

Abstract
Background:
Childhood acute lymphoblastic leukemia (ALL) is characterized by recurrent genetic aberrations. The identification of those abnormalities is clinically important because they are considered significant risk-stratifying markers.
Aims:
There are insufficient data of cytogenetic profiles in Saudi Arabian patients with childhood ALL leukemia. We have examined a cohort of 110 cases of ALL to determine the cytogenetic profiles and prevalence of FLT3 mutations and analysis of the more frequently observed abnormalities and its correlations to other biologic factors and patient outcomes and to compare our results with previously published results.
Materials and methods:
Patients—We reviewed all cases from 2007 to 2016 with an established diagnosis of childhood ALL. Of the 110 patients, 98 were B-lineage ALL and 12 T-cell ALL. All the patients were treated by UKALL 2003 protocol and risk stratified according previously published criteria. Cytogenetic analysis—Chromosome banding analysis and fluorescence in situ hybridization were used to detect genetic aberrations. Analysis of FLT3 mutations—Bone marrow or blood samples were screened for FLT3 mutations (internal tandem duplications, and point mutations, D835) using polymerase chain reaction methods.
Result:
Cytogenetic analysis showed chromosomal anomalies in 68 out of 102 cases with an overall incidence 66.7%. The most frequent chromosomal anomalies in ALL were hyperdiploidy, t(9;22), t(12;21), and MLL gene rearrangements. Our data are in accordance with those published previously and showed that FLT3 mutations are not common in patients with ALL (4.7%) and have no prognostic relevance in pediatric patients with ALL. On the contrary, t(9;22), MLL gene rearrangements and hypodiploidy were signs of a bad prognosis in childhood ALL with high rate of relapse and shorter overall survival compared with the standard-risk group (P = .031).The event-free survival was also found to be worse (P = .040).
Conclusions:
Our data are in accordance with those published previously, confirming the overall frequency of cytogenetic abnormalities and their prognostic relevance.
Keywords: acute lymphoblastic leukemia, cytogenetic abnormalities, FLT3, outcome
Background
Acute lymphoblastic leukemia (ALL) is the most common malignancy of childhood, occurring in ~77% of childhood leukemias and ~25% of all childhood cancers.1 Childhood ALL is characterized by recurrent clonal chromosomal abnormalities in ~80% of ALL. These recurrent clonal chromosomal abnormalities include both numerical and structural changes.2 The identification of these chromosomal aberrations is clinically important because they are considered significant risk-stratifying markers, predictors of clinical prognosis, and essential for therapeutic planning, implementation of targeted therapy, and sensitive monitoring of treatment response.3 Recent studies have helped to understand the genetic basis of clonal evolution and relapse and the role of genetic variants in leukemogenesis.3
Fluorescence in situ hybridization (FISH) studies complement the diagnostic karyotype by providing a higher resolution of analysis with clarification of rearrangements observed by G-banding and identification of cryptic abnormalities not observed by karyotyping.4,5 Although many chromosomal abnormalities have been described in ALL, approximately 30% of pediatric patients with ALL do not have cytogenetic abnormalities of clinical significance.6
UKALL is a chemotherapy protocol for children diagnosed with ALL above 1 year of age. The chemotherapy starts with induction, central nervous system (CNS)–directed consolidation, intermaintenance, delayed intensification, and maintenance.7 Initially, eligible patients will be stratified into 3 risk groups: standard risk, intermediate risk, and high risk, based on published criteria in the UKALL 2003 protocol.8 Currently, there are significant improvements in outcome for pediatric ALL; however, therapy fails in ~25% of patients. These failures often occur unpredictably in patients with a favorable prognosis and good cytogenetics at diagnosis.6,9
Thus far, insufficient data exist regarding the cytogenetic profile and FLT3 mutational analysis in childhood patients with ALL from Saudi Arabia.
In this retrospective study, we have examined a cohort of 110 patients with ALL to determine cytogenetic profile and FLT3 mutational analysis as well as the analysis of the more frequently observed abnormalities associated with ALL with biological and clinical correlations to assess patient outcome.
Material and Methods
Patients
We reviewed all cases from 2007 to 2016 with a confirmed diagnosis of childhood ALL. The diagnosis in all cases was established based on morphology, flow cytometric analysis, immunohistochemistry, and genetic studies. This study included patients between 1 and 14 years of age as this is considered as the pediatric age group in Saudi Arabia. Patients were excluded if they were under 1 year of age or had insufficient data or because cytogenetic studies were not performed in some cases. The medical records were reviewed for epidemiologic, demographic, and laboratory data including immunophenotype, cytogenetic, and molecular analysis in addition to patient outcome variables such as remission status or relapsed disease, chemotherapeutic responses, follow-up, and morbidity.
The patients with ALL were treated according to UKALL 2003 protocol. Patients were classified into 3 risk groups based on prognostic factors such as age, sex, white blood cell count at diagnosis, immunophenotype, and cytogenetic findings according to published criteria.8
At the end of induction, the bone marrow (BM) aspirate was assessed for remission status. Patients were considered to be in remission if there were <5% blasts in the BM with evidence of trilineage recovery in addition to resolution of all sites of extramedullary disease. The Departmental Research Ethics Committee has approved this study.
Cytogenetic analysis
Standard cytogenetic preparations were made from BM and/or peripheral blood (PB). Briefly, BM aspirate and PB were mixed with specific complete medium for BM supplied in ready-to-use test tubes (Chromosome Kit M; Euroclone, Pero, Italy) with a final concentration of 2 × 106 to 4 × 106 nucleated cells per milliliter. The culture was incubated overnight at 37°C. Standard harvesting procedures were used: colcemid (0.1-mL Kmax Colcemid Solution, Gibco, Grand Island, NY, USA) was added to the culture for 20 minutes, followed by a 0.075-mol/L concentration of potassium chloride for 30 minutes at 37°C. The fixation procedure consisted of 3 changes of methanol/glacial acetic acid (3:1). After slide preparation, slides were placed in a 60°C oven overnight, followed by GTG banding. At least 20 metaphases were analyzed using Giemsa-trypsin staining. Karyotypes were interpreted and reported according to the International System for Cytogenetic Nomenclature.10 Fluorescence in situ hybridization was performed according to the manufacturer’s instructions. At least 100 interphase nuclei were analyzed for each case, and if needed, the metaphases were also analyzed. The probes used to detect ALL-specific abnormalities in our institute are BCR-ABL translocation t(9;22), RUNX1-ETV6 translocationt(12;21), MLL gene rearrangements (11q23) and MYC (8q24), TCF3/PBX1 t(1;19), and p16 deletions.
Analysis of FLT3 mutations
Analysis of FLT3-ITD mutation
DNA was extracted using QIAamp DNA Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instruction. Polymerase chain reaction (PCR) reaction was composed of 200 ng of DNA, 50-mM KCL, 10-mMTris-HCL, pH: 8.3, 1.5-mM MgCL2, 0.001% (wt/vol) gelatin, 200-μM deoxynucleotide triphosphates, 0.4-μM of each published primer,11 and 1 U of gold Taq polymerase, in a volume of 50 μL. The PCR consisted of an initial incubation step at 95°C for 10 minutes followed by 35 cycles at 94°C for 30 seconds, 57°C for 60 seconds, and 72°C for 90 seconds and final extension step at 72°C for 10 minutes.
Analysis of the FLT3-D835 mutation
Polymerase chain reaction was set up as above using published primers.12 Polymerase chain reaction product was digested with EcoRV (Promega, Madison, USA) at 37°C for 2 hours. The digestion products were separated on a 3% agarose gel. Incomplete digestion indicated the presence of a mutant gene.
Statistical Analysis
The Kaplan-Meier technique was used to analyze the probability of overall survival (OS) and event-free survival (EFS). Overall survival was calculated from time of diagnosis to death, and EFS was calculated from time of diagnosis to death. Relapse was indicated by the presence of disease at any site or evidence of persistent leukemia (morphologically persistent BM disease as greater than or equal to 5% lymphoblasts or molecular evidence of minimal residual disease at the end of induction). Continuous variables, such as white blood cell count and hemoglobin, were compared using t test. Differences between mean values were considered as significant at P < .05. Complete remission was defined as restoration of normal hematopoiesis with less than 5% blast cells in the BM and PB neutrophil count equal to or greater than 1.5 × 109/L with a platelet count of more than 100 × 109/L.
Results
The patient characteristics included in the study are summarized in Table 1. Of the 110 patients, 98 were B-lineage ALL (89%), and 12 were T-cell ALL (11%). The range of age was 1 to 14 years with median of 5.9 years. In total, 66 of the patients were men (60%) and 44 were women (40%). Five patients (4.5%) had CNS disease documented at diagnosis. Most of the patients (80%) were within good-risk age category (<10 years). In all, 2 patients (2/66) had testicular relapse only without BM relapse, and 5 patients (4.5%) had isolated CNS relapse, whereas 3 patients (2.7%) had both CNS and BM relapses.
Table 1.
Patient characteristics and outcome and FLT3 mutations.
| Parameter | ALL (n = 110) |
|---|---|
| Male:female | 66:44 |
| Median age, y | 5.9 |
| Median WBC count (×109/L) | 9800 |
| Median BM blasts, % | 70 |
| Risk groups | |
| High | 28 |
| Intermediate | 35 |
| Standard | 57 |
| Remission status | |
| CR1 | 81 |
| CR2 | 8 |
| CR3 | 5 |
| Refractory | 6 |
| Outcome | |
| Alive | 90 |
| Died | 16 |
| Lost follow-up | 4 |
| 5-y overall survival rate, % | 83.4 |
| FLT3 mutation rate (n = 64) | |
| Flt3-ITD | 1/64 (1.6 %) |
| Flt3-D835 | 2/64 (3.1 %) |
| Total | 3/64 (4.7 %) |
Abbreviations: ALL, acute lymphoblastic leukemia; BM, bone marrow; ITD, internal tandem duplication; WBC, white blood cell.
The cytogenetic abnormalities detected in our study are summarized in Table 2.
Table 2.
Cytogenetic abnormalities detected in the study.
| Karyotype | 33 normal and 20 abnormal |
|---|---|
| Available for FISH | n = 100 |
| t(9;22) | 10 (10%) |
| t(12;21) | 10 (10%) |
| MLL | (5%) |
| MYC | 3 (3%) |
| t(1;19) | 2 (2%) |
| +21 | 8 (8 %) |
| del 12p | 4 (4%) |
| +9 | 4 (4%) |
| +8 | 2 (2%) |
| +19 | 2 (2%) |
| +11 | 2 (2%) |
| del(6) del(6)(q21q26) | 2 (2%) |
| +X | 3 (3%) |
| Hyperdiploid | 16 (16%) |
| Other abnormalities | 19 (19%) |
Abbreviation: FISH, fluorescence in situ hybridization.
Fluorescence in situ hybridization was available in 100 of 110 cases (90.9%), whereas karyotype was available in only 53 of all cases (48.2%) because of various reasons such as a low number of analyzed metaphases, failed cytogenetic studies, lack of vital cells in the sample (necrotic cells), clotting of the sample, or the use of an unsuitable anticoagulant. Two cases had karyotype results only without FISH results. A high-hyperdiploid karyotype was defined as the presence of 51 to 67 chromosomes.
Cytogenetic analysis showed chromosomal anomalies including numerical and/or structural changes in 68 out of 102 cases available with overall incidence 66.7%.
The most frequent chromosomal anomalies in patients tested were hyperdiploidy, t(9;22), and t(12;21). Cytogenetic studies detected hyperdiploidy, BCR-ABL1 translocation, RUNX1-ETV6 translocation, and MLL gene rearrangements in 16%, 10%, 10%, and 5% of the patients tested, respectively. Gain of chromosome 21 frequently occurred (8%). Additional common chromosomal abnormalities which were observed included the following: chromosome 9 (4%), del 12p (4%), and X chromosome (3%).
Gene rearrangements were identified by FISH in 13 (39.4%) patients with normal karyotype. Rare chromosomal abnormalities detected included a patient with B-ALL and eosinophilia which was found positive for t(5;14). Another case of T-ALL was positive for t(9;22) confirmed by both FISH and PCR. Infrequent aberrations such as MLL deletion, ABL deletion, and TEL/AML1 fusion with AML1 deletion were also observed.
Conventional cytogenetic study was available in a limited number of cases (53 cases), and thus, the correlation with FISH analysis and other factors was not possible. Due to the presence of multiple cytogenetic abnormalities resident in a particular patient, the patient was enumerated multiple times. We were also unable to analyze the relationship between the outcomes of each genetic subgroup due to the limited number of patients in each genetic subgroup. The estimated OS at 5 years was 83.4%, and disease-free survival (DFS) was 80.1% with EFS of 71.5% (Figure 1). There was a significantly worse OS for the high-risk (HR) patients (70.3%) compared with 90.4% for standard-risk (SR) patients at 5 years (P < .001) (Figure 2A and B). The OS for patients with B-ALL was significantly higher than for T-ALL (86.0% vs 67.6%; P = .017) (Figure 3).
Figure 1.
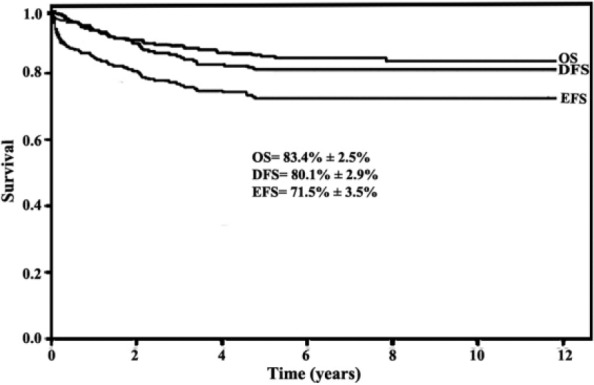
Survival estimates for patients with acute lymphoblastic leukemia (ALL): overall survival (OS), disease-free survival (DFS), and event-free survival (EFS); OS at 5 years was 83.4%, and DFS was 80.1% with EFS of 71.5%.
Figure 2.
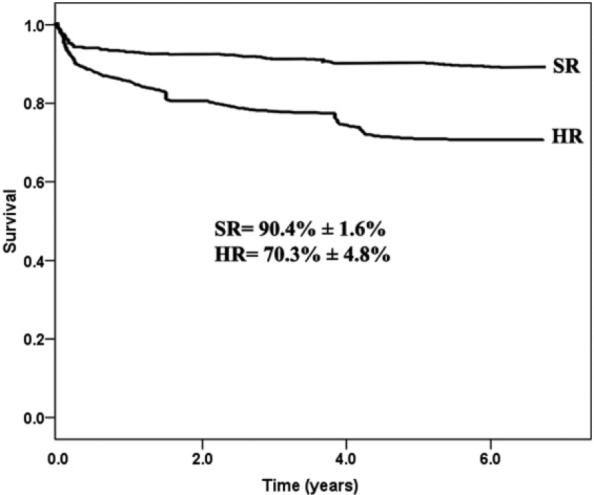
Comparisons of the overall survival between the high-risk (HR) and standard-risk (SR) patients with B-ALL (70.3% for HR vs 90.4% for SR, P < .001). ALL indicates acute lymphoblastic leukemia.
Figure 3.
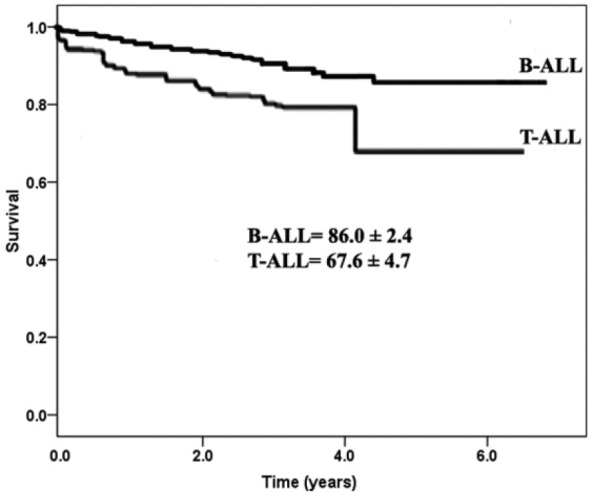
Comparisons of the overall survival (OS) between B-ALL and T-ALL: the OS for patients with B-ALL was significantly higher than for T-ALL (86.0% vs 67.6%; P = .017). ALL indicates acute lymphoblastic leukemia.
Discussion
Acute lymphoblastic leukemia is the most common childhood cancer, and despite cure rates exceeding 90% in children, it remains an important cause of mortality in children. Recent studies have helped to understand the genetic basis of clonal evolution and relapse in both T-ALL and B-ALL. Accurate, comprehensive identification of the full range of genetic alterations in ALL is important for diagnosis, risk stratification, implementation of targeted therapy, sensitive monitoring of treatment response, and monitoring of minimal residual disease in childhood ALL.2 Many chromosomal rearrangements have been described, and their associations with clinical, biologic, and prognostic features are well-documented. It is known that t(9;22), 11q23 abnormalities, and hypodiploidy confer a poor prognosis, and t(12;21) and hyperdiploidy are associated with a favorable outcome.13
In this study, we established the approximate incidences of gene rearrangements in childhood ALL in the Saudi Arabian population. The t(9;22) (q34;q11) (BCR/ABL1) also known as the Philadelphia chromosome (PH positive) was observed in 10% of patients in our cohort. This incidence is considered high when compared with those reported by others (reference of the others) who observed t(9;22) in about 2% to 5% of children with ALL.3,14–16 This result also differs from a local study17 which reported 4.2% BCR-ABL1 positivity among 429 childhood ALL. The difference may be attributed to geographic, ethnic, and socioeconomic differences within the region which have been shown to impact on genetic and phenotypic subtypes of ALL. The limited patient numbers in our study may also be contributing to the differences in data. An interesting finding was the detection of t(9;22) in a case of de novo T-ALL without history of previous chronic myeloid leukemia. This rare observation was confirmed by PCR to be consistent with the BCR/ABL p190 kD mutation which is common in ALL. To our knowledge, this observation has not been widely reported in the literature.18 The presence of the Ph chromosome has been linked to an inferior outcome in pediatric ALL and an increased risk of relapse.19 Allogeneic BM transplantation is considered a curative therapy for this group of patients.20
The t(12;21) (p13;q22) [ETV6-RUNX1] which is detectable only by FISH was observed in 10% in our cohort study and is low compared with those reported by international and local studies who detected this abnormality in ~25% and ~21% of children with B-lineage leukemia, respectively.2,17,21 A regional study which included 300 children with ALL, however, reported an incidence of 12% of this tranlocation22 reflecting the findings in our study. The difference in the results detected between our study and national and regional studies is most likely due to the limited quantity of patients in our cohort (110 cases compared with 214 and 300 cases in other studies, respectively).
Children with high-hyperdiploid ALL respond well to standard chemotherapy regimens and show a superior outcome when compared with their nonhyperdiploid patients.7,23 A hyperdiploid karyotype is detected in 16% of our patients, which is low compared with those reported by international and local studies who reported about 30% to 40% and 24% of children with B-ALL, respectively.2,17,21 The gain of chromosome 21 detected in 8% of our patients as well as detection of chromosomes 9 (4%) and X chromosome (3%) is similar to that reported in other studies.24,25
The presence of MLL gene rearrangements signifies a bad prognosis in childhood ALL. Translocations of MLL are the most common rearrangements detected, whereas deletions are rarely seen.26 The frequency of 11q23/MLL gene rearrangement in this study was 5%, and this finding is similar to previously published studies.26,27 However, it is increased compared with some international and national studies.17,28 Of note, all cases of MLL gene rearrangement were detected by FISH and not by conventional G-banding analysis.
The chromosomal translocation t(1;19)(q23;p13) (TCF3/PBX1) and its variant form der(19) t(1;19) found in 3-5% of ALL results in the expression of the E2A-PBX1 fusion transcript.29 In our study, the frequency of this cytogenetic abnormality was 2% among tested patients which is similar to results published in Western and local studies.17,28,29
Hypodiploidy has unfavorable prognostic implications in patients with B-ALL, and therefore, identification of hypodiploidy is important.28 In our study, we detect 2 cases with hypodiploidy, both were women with B-ALL, one case was a 4-year-old with a hypodiploid karyotype (44,XX,−4,−8) where the other case was a 10-year-old with normal karyotype (46,XX) and detected only by FISH.
Although the overall frequency of prognostically relevant, common genetic lesions (RUNX1-ETV6, E2A-PBX1, BCR-ABL1, MLL rearrangements, and hyperdiploidy) in our study is different from international, regional, and national studies, it has been found to be similar to those reported from most Western, regional, and local studies17,22,30,31 regarding its impact on patient outcome. This is evident by a significantly worse survival outcome for the HR patients (71.3%) compared with (92.4%) for SR patients at 5 years (P < .001).
The survival outcomes of our patients were comparable with those reported by local, regional, and Western studies.17,22,30–33 At the national level, 2 large studies from Saudi Arabia were published: one study reported 5-year OS, EFS, and DFS rates of 86.9%, 73.1%, and 79.1%, respectively17 and other study reported 5-year OS, EFS, and DFS at 84.7%, 77.0%, and 81.4%, respectively.30 Both these results compared well with our own estimates. Similarly, at the regional level, 2 studies have reported survival outcomes similar to those observed in our study. The first one reported 5-year OS and EFS rates of 85% and 73%,32 whereas the other study reported rates of 89% and 80%, respectively.22 Our results are comparable with most of the published studies with the OS rate remaining inferior to those reported by the leading leukemia cooperative groups reporting survival rates exceeding 90%.31,33
FLT3 gene mutations, particularly internal tandem duplication (ITD) in AML, are well established as the most frequent somatic alterations in AML. A poor prognosis is associated with FTL3 gene mutations in AML and they are found in approximately 5% to 15% of children and 25% to 35% of adults with AML.34,35 As expected, both FLT3 mutations (ITDs and point mutations, D835) not common among patients with ALL in Saudi Arabia occurring with a low overall incidence at 4.7% (3/64, 4.7%). In addition, this abnormality has no prognostic relevance and does not affect clinical outcome in these patients with ALL. These findings concur with previously published international and local reports.36–38 This preliminary study, although limited sample size of our cohort, suggested that the incidence of FLT3 gene mutations most probably is low in Saudi pediatric patients with ALL. A further study with larger number of patient samples is necessary to confirm the findings and assessment of the prognostic value of FLT3 mutations among pediatric patients with ALL.
Conclusions
We established the incidences of gene rearrangements in patients with childhood ALL from Saudi Arabia. Cytogenetic abnormalities occur commonly. The combination of conventional cytogenetic and FISH methods improves the detection rate of genetic abnormalities in childhood ALL. Our data are in accordance with those published and confirm that the overall frequency of cytogenetic abnormalities and their prognostic relevance were comparable with those reported in the literature. FLT3 mutations not common among Saudi ALL the data derived from this study have shown that the clinical characteristics and treatment outcome of our patients are similar to results published in Western, regional, and national studies.
Acknowledgments
This study is conducted under the auspices of the Saudi Arabian Pediatric Hematology/Oncology Society (SAPHOS).
Footnotes
Author Contributions: Conceived and designed the experiments: GE, NA, YE, MAS, OA. Analyzed the data: GE, HE, IB, MAW, EA. Wrote the first draft of the manuscript: GE, NA, FA, YE. Contributed to the writing of the manuscript: EA, FA, MAW, OAS, IB. Agree with manuscript results and conclusions: All authors. Jointly developed the structure and arguments for the paper: GE, MAS, IB, MA. Made critical revisions and approved final version: GE, OA, MA, EA, OAS, HE. All authors reviewed and approved of the final manuscript.
Peer review:Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 425 words, excluding any confidential comments to the academic editor.
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by SANAD Children’s Cancer Support Association and SANAD Research Grants Program (grant number: RGP-2015-06).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1. Ma H, Sun H, Sun X. Survival improvement by decade of patients aged 0-14 years with acute lymphoblastic leukemia: a SEER analysis. Sci Rep. 2014;4:4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harrison CJ, Moorman AV, Barber KE, et al. Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol. 2005;129:520. [DOI] [PubMed] [Google Scholar]
- 3. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35:975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vance GH. Detection of recurrent cytogenetic abnormalities in acute lymphoblastic and myeloid leukemias using fluorescence in situ hybridization. Methods Mol Biol. 2013;999:79–91. [DOI] [PubMed] [Google Scholar]
- 5. Liu Q, Jiang H, Sun HJ, Song YJ, Bao LM. [Cytogenetic analysis of childhood acute lymphoblastic leukemia]. Zhonghua Xue Ye Xue Za Zhi. 2012;33:282–285. [PubMed] [Google Scholar]
- 6. Dawson AJ, Yanofsky R, Vallente R, et al. Array comparative genomic hybridization and cytogenetic analysis in pediatric acute leukemias. Curr Oncol. 2011;18:e210–e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hann I, Vora A, Harrison G, et al. Determinants of outcome after intensified therapy of childhood lymphoblastic leukaemia: results from Medical Research Council United Kingdom acute lymphoblastic leukaemia XI protocol. Br J Haematol. 2001;113:103–114. [DOI] [PubMed] [Google Scholar]
- 8. Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14:18–24. [DOI] [PubMed] [Google Scholar]
- 9. Kearney L, Horsley SW. Molecular cytogenetics in haematological malignancy: current technology and future prospects. Chromosoma. 2005;114:286–294. [DOI] [PubMed] [Google Scholar]
- 10. Mitelman F. ISCN: An International System for Human Cytogenetic Nomenclature. Basel, Switzerland: Karger; 1995. [Google Scholar]
- 11. Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- 12. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. [DOI] [PubMed] [Google Scholar]
- 13. Ma SK, Wan TS, Chan LC. Cytogenetics and molecular genetics of childhood leukemia. Hematol Oncol. 1999;17:91–105. [DOI] [PubMed] [Google Scholar]
- 14. Ribeiro RC, Abromowitch M, Raimondi SC, Murphy SB, Behm F, Williams DL. Clinical and biologic hallmarks of the Philadelphia chromosome in childhood acute lymphoblastic leukemia. Blood. 1987;70:948. [PubMed] [Google Scholar]
- 15. Braoudaki M, Tzortzatou-Stathopoulou F. Clinical cytogenetics in pediatric acute leukemia: an update. Clin Lymphoma Myeloma Leuk. 2012;12:230–237. [DOI] [PubMed] [Google Scholar]
- 16. Moorman AV, Harrison CJ, Buck GA, et al. ; Adult Leukaemia Working Party, Medical Research Council/National Cancer Research Institute. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109:3189. [DOI] [PubMed] [Google Scholar]
- 17. Al-Sudairy R, Al-Nasser A, Alsultan A, et al. Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia in Saudi Arabia: a multi-institutional retrospective national collaborative study. Pediatr Blood Cancer. 2014;61:74–80. [DOI] [PubMed] [Google Scholar]
- 18. Raanani P, Trakhtenbrot L, Rechavi G, et al. Philadelphia-chromosome-positive T-lymphoblastic leukemia: acute leukemia or chronic myelogenous leukemia blastic crisis. Acta Haematol. 2005;113:181–189. [DOI] [PubMed] [Google Scholar]
- 19. Bhojwani D, Howard SC, Pui CH. High-risk childhood acute lymphoblastic leukemia. Clin Lymphoma Myeloma. 2009;9:S222–S230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harrison CJ, Foroni L. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Rev Clin Exp Hematol. 2002;6:91–113. [DOI] [PubMed] [Google Scholar]
- 21. Moorman AV, Ensor HM, Richards SM, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11:429. [DOI] [PubMed] [Google Scholar]
- 22. Halalsheh H, Abuirmeileh N, Rihani R, Bazzeh F, Zaru L, Madanat F. Outcome of childhood acute lymphoblastic leukemia in Jordan. Pediatr Blood Cancer. 2011;57:385–391. [DOI] [PubMed] [Google Scholar]
- 23. Heerema NA, Sather HN, Sensel MG, et al. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (>50 chromosomes). J Clin Oncol. 2000;18:1876–1887. [DOI] [PubMed] [Google Scholar]
- 24. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030. [DOI] [PubMed] [Google Scholar]
- 25. Raimondi SC, Pui CH, Hancock ML, Behm FG, Filatov L, Rivera GK. Heterogeneity of hyperdiploid (51-67) childhood acute lymphoblastic leukemia. Leukemia. 1996;10:213. [PubMed] [Google Scholar]
- 26. Goud KI, Dayakar S, Prasad S, Rao KN, Shaik A, Vanjakshi S. Molecular diagnosis of lymphoblastic leukemia. J Can Res Ther. 2013;9:493–496. [DOI] [PubMed] [Google Scholar]
- 27. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41. [DOI] [PubMed] [Google Scholar]
- 28. Loghavi S, Kutok JL, Jorgensen JL. B-acute lymphoblastic leukemia/lymphoblastic lymphoma. Am J Clin Pathol. 2015;144:393–410. [DOI] [PubMed] [Google Scholar]
- 29. Devaraj PE, Foroni L, Janossy G, Hoffbrand AV, Secker-Walker LM. Expression of the E2A-PBX1 fusion transcripts in t(1;19)(q23;p13) and der(19)t(1;19) at diagnosis and in remission of acute lymphoblastic leukemia with different B lineage immunophenotypes. Leukemia. 1995;9:821–825. [PubMed] [Google Scholar]
- 30. Jastaniah W, Elimam N, Abdalla K, et al. Identifying causes of variability in outcomes in children with acute lymphoblastic leukemia treated in a resource-rich developing country. Pediatr Blood Cancer. 2015;62:945–950. [DOI] [PubMed] [Google Scholar]
- 31. Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia from 1990-2005: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tantawy AA, El-Rashidy FH, Ragab IA, Ramadan OA, El-Gaafary MM. Outcome of childhood acute Lymphoblastic leukemia in Egyptian children: a challenge for limited health resource countries. Hematology. 2013;18:204–210. [DOI] [PubMed] [Google Scholar]
- 33. Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yokota S, Kiyoi H, Nakao M, et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies A study on a large series of patients and cell lines. Leukemia. 1997;11:1605–1609. [DOI] [PubMed] [Google Scholar]
- 35. Meshinchi S, Woods WG, Stirewalt DL, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001;97:89–94. [DOI] [PubMed] [Google Scholar]
- 36. Xu B, Li L, Tang JH, Zhou SY. Detection of FLT3 gene and FLT3/ITD mutation by polymerase chain reaction-single-strand conformation polymorphism in patients with acute lymphoblastic leukemia. Di Yi Jun Yi Da Xue Xue Bao. 2005;25:1207–1210. [PubMed] [Google Scholar]
- 37. Xu F, Taki T, Yang HW, et al. Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukaemia as well as acute myeloid leukaemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukaemia in children. Br J Haematol. 1999;105:155–162. [PubMed] [Google Scholar]
- 38. Elyamany G, Awad M, Alsuhaibani O, et al. FLT3 internal tandem duplication and D835 mutations in patients with acute lymphoblastic leukemia and its clinical significance. Mediterr J Hematol Infect Dis. 2014;6:e.2014038. [DOI] [PMC free article] [PubMed] [Google Scholar]