., 2014; Wilhelm et al., 2014). The proteomic analysis of tissue specimens is, however, hindered by their heterogeneity, which alters protein abundance dynamic range. Furthermore, the presence of stromal and infiltrating cells adds another layer of complexity by hampering accurate protein quantitation of target epithelial tumor cells (Kondo, 2014). To address this issue, LCM offers a robust cell sub‐population enrichment technique, allowing accurate downstream analysis of morphologically heterogeneous specimens (Emmert‐buck et al., 1996; Vogel et al., 2007). Genomic and proteomic analyses of LCM derived material showed the feasibility of this technique in molecular profiling studies and pointed out its efficacy in studying disease associated signaling pathways when compared to whole tissue analyses (Cheng and Zhang, 2013; Sereni et al., 2015; Xu, 2010; F. Yang et al., 2006). LCM yields sub‐microgram protein amounts due to the fact that only a limited number of cells can be dissected from each sample. In the light of this, coupling LCM enrichment to chemical labeling methods would require extensive sample preparation and workflow optimization, which would be unsuitable in the analysis of large sample sets. Label‐fee quantification (LFQ) software algorithms have demonstrated to be accurate tools in the quantitation of proteins, allowing high yield identification and reliable quantitation of measured peptides even from minute amount of analyzed specimens (Cox and Mann, 2008; Megger et al., 2013). We have optimized a tissue proteomic pipeline for biomarker discovery coupling LCM cell enrichment to high resolution LC‐MS and LFQ, capable of quantifying more than 3000 proteins from only 4000 dissected epithelial cells (Braakman et al., 2012; Liu et al., 2012). Using this workflow, we recently developed and validated a prognostic protein signature for triple negative breast cancer (Liu et al., 2014). Despite our workflow has demonstrated to be a robust methodology for the discovery of cancer biomarkers, application of shotgun proteomics in clinical diagnostics remains problematic due to the extensive and time consuming sample preparation required. In this perspective, IHC or selected reaction monitoring/multiple reaction monitoring (SRM/MRM) MS may be more suitable biomarker verification techniques that do not require extensive method optimization or sample preparation (Whiteaker et al., 2011). Although antibody specificity and lack of accurate quantitation remain important issues, IHC still remains a major technique in clinical diagnostics and significantly requires less amount of optimization time in comparison to ELISA or even SRM/MRM MS.
In this study we describe the development of a predictive protein signature for tamoxifen resistance in ER positive breast cancer by coupling LCM tumor cell enrichment and high resolution LC‐MS in the analysis of independent training and test patient cohorts. We also provide further validation by IHC analysis of signature proteins on an independent panel of paraffin‐embedded tissues captured in a tissue micro‐array (TMA).
2. Materials and methods
2.1. Sample sets
From an initial selection of 200 tissues collected from patients that received tamoxifen as first line therapy we excluded tissues with a low percentage of tumor cells (i.e. <40%; n = 88; Figure 1). A total of 112 ER positive fresh frozen primary breast cancer tissue samples were then included in our sets: 56 from Erasmus MC University Medical Center (EMC), Rotterdam (years of surgery: 1981–1994), 41 from the National Cancer Institute – Antoni van Leeuwenhoek hospital (NKI‐AVL), Amsterdam (1980–1996), and 15 from Radboud University Medical Center (RadboudUMC), Nijmegen (1991–1996; Table 1). EMC derived samples constituted the training set, while NKI‐AVL and RadboudUMC provided an independent external test set. ER positivity in tumor cytosols was assessed by quantitative biochemical assays (EMC), reverse‐transcriptase quantitative polymerase chain reaction (RadboudUMC), or IHC (NKI‐AVL). All patients underwent surgery of their primary tumor (conservative or non‐conservative), developed recurrent disease, and were treated with tamoxifen as first line therapy. Due to lack of response data for a subset of specimens, treatment outcome was defined based on time to progression (TTP): disease progression ≤6 months and >6 months after start of first line tamoxifen administration were defined as poor and good outcome, respectively. The training set comprised 24 and 32 patients who showed good and poor outcome upon tamoxifen treatment, respectively. The test set included tumors of 41 good and 15 poor outcome patients. The NKI‐AVL cohort did not contain stage IV tumors, while such specimens were found in the EMC and RadboudUMC sets. In addition, 2 tumor tissues of which clinical follow up information was not available were used as LCM and whole tissue lysate (WTL) controls. For biological replicates, both tumor tissues were subjected to 4 rounds of LCM. Of one of these, a WTL was prepared from one sample and digested in triplicate.
Figure 1.
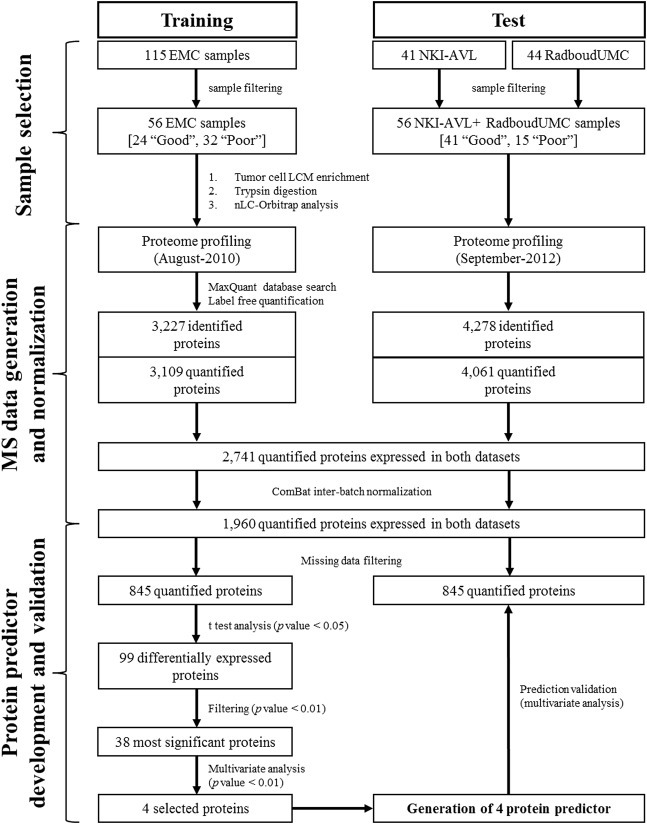
Data analysis flow‐chart and development of predictor for tamoxifen treatment outcome. Patients were divided into two independent cohorts and separately measured by LC‐MS. Proteomic data from training and test sets were analyzed separately in MaxQuant. Identified proteins were filtered for reversed sequences and for Posterior Error Probability score (PEP < 0.05), intensities of commonly expressed proteins were normalized using ComBat algorithm to minimize batch effects, and filtered for missing data (10 minimum observations for global proteomic analysis and allowing 30% and 0% missing data in training and test set respectively for predictor generation). Student t test (p value < 0.05) was then used to assess differences in protein expression levels between good and poor outcome patients. A multivariate regression model was used to obtain an optimal list of 4 proteins to be tested as a predictor of tamoxifen treatment outcome: CGN, G3BP2, PDCD4 and OCIAD1. The 4‐protein signature was confirmed in an external test set. Acronyms: EMC = Erasmus MC, University Medical Center; NKI‐AVL=Netherlands Cancer Institute‐ Antoni van Leeuwenhoek hospital; RadboudUMC = Radboud University Medical Center.
Table Table 1.
Patient and tumor characteristics.
| Traininga | Testa | ||
|---|---|---|---|
| EMC | NKI‐AVL | RadboudUMC | |
| All patients | 56 (100) | 41 (100) | 15 (100) |
| Age | |||
| ≤ 55 years | 15 (27) | 12 (29) | 1 (7) |
| > 55 years | 41 (73) | 29 (71) | 14 (93) |
| Menopausal status | |||
| Premenopausal | 10 (18) | 11 (27) | 0 (0) |
| Postmenopausal | 46 (82) | 30 (63) | 15 (100) |
| Tumor size | |||
| T1 (≤2 cm) | 12 (21) | 20 (49) | 5 (33) |
| T2 (2–5 cm) + Tx | 40 (72) | 19 (46) | 9 (60) |
| T3 (>5 cm) + T4 | 4 (7) | 2 (5) | 1 (7) |
| Tumor differentiationb | |||
| Good/Moderate | 13 (59) | 29 (71) | 8 (53) |
| Poor | 33 (23) | 12 (29) | 4 (27) |
| Unknown | 10 (18) | 0 (0) | 3 (20) |
| Disease free interval | |||
| ≤ 12 months | 24 (43) | 4 (10) | 5 (33) |
| > 12 months | 32 (57) | 37 (90) | 10 (67) |
| PgRc | |||
| Negative | 9 (16) | 17 (41) | 11 (73) |
| Positive | 44 (79) | 24 (59) | 4 (27) |
| Involved lymph nodes | |||
| 0 | 31 (55) | 24 (58) | 6 (40) |
| ≥ 1 | 20 (36) | 16 (39) | 7 (47) |
| unknown | 5 (9) | 1 (3) | 2 (13) |
| Dominant site of relapse | |||
| Loco‐regional | 8 (14) | 4 (10) | 0 (0) |
| Bone | 26 (46) | 12 (29) | 6 (40) |
| Visceral | 13 (24) | 6 (15) | 9 (60) |
| Bone and other | 9 (16) | 14 (34) | 0 (0) |
| Unknown | 0 (0) | 5 (12) | 0 (0) |
Acronym: PgR: progesterone receptor.
Data are reported as number (percentage).
Histopathological characteristics were evaluated by local pathologists, according to standard clinical practice at time of sample collection.
Missing data not reported.
In addition, a total of 447 formalin‐fixed and paraffin‐embedded tissues collected from EMC and regional hospitals were comprised in a tissue micro‐array. For further analyses, we included only ER positive tumors and patients who did not receive hormonal adjuvant therapy. Patients with a revised histology that showed no tumor, or patients with a progression within 3 weeks were excluded as well, leading to a total of 408 ER positive tissues from patients treated with tamoxifen as first‐line therapy for recurrent disease (Supplemental Table 1). Response data were collected according to the standard International Union Against Cancer criteria (Hayward and Carbone, 1977). In this set, 11 (2.7%) and 51 (12.5%) patients respectively showed complete (CR) and partial remission (PR). Two hundred and five (50.3%) patients showed no change (NC) of disease, of whom 170 (41.7%) showed NC >6 months (defined as stable disease, SD) while 35 (8.6%) showed NC ≤6 months after start of therapy. Progressive disease (PD) was observed in 141 (34.6%) patients. Clinical benefit was defined as CR + PR + SD patients (n = 232; 57%), while objective response was defined as CR + PR only (n = 62; 15%). This retrospective study used coded primary tumor tissues, in accordance with the Code of Conduct of the Federation of Medical Scientific Societies in the Netherlands (http://www.federa.org/codes‐conduct). Reporting Recommendations for Tumor Marker Prognostic Studies were followed where possible (Altman et al., 2012).
2.2. Laser capture microdissection
All tissue samples were cut into 8 μm cryo‐sections, and collected on UV‐sterilized polyethylene naphthalate (PEN) coated glass slides (Carl Zeiss Microsystems GmbH, Göttingen, Germany) for downstream LCM. In addition, 5 μm sections were collected on regular glass slides and stained with hematoxylin and eosin dyes for histological evaluation. Sections on PEN slides were dehydrated with 95% ethanol and immediately stored at −80 °C, until further processing. Prior to LCM, PEN slides were thawed at room temperature and subsequently stained with hematoxylin as follows: distilled water, hematoxylin, distilled water, 50% ethanol, 70% ethanol, 95% ethanol, 100% ethanol, 100% ethanol. During dehydration steps Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific Inc, Rockford, IL, USA) at a 1:100 v/v concentration was added in order to prevent proteolytic degradation of proteins. An area of approximately 500,000 μm2 (∼4000 tumor cells) was collected from each tissue using a photo‐activated localization microscopy Micro Beam device and gathered in an opaque adhesive cap (Carl Zeiss Microsystems GmbH, Göttingen, Germany). A volume of 20 μl of 0.1% w/v Rapigest surfactant (Waters Corporation, Milford, MA, USA) in 50 mM ammonium bicarbonate solution was used to transfer the collected LCM samples into LoBind™ Eppendorf tubes (Eppendorf AG, Hamburg, Germany). Tissue containing buffer was immediately frozen after collection and stored at −80 °C.
2.3. Protein digestion
LCM collected material was disrupted in a horn sonifier bath using an Ultrasonic Disruptor Sonifier II (Bransons Utrasonics, Danbury, CT, USA) at 70% amplitude. Proteins were denatured at 95 °C, reduced with 100 mM DTT for 30 min at room temperature, and alkylated in the dark with 300 mM iodoacetamide for 30 min at room temperature. Samples were then digested for 4 h at 37 °C after addition of MS grade trypsin at a 1:4 enzyme‐protein ratio (i.e. 100 ng/μl). Samples were acidified with TFA, and spun down at 14,000 RPM. Supernatants were collected and transferred to HPLC vials (Sigma–Aldrich Corporation, St. Louis, MO, USA).
2.4. High resolution MS
Mass spectrometry measurements were performed with a nano liquid chromatography system (Ultimate 3000, Dionex, Amsterdam, The Netherlands) coupled online to a linear Ion Trap – Orbitrap XL™ mass spectrometer (Thermo Electron, Bremen, Germany). Samples were first loaded on a trap column (PepMap C18, 300 μm ID × 5 mm length, 5 μm particle size, 100 Å pore size; Dionex), then washed and desalted in 0.1% TFA acidified water. Trap column and analytical column (PepMap C18, 75 μm ID × 50 cm, 3 μm particle size and 100 Å pore size; Dionex) were then coupled and peptides were eluted in a 3 h binary gradient (flow: 300 nl/min; mobile phase A: 2% acetonitrile and 0.1% formic acid in H2O; mobile phase B: 80% acetonitrile and 0.08% formic acid). Gradient was run as follows: 0%–25% mobile phase B for 2 h, increase to 50% mobile phase B in 1 h. For ESI, metal‐coated nano ESI emitters (New Objective, Woburn, MA) were used and a spray voltage of 1.6 kV was applied. High‐resolution scan was acquired from 400 to 1800 Th and was used for MS detection. Automatic gain was set at 106 ions and lock mass was set at 445.120025 u protonated with (Si(CH3)2O)6. The 5 most intense peaks in full scan were selected and fragmented by collision induced dissociation (CID) applying 35% normalized collision energy and detected in the ion trap. Ions falling out of the ±5 ppm window or for which precursor intensity fell below 1.5 signal‐to‐noise ratio during 10 scans were excluded.
2.5. Protein identification and quantification
A total of 112 samples were analyzed by LTQ‐Orbitrap XL™ MS, together with 4 biological LCM replicates of control samples, and of which one was measured with a triplicate of its matching WTL. MS spectra of the training and test cohorts were generated and analyzed separately with a time interval of two years. Orbitrap.RAW files derived from MS analyses were imported and analyzed in MaxQuant (version 1.2.2.5) (Cox and Mann, 2008), using Andromeda peptide search engine (Cox et al., 2011). Analysis of spectra was performed using the following options: acetylation of the N‐terminus and oxidation of methionine were selected as variable modifications, multiplicity was set to 1. FASTA file used for protein search was UniProt‐SwissProt human canonical database (version 2012‐09, human canonical proteome; 20.243 identifiers). Minimal peptide length was set to 7 amino acids, match between runs and LFQ options were selected and kept as default. Other options were kept as default (e.g. fixed peptide modifications: carbamidomethylation; false discovery rate = 0.01). For further data analysis, “ProteinGroups.txt” file was imported into Microsoft Excel and protein identifiers were filtered based on PEP score (cutoff <0.05). Contaminants and reversed sequences were excluded. LFQ intensities for each sample were selected and each value was Log10 transformed. Protein intensities from training and test sets were then normalized using ComBat (Johnson et al., 2007) algorithm in R free software, allowing 10 minimum observations for whole dataset analysis. A second protein list was generated allowing 30% missing data points in the training set and none in the test set for predictor development. LCM and WTL control samples were not included in the ComBat normalization procedure due to the lower amount of identified and quantified proteins. Coefficients of variations of Log10 transformed MS data were calculated according to the following formula (Bland and Altman, 1996):
Pearson correlation coefficients between measurements of LCM and WTL replicates were calculated in Perseus (Max Planck Institute for Biochemistry, Muenchen, Germany). The MS proteomic data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaíno et al., 2013) with dataset identifiers PXD000484 and PXD000485.
2.6. Tissue micro‐array
TMA was prepared using an ATA 27 (Beecher Instruments, Sun Prairie, WI, USA). 408 paraffin‐embedded primary, ER positive breast cancer tissues derived from patients treated with first line tamoxifen upon recurrence were used to prepare the TMA. Tissue cores of 0.6 mm were taken from each tissue paraffin block and transferred in triplicate into a TMA recipient block. For each tumor tissue sample, three different areas of the tumor were taken as biological replicates. TMA slides were digitalized and analyzed using Slidepath software (Leica Microsystems, Solms, Germany).
2.7. Immunohistochemistry
Paraffin‐embedded tissues on glass slides were de‐paraffinized at 60 °C, and remnants of paraffin were removed by sequential washings in xylene (3 × 5 min). Re‐hydration was performed by washings through decreasing concentrations of ethanol following with distilled water as follows: 100% ethanol (1 × 5 min, 2 × 2 min), 70% ethanol (1 × 2 min), 50% ethanol (1 × 2 min), distilled water (1 × 2 min). Slides were then incubated at 95 °C for 40 min in DAKO (Agilent Technologies Inc, Santa Clara, CA, USA) antigen retrieval solution (pH 6) diluted 1:10 in MilliQ water, cooled down to room temperature and washed with PBS buffer 3 times for 5 min. Blocking solution consisting of 5% BSA in PBS was added to the slides and incubated for 30 min. Primary antibodies were diluted in DAKO Antibody Diluent, added to each slide and incubated for 1 h at room temperature. Slides were then washed with PBS, and DAKO Envision® secondary antibody (Goat anti‐Mouse‐HRP and G anti‐R‐HRP, 100 μl per slide) solution was added to each slide and incubated for 45 min at room temperature. A washing cycle with PBS was performed for 5 min and a 1:15 solution of DAB+ chromogen in antibody diluent was added, following incubation in the dark for 10 min. Slides were then washed in tap water for 5 min, stained with hematoxylin/eosin for 1 min each and dehydrated again through sequential washings in 50%–70%–100% ethanol and xylene of 5 min each. Cover glasses were mounted with Pertex and slides were left to dry. TMA slides were stained for Programmed Cell Death 4 (PDCD4) protein (1:200), OCIAD1 (1:800), G3BP2 (1:50), and CGN (1:25). Anti‐PDCD4 mouse monoclonal (id: LS‐B2949; clone K4C1) and anti‐OCIAD1 rabbit polyclonal (id: LS‐B5046) antibodies were purchased from Lifespan Technologies (Lifespan technologies Inc, Seattle, WA, USA), anti‐G3BP2 rabbit polyclonal (id: NBP1‐82976) antibody was purchased from Novus Biologicals (Novus Biologicals LLC, 8100 Littleton, CO, USA), and anti‐CGN rabbit polyclonal (HPA027657) antibody was purchased from Sigma.
2.8. IHC staining analysis
Data from scored tissues were filtered for missing data and adjuvant endocrine therapy, leading to a final list of 294 tissue samples. PDCD4 antibody stained tissues were separately scored for nuclear and cytoplasmic staining intensity (categories: negative, weak, moderate, strong) and percentage of stained tumor cells (categories: 0%, 1–10%, 11–20%, 21–30%, 31–40%, 41–50%, 51–60%, 71–80%, 81–90%, 91–100%). CGN, and OCIAD1 stained tissues were scored based on intensity parameters only, while G3BP2 scoring included quantity levels as well. TMA was scored by two independent researchers, and the average, consolidated scores of triplicate cores were used for statistical analysis. Due to the fact that PDCD4 cytoplasmic and nuclear stainings were co‐expressed in the evaluated TMA cores, these were merged in order to assess total protein levels. PDCD4 nuclear and cytoplasmic scores were numerically transformed and merged into a histo‐score (Supplemental Table 2) according to formula:
Histo‐score cutoff (i.e. 30) reflective of weak vs strong protein expression was used to stratify patient groups: low and high PDCD4 protein expressing tumors displayed a histo‐score below (<) or above (≥) the cutoff (Supplemental Table 3). PDCD4 cytoplasmic quantity was ranging only from 80% to 90% so it was not included in the histo‐score calculation formula.
2.9. Statistical analysis
Differences in clinical parameters between training and test sets were evaluated by Mann–Whitney U and Pearson χ2 tests (two sided tests). Commonly expressed proteins between the two LCM sets and proteins quantified in WTL sample replicates were annotated through DAVID (Huang and Lempicki, 2008; Huang et al., 2009) for organelle distribution using Swissprot keyword database. Average abundance levels of these proteins in all 112 measured samples were used to generate a waterfall plot of protein abundance distribution.
Protein list used for predictor development was tested for protein differential abundance between patient groups through Student's t‐test (two sided, unequal variances assumed). Hierarchical clustering was performed on all quantified and differentially expressed proteins (t test p value < 0.05), respectively (complete linkage; distance metric: correlation‐uncentered). Significant proteins in the training set were submitted along with their fold changes and t test p values to Ingenuity Pathway Analysis (IPA) network analysis with the following settings: Data sources: all; Confidence: high (predicted) and experimentally observed; species: human. Network was plotted using path designer (Ingenuity Systems, Redwood City, CA, USA).
In order to rule out possible indiscriminative identifiers, the protein predictor was developed selecting the 38 most significant proteins (univariate p value < 0.01) in the training set and a Cox regression multivariate analysis was performed with a step‐down procedure, which involved iteratively removing the least significant proteins (multivariate p value ≥ 0.01) until all remaining proteins in the model showed a multivariate p value < 0.01. Each protein score (t value) was then multiplied by its abundance, and values were then summed for all proteins to obtain a patient score, which was then coupled to outcome data. Each patient score was plotted in a receiver operating characteristic (ROC) curve. Youden index (max of J = Sensitivity + Specificity−1) was set as cutoff in the training set and used to categorize patients in the test set. Log‐rank tests on the survival curves of predicted groups were performed to assess significance of prediction. Association of predictor proteins to TTP was assessed through Cox regression, correcting for patient and tumor characteristics. IHC stainings were used to test for association with TTP, clinical benefit and objective response in combination with clinical parameters by Cox and logistic regression analyses, respectively. Co‐variables that were found not significant in univariate regression analyses were excluded from multivariate models. Cox regression and logistic regression analyses, hazard ratios, odds ratios and confidence intervals were calculated in Stata (version 13.1; Stata Corp, College Station, TX, USA).
3. Results
3.1. Analysis of patient cohorts
One hundred and twelve ER positive primary breast tumor tissues, of which 56 comprised the training set and another 56 the test set, were processed according to our tissue proteomics workflow (Braakman et al., 2012; Liu et al., 2012) and analyzed through high resolution MS.
Analysis of tumor and patient characteristics between the training and test sets showed that age and menopausal status at start of tamoxifen therapy, lymph node status, and tumor size were not significantly different. The test set contained a higher proportion of poorly differentiated tumors (Pearson's χ2 = 21.19, p value < 0.001) compared to the training set. Furthermore, patients in the test cohort had a median disease free interval (DFI) of 51.4 months (range: 0–195 months), which was significantly longer (Mann–Whitney U = −3.814, p value < 0.001) than for patients in the training set (median: 16.4 months, range: 0–90.8 months). This can be attributed to the lack of stage IV tumors in the NKI‐AVL cohort, which possibly contributed to the difference in DFI and grade between training and test set.
3.2. MS analysis of ER positive breast cancer
LCM discovery and test samples were analyzed along with 8 LCM replicates from 2 separate control tissues, and 3 technical replicates of a control WTL. A total of 2215 proteins were quantified in LCM control samples, and 1320 proteins in the WTL sample replicates, with only 852 proteins quantified in both LCM and WTL controls. Pearson correlation coefficients between LCM samples ranged from 0.92 to 0.97 while it ranged from 0.96 to 0.97 between WTL measurements (Supplemental Figure 1A). Hierarchical clustering of LCM and WTL controls showed grouping according to sample origin without miss‐classifications (Supplemental Figure 1B). Median coefficients of variation of biological and technical replicates were 16.05% (interquartile range, IQR: 10.77–24.56) and 20.35% (IQR: 11.55–34.28), respectively. Reproducibility of MS measurements was defined as acceptable given the low number of control samples replicate measurements.
A total of 3227 proteins were identified in the training set, of which 3109 were quantified by LFQ. In the test set, 4278 proteins were identified and 4061 proteins were quantified. LFQ intensity values of 2741 proteins commonly expressed between the training and test set were normalized for batch differences and filtered for missing data to generate two protein lists: a 1960 protein list (10 minimum observations; Supplemental Table 4) for general proteome analysis and an 845 protein list for predictor development (30% missing data in training set and 0% missing data in test set; Supplemental Table 5). From the analysis of 1960 expressed proteins, a wide distribution of protein abundances was observed over 3 orders of magnitude (Figure 2A). Interferon signaling related (e.g. IFI16, IFIT5) and chaperone associated proteins (e.g. DNAJC7, BAG1) displayed low overall abundance (Figure 2B), while luminal epithelial specific (e.g. KRT18), metabolism related (e.g. PKM, ATP5A1), and heat‐shock (e.g. HSPD1, HSPB1) proteins were found to be highly abundant (Figure 2C). In the training set, CV was 14.10% (IQR: 10.22–18.78), whereas it was 13.86% (IQR: 10.33–18.57) in the test set.
Figure 2.
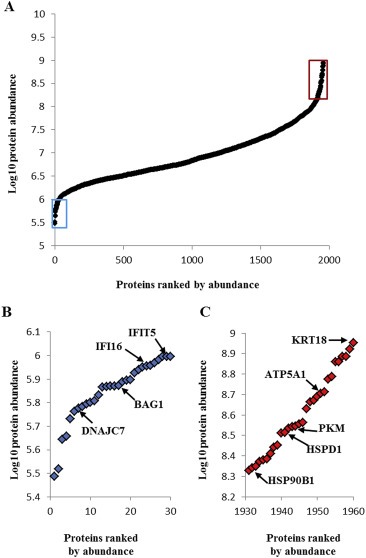
Protein abundance levels in 112 ER positive breast cancer samples. The waterfall plot shows mean protein abundance distribution of 1.960 commonly expressed proteins. The mean abundance of each quantified protein was calculated and plotted. The 30 least (blue) and most (red) abundant proteins are boxed in panel (A) and enlarged in panel (B) and (C), respectively.
DAVID based annotation for subcellular compartment showed that in the 112 breast cancer tissues the majority of expressed proteins belonged to the nuclear (25.76%) and cytoplasmic (56.38%) compartments while the endoplasmic reticulum (9.54%), Golgi apparatus (6.43%), mitochondria (12.65%), plasma membrane (7.50%), and the extracellular matrix (1.84%) comprised a lower amount of proteins. The smallest group consisted of plasma proteins (0.46%; Figure 3A). The distribution of intensity levels of the 1320 proteins quantified in the WTL control sample showed a similar dynamic range but with increased variation, probably due to exclusion from the normalization procedure (Figure 3B). Annotation for cellular compartments showed a similar distribution of the 1320 identified proteins into subcellular compartments compared to the 112 tissue set but with a notable enrichment of extracellular matrix (e.g. COL1A1) and plasma proteins (e.g. APOA1), which represented 7.19% and 6.89% of all quantified proteins in this set, respectively. The minor contribution of extracellular matrix and plasma proteins in the LCM samples suggests that LCM indeed resulted in highly enriched epithelial tumor cell fractions.
Figure 3.
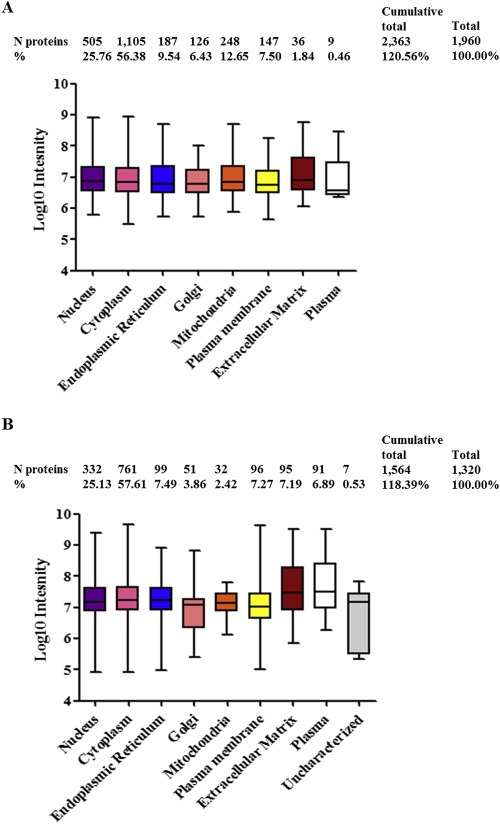
Protein compartmentalization and abundance correlation analysis. Panel shows quantified protein abundance range per subcellular compartment in the LCM enriched 112 ER positive tumors (A) and in WTL control replicates (B). Number of proteins per compartment and percentages are displayed above the dot plot.
Distribution of intensities of organelle specific proteins showed comparable average levels of expression, therefore showing that all cell compartments were quantified. In the LCM annotated set several proteins showed multiple organelle localization. The nuclear and cytoplasmic compartments showed the highest degree of overlap with 249 (12.70%) proteins, mostly represented by proteasome subunits (e.g. PSME3) and proteins involved in RNA binding (e.g. RBM3, HNRNPA1). A small number of multi‐compartmentalized proteins was constituted by vesicular transport components between the endoplasmic reticulum and the Golgi apparatus (n = 39; 1.99%) such as SEC23A. A subset of proteins was also found co‐localized in both Golgi and cytoplasm (e.g. SEC24D). The remaining compartments showed expression of locally specific proteins (median overlap: 0.59% of total) such as oxidative chain proteins in the mitochondrion (e.g. UQCRC1) or DNA replication and repair involved proteins in the nucleus (e.g. FEN1). These data indicate the capability of LC‐MS coupled to LCM enrichment to assess protein abundances throughout all cellular compartments from minute amounts of epithelial tumor tissues.
3.3. Analysis of differentially expressed proteins
Due to the fact that the 1960 proteins did not clearly discriminate patient groups (Supplemental Figure 2), a more stringent filter for missing values was therefore applied and candidate proteins were selected based on their differential abundance between patient groups. On the panel of filtered 845 quantified proteins, a Student's t test was performed to identify 99 proteins that were differentially abundant between good and poor outcome patients in the training set (p value < 0.05). Of these, 50 proteins were found upregulated in the poor outcome group and 49 displayed higher expression in the good outcome group (Supplemental Table 6). In order to define molecular interaction networks between significant molecules, network analysis in IPA was performed. The network that displayed the most hits comprised proteins involved in cell growth and proliferation and cell death and survival, such as CDC37 (upregulated in poor outcome) and PDCD4 (upregulated in good outcome; Supplemental Figure 3). Several molecules included in the network that were found upregulated in the poor outcome patient group were involved in integrin‐linked kinase signaling (e.g. ITGB1, CFL1), a key pathway in cell migration and proliferation, protein translation (e.g. EIF4G1), and DNA mismatch repair (e.g. MSH2). The proteins found upregulated in the good outcome group and comprised in this network were involved in cell cycle (e.g. KRT18) and cell growth (e.g. NOP58). Although not present among the significant proteins, Akt and MAPK pathways constituted the focal point of the network, suggesting their activation based on their interactors expression levels. IPA analysis showed that differentially expressed proteins were involved in cell growth and proliferation and suggested that actors involved in such pathways may have a key role in tamoxifen resistance.
3.4. Development of a protein signature predictive of tamoxifen therapy outcome
Based on the 99 differentially abundant proteins, hierarchical clustering separated the two patient groups (Figure 4A): 20 out of 28 predicted good outcome patients were correctly classified as “Good”, while 24 out of 28 predicted poor outcome patients correctly grouped in the “Poor” cluster. After more stringent filtering (p value < 0.01) 38 proteins remained, which were included in a multivariate Cox regression model. Using a step‐down approach, we identified a 4‐protein signature that best predicted outcome to tamoxifen treatment. The signature comprised the following proteins: programmed cell death 4 (PDCD4; t test p value < 0.001), Cingulin (CGN; t test p value = 0.006), ovarian carcinoma immuno‐reactive antigen domain containing protein 1 (OCIAD1; t test p value < 0.001) and Ras‐GTPase activating protein‐binding protein 2 (G3BP2; t test p value < 0.001; Table 2 and Table 3). Based on LFQ intensity levels, OCIAD1, CGN and PDCD4 showed a relatively high abundance in good outcome patients, while G3BP2 was more highly abundant in the poor outcome group (Figure 4B). Next, patient scores of the 4‐protein predictor were plotted in a ROC curve to select a cut‐off with the highest sensitivity and specificity at predicting poor outcome (J = 0.740, area under the curve = 0.93, sensitivity = 90.6%, specificity = 83.3%; Figure 5A). The 4‐protein predictor was then validated in the test cohort through Cox regression and Kaplan–Meier analyses. In both Cox univariate and multivariate regression analysis for TTP, the 4‐protein signature was significantly correlated with outcome of tamoxifen therapy (HR = 2.44; 95% CI = 1.30 to 4.54; p value = 0.006) and multivariate (HR = 2.17; 95% CI = 1.15 to 4.17; p value = 0.017) regression analyses corrected for traditional predictive factors (Table 4). In Kaplan Meier analysis, patients with predicted poor outcome had significantly shorter TTP compared to those with a predicted good outcome (HR = 2.32; 95% CI = 1.29 to 4.17; Log‐rank p value = 0.004; Figure 5B). In the test set, sensitivity, specificity, positive predicted value (PPV), and negative predicted value (NPV) in predicting poor outcome patients were 86.7%, 41.5%, 35.1% and 89.5%, respectively.
Figure 4.
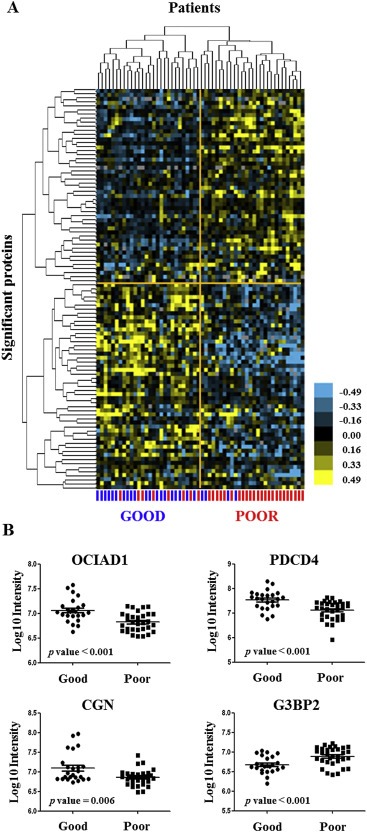
Hierarchical clustering and differential protein abundance of 4‐protein predictor. Samples in the training set (n = 56) were hierarchically clustered based on 99 differentially abundant proteins (t test p value < 0.05). Log10 intensities of differentially abundant proteins constituting the predictor for tamoxifen treatment outcome are shown in scatter dot plots. Eight poor and four good outcome patients were misclassified (A). Three out of four proteins, CGN (Uniprot accession number: Q9P2M7; p value = 0.006), OCIAD1 (Uniprot accession number: Q9NX40; p value < 0.001) and PDCD4 (Uniprot accession number: Q53EL6; p value < 0.001), had higher abundance in patients with good outcome, whereas G3BP2 (Uniprot accession number Q9UN86; p value < 0.001) was found more highly expressed in the poor outcome patient group (B).
Table Table 2.
LFQ based identification of 4 proteins in discovery and validation sets.
| Protein ID | Gene name | Molecular weight (kDa) | Peptides/Unique peptidesa | Sequence coverage/Unique sequence coverage (%/%)b | PEP scorec | |||
|---|---|---|---|---|---|---|---|---|
| Training | Test | Training | Test | Training | Test | |||
| Q9P2M7 | CGN | 136.380 | 22/22 | 40/40 | 22.8/22.8 | 39.5/39.5 | 3.63E‐133 | 7.88E‐268 |
| Q9UN86 | G3BP2 | 54.120 | 5/5 | 8/7 | 16.4/16.4 | 22.0/19.5 | 1.61E‐34 | 5.85E‐84 |
| Q9NX40 | OCIAD1 | 27.626 | 8/8 | 9/9 | 32.2/32.2 | 32.7/32.7 | 5.35E‐146 | 2.59E‐247 |
| Q53EL6 | PDCD4 | 51.735 | 19/19 | 18/18 | 49.3/49.3 | 47.8/47.8 | 1.24E‐225 | 3.23E‐281 |
Ratio between peptides and unique peptides associated to each predictor protein.
Peptides/Unique peptides sequence coverage of each protein sequence.
PEP: represents an estimation of a false identification.
Table Table 3.
Information on the 4 proteins constituting the predictor for tamoxifen therapy outcome.
| Gene name | GO cellular component | Protein name | Student t | p value |
|---|---|---|---|---|
| CGN | Cell junction | Cingulin | 3.13 | 0.006 |
| G3BP2 | Cytoplasm | Ras GTPase‐activating protein‐binding protein 2 | 3.50 | <0.001 |
| OCIAD1 | Endosome, Mitochondrion | Ovarian carcinoma immunogenic antigen domain‐containing protein 1 | 4.15 | <0.001 |
| PDCD4 | Cytoplasm, Nucleus | Programmed cell death protein 4 | 3.99 | <0.001 |
Figure 5.
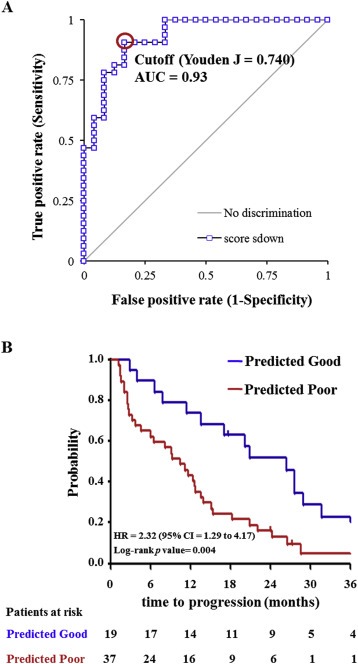
ROC curve of the training set and Kaplan–Meier curves for TTP as a function of predicted outcome in patients in the test set. Patient outcome scores from the training set were calculated based on abundance levels of the 4 predictor proteins and protein weights (i.e. Student t value). The ROC curve was generated and Youden maximum (J = 0.740) was chosen as the best discriminatory cutoff (A). Patient scores were subsequently calculated for patients in the test set, survival curves were generated for the predicted groups and differences were assessed with the Log‐rank test (B). Acronym: AUC: area under the curve; HR: hazard ratio; CI: confidence interval.
Table Table 4.
Univariate and multivariate Cox regression analysis for time to progression.
| Factors | Hazard ratio | Univariate 95% CI | p value | Hazard ratio | Multivariate 95% CI | p value |
|---|---|---|---|---|---|---|
| 4 protein predictor score | ||||||
| High | 1.00 | 1.00 | ||||
| Low | 2.44 | 1.30–4.54 | 0.006 | 2.17 | 1.15–4.17 | 0.017 |
| Age | ||||||
| ≤55 years | 1.00 | 1.00 | ||||
| >55 years | 0.44 | 0.23–0.86 | 0.017 | 0.55 | 0.28–1.08 | 0.083 |
| Disease free interval | ||||||
| ≤12 months | 1.00 | |||||
| >12 months | 0.63 | 0.30–1.31 | 0.213 | |||
| Dominant site of relapse | Overall p | |||||
| Loco‐regional | 1.00 | 0.270 | ||||
| Bone | 0.89 | 0.30–2.68 | ||||
| Visceral | 0.68 | 0.22–2.09 | ||||
| Bone and other | 0.45 | 0.14–1.40 | ||||
| PgR | ||||||
| Negative | 1.00 | |||||
| Positive | 0.57 | 0.32–1.00 | 0.052 | |||
Acronym: PgR: progesterone receptor.
3.5. Immunohistochemical assessment of PDCD4 expression and correlation with TTP
While our tissue proteomics pipeline proved to be successful in identifying and validating the 4‐protein predictor, this technology is not yet readily available in a clinical setting. Therefore, we assessed protein expression of PDCD4, G3BP2, CNG, and OCIAD1 through IHC, a technology that is routinely used in diagnostic laboratories, in an independent set of formalin‐fixed paraffin‐embedded breast cancer tissues incorporated in a TMA. Normal breast epithelium (i.e. acini and ducts) and leukocytes displayed expression of all markers except for CGN, which stained the myoepithelial cell layer only. Blood vessels displayed expression of all 4 proteins, while overall low to negative staining was displayed in the stromal compartment. Examples of comparative IHC analysis of normal breast tissue, blood vessels, leucocytes and breast carcinoma cells are displayed in Supplemental Figure 4A–B. Strong PDCD4 staining (histo‐score ≥ 30) was found to be significantly associated with longer TTP in univariate (HR = 0.75; 95% CI = 0.59 to 0.96; p value = 0.020) and multivariate Cox regression analysis (HR = 0.72; 95% CI = 0.57 to 0.92; p value = 0.009) corrected for traditional predictive factors (Table 5). PDCD4 stained tissues showing both low or high protein expression and the Kaplan–Meier curve for TTP as a function of the PDCD4 histo‐score are shown in Figure 6A and Figure 6B, respectively. In logistic regression analyses for clinical benefit or objective response, PDCD4 levels (histo‐score ≥ 30 vs. < 30) were not significantly associated with the type of response (data not shown). OCIAD1, CGN and G3BP2 stainings showed strong intensities and high quantities of stained tumor cells in the vast majority of specimens. The limited dynamic range in staining intensities proved insufficient to find a significant association of CGN, OCIAD1 and G3BP2 levels with TTP, clinical benefit or objective response (data not shown).
Table Table 5.
Univariate and multivariate Cox regression analysis for time to progression.
| Factors | Hazard ratio | Univariate 95% CI | p value | Hazard ratio | Multivariate 95% CI | p value |
|---|---|---|---|---|---|---|
| PDCD4 | ||||||
| Low | 1.00 | 1.00 | ||||
| High | 0.75 | 0.59–0.96 | 0.020 | 0.72 | 0.57–0.92 | 0.009 |
| Age | ||||||
| ≤55 | 1.00 | 1.00 | ||||
| >55 | 0.58 | 0.45–0.70 | <0.001 | 0.52 | 0.40–0.67 | <0.001 |
| Disease free interval | ||||||
| ≤12 months | 1.00 | 1.00 | ||||
| >12 months | 0.73 | 0.54–0.99 | 0.042 | 0.63 | 0.46–0.87 | 0.004 |
| Dominant site of relapse | Overall p | |||||
| Loco‐regional | 1.00 | 0.310 | ||||
| Bone | 1.39 | 0.91–2.10 | ||||
| Visceral | 1.15 | 0.73–1.81 | ||||
| Bone and other | 1.38 | 0.89–2.13 | ||||
| PgR | ||||||
| Negative | 1.00 | |||||
| Positive | 0.77 | 0.59–1.01 | 0.062 | |||
Acronym: PgR: progesterone receptor.
Figure 6.
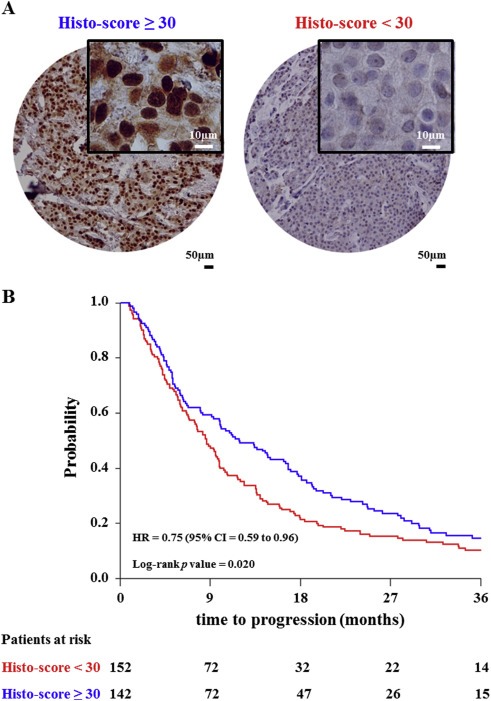
PDCD4 immunohistochemical staining of tissue micro‐array. Tissue cores showed two different staining patterns that have been evaluated by histo‐score (i.e. Histo‐score < 30 and ≥ 30), representing low and high PDCD4 protein expression (A). Patients were categorized according to histo‐score cutoff and TTP was plotted as a Kaplan–Meier curve. The Log‐rank test was used to test for differences in TTP between the two survival curves (B). Acronym: HR: hazard ratio; CI: confidence interval.
4. Discussion
About half of the recurrent ER positive breast cancer patients treated with tamoxifen show intrinsic resistance to the drug. Despite many studies describing several mechanisms associated to tamoxifen resistance and a large amount of markers associated to patient hormonal treatment outcome, there is no molecular predictor available in the clinic (Chung and Baxter, 2012; Droog et al., 2013). Furthermore, the search for biomarkers in the analysis of clinical specimen is often hindered by tissue heterogeneity, which complicates accurate measurement of tumor protein abundance. In the light of this, tissue enrichment technologies offer an invaluable tool to quantify the proteome of specific cell subpopulations. Though mechanisms of resistance encompass not only a plethora of molecular mechanisms, but also different cell types as stromal ones (den Boon et al., 2015; Jung et al., 2015), analysis of whole tissue specimens would suffer from “signal dilution” derived from protein differential expression in heterogeneous tissues. Furthermore, analysis of microdissected stroma is hindered by the presence of high‐abundance proteins (e.g. collagen family) and often needs additional protein separation methods. In this perspective, we have focused only on the epithelial tumor markers involved in tamoxifen resistance. Having successfully coupled LCM tissue enrichment with high resolution MS in a biomarker discovery pipeline (Braakman et al., 2012, 2012, 2014), we have here developed and validated a 4‐protein signature predicting outcome to tamoxifen treatment in an independent set of ER positive recurrent breast cancer.
Despite the low amount of material derived from tissue enrichment compared to whole tissue specimens, a higher number of proteins was identified and quantified in our LCM samples (training and test sets, and controls) compared to the WTL control, suggesting interference from highly abundant proteins (e.g. collagen family) in the latter. Furthermore, from our global proteomic analysis of our combined training and test sets we showed that plasma and stromal proteins contamination was minimized in the LCM derived material while proteins expressed in subcellular compartments were enriched. This allowed us to take a unique snapshot of protein abundance of breast cancer epithelial tissue and to derive markers specifically involved in tumor cell treatment resistance pathways. From a subset of commonly expressed proteins in our 112 ER‐positive breast cancer tissues we developed and validated a protein signature comprising PDCD4, CGN, OCIAD1 and G3BP2, which was capable of predicting tamoxifen treatment outcome in the test set with 86.7% sensitivity, 41.4% specificity, 35.1% PPV and 89.5% NPV and independently from traditional predictive parameters.
The selection of a large cohort of hormonal‐treatment naive patients allowed us to assess tumor protein abundance directly related to first line tamoxifen treatment without any expression changes derived from previous therapies. Furthermore, the availability of an in‐house training and a multi‐center test set enabled us to test the robustness of our predictor in a heterogeneous set of samples, reflective of differences in pathological evaluation and standard of care among medical centers. While our in‐house training set showed almost equal distribution of patient groups, the multi‐center cohort comprised a high number of good outcome patients, which could be explained by different grading systems used in local hospitals. To transfer our findings more easily to a clinical setting, we also performed IHC staining on an independent cohort of ER‐positive breast cancer tissues, which confirmed PDCD4 to be an independent predictive marker of tamoxifen sensitivity. Nevertheless, the MS based 4‐protein signature was a stronger predictor than the single marker PDCD4, emphasizing the potential of proteomic technologies in the dissection of tumor molecular pathways. Still, introduction of high resolution MS in routine clinical diagnostics remains problematic due to extensive and laborious sample preparation and relatively high costs. On the other side, targeted MS methods offer an accurate tool to detect and quantitate target analytes (i.e. peptides or metabolites) from biological specimens at a relatively lower cost, sample processing and measurement times (Grebe and Singh, 2011; Yassine et al., 2013), and would therefore constitute a more eligible technique for clinical introduction.
Pathways analysis on differentially expressed proteins showed how cell growth and proliferation pathways are key components in tamoxifen therapy response and resistance. Akt and MAPK, although not present among differentially expressed proteins, constituted the center of the molecular interaction network, showing how cell cycle progression through estrogen‐independent mechanisms can overcome tamoxifen treatment. Activation of Akt signaling has been linked to tamoxifen resistance in previous studies (Clark et al., 2002; Klinge, 2015; Nass and Kalinski, 2015), but other molecular mechanisms may be involved. In the light of this, the 4 protein signature not only is capable of discriminating patients that manifested good and poor outcome to tamoxifen treatment, but may also pinpoint other molecular mechanisms of resistance. PDCD4 is an inhibitor of protein translation, which functions both in the nucleus and the cytoplasm (Lankat‐Buttgereit and Göke, 2009). This protein has already been described as a tumor suppressor capable of inhibiting protein synthesis and gene expression by preventing the interaction of eukaryotic initiation factor (eIF) 4A1 and eIF4G, and by binding to target gene transcripts (e.g. MAP4K) in the nucleus, respectively (Biyanee et al., 2014; H. Yang et al., 2006). The nuclear localization of PDCD4 is attributed to Akt phosphorylation in a PI3K‐dependent manner (Palamarchuk et al., 2005). PDCD4 levels have also been negatively correlated to increased expression of miR‐21 in MCF‐7 cells after tamoxifen treatment (Klinge et al., 2010; Manavalan et al., 2011). CGN is involved in tight junction formation and it has been described as a potential epithelial differentiation marker in human neoplasias (Citi et al., 1991; Paschoud et al., 2007). Together with Paracingulin, CNG controls the expression of GATA‐4, contributing to down‐regulation of RhoA in cells, a key regulator of cell cycle progression that displays its function through cytoskeletal re‐organization (Guillemot et al., 2013). OCIAD1 expression has been suggested as a thyroid cancer biomarker and has been correlated to distant metastasis formation, since it was found overexpressed in metastatic ovarian cancer by MS analysis (Sengupta et al., 2008; Yang et al., 2012). Recent studies have demonstrated that OCIAD1 directly interacts with STAT3 and aids in its activation, though whether this leads to activation of the tumor suppressor pathway or the oncogenic one still remains unclear (Lee et al., 2012; Musteanu et al., 2010; Sinha et al., 2013). G3BP2 has been shown to be involved in stress granule formation along with its relative G3BP1, as well as in mRNA binding and gene expression regulation. G3BP1 protein has been shown to have a distinct role in breast cancer cell proliferation by stabilizing mRNA molecules, but its homologue G3BP2 was not associated to any of these characteristics, keeping the function of this protein still ambiguous (Kociok et al., 1999; Matsuki et al., 2013; Winslow et al., 2013). With the exception of OCIAD1, no studies observed a correlation between levels of PDCD4, G3BP2, or CGN and patient survival or therapy response in clinical cancers; nonetheless these markers may play a role in the type of response to tamoxifen in breast cancer. The anti‐proliferative effects of PDCD4 and CNG may have a synergistic role with the anti‐estrogenic action of tamoxifen, which results in the block of cell proliferation. Due to its relatively high expression in good outcome patients, OCIAD1 may activate the tumor suppressor role of STAT3 in ER positive breast cancer patients, further inhibiting proliferation. On the other hand, expression of G3BP2 could actually counteract tamoxifen action by stabilizing mRNAs of estrogen‐responsive elements as well as the ones of ER unrelated growth factors.
5. Conclusions
We hereby demonstrate that LCM coupled to high resolution LC‐MS not only enables the proteomic analysis of pure cell subpopulations, but it also provides a powerful tool for biomarker discovery studies. This allowed us to delve into the breast cancer proteome and to generate and validate a signature predictive of tamoxifen therapy outcome in recurrent ER‐positive breast cancer. In addition, a technical validation through IHC verified that PDCD4 is an independent marker associated with good outcome patients, although it is difficult to distinguish small changes in protein expression by IHC. Despite the fact that shotgun LC‐MS coupled to LCM based cell enrichment has shown to be a robust tool for biomarker discovery, time‐consuming sample preparation and relatively high costs may hinder its introduction into a clinical setting. In the light of this, targeted LC‐MS methods such as multiple reaction monitoring would be suited to fill this gap, given the fact that accurate quantification of target analytes can be performed at lower costs with reasonable optimization times and in a multiplexed fashion.
Financial support
This study was supported by the Dutch Cancer Society (KWF), EMCR2009‐4319 and the CTMM‐Breast Care project 030‐104‐06.
Conflict of interest
The authors declare no conflict of interest regarding this work.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
The authors wish to thank all collaborating local hospitals for providing formalin‐fixed paraffin‐embedded tissues, and Renée Foekens and Anita Trapman‐Jansen for assembling the TMA. Marion Meijer van Gelder is thanked for clinical data management. Proteomics data deposition to the ProteomeXchange consortium was supported by the PRIDE team, EBI, with dataset identifiers PXD000484 and PXD000485.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.07.004.
Tommaso De Marchi, Liu Ning Qing, Stingl Cristoph, Timmermans Mieke A., Smid Marcel, Look Maxime P., Tjoa Mila, Braakman Rene B.H., Opdam Mark, Linn Sabine C., Sweep Fred C.G.J., Span Paul N., Kliffen Mike, Luider Theo M., Foekens John A., Martens John W.M., Umar Arzu, (2016), 4‐protein signature predicting tamoxifen treatment outcome in recurrent breast cancer, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.07.004.
Contributor Information
Tommaso De Marchi, Email: t.demarchi@erasmusmc.nl.
Ning Qing Liu, Email: n.liu@ncmls.ru.nl.
Cristoph Stingl, Email: c.stingl@erasmusmc.nl.
Mieke A. Timmermans, Email: a.timmermans@erasmusmc.nl
Marcel Smid, Email: m.smid@erasmusmc.nl.
Maxime P. Look, Email: m.look@erasmusmc.nl
Mila Tjoa, Email: milatjoa@hotmail.com.
Rene B.H. Braakman, Email: r.braakman@erasmusmc.nl
Mark Opdam, Email: m.opdam@nki.nl.
Sabine C. Linn, Email: s.linn@nki.nl
Fred C.G.J. Sweep, Email: f.sweep@labgk.umcn.nl
Paul N. Span, Email: P.Span@rther.umcn.nl
Mike Kliffen, Email: KliffenM@maasstadziekenhuis.nl.
Theo M. Luider, Email: t.luider@erasmusmc.nl
John A. Foekens, Email: j.foekens@erasmsumc.nl
John W.M. Martens, Email: j.martens@erasmusmc.nl
Arzu Umar, Email: a.umar@erasmsumc.nl.
References
- Altman, D.G. , McShane, L.M. , Sauerbrei, W. , Taube, S.E. , 2012. Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): explanation and elaboration. PLoS Med. 9, e1001216 10.1371/journal.pmed.1001216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelen, K. , Zwart, W. , Linn, S.C. , 2012. Can predictive biomarkers in breast cancer guide adjuvant endocrine therapy?. Nat. Rev. Clin. Oncol. 9, 529–541. 10.1038/nrclinonc.2012.121 [DOI] [PubMed] [Google Scholar]
- Biyanee, A. , Ohnheiser, J. , Singh, P. , Klempnauer, K.-H. , 2014. A novel mechanism for the control of translation of specific mRNAs by tumor suppressor protein Pdcd4: inhibition of translation elongation. Oncogene. 1–9. 10.1038/onc.2014.83 [DOI] [PubMed] [Google Scholar]
- Bland, J. , Altman, D. , 1996. Measurement error proportional to the mean. BMJ Br. Med. J. 313, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman, R.B.H. , Tilanus-Linthorst, M.M. , Liu, N.Q. , Stingl, C. , Dekker, L.J.M. , Luider, T.M. , Martens, J.W.M. , Foekens, J.A. , Umar, A. , 2012. Optimized nLC-MS workflow for laser capture microdissected breast cancer tissue. J. Proteomics. 75, 2844–2854. 10.1016/j.jprot.2012.01.022 [DOI] [PubMed] [Google Scholar]
- Cardoso, F. , Harbeck, N. , Fallowfield, L. , Kyriakides, S. , Senkus, E. , 2012. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 23, (Suppl. 7) vii11–vii19. 10.1093/annonc/mds232 [DOI] [PubMed] [Google Scholar]
- Cheng, L. , Zhang, S. , 2013. Laser-assisted microdissection in translational research: theory, technical considerations, and future applications. Appl. Immunohistochem. Mol. Morphol. 21, 31–47. [DOI] [PubMed] [Google Scholar]
- Chung, L. , Baxter, R.C. , 2012. Breast cancer biomarkers: proteomic discovery and translation to clinically relevant assays. Expert Rev. Proteomics. 9, 599–614. 10.1586/epr.12.62 [DOI] [PubMed] [Google Scholar]
- Citi, S. , Amorosi, A. , Franconi, F. , 1991. Cingulin, a specific protein component of tight junctions, is expressed in normal and neoplastic human epithelial tissues. Am. J. Pathol. 138, 781–789. [PMC free article] [PubMed] [Google Scholar]
- Clark, A.S. , West, K. , Streicher, S. , Dennis, P.A. , 2002. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 1, 707–717. [PubMed] [Google Scholar]
- Cox, J. , Mann, M. , 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- Cox, J. , Neuhauser, N. , Michalski, A. , Scheltema, R.A. , Olsen, J.V. , Mann, M. , 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805. 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- Den Boon, J.A. , Pyeon, D. , Wang, S.S. , Horswill, M. , Schiffman, M. , Sherman, M. , Zuna, R.E. , Wang, Z. , Hewitt, S.M. , Pearson, R. , Schott, M. , Chung, L. , He, Q. , Lambert, P. , Walker, J. , Newton, M.A. , Wentzensen, N. , Ahlquist, P. , 2015. Molecular transitions from papillomavirus infection to cervical precancer and cancer: role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. 10.1073/pnas.1509322112 201509322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabovich, A.P. , Martínez-Morillo, E. , Diamandis, E.P. , 2014. Toward an integrated pipeline for protein biomarker development. Biochim. Biophys. Acta. 10.1016/j.bbapap.2014.09.006 [DOI] [PubMed] [Google Scholar]
- Droog, M. , Beelen, K. , Linn, S. , Zwart, W. , 2013. Tamoxifen resistance: from bench to bedside. Eur. J. Pharmacol. 1–11. 10.1016/j.ejphar.2012.11.071 [DOI] [PubMed] [Google Scholar]
- Emmert-buck, M.R. , Bonner, R.F. , Smith, P.D. , Chuaqui, R.F. , Zhuang, Z. , Goldstein, S.R. , Weiss, R.A. , Liotta, L.A. , 1996. Laser capture microdissection. Science. 274, 8–11. [DOI] [PubMed] [Google Scholar]
- Grebe, S. , Singh, R. , 2011. LC-MS/MS in the clinical laboratory–where to from here?. Clin. Biochem. Rev. 32, 5–31. [PMC free article] [PubMed] [Google Scholar]
- Guillemot, L. , Spadaro, D. , Citi, S. , 2013. The junctional proteins cingulin and paracingulin modulate the expression of tight junction protein genes through GATA-4. PLoS One. 8, e55873 10.1371/journal.pone.0055873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward, J. , Carbone, P. , 1977. Assessment of response to therapy in advanced breast cancer. Cancer. 39, 1289–1294. [DOI] [PubMed] [Google Scholar]
- Huang, B.D.W. , Lempicki, R. , 2008. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 1–43. [DOI] [PubMed] [Google Scholar]
- Huang, D.W. , Sherman, B.T. , Lempicki, R.A. , 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, W.E. , Li, C. , Rabinovic, A. , 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 8, 118–127. 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- Jung, Y.Y. , Lee, Y.K. , Koo, J.S. , 2015. Expression of cancer-associated fibroblast-related proteins in adipose stroma of breast cancer. Tumor Biol. 10.1007/s13277-015-3594-9 [DOI] [PubMed] [Google Scholar]
- Kim, M.-S. , Pinto, S.M. , Getnet, D. , Nirujogi, R.S. , Manda, S.S. , Chaerkady, R. , Madugundu, A.K. , Kelkar, D.S. , Isserlin, R. , Jain, S. , Thomas, J.K. , Muthusamy, B. , Leal-Rojas, P. , Kumar, P. , Sahasrabuddhe, N.A. , Balakrishnan, L. , Advani, J. , George, B. , Renuse, S. , Selvan, L.D.N. , Patil, A.H. , Nanjappa, V. , Radhakrishnan, A. , Prasad, S. , Subbannayya, T. , Raju, R. , Kumar, M. , Sreenivasamurthy, S.K. , Marimuthu, A. , Sathe, G.J. , Chavan, S. , Datta, K.K. , Subbannayya, Y. , Sahu, A. , Yelamanchi, S.D. , Jayaram, S. , Rajagopalan, P. , Sharma, J. , Murthy, K.R. , Syed, N. , Goel, R. , Khan, A.A. , Ahmad, S. , Dey, G. , Mudgal, K. , Chatterjee, A. , Huang, T.-C. , Zhong, J. , Wu, X. , Shaw, P.G. , Freed, D. , Zahari, M.S. , Mukherjee, K.K. , Shankar, S. , Mahadevan, A. , Lam, H. , Mitchell, C.J. , Shankar, S.K. , Satishchandra, P. , Schroeder, J.T. , Sirdeshmukh, R. , Maitra, A. , Leach, S.D. , Drake, C.G. , Halushka, M.K. , Prasad, T.S.K. , Hruban, R.H. , Kerr, C.L. , Bader, G.D. , Iacobuzio-Donahue, C.A. , Gowda, H. , Pandey, A. , 2014. A draft map of the human proteome. Nature. 509, 575–581. 10.1038/nature13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge, C.M. , 2015. miRNAs regulated by estrogens, tamoxifen, and endocrine disruptors and their downstream gene targets. Mol. Cell. Endocrinol. 10.1016/j.mce.2015.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge, C.M. , Riggs, K.A. , Wickramasinghe, N.S. , Emberts, C.G. , McConda, D.B. , Barry, P.N. , Magnusen, J.E. , 2010. Estrogen receptor alpha 46 is reduced in tamoxifen resistant breast cancer cells and re-expression inhibits cell proliferation and estrogen receptor alpha 66-regulated target gene transcription. Mol. Cell. Endocrinol. 323, 268–276. 10.1016/j.mce.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kociok, N. , Esser, P. , Unfried, K. , Parker, F. , Schraermeyer, U. , Grisanti, S. , Toqué, B. , Heimann, K. , 1999. Upregulation of the RAS-GTPase activating protein (GAP)-binding protein (G3BP) in proliferating RPE cells. J. Cell. Biochem. 74, 194–201. [PubMed] [Google Scholar]
- Kondo, T. , 2014. Inconvenient truth: cancer biomarker development by using proteomics. Biochim. Biophys. Acta. 1844, 861–865. 10.1016/j.bbapap.2013.07.009 [DOI] [PubMed] [Google Scholar]
- Lankat-Buttgereit, B. , Göke, R. , 2009. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol. Cell. 101, 309–317. 10.1042/BC20080191 [DOI] [PubMed] [Google Scholar]
- Lee, J. , Kim, J.C.K. , Lee, S.-E. , Quinley, C. , Kim, H. , Herdman, S. , Corr, M. , Raz, E. , 2012. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 287, 18182–18189. 10.1074/jbc.M111.328831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, N.Q. , Braakman, R.B.H. , Stingl, C. , Luider, T.M. , Martens, J.W.M. , Foekens, J.A. , Umar, A. , 2012. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J. Mammary Gland Biol. Neoplasia. 17, 155–164. 10.1007/s10911-012-9252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, N.Q. , Stingl, C. , Look, M.P. , Smid, M. , Braakman, R.B.H. , De Marchi, T. , Sieuwerts, A.M. , Span, P.N. , Sweep, F.C.G.J. , Linderholm, B.K. , Mangia, A. , Paradiso, A. , Dirix, L.Y. , Van Laere, S.J. , Luider, T.M. , Martens, J.W.M. , Foekens, J. a , Umar, A. , 2014. Comparative proteome analysis revealing an 11-protein signature for aggressive triple-negative breast cancer. J. Natl. Cancer Inst. 10.1093/jnci/djt376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan, T.T. , Teng, Y. , Appana, S.N. , Datta, S. , Kalbfleisch, T.S. , Li, Y. , Klinge, C.M. , 2011. Differential expression of microRNA expression in tamoxifen-sensitive MCF-7 versus tamoxifen-resistant LY2 human breast cancer cells. Cancer Lett. 313, 26–43. 10.1016/j.canlet.2011.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki, H. , Takahashi, M. , Higuchi, M. , Makokha, G.N. , Oie, M. , Fujii, M. , 2013. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells. 18, 135–146. 10.1111/gtc.12023 [DOI] [PubMed] [Google Scholar]
- Megger, D. , Bracht, T. , Meyer, H. , Sitek, B. , 2013. Label-free quantification in clinical proteomics. Biochim. Biophys. Acta. 1834, 1581–1590. [DOI] [PubMed] [Google Scholar]
- Milani, A. , 2014. Overcoming endocrine resistance in metastatic breast cancer: current evidence and future directions. World J. Clin. Oncol. 5, 990 10.5306/wjco.v5.i5.990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musteanu, M. , Blaas, L. , Mair, M. , Schlederer, M. , Bilban, M. , Tauber, S. , Esterbauer, H. , Mueller, M. , Casanova, E. , Kenner, L. , Poli, V. , Eferl, R. , 2010. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology. 138, 1003–1011. 10.1053/j.gastro.2009.11.049 e1–5 [DOI] [PubMed] [Google Scholar]
- Nass, N. , Kalinski, T. , 2015. Tamoxifen resistance: from cell culture experiments towards novel biomarkers. Pathol. Res. Pract. 211, 189–197. 10.1016/j.prp.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Palamarchuk, A. , Efanov, A. , Maximov, V. , Aqeilan, R.I. , Croce, C.M. , Pekarsky, Y. , 2005. Akt phosphorylates and regulates Pdcd4 tumor suppressor protein. Cancer Res. 65, 11282–11286. 10.1158/0008-5472.CAN-05-3469 [DOI] [PubMed] [Google Scholar]
- Paschoud, S. , Bongiovanni, M. , Pache, J.-C. , Citi, S. , 2007. Claudin-1 and claudin-5 expression patterns differentiate lung squamous cell carcinomas from adenocarcinomas. Mod. Pathol. 20, 947–954. 10.1038/modpathol.3800835 [DOI] [PubMed] [Google Scholar]
- Sengupta, S. , Michener, C.M. , Escobar, P. , Belinson, J. , Ganapathi, R. , 2008. Ovarian cancer immuno-reactive antigen domain containing 1 (OCIAD1), a key player in ovarian cancer cell adhesion. Gynecol. Oncol. 109, 226–233. 10.1016/j.ygyno.2007.12.024 [DOI] [PubMed] [Google Scholar]
- Sereni, M.I. , Baldelli, E. , Gambara, G. , Deng, J. , Zanotti, L. , Bandiera, E. , Bignotti, E. , Ragnoli, M. , Tognon, G. , Ravaggi, A. , Meani, F. , Memo, M. , Angioli, R. , Liotta, L. a , Pecorelli, S.L. , Petricoin, E. , Pierobon, M. , 2015. Functional characterization of epithelial ovarian cancer histotypes by drug target based protein signaling activation mapping: implications for personalized cancer therapy. Proteomics. 15, 365–373. 10.1002/pmic.201400214 [DOI] [PubMed] [Google Scholar]
- Sinha, A. , Khadilkar, R.J. , S, V.K. , Roychowdhury Sinha, A. , Inamdar, M.S. , 2013. Conserved regulation of the Jak/STAT pathway by the endosomal protein asrij maintains stem cell potency. Cell Rep. 4, 649–658. 10.1016/j.celrep.2013.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno, J.A. , Côté, R.G. , Csordas, A. , Dianes, J.A. , Fabregat, A. , Foster, J.M. , Griss, J. , Alpi, E. , Birim, M. , Contell, J. , O'Kelly, G. , Schoenegger, A. , Ovelleiro, D. , Pérez-Riverol, Y. , Reisinger, F. , Ríos, D. , Wang, R. , Hermjakob, H. , 2013. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069. 10.1093/nar/gks1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, A. , Lorenz, K. , Horneffer, V. , Hüttmann, G. , von Smolinski, D. , Gebert, A. , 2007. Mechanisms of laser-induced dissection and transport of histologic specimens. Biophys. J. 93, 4481–4500. 10.1529/biophysj.106.102277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker, J.R. , Lin, C. , Kennedy, J. , Hou, L. , Trute, M. , Sokal, I. , Yan, P. , Schoenherr, R.M. , Zhao, L. , Voytovich, U.J. , Kelly-Spratt, K.S. , Krasnoselsky, A. , Gafken, P.R. , Hogan, J.M. , Jones, L.A. , Wang, P. , Amon, L. , Chodosh, L.A. , Nelson, P.S. , McIntosh, M.W. , Kemp, C.J. , Paulovich, A.G. , 2011. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 29, 625–634. 10.1038/nbt.1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm, M. , Schlegl, J. , Hahne, H. , Gholami, A.M. , Lieberenz, M. , Savitski, M.M. , Ziegler, E. , Butzmann, L. , Gessulat, S. , Marx, H. , Mathieson, T. , Lemeer, S. , Schnatbaum, K. , Reimer, U. , Wenschuh, H. , Mollenhauer, M. , Slotta-Huspenina, J. , Boese, J.-H. , Bantscheff, M. , Gerstmair, A. , Faerber, F. , Kuster, B. , 2014. Mass-spectrometry-based draft of the human proteome. Nature. 509, 582–587. 10.1038/nature13319 [DOI] [PubMed] [Google Scholar]
- Winslow, S. , Leandersson, K. , Larsson, C. , 2013. Regulation of PMP22 mRNA by G3BP1 affects cell proliferation in breast cancer cells. Mol. Cancer. 12, 156 10.1186/1476-4598-12-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, B.J. , 2010. Combining laser capture microdissection and proteomics: methodologies and clinical applications. Proteomics Clin. Appl. 4, 116–123. 10.1002/prca.200900138 [DOI] [PubMed] [Google Scholar]
- Yang, F. , Foekens, J.A. , Yu, J. , Sieuwerts, A.M. , Timmermans, M. , Klijn, J.G. , Atkins, D. , Wang, Y. , Jiang, Y. , 2006. Laser microdissection and microarray analysis of breast tumors reveal ER-alpha related genes and pathways. Oncogene. 25, 1413–1419. 10.1038/sj.onc.1209165 [DOI] [PubMed] [Google Scholar]
- Yang, H. , Matthews, C. , Clair, T. , 2006. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol. Cell. Biol. 10.1128/MCB.26.4.1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, A.-H. , Chen, J.-Y. , Lee, C.-H. , Chen, J.-Y. , 2012. Expression of NCAM and OCIAD1 in well-differentiated thyroid carcinoma: correlation with the risk of distant metastasis. J. Clin. Pathol. 65, 206–212. 10.1136/jclinpath-2011-200416 [DOI] [PubMed] [Google Scholar]
- Yassine, H. , Borges, C.R. , Schaab, M.R. , Billheimer, D. , Stump, C. , Reaven, P. , Lau, S.S. , Nelson, R. , 2013. Mass spectrometric immunoassay and MRM as targeted MS-based quantitative approaches in biomarker development: potential applications to cardiovascular disease and diabetes. Proteomics Clin. Appl. 7, 528–540. 10.1002/prca.201200028 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data