

Abstract
Background:Chk1 inhibitors are currently under clinical evaluation as single agents and in combination with cytotoxic chemotherapy. Understanding determinants of sensitivity and novel combinations is critical for further clinical development.
Potentiation of mTOR inhibitor cytotoxicity by the Chk1 inhibitor V158411 was determined in p53 mutant colon cancer cells. DNA damage response, expression levels of repair proteins, cell cycle effects and the contribution of alternative DSB repair pathways were further evaluated by western blotting and high content analysis.
Results:mTOR inhibitors AZD8055, RAD‐001, rapamycin and BEZ235 induced synergistic cytotoxicity with the Chk1 inhibitor V158411 in p53 mutant colon cancer cells. Reduced FANCD2, RAD51 and RPA70, core proteins in homologous recombination repair (HRR) and interstrand crosslink repair (ICLR), following inhibition of mTOR was associated with increased V158411 induced DSBs and caspase 3‐independent cell death. Dual mTOR and Chk1 inhibition activated DNA‐PKcs. Cells defective in DNA‐PKcs exhibited increased resistance to V158411 with Chk1 expression closely correlated to DNA‐PKcs expression in various types of cancer.
Conclusions:Down regulation of proteins involved in HRR or ICLR by mTOR inhibitors is associated with increased sensitivity of human tumours to Chk1 inhibitors such as V158411. High levels of DNA‐PKcs may be a potential biomarker to stratify patients to Chk1 inhibitor therapy alone or in combination with mTOR inhibitors.
Keywords: Chk1, mTOR, DNA-PKcs, V158411, Double strand break repair
Highlights
mTOR inhibitors induce synergistic cytotoxicity with V158411.
mTOR inhibition increases V158411 induced DNA DSBs.
Mechanism through regulation of HR and ICL repair proteins by mTOR.
DNA‐PKcs overexpressing cells were more sensitive to Chk1 inhibition.
Correlation between CHEK1 and PRKDC (DNA‐PKcs) mRNA levels in human tumour samples.
1. Introduction
The DNA damage response (DDR) is the result of adaptation to the high level of DNA damage sustained by the genome from endogenous and environmental sources on a daily basis. Activation of the DDR results in a number of cellular responses including checkpoint activation and cell cycle arrest, initiation of DNA repair, regulation of transcription, and apoptosis (Bucher and Britten, 2008; Dai and Grant, 2010; Liu et al., 2000; Smith et al., 2010). The checkpoint kinase Chk1 is a central, key component of the DDR and is activated by phosphorylation on serine 317 and serine 345 by the ATR and ATM kinases (Niida et al., 2007; Tapia‐Alveal et al., 2009) and auto‐phosphorylation on serine 296 (Ng et al., 2004). Activation is in response to DNA damage in the form of single‐stranded regions of DNA and double‐strand breaks (DSB) induced by both endogenous (e.g. DNA replication stress) or exogenous (e.g. genotoxic agents or radiation) sources. Chk1 activation results in the inhibition/degradation of the Cdc25 family of phosphatases. Cell cycle progression is therefore prevented through the maintenance of inhibitory phosphorylation (Y15/T14) on Cdk1 and Cdk2. Biochemical and genetic studies have demonstrated Chk1 to be essential and indispensable for the S‐ and G2/M checkpoints (Cho et al., 2005; Liu et al., 2000). The G1/S checkpoint is dysfunctional in most human cancers, e.g. by p53 mutation or functional inactivation (Massague, 2004), rendering cancer cells reliant on Chk1/Chk2 for checkpoint activation in the presence of endogenous or exogenous DNA damage.
With the aim of selectively exploiting tumour‐specific G1 checkpoint dysfunction, Chk1 inhibitors have been developed (reviewed in (Chen et al., 2012; Garrett and Collins, 2011)). The pre‐clinical and clinical development of these inhibitors has focussed on their ability to potentiate the cytotoxicity of genotoxic chemotherapy drugs (such as gemcitabine, irinotecan or cisplatin) or ionising radiation. All of these agents induce DNA damage and activate the DDR resulting in cell cycle arrest. Inhibition of Chk1 following genotoxic stress induced by these agents results in checkpoint abrogation, inhibition of DNA repair and induction of cell death in cells, particularly in those with a defective p53 response. This approach is currently being evaluated in the clinic in a range of Phase I and II trials.
In addition to its role in the DDR, Chk1 has been demonstrated to be important for replication origin firing (Ge and Blow, 2010; Maya‐Mendoza et al., 2007; Petermann et al., 2010), high rates of replication fork progression and replication fork stabilization (Smith‐Roe et al., 2013; Syljuasen et al., 2005). This evidence implicating a role for Chk1 in the cell‐cycle and DNA replication in the absence of exogenous DNA damage suggests combining a Chk1 inhibitor with alternative, molecularly targeted therapeutic agents may be a rational therapeutic option. Synergism with Chk1 inhibitors (UCN‐01, LY2603618 and PF‐477736) or a pan Chk1/Chk2 inhibitor (AZD7762) has so far been observed with MEK inhibitors in glioblastoma (Tang et al., 2012a) and cytokinetically quiescent multiple myeloma (Pei et al., 2011); with PARP inhibitors in breast cancer (Mitchell et al., 2010; Shibata et al., 2011; Tang et al., 2012a); with Src family kinase inhibitors in glioblastoma (Tang et al., 2012b), multiple myeloma (Dai et al., 2008, 2011) and breast cancer (Mitchell et al., 2011); with farnesyl transferase inhibitors in leukaemia and myeloma (Dai et al., 2005); and with the mTOR inhibitor rapamycin in leukaemia (Hahn et al., 2005). Chk1 inhibitors have demonstrated single agent activity in cancers harbouring defects in DNA repair pathways or with high levels of replicative stress including neuroblastoma (Cole et al., 2011), melanoma (Brooks et al., 2013), leukaemia and lymphoma (Bryant et al., 2014b; Ferrao et al., 2012; Murga et al., 2011), breast cancer (Bryant et al., 2014a; Shibata et al., 2011), and cell lines defective in components of the Fanconi's anaemia DNA repair pathway (Chen et al., 2009).
V158411 is a potent, cell selective inhibitor of Chk1 that potentiates the cytotoxicity of a range of DNA damaging cancer therapeutic agents, such as gemcitabine and camptothecin, in p53 mutant cancer cell lines (Rawlinson and Massey, 2014). We report here for the first time the remarkable synergy between V158411 and a variety of mTOR inhibitors in the cytotoxicity to models of human solid tumours and the surprising observation that high expression of DNA‐PKcs, a key component of the DNA DSB repair pathway, non‐homologous end‐joining (NHEJ), confers sensitivity to V158411.
2. Materials and methods
2.1. Cell lines and cell culture
HT29, SW620 and Colo205 cell lines were purchased from the American Type Culture Collection (ATCC), established as a low passage cell bank and then routinely passaged in our laboratory for less than 3 months after resuscitation. These were routinely cultured in DMEM containing 10% FCS and 1% penicillin/streptomycin at 37 °C in a normal humidified atmosphere supplemented with 5% CO2. AA8, V3 and xrs‐6 cells were obtained from Keith Caldecott at the Genome stability unit at University of Sussex. M059J, DNA‐PKcs‐deficient human glioblastoma cells (Anderson et al., 2001), were grown in DMEM supplemented with 10% FBS. M059J‐Fus‐1 (M059J transfected with a portion of chromosome 8 carrying the DNA‐PKcs gene; (Virsik‐Kopp et al., 2004)) cells were cultured in media with 400 μg/ml G418. Cells were authenticated by STR profiling (LGC Standards, Teddington UK).
2.2. Compounds
Solid stocks were purchased from the indicated suppliers and prepared as concentrated stock solutions in the appropriate solvent: gemcitabine (Apin Chemicals Inc), 20 mM in H2O; BEZ235 (Selleckchem), 2 mM in DMSO; rapamycin (LC Laboratories), 5 mM in DMSO; AZD6244 (Selleckchem), 20 mM in DMSO; sorafenib (Selleckchem), 20 mM in DMSO; sunitinib (Selleckchem), 20 mM in DMSO; AZD8055 (Selleckchem), 5 mM in DMSO; RAD‐001 (Selleckchem), 5 mM in DMSO; BEZ235 (Selleckchem), 2 mM in DMSO. V158411 was from Vernalis Research and prepared as a 20 mM DMSO stock. Compounds were serially diluted in DMSO to 500× or 1000× then to 5× or 10× in complete media before addition to cells to yield a 1× final concentration.
2.3. Potentiation assays
5000 cells per well were seeded in 96‐well plates and incubated overnight. Cells were treated with a 10‐point titration of cytotoxic chemotherapeutic agent in the presence of a fixed concentration of Chk1 inhibitor for 72 h. The effect on cell proliferation was determined using CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS, Promega) and read on a Victor plate reader (PerkinElmer).
2.4. Anchorage independent growth assays
1500 cells/well in 0.4% low melting point agarose (SeaPlaque, Lonza) in complete media were plated on to 96‐well plates coated with 0.8% low melting point agarose in complete media. Wells were subsequently overlaid with complete media containing cytotoxic chemotherapeutic agents and Chk1 inhibitor. Following incubation for 168 h, cell viability was determined using CellTiter Blue (Promega) and fluorescence determined using a Victor plate reader (PerkinElmer).
2.5. Spheroid growth assays
Multi‐cellular tumour spheroid assays were preformed essentially as described previously. 1000 HT29 cells/well were seeded in 96‐well round bottomed ultra‐low attachment microplates (Corning Costar), centrifuged at 1000× g for 3 min and spheroids formed for 72 h. Spheroid cell viability after incubation for 168 h was determined using CellTiter‐Glo Luminescent Cell Viability Assay (Promega).
2.6. Clonogenic survival assay
50 to 10,000 cells were plated per well of a 6 well plate and allowed to attach for 24 h. Cells were subsequently treated with V158411 for 24 h then media removed, cells washed and fresh, drug free media added. Cells were subsequently incubated for 8–21 days then colonies fixed with Carnoy's fixative and stained with 1% crystal violet. Colonies were counted on a G‐BOX (Syngene) with viable colonies determined as those containing >50 cells.
2.7. Calculation of drug synergy
Combination Index (CI) values were calculated using CalcuSyn software (Biosoft, Cambridge, UK) based on the median‐effect principle of Chou and Talay (Chou, 2006, 2010) with a constant‐ratio design for the combination assay.
2.8. Immunoblotting
Antibodies against Chk1, pChk1 (S317), pChk1 (S345), pChk2 (T68), pChk2 (S516), Chk2, pH2AX (S139), PARP, cleaved PARP, cleaved caspase‐3, p70S6K (T389), p4EBP1 (S65), p4EBP1 (T37/46), pRPS6 (S240/244), LC3B, pAKT (S473), pMEK1/2 (S217/221), pJNK (T183/Y185), DNA‐PKcs, pDNA‐PKcs (S2056), and RPA70 were purchased from Cell Signaling Technologies, caspase‐2 from Emdmillipore and pChk1 (S296), FANCD2, FANCF and RAD51 from Abcam.
Cells were washed once with PBS and lysed in RIPA buffer containing protease and phosphatase inhibitor cocktails (Roche). Protein concentration was determined using a BCA kit (Pierce). Equal amounts of lysate were separated by SDS‐PAGE and western blot analysis conducted using the antibodies indicated above. Densitometric analysis was conducted with ImageJ software (NIH) and normalized to actin expression levels.
2.9. High‐content cell cycle analysis
Cells were labelled with 10 μM EdU for 1 h immediately prior to fixation with 3.7% paraformaldehyde in PBS at room temperature for 15 min. Cells were washed twice in PBS then twice in 3% BSA in PBS before permeabilisation with 0.5% Triton X100 in PBS for 20 min at room temperature. Cells were washed twice with 3% BSA in PBS before incorporated EdU was labelled with Alexa488 using a Click‐iT EdU labelling kit (LifeTechnologies). Following blocking for 30 min with 5% normal goat serum in PBS, cells were incubated with an anti‐pHH3 (S10) primary antibody (#9706, Cell Signaling Technologies) diluted 1:400 in antibody dilution buffer (1% BSA, 0.3% Triton X100 in PBS) at 4 °C for 16 h. Cells were washed with PBS then incubated with an Alexa546‐labelled secondary antibody (1:500, LifeTechnologies) and Hoechst 33342 (1 μg/ml) in antibody dilution buffer at room temperature for 60 min. Following washing with PBS, cells were imaged with an Operetta high content imaging system (PerkinElmer) at 10× magnification and analysed using Harmony software (PerkinElmer).
2.10. Mitotic index
Cells were fixed in 3.7% paraformaldehyde in PBS at room temperature for 15 min, washed with PBS, blocked with 5% normal goat serum in 0.3% Triton X100 in PBS for 1 h room temperature then incubated with an anti‐pHH3 (S10) primary antibody (#9706, Cell Signaling Technologies) diluted 1:400 in antibody dilution buffer at 4 °C for 16 h. Cells were washed with PBS then incubated with an Alexa546‐labelled secondary antibody (1:500, LifeTechnologies) and Hoechst 33342 (1 μg/ml) in antibody dilution buffer at room temperature for 60 min. Following washing with PBS, cells were imaged with an Operetta high content imaging system (PerkinElmer) at 10× magnification and analysed using Harmony software (PerkinElmer).
2.11. pH2AX high content analysis
Following compound treatment, cells were fixed in 3.7% paraformaldehyde in PBS at room temperature for 15 min, washed with PBS, blocked with 5% normal goat serum in 0.3% Triton X100 in PBS for 1 h room temperature then incubated with an anti‐pH2AX (S139) primary antibody (#9718, Cell Signaling Technologies) diluted 1:800 in antibody dilution buffer at 4 °C for 16 h. Cells were washed with PBS then incubated with an Alexa647‐labelled secondary antibody (1:500, LifeTechnologies) and Hoechst 33342 (1 μg/ml) in antibody dilution buffer at room temperature for 60 min. Following washing with PBS, cells were imaged with an Operetta high content imaging system (PerkinElmer) at 10× magnification and analysed using Harmony software (PerkinElmer).
2.12. High content analysis of cytotoxicity and apoptosis
2.12.1. Apoptosis
Following compound treatment, cells were fixed in 3.7% paraformaldehyde in PBS at room temperature for 15 min, washed with PBS, blocked with 5% normal goat serum in 0.3% Triton X100 in PBS for 1 h room temperature then incubated with an anti‐cleaved caspase‐3 primary antibody (#9664, Cell Signaling Technologies) diluted 1:400 in antibody dilution buffer at 4 °C for 16 h. Cells were washed with PBS then incubated with an Alexa647‐labelled secondary antibody (1:500, LifeTechnologies) and Hoechst 33342 (1 μg/ml) in antibody dilution buffer at room temperature for 60 min. Following washing with PBS, cells were imaged with an Operetta high content imaging system (PerkinElmer) at 10× magnification and analysed using Harmony software (PerkinElmer).
2.12.2. Dead cell determination
Unfixed cells were stained with NucGreen DEAD ready probes reagent (LifeTechnologies) and Hoechst 33342 for 60 min at 37 °C then fixed with 3.7% paraformaldehyde in PBS for 15 min. Dead cells were imaged using an Operetta high content imaging system with the Hoechst and Alexa488 filter sets and 10× objective.
2.12.3. Mitochondrial membrane potential (MOMP)
Unfixed cells were simultaneously labelled with Miototracker Orange CMTMRos (0.5 μM, LifeTechnologies), Mitotracker Deep Red (0.5 μM, LifeTechnologies) and Hoechst 33342 for 60 min at 37 °C. Cells were washed, fixed in 3.7% paraformaldehyde and imaged using an Operetta high content imaging system with the Hoechst, Alexa546 and Alexa647 filter sets and 20× objective. MOMP was calculated as the ratio between Alexa546 and Alexa647 total cytoplasmic cell staining.
2.12.4. Nuclear abnormalities
Following compound treatment, cells were fixed with 3.7% paraformaldehyde then labelled with Hoechst 33342 for 45 min at room temperature. Nuclei were imaged using an Operetta high content imaging system with the Hoechst filter sets and 10× objective.
2.13. Analysis of CHEK1 and PRKDC gene expression in tumour samples
CHEK1 and PRKDC gene expression levels were compared from the following datasets from the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/projects/geo/): breast (GSE29431 and GSE65194), ovarian (GSE26712), NSCLC (GSE18842), HCC (GSE6764) and colon (GSE13294). Data for individual genes of interest was analysed using the online GEO2R tool. Background signal intensity was subtracted and data normalised to the housekeeping gene HPRT1. Where more than one probe was associated with a single gene the average expression across the probes for the gene of interest was used.
2.14. Statistical analysis
Results were analysed using a Student's t‐Test tool within the data analysis package provided by Microsoft Excel.
3. Results
3.1. V158411 exhibits synergistic combinatorial activity with mTOR inhibitors in colon cancer cell lines
In a small, preliminary screen of kinase inhibitors, we identified combinatorial activity between the Chk1 inhibitor V158411 and the mTOR inhibitor rapamycin in the p53 mutant colon cancer cell lines HT29, SW620 and Colo205 growing anchorage dependently, anchorage independently or as multi‐cellular tumour spheroids (Figure S1A). V158411 reduced the rapamycin GI50 by between 3.2 and 9.0‐fold in all the models tested with the exception of Colo205 cells growing anchorage independently (Figure 1A). We therefore quantified the degree of synergy between V158411 and additional mTOR inhibitors including the ATP‐competitive, mTORC1/2 selective inhibitor AZD8055 (Chresta et al., 2010), the mTORC1 rapalog inhibitors rapamycin and RAD‐001, and the ATP‐competitive, PI3‐kinase/mTOR inhibitor BEZ235 (Maira et al., 2008) using Bliss analysis (Bliss, 1939). This compares the effect of two agents when combined with the theoretical effect if the two agents are additive. Synergy was observed between all four inhibitors of mTOR and V158411 (Figure S1B). The concentration range of mTOR inhibitor over which synergy was observed was quite broad. In comparison however, synergy was observed at a single concentration of V158411, 0.4 μM, suggesting that mTOR inhibition potentiated the cytotoxicity of V158411 not vice versa. Likewise, determination of synergy using the median‐effect principle of Chou and Talay revealed synergy between V158411 and AZD8055, RAD‐001 and BEZ235 (CI values < 1.0; Figure S1C). Synergy was more marked at lower effective dose combinations. A CI value for the combination of V158411 with rapamycin could not be calculated due to the shape of the rapamycin single agent dose response curve. Treatment of HT29 cells with V158411 in combination with an mTOR inhibitor or gemcitabine for 72 or 168 h significantly (P < 0.05) increased growth inhibition compared to each agent alone (Figure 1B).
Figure 1.
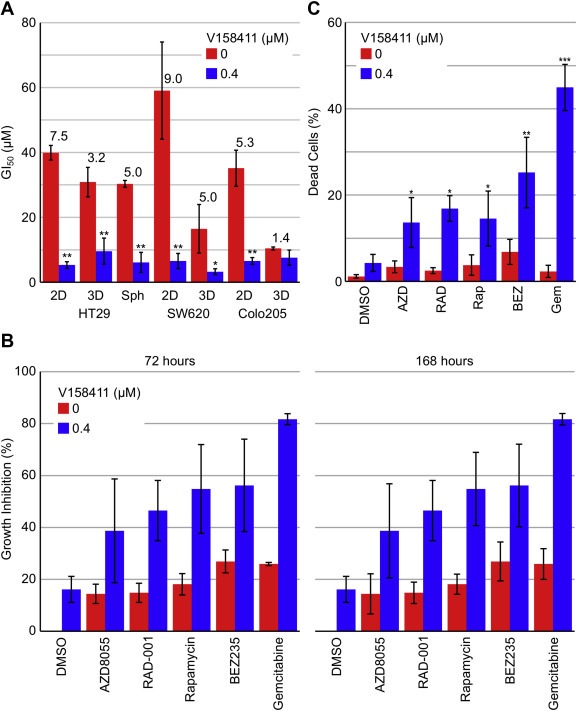
The Chk1 inhibitor V158411 and the mTOR inhibitors AZD8055, RAD‐001, rapamycin and BEZ235 demonstrate synergistic combinatorial activity in colon cancer cell lines. (A) GI50 and combination GI50 (cGI50) values in HT29, SW620 and Colo205 cancer cells grown anchorage dependently (2D), anchorage independently (3D) or as multicellular tumour spheroids (Sph) following 72 or 168 h treatment with rapamycin and 400 nM V158411. Pf values are indicated above the bars and were calculated from the average GI50 and cGI50 where Pf equals average GI50/average cGI50. *, P < 0.05; **, P < 0.01. (B) Growth inhibition values for 0.04 μM AZD8055, RAD‐001, rapamycin or BEZ235, or 0.01 μM gemcitabine in combination with 0 or 0.4 μM V158411 for 72 or 168 h. Values are the average of 3 determinations ± SD. (C) HT29 cells were treated with 0.1 μM AZD8055, RAD‐001, rapamycin, BEZ235 or gemcitabine in combination with 0 or 0.4 μM V158411. Loss of membrane integrity was assessed using NucGreen DEAD stain by high content analysis following 72 h compound treatment.
The mechanism by which mTOR inhibition enhanced V158411 induced growth inhibition was further evaluated. Treatment of HT29 cells with V158411 and an mTOR inhibitor resulted in an increase in cell death as measured by loss of membrane integrity (Figure 1C) and an increase in mitochondrial mass and a decrease in mitochondrial membrane potential (Figure S2A) and was independent of caspase‐3 activation (Figure S2B and S2C). Combination treatment increased the fraction of cells (P < 0.01) with abnormal nuclei (Figure S2D and S2E) compared to single agent treatment. These abnormal nuclei tended to demonstrate altered nuclear morphology, increased nuclear intensity and increased variability in intra‐nuclei staining.
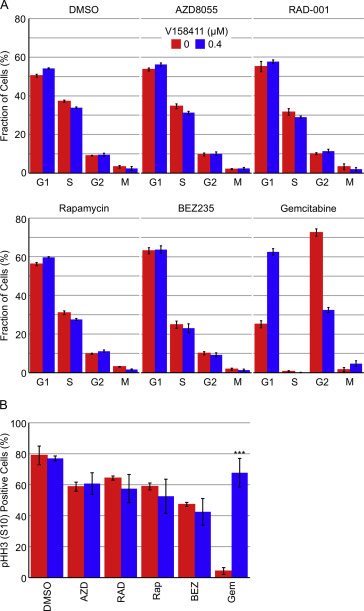
Inhibition of Chk1 or mTOR for 48 h did not substantially alter the cell cycle profile of HT29 cells. The cell cycle profiles of HT29 cells treated with an mTOR inhibitor plus V158411 were not different from HT29 cells treated with mTOR inhibitor alone (Figure S3A). In comparison, treatment with the antimetabolite/nucleoside analogue, gemcitabine, induced an increase in cells with large nuclei and cell cycle arrest in G2. In combination with V158411, G2 cell cycle arrest was abolished and cells passed through mitosis and accumulated in G1. To further understand the cell cycle effects of V158411/mTOR inhibitor combination treatment, we utilised a nocodazole‐trap mitotic index assay. Treatment of HT29 cells with gemcitabine resulted in cell cycle arrest and prevention of entry into mitosis. This block to mitotic entry induced by gemcitabine is abrogated when cells are subsequently treated with V158411 (Figure S3B). Cells treated with gemcitabine then V158411 enter mitosis where they are subsequently trapped by nocodazole with approximately 68% of the cells now pHH3 (S10) positive. Treatment of HT29 cells with V158411 alone did not prevent cells accumulating in mitosis in the presence of nocodazole. However, mTOR inhibitors as single agents reduced the number of cells accumulating in mitosis by 20–30%. In combination with V158411 this block to mitotic entry induced by mTOR inhibition was not reversed (Figure S3B).
3.2. mTOR/DDR signalling pathway changes
In combination with DNA damaging cytotoxic agents such as gemcitabine or camptothecin, V158411 increases the activation of the DDR (as determined by increased phosphorylation of Chk1 at S317 and S345 and Chk2 at T68) and increased phosphorylation of H2AX at S139 indicative of increased DNA DSB and/or collapsed replication forks. Treatment of HT29 cells with all four mTOR inhibitors decreased phosphorylation of 70S6K, RPS6 and 4EBP1, as expected, and in the case of the two rapalogs, activated AKT through the known feedback loop (Figure 2A). V158411 as a single agent did not substantially affect the mTOR signalling pathway. Exposure of HT29 cells to an mTOR inhibitor alone did not result in DDR activation or increased DNA DSB (Figure 2B) and the combination of an mTOR inhibitor with V158411 (i) did not affect checkpoint activation by V158411 alone or (ii) further affect signalling events downstream of mTOR that may modulate the response of cells other than a modest attenuation of the inhibition of RPS6 phosphorylation. There was no increase in the induction of autophagy or changes to the AKT feedback loop. We observed no changes to MEK or JNK phosphorylation, both of which have been suggested to be modulated by Chk1/mTOR inhibitor combination treatment.
Figure 2.
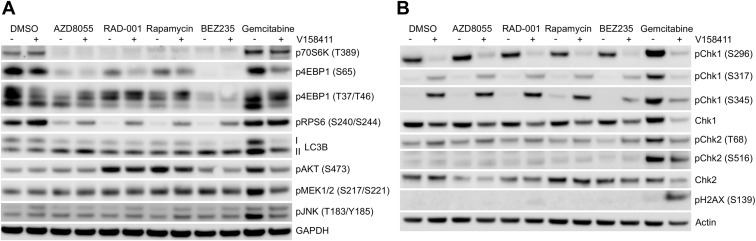
mTOR/DDR signalling pathway changes. HT29 cells were treated with 0.1 μM of the mTOR inhibitors AZD8055, RAD‐001, rapamycin or BEZ235, or gemcitabine in combination with 0 or 0.4 μM of the Chk1 inhibitor V158411 for 48 h. Expression levels of proteins involved in mTOR signalling (A) or DNA damage signalling (B) were determined by immunoblotting.
3.3. mTOR inhibition increases V158411 induced DNA double strand breaks
Cytotoxic chemotherapeutic agents such as gemcitabine and camptothecin induce DNA damage as evidenced by an increase in phosphorylation of H2AX at S139 (γH2AX); a marker of stalled replication forks and DNA DSB. Inhibition of Chk1 with small molecule inhibitors such as V158411 result in increased H2AX phosphorylation. Treatment of HT29 cells with gemcitabine but not an mTOR inhibitor caused a time dependent increase in the fraction of cells staining positive for γH2AX. V158411, as a single agent, induced γH2AX in a small fraction of cells; 8% and 7% following 24 or 48 h treatment respectively. However, V158411 increased the number of gemcitabine‐induced γH2AX positive cells to from 42% to 93% (P < 0.01) (Figures 3A and S4). Combination of V158411 with an mTOR inhibitor significantly increased the fraction of cells staining positive for γH2AX by 2–3 fold (P < 0.01) with RAD‐001 exhibiting the greatest potentiation of γH2AX (Figure 3A).
Figure 3.
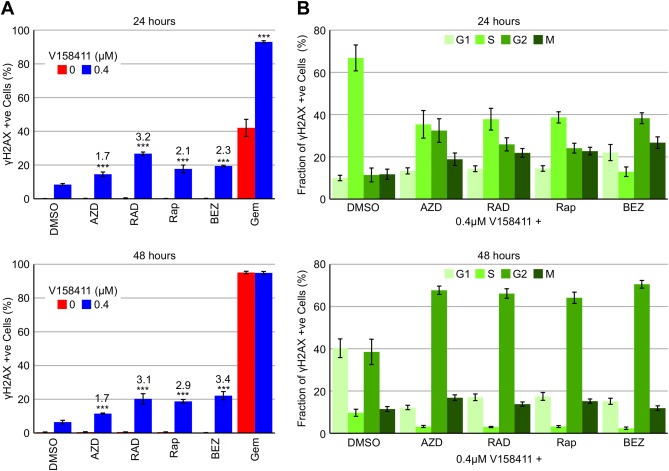
Inhibition of mTOR increases V158411 induced DNA DSB. HT29 cells were treated with 0.1 μM of the mTOR inhibitors AZD8055, RAD‐001, rapamycin or BEZ235, or gemcitabine in combination with 0 or 0.4 μM of the Chk1 inhibitor V158411 and pH2AX (S139) expression determined by high content analysis. (A) The fraction of γH2AX positive cells was determined by high content image analysis with Harmony software. The fold increase in γH2AX positive nuclei compared to V158411 + DMSO treatment alone is indicated above the bars. ***, P < 0.001. (B) Cell cycle phase of γH2AX positive nuclei was determined by high content image analysis using Harmony software following counterstaining for EdU and pHH3 (S10). Values are the average of 4 determinations ± SD.
To further understand the combinatorial activity of V158411 with an mTOR inhibitor, we utilised DNA (throughout the cell cycle), EdU (S‐phase specific) and pHH3 (S10) (mitosis specific) staining with high content analysis to evaluate the cell cycle distribution of γH2AX positive cells induced by V158411 and/or mTOR inhibitor treatment. Following 24 h exposure to 0.4 μM V158411 γH2AX positive cells were induced predominantly (67%) in the S‐phase of the cell cycle. Following 48 h treatment, only 10% of S‐phase cells were γH2AX positive, while approximately 40% of G1 and G2 cells were γH2AX positive (Figure 3B). Co‐treatment with an mTOR inhibitor profoundly altered the cell cycle distribution of γH2AX positive cells such that after 24 h the fraction of γH2AX positive cells in S‐phase was decreased approximately 2–3 fold compared to V158411 alone with a concomitant increase in γH2AX positive G2 and M cells. This was particularly pronounced for BEZ235 with the fraction of γH2AX positive cells in S, G2 and M being 13, 38 and 27% respectively. A longer co‐treatment of 48 h had an even greater effect on the cell cycle distribution of γH2AX positive cells with <5% of the γH2AX positive cells in S‐phase, the vast majority (≈60%) in G2 and smaller fractions (≈15% each) in G2 and M.
3.4. Inhibition of mTOR decreased the expression of key proteins involved in homologous recombination and intra‐strand crosslink repair
A previous study has demonstrated a key role for mTOR in the regulation of FANCD2 gene expression through mTORC1‐S6K where inhibition of mTOR with AZD8055 down regulated FANCD2 expression in paediatric rhabdomyosarcoma (Shen et al., 2013). Cell lines with mutations in FANCA, FANCG or FANCD2 were identified as hypersensitive to pharmacological inhibition of Chk1 compared to their paired isogenic corrected cells (Chen et al., 2009). Treatment of HT29 cells with V158411 or an mTOR inhibitor decreased protein expression levels of FANCD2, FANCF, RAD51 and RPA70 (Figure 4A and 4B). In cells treated with a combination of V158411 and mTOR inhibitor, the expression levels of FANCD2, FANCF, RAD51 and RPA70 were generally reduced further than that induced by the mTOR inhibitor or V158411 alone. Inhibition of Chk1 but not mTOR activated DNA‐PKcs as determined by increased autophosporylation of DNA‐PKcs on S2056. The combination of Chk1 and mTOR inhibition increased pDNA‐PKcs (S2056) by 3–7‐fold compared to Chk1 inhibition alone (Figure 4C and 4D). This was coupled with an increase in phosphorylation of RPA32 on serine 4/8 (Figure 4B).
Figure 4.
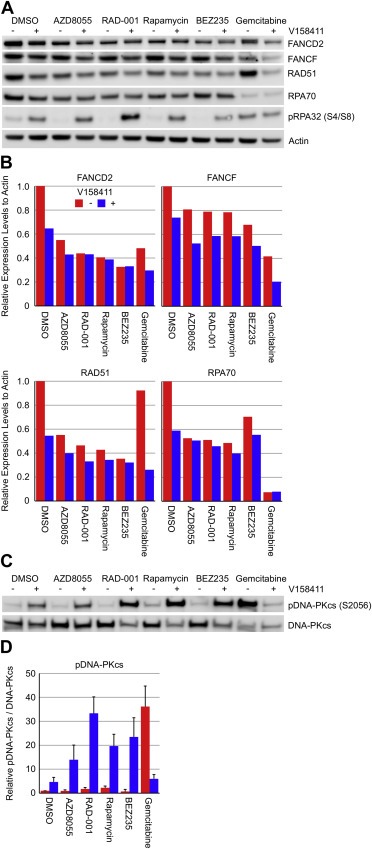
Inhibition of Chk1 in combination with mTOR inhibition modulates key proteins involved in homologous recombination and intra‐strand crosslink repair. HT29 cells were treated with 0.1 μM of the mTOR inhibitors AZD8055, RAD‐001, rapamycin or BEZ235, or gemcitabine in combination with 0 or 0.4 μM of the Chk1 inhibitor V158411 for 48 h. (A) Expression levels of proteins involved in DNA damage signalling and repair were determined by immunoblotting. (B) Densitometric analysis of FANCD2, FANCF, RAD51 or RPA70 expression levels were determined. Expression was normalised to actin protein levels. (C) Expression levels of pDNA‐PKcs and DNA‐PKcs were determined by immunoblotting and (D) quantified by densitometric analysis. Values are the average of 3 independent determinations ± SD.
3.5. DNA‐PKcs but not non‐homologous end joining confers sensitivity to Chk1 inhibition
Down regulation of proteins involved in HRR was associated with increased sensitivity to the Chk1 inhibitor V158411. The role of the non‐homologous end joining (NHEJ) DNA DSB repair pathway in Chk1 inhibitor sensitivity was evaluated. We utilised the parental Chinese hamster ovary AA8 cell line and the DNA‐PKcs mutant and Ku80 mutant derivatives V3 and xrs‐6 to assess the role of DNA‐PKcs in sensitivity to Chk1 inhibition. Surprisingly, the V3 cells were more resistant to V158411 whilst the xrs‐6 cells were more sensitive than the parental cells (Figure 5A). Since both Ku80 and DNA‐PKcs are components of NHEJ the differential sensitivity of the V3 and xrs‐6 cells implied a specific role for DNA‐PKcs. To investigate the role of DNA‐PKcs in Chk1 inhibitor sensitivity further we used the DNA‐PKcs defective M059J human glioblastoma cell line and M059J‐Fus1 where DNA‐PKcs is over‐expressed by complementation with a fragment of chromosome 8 containing PRKDC. Expression of DNA‐PKcs in the defective M059J cells profoundly increased their sensitivity to V158411 (Figure 5B) with the M059J‐Fus1 cells being around 10‐fold more sensitive to V158411 than the M059J cells.
Figure 5.
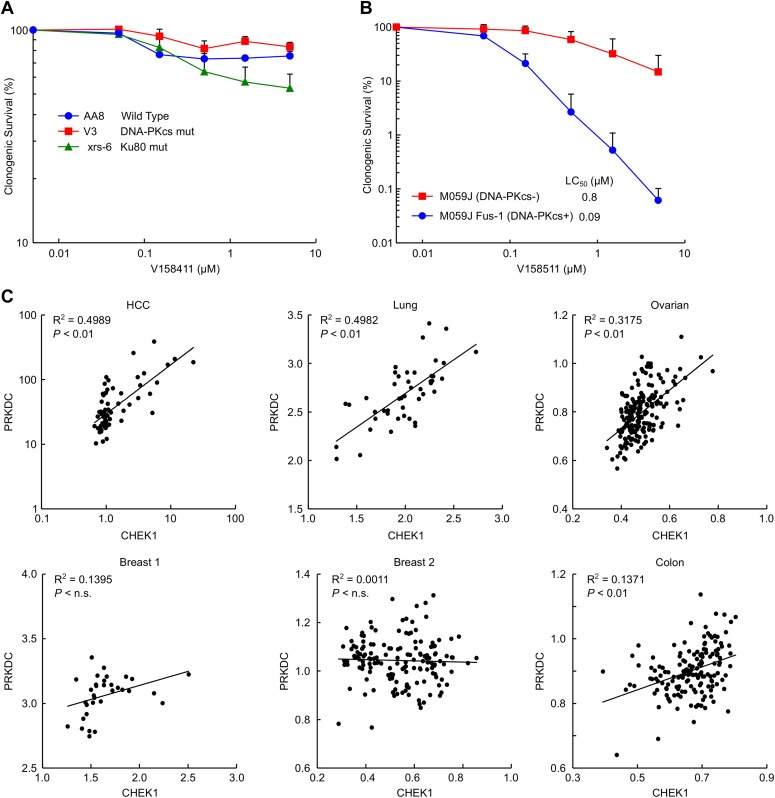
DNA‐PKcs expression but not non‐homologous end joining confers sensitivity to Chk1 inhibition. (A) Parental CHO‐AA8 or the DNA‐PKcs defective V3 or Ku80 defective xrs‐6 variants were treated with the Chk1 inhibitor V158411 for 24 h and clonogenic survival determined. Values are the average of ≥2 replicates in 3 independent experiments ± SD. (B) DNA‐PKcs defective M059J cells and the DNA‐PKcs corrected variant M059J‐Fus1 were treated with V158411 for 24 h and clonogenic survival determined. Values are the average of duplicate measurements in 3 independent experiments ± SD. (C) The correlation between PRKDC (DNA‐PKcs) and CHEK1 (Chk1) mRNA expression levels was determined from publicly available datasets in HCC, lung, ovarian, breast, and colon tumours.
An analysis of publicly available gene expression datasets of mRNA levels from patient derived material available on the Gene Expression Omnibus (GEO) database revealed a good correlation between CHEK1 and PRKDC mRNA expression levels in lung (R2 0.50, P < 0.01) and HCC (R2 0.50, P < 0.01) tumours with a slightly less robust correlation seen in ovarian cancer (R2 0.32, P < 0.01), a weak correlation in colon (R2 0.14, P < 0.01) cancer, but not breast (R2 0.14 and 0.001, n.s.) cancer (Figure 5C).
4. Discussion
The primary focus of Chk1 inhibitor development has been the targeted chemopotentiation of DNA damaging cytotoxic drugs, particularly in p53 dysfunctional cells. Previous studies have also demonstrated potential combination activity with molecularly targeted agents in a variety of tumour types. One serious limitation of these studies was the focus on the non‐selective Chk1 kinase inhibitors UCN‐01 and AZD7762 (Zabludoff et al., 2008). Here we utilised the selective Chk1 inhibitor V158411 to screen a limited set of kinase inhibitors and identified clear synergy with rapamycin in three colon cancer cell lines. Synergy was further noted with additional mTOR inhibitors AZD8055 and RAD‐001 and the pan‐PIKK inhibitor BEZ235 (Mukherjee et al., 2012). The limited concentration range of V158411 but not the mTOR inhibitor over which synergy was observed suggested that the mTOR inhibitor potentiated the cytotoxicity of the Chk1 inhibitor and not vice versa. Inhibition of mTOR in combination with V158411 resulted in increased caspase 3‐independent cell death via increased DNA DSBs that was associated with decreased expression of proteins involved in homologous recombination repair (HRR) and interstrand crosslink repair (ICLR).
Fanconi's anaemia (FA) deficient cell lines are hypersensitive to cell killing by Chk1 inhibitors or Chk1 targeted siRNA compared to their matched isogenic FA proficient cell line (Chen et al., 2009) and suggests that Chk1 and FA collaborate to maintain genomic integrity. Inhibition of mTOR is classically associated with increased radio‐ and chemosensitisation but the mechanism has been poorly understood. Two recent studies, however, have demonstrated that mTOR inhibitors can down regulate FANCD2 protein levels leading to increased DNA damage and cytotoxicity in combination with cytotoxic drugs (Shen et al., 2013; Guo et al., 2014). Both studies suggested that dual inhibition of mTORC1 and mTORC2 by ATP competitive kinase inhibitors rather than mTORC1 alone by rapalogs induces greater potentiation of DNA damage. However, in our study, we found little difference in potentiation of V158411 cytotoxicity between the rapalogs rapamycin and RAD‐001, and AZD8055 suggesting that mTORC1 and potentially not mTORC2 activity was important for regulating levels of proteins involved in HRR and ICLR and sensitivity to V158411. mTORC1 is regulated by environmental cues such as oxygen tension, amino acid levels, cellular stress, and growth factor and oncogenic signalling with the mTOR signalling axis dysregulated in many cancer types. Activation of mTORC1 results in the potential for increased replication stress and an increased reliance on the DDR to maximise cellular survival.
The checkpoint kinase Chk1 is a key component of the DDR and essential for the cellular response to exogenously induced DNA damage, particularly in cells lacking G1 checkpoint control, e.g., due to p53 mutation. Chk1 plays a critical role in unperturbed DNA replication through the control of replication origin firing, replication fork stabilisation and potentially reinitiation of DNA synthesis via translesion synthesis (Gonzalez Besteiro and Gottifredi, 2015). Inhibition of Chk1 therefore has the potential to increase replication stress and an associated increase in DNA damage. Chk1 is involved in controlling the timely activation of replication origins and Chk1 depletion or inhibition results in an aberrant increase in replication origin activation. This loss of controlled replication origin firing results in increased replication blockage. Evidence suggests a role for Chk1 in the protection of stalled forks from collapse and cleavage. The absence or inhibition of Chk1 results in increased cleavage of DNA replication intermediates by the Mus81‐Eme1 endonuclease (Forment et al., 2011; Murfuni et al., 2013) or the Mre11 nuclease (Thompson et al., 2012) and an increase in replication‐associated DSBs. We found that inhibition of Chk1 with V158411 increased the number of S‐phase cells with pan‐nuclear γH2AX staining indicative of increased S‐phase DSBs. Chk1 plays a critical role in promoting the repair of DSBs by the error‐free HRR pathway during S‐phase. Interaction with, and the subsequent phosphorylation of, RAD51 on T309 by Chk1 following replication stress serves as a critical signal for the recruitment of RAD51 to DNA damage foci (Bahassi et al., 2008). Replication forks that have irreversibly stalled are processed to DSBs and then repaired by the HRR pathway, a process promoted by Chk1 (Sorensen et al., 2005). Inhibition of Chk1 therefore increases the replication induced DNA damage load and increases the cells reliance on the HRR and ICLR pathways to maintain genomic stability and cell viability.
We found that various mTOR inhibitors decreased expression of HRR associated proteins and increased the number of V158411‐induced γH2AX positive nuclei, indicative of increased replication fork collapse. Increased numbers of these γH2AX positive cells appeared to have transitioned from S‐phase into G2 suggesting greater abrogation of checkpoint activation. Additionally, Chk1 may positively regulate TLS in unperturbed DNA replication thereby reducing prolonged stalling, and the subsequent cleavage, of replication forks.
Inhibition of mTOR in combination with V158411 increased phosphorylation and activation of DNA‐PKcs. Cells deficient in DNA‐PKcs were more resistant to Chk1 inhibitor induced cytotoxicity than those with high DNA‐PKcs expression. The error‐prone NHEJ repair pathway competes with HRR for the repair of DSBs (Allen et al., 2003; Delacote and Lopez, 2008; Mao et al., 2008) and depletion of DNA‐PKcs can restore HRR function and reverse PARP inhibitor sensitivity in BRCA mutant cells (Patel et al., 2011). Reduced HRR capacity following mTOR inhibition shifts repair of Chk1 inhibitor‐induced replicative damage to lower fidelity NHEJ (Bunting and Nussenzweig, 2013) resulting in reduced cellular viability (Figure 6). We identified a clear relationship between Chk1 and DNA‐PKcs expression levels in various human cancers. This potentially suggests, at least in some tumour types, Chk1 promoted HRR is necessary to counterbalance DNA‐PKcs driven NHEJ in order to maintain genomic viability in response to replicative stress. DNA‐PKcs therefore plays a clear role in reducing cell viability in response to Chk1 inhibitor induced replication damage.
Figure 6.
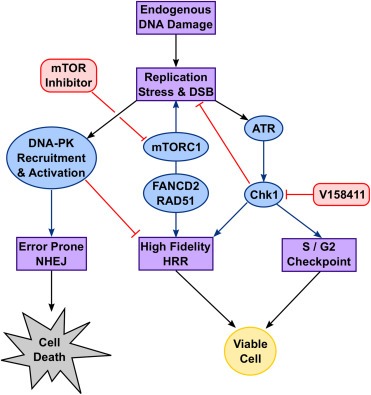
Model of the involvement of mTOR and DNA‐PKcs in DNA damage response signalling.
In response to Chk1 inhibitor induced replicative stress, we demonstrate that DNA‐PKcs defective cells exhibit greater resistance to Chk1 inhibition. This is in direct contrast to genotoxin induced replication stress and DNA damage, for example by hydroxyurea, camptothecin or etoposide, where cells with DNA‐PKcs defects do not induce RPA32 phosphorylation and exhibit an aberrant checkpoint function. DNA‐PKcs deficient cells have been shown to be more sensitive to exogenous damage induced replication stress (Ashley et al., 2014; Liu et al., 2012; Vidal‐Eychenie et al., 2013). These differences suggest potentially subtle differences in either DNA substrates or the temporal activation of Chk1 versus DNA‐PKcs between endogenous and exogenous damage induced replication fork arrest.
In conclusion, mTOR inhibitors that down regulate proteins involved in HRR and ICLR have the potential to increase the activity of Chk1 inhibitors such as V158411 against human tumours. Additionally, tumours with high levels of DNA‐PKcs, may demonstrate increased sensitivity to Chk1 inhibitor therapy, potentially through suppression of HRR. DNA‐PKcs is quantifiable by IHC suggesting it may represent a feasible biomarker to stratify patients for single‐agent CHK1 inhibitor therapy versus the combination with an mTOR inhibitor.
Supporting information
The following are the supplementary data related to this article:
Figure S1 V158411 and mTOR inhibition demonstrate synergistic combinatorial activity. (A) Comparison of potentiation factors (Pf) in HT29, SW620 and Colo205 cancer cells grown anchorage dependently (2D), anchorage independently (3D) or as multicellular tumour spheroids (Sph) following 72 or 168 h treatment with kinase inhibitors and 0.4 μM V158411. Pf values were calculated from the average GI50 and combination GI50 (cGI50) of at least three determinations where Pf equals average GI50 / average cGI50. Grey, Pf > 2.0, P < 0.05. (B) HT29 cells were treated with indicated concentrations of V158411 and AZD8055, RAD‐001, rapamycin or BEZ235 for 72 h and cell viability determined. Synergy was calculated using Bliss analysis as previously described and is the average of 3 determinations. (C) Combination Index (CI) values were calculated using constant drug ratio following 72 h treatment.
{kind=link}
Figure S2 V158411 and mTOR inhibition increase cell death in a caspase‐3 independent fashion. HT29 cells were treated with 0.1 μM AZD8055, RAD‐001, rapamycin, BEZ235 or gemcitabine in combination with 0 or 0.4 μM V15841. (A) Mitochondrial membrane potential was assessed using mitotracker orange and red dye ratio by high content analysis following 72 h compound treatment. Apoptosis after 48 h as determined by (B) immunoblotting for cleaved PARP and cleaved caspase‐3 or (C) by high content analysis following staining for cleaved caspase‐3. (D) Example images demonstrating nuclear abnormalities following 24 h treatment with the indicated combinations of compound. (E) Nuclear abnormalities, based on nuclear size, shape and intra‐nuclear Hoechst staining variability, following 24 h treatment were determined by high content analysis.
{kind=link}
Figure S3 Cell cycle perturbations in HT29 cells following simultaneous administration of V158411 and mTOR inhibitors. (A) HT29 cells were treated with a combination of 0.04 μM AZD8055, RAD‐001, rapamycin or BEZ235, or 0.01 μM gemcitabine and 0 or 0.4 μM V158411 for 48 h. Cell cycle profiles were determined using high content analysis. (B) HT29 cells were treated with 0.04 μM AZD8055, RAD‐001, rapamycin or BEZ235, or 0.01 μM gemcitabine for 24 h followed by 0 or 0.4 μM V158411 plus 0.3 μM nocodazole for a further 24 h. The fraction of pHH3 (S10) positive cells was determined by high content analysis.
{kind=link}
Figure S4 Dual inhibition of mTOR and V158411 increases nuclear γH2AX. HT29 cells were treated with 0.1 μM AZD8055, RAD‐001, rapamycin, BEZ235 or gemcitabine in combination with 0 or 0.4 μM V158411 and pH2AX (S139) expression determined by high content analysis. Example images following 48 h treatment demonstrating nuclei (blue) and γH2AX positive nuclei (red).
{kind=link}
Acknowledgements
AJM is an employee and stock option holder of Vernalis Research. We gratefully acknowledge a generous donation from the Sir Bobby Robson Foundation (to PS).
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.08.004.
Massey Andrew J., Stephens Peter, Rawlinson Rebecca, McGurk Lauren, Plummer Ruth, Curtin Nicola J., (2016), mTORC1 and DNA‐PKcs as novel molecular determinants of sensitivity to Chk1 inhibition, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.08.004.
Contributor Information
Andrew J. Massey, Email: a.massey@vernalis.com
Nicola J. Curtin, Email: nicola.curtin@ncl.ac.uk
References
- Allen, C. , Halbrook, J. , Nickoloff, J.A. , 2003. Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. 1, 913–920. [PubMed] [Google Scholar]
- Anderson, C.W. , Dunn, J.J. , Freimuth, P.I. , Galloway, A.M. , Allalunis-Turner, M.J. , 2001. Frameshift mutation in PRKDC, the gene for DNA-PKcs, in the DNA repair-defective, human, glioma-derived cell line M059J. Radiat. Res. 156, 2–9. [DOI] [PubMed] [Google Scholar]
- Ashley, A.K. , Shrivastav, M. , Nie, J. , Amerin, C. , Troksa, K. , Glanzer, J.G. , Liu, S. , Opiyo, S.O. , Dimitrova, D.D. , Le, P. , Sishc, B. , Bailey, S.M. , Oakley, G.G. , Nickoloff, J.A. , 2014. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair (Amst). 21, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahassi, E.M. , Ovesen, J.L. , Riesenberg, A.L. , Bernstein, W.Z. , Hasty, P.E. , Stambrook, P.J. , 2008. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 27, 3977–3985. [DOI] [PubMed] [Google Scholar]
- Bliss, C.I. , 1939. The toxicity of poisons applied jointly. Ann. Appl. Biol. 26, 585–615. [Google Scholar]
- Brooks, K. , Oakes, V. , Edwards, B. , Ranall, M. , Leo, P. , Pavey, S. , Pinder, A. , Beamish, H. , Mukhopadhyay, P. , Lambie, D. , Gabrielli, B. , 2013. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene. 32, 788–796. [DOI] [PubMed] [Google Scholar]
- Bryant, C. , Rawlinson, R. , Massey, A.J. , 2014. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer. 14, 570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, C. , Scriven, K. , Massey, A.J. , 2014. Inhibition of the checkpoint kinase Chk1 induces DNA damage and cell death in human Leukemia and Lymphoma cells. Mol. Cancer. 13, 147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher, N. , Britten, C.D. , 2008. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer. 98, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting, S.F. , Nussenzweig, A. , 2013. End-joining, translocations and cancer. Nat. Rev. Cancer. 13, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.C. , Kennedy, R.D. , Sidi, S. , Look, A.T. , D'Andrea, A. , 2009. CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol. Cancer. 8, 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T. , Stephens, P.A. , Middleton, F.K. , Curtin, N.J. , 2012. Targeting the S and G2 checkpoint to treat cancer. Drug Discov. Today. 17, 194–202. [DOI] [PubMed] [Google Scholar]
- Cho, S.H. , Toouli, C.D. , Fujii, G.H. , Crain, C. , Parry, D. , 2005. Chk1 is essential for tumor cell viability following activation of the replication checkpoint. Cell Cycle. 4, 131–139. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58, 621–681. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , 2010. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Chresta, C.M. , Davies, B.R. , Hickson, I. , Harding, T. , Cosulich, S. , Critchlow, S.E. , Vincent, J.P. , Ellston, R. , Jones, D. , Sini, P. , James, D. , Howard, Z. , Dudley, P. , Hughes, G. , Smith, L. , Maguire, S. , Hummersone, M. , Malagu, K. , Menear, K. , Jenkins, R. , Jacobsen, M. , Smith, G.C. , Guichard, S. , Pass, M. , 2010. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 70, 288–298. [DOI] [PubMed] [Google Scholar]
- Cole, K.A. , Huggins, J. , Laquaglia, M. , Hulderman, C.E. , Russell, M.R. , Bosse, K. , Diskin, S.J. , Attiyeh, E.F. , Sennett, R. , Norris, G. , Laudenslager, M. , Wood, A.C. , Mayes, P.A. , Jagannathan, J. , Winter, C. , Mosse, Y.P. , Maris, J.M. , 2011. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. U.S.A. 108, 3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Chen, S. , Pei, X.Y. , Almenara, J.A. , Kramer, L.B. , Venditti, C.A. , Dent, P. , Grant, S. , 2008. Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells. Blood. 112, 2439–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Chen, S. , Shah, R. , Pei, X.Y. , Wang, L. , Almenara, J.A. , Kramer, L.B. , Dent, P. , Grant, S. , 2011. Disruption of Src function potentiates Chk1-inhibitor-induced apoptosis in human multiple myeloma cells in vitro and in vivo. Blood. 117, 1947–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Grant, S. , 2010. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 16, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Rahmani, M. , Pei, X.Y. , Khanna, P. , Han, S.I. , Mitchell, C. , Dent, P. , Grant, S. , 2005. Farnesyltransferase inhibitors interact synergistically with the Chk1 inhibitor UCN-01 to induce apoptosis in human leukemia cells through interruption of both Akt and MEK/ERK pathways and activation of SEK1/JNK. Blood. 105, 1706–1716. [DOI] [PubMed] [Google Scholar]
- Delacote, F. , Lopez, B.S. , 2008. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle. 7, 33–38. [DOI] [PubMed] [Google Scholar]
- Ferrao, P.T. , Bukczynska, E.P. , Johnstone, R.W. , McArthur, G.A. , 2012. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene. 31, 1661–1672. [DOI] [PubMed] [Google Scholar]
- Forment, J.V. , Blasius, M. , Guerini, I. , Jackson, S.P. , 2011. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS One. 6, e23517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett, M.D. , Collins, I. , 2011. Anticancer therapy with checkpoint inhibitors: what, where and when?. Trends Pharmacol. Sci. 32, 308–316. [DOI] [PubMed] [Google Scholar]
- Ge, X.Q. , Blow, J.J. , 2010. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 191, 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Besteiro, M.A. , Gottifredi, V. , 2015. The fork and the kinase: a DNA replication tale from a CHK1 perspective. Mutat. Res. Rev. Mutat. Res. 763, 168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, F. , Li, J. , Zhang, S. , Du, W. , Amarachintha, S. , Sipple, J. , Phelan, J. , Grimes, H.L. , Zheng, Y. , Pang, Q. , 2014. mTOR kinase inhibitor sensitizes T-cell lymphoblastic leukemia for chemotherapy-induced DNA damage via suppressing FANCD2 expression. Leukemia. 28, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, M. , Li, W. , Yu, C. , Rahmani, M. , Dent, P. , Grant, S. , 2005. Rapamycin and UCN-01 synergistically induce apoptosis in human leukemia cells through a process that is regulated by the Raf-1/MEK/ERK, Akt, and JNK signal transduction pathways. Mol. Cancer Ther. 4, 457–470. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Guntuku, S. , Cui, X.S. , Matsuoka, S. , Cortez, D. , Tamai, K. , Luo, G. , Carattini-Rivera, S. , DeMayo, F. , Bradley, A. , Donehower, L.A. , Elledge, S.J. , 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Opiyo, S.O. , Manthey, K. , Glanzer, J.G. , Ashley, A.K. , Amerin, C. , Troksa, K. , Shrivastav, M. , Nickoloff, J.A. , Oakley, G.G. , 2012. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 40, 10780–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira, S.M. , Stauffer, F. , Brueggen, J. , Furet, P. , Schnell, C. , Fritsch, C. , Brachmann, S. , Chene, P. , De, P.A. , Schoemaker, K. , Fabbro, D. , Gabriel, D. , Simonen, M. , Murphy, L. , Finan, P. , Sellers, W. , Garcia-Echeverria, C. , 2008. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863. [DOI] [PubMed] [Google Scholar]
- Mao, Z. , Bozzella, M. , Seluanov, A. , Gorbunova, V. , 2008. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 7, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague, J. , 2004. G1 cell-cycle control and cancer. Nature. 432, 298–306. [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza, A. , Petermann, E. , Gillespie, D.A. , Caldecott, K.W. , Jackson, D.A. , 2007. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 26, 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, C. , Hamed, H.A. , Cruickshanks, N. , Tang, Y. , Bareford, M.D. , Hubbard, N. , Tye, G. , Yacoub, A. , Dai, Y. , Grant, S. , Dent, P. , 2011. Simultaneous exposure of transformed cells to SRC family inhibitors and CHK1 inhibitors causes cell death. Cancer Biol. Ther. 12, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, C. , Park, M. , Eulitt, P. , Yang, C. , Yacoub, A. , Dent, P. , 2010. Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in carcinoma cells. Mol. Pharmacol. 78, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, B. , Tomimatsu, N. , Amancherla, K. , Camacho, C.V. , Pichamoorthy, N. , Burma, S. , 2012. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 14, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murfuni, I. , Basile, G. , Subramanyam, S. , Malacaria, E. , Bignami, M. , Spies, M. , Franchitto, A. , Pichierri, P. , 2013. Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet. 9, e1003910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga, M. , Campaner, S. , Lopez-Contreras, A.J. , Toledo, L.I. , Soria, R. , Montana, M.F. , D'Artista, L. , Schleker, T. , Guerra, C. , Garcia, E. , Barbacid, M. , Hidalgo, M. , Amati, B. , Fernandez-Capetillo, O. , 2011. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 18, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, C.P. , Lee, H.C. , Ho, C.W. , Arooz, T. , Siu, W.Y. , Lau, A. , Poon, R.Y. , 2004. Differential mode of regulation of the checkpoint kinases CHK1 and CHK2 by their regulatory domains. J. Biol. Chem. 279, 8808–8819. [DOI] [PubMed] [Google Scholar]
- Niida, H. , Katsuno, Y. , Banerjee, B. , Hande, M.P. , Nakanishi, M. , 2007. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol. Cell Biol. 27, 2572–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A.G. , Sarkaria, J.N. , Kaufmann, S.H. , 2011. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, X.Y. , Dai, Y. , Youssefian, L.E. , Chen, S. , Bodie, W.W. , Takabatake, Y. , Felthousen, J. , Almenara, J.A. , Kramer, L.B. , Dent, P. , Grant, S. , 2011. Cytokinetically quiescent (G0/G1) human multiple myeloma cells are susceptible to simultaneous inhibition of Chk1 and MEK1/2. Blood. 118, 5189–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann, E. , Woodcock, M. , Helleday, T. , 2010. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. U.S.A. 107, 16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlinson, R. , Massey, A.J. , 2014. gammaH2AX and Chk1 phosphorylation as predictive pharmacodynamic biomarkers of Chk1 inhibitor-chemotherapy combination treatments. BMC Cancer. 14, 483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, C. , Oswald, D. , Phelps, D. , Cam, H. , Pelloski, C.E. , Pang, Q. , Houghton, P.J. , 2013. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res. 73, 3393–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata, H. , Miuma, S. , Saldivar, J.C. , Huebner, K. , 2011. Response of subtype-specific human breast cancer-derived cells to poly(ADP-ribose) polymerase and checkpoint kinase 1 inhibition. Cancer Sci. 102, 1882–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. , Tho, L.M. , Xu, N. , Gillespie, D.A. , 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112. [DOI] [PubMed] [Google Scholar]
- Smith-Roe, S.L. , Patel, S.S. , Zhou, Y. , Simpson, D.A. , Rao, S. , Ibrahim, J.G. , Cordeiro-Stone, M. , Kaufmann, W.K. , 2013. Separation of intra-S checkpoint protein contributions to DNA replication fork protection and genomic stability in normal human fibroblasts. Cell Cycle. 12, 332–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, C.S. , Hansen, L.T. , Dziegielewski, J. , Syljuasen, R.G. , Lundin, C. , Bartek, J. , Helleday, T. , 2005. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201. [DOI] [PubMed] [Google Scholar]
- Syljuasen, R.G. , Sorensen, C.S. , Hansen, L.T. , Fugger, K. , Lundin, C. , Johansson, F. , Helleday, T. , Sehested, M. , Lukas, J. , Bartek, J. , 2005. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell Biol. 25, 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Y. , Dai, Y. , Grant, S. , Dent, P. , 2012. Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol. Ther. 13, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Y. , Hamed, H.A. , Poklepovic, A. , Dai, Y. , Grant, S. , Dent, P. , 2012. Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in mammary tumors. Mol. Pharmacol. 82, 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Alveal, C. , Calonge, T.M. , O'Connell, M.J. , 2009. Regulation of chk1. Cell Div. 4, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, R. , Montano, R. , Eastman, A. , 2012. The Mre11 nuclease is critical for the sensitivity of cells to Chk1 inhibition. PLoS One. 7, e44021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Eychenie, S. , Decaillet, C. , Basbous, J. , Constantinou, A. , 2013. DNA structure-specific priming of ATR activation by DNA-PKcs. J. Cell Biol. 202, 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virsik-Kopp, P. , Rave-Frank, M. , Hofman-Huther, H. , Schmidberger, H. , 2004. Role of DNA-dependent protein kinase in the process of radiation-induced aberration formation. Int. J. Radiat. Biol. 80, 125–133. [DOI] [PubMed] [Google Scholar]
- Zabludoff, S.D. , Deng, C. , Grondine, M.R. , Sheehy, A.M. , Ashwell, S. , Caleb, B.L. , Green, S. , Haye, H.R. , Horn, C.L. , Janetka, J.W. , Liu, D. , Mouchet, E. , Ready, S. , Rosenthal, J.L. , Queva, C. , Schwartz, G.K. , Taylor, K.J. , Tse, A.N. , Walker, G.E. , White, A.M. , 2008. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 7, 2955–2966. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Figure S1 V158411 and mTOR inhibition demonstrate synergistic combinatorial activity. (A) Comparison of potentiation factors (Pf) in HT29, SW620 and Colo205 cancer cells grown anchorage dependently (2D), anchorage independently (3D) or as multicellular tumour spheroids (Sph) following 72 or 168 h treatment with kinase inhibitors and 0.4 μM V158411. Pf values were calculated from the average GI50 and combination GI50 (cGI50) of at least three determinations where Pf equals average GI50 / average cGI50. Grey, Pf > 2.0, P < 0.05. (B) HT29 cells were treated with indicated concentrations of V158411 and AZD8055, RAD‐001, rapamycin or BEZ235 for 72 h and cell viability determined. Synergy was calculated using Bliss analysis as previously described and is the average of 3 determinations. (C) Combination Index (CI) values were calculated using constant drug ratio following 72 h treatment.
Figure S2 V158411 and mTOR inhibition increase cell death in a caspase‐3 independent fashion. HT29 cells were treated with 0.1 μM AZD8055, RAD‐001, rapamycin, BEZ235 or gemcitabine in combination with 0 or 0.4 μM V15841. (A) Mitochondrial membrane potential was assessed using mitotracker orange and red dye ratio by high content analysis following 72 h compound treatment. Apoptosis after 48 h as determined by (B) immunoblotting for cleaved PARP and cleaved caspase‐3 or (C) by high content analysis following staining for cleaved caspase‐3. (D) Example images demonstrating nuclear abnormalities following 24 h treatment with the indicated combinations of compound. (E) Nuclear abnormalities, based on nuclear size, shape and intra‐nuclear Hoechst staining variability, following 24 h treatment were determined by high content analysis.
Figure S3 Cell cycle perturbations in HT29 cells following simultaneous administration of V158411 and mTOR inhibitors. (A) HT29 cells were treated with a combination of 0.04 μM AZD8055, RAD‐001, rapamycin or BEZ235, or 0.01 μM gemcitabine and 0 or 0.4 μM V158411 for 48 h. Cell cycle profiles were determined using high content analysis. (B) HT29 cells were treated with 0.04 μM AZD8055, RAD‐001, rapamycin or BEZ235, or 0.01 μM gemcitabine for 24 h followed by 0 or 0.4 μM V158411 plus 0.3 μM nocodazole for a further 24 h. The fraction of pHH3 (S10) positive cells was determined by high content analysis.
Figure S4 Dual inhibition of mTOR and V158411 increases nuclear γH2AX. HT29 cells were treated with 0.1 μM AZD8055, RAD‐001, rapamycin, BEZ235 or gemcitabine in combination with 0 or 0.4 μM V158411 and pH2AX (S139) expression determined by high content analysis. Example images following 48 h treatment demonstrating nuclei (blue) and γH2AX positive nuclei (red).