

Abstract
Neuroblastoma is an embryonal pediatric tumor that originates from the developing sympathetic nervous system and shows a broad range of clinical behavior, ranging from fatal progression to differentiation into benign ganglioneuroma. In experimental neuroblastoma systems, retinoic acid (RA) effectively induces neuronal differentiation, and RA treatment has been therefore integrated in current therapies. However, the molecular mechanisms underlying differentiation are still poorly understood. We here investigated the role of transcription factor activating protein 2 beta (TFAP2B), a key factor in sympathetic nervous system development, in neuroblastoma pathogenesis and differentiation. Microarray analyses of primary neuroblastomas (n = 649) demonstrated that low TFAP2B expression was significantly associated with unfavorable prognostic markers as well as adverse patient outcome. We also found that low TFAP2B expression was strongly associated with CpG methylation of the TFAP2B locus in primary neuroblastomas (n = 105) and demethylation with 5‐aza‐2′‐deoxycytidine resulted in induction of TFAP2B expression in vitro, suggesting that TFAP2B is silenced by genomic methylation. Tetracycline inducible re‐expression of TFAP2B in IMR‐32 and SH‐EP neuroblastoma cells significantly impaired proliferation and cell cycle progression. In IMR‐32 cells, TFAP2B induced neuronal differentiation, which was accompanied by up‐regulation of the catecholamine biosynthesizing enzyme genes DBH and TH, and down‐regulation of MYCN and REST, a master repressor of neuronal genes. By contrast, knockdown of TFAP2B by lentiviral transduction of shRNAs abrogated RA‐induced neuronal differentiation of SH‐SY5Y and SK‐N‐BE(2)c neuroblastoma cells almost completely. Taken together, our results suggest that TFAP2B is playing a vital role in retaining RA responsiveness and mediating noradrenergic neuronal differentiation in neuroblastoma.
Keywords: TFAP2B, Neuroblastoma, Differentiation, Prognostic marker, Retinoic acid
Highlights
Low TFAP2B transcript levels are associated with unfavorable prognostic markers and poor outcome in neuroblastoma.
TFAP2B may be silenced by promoter methylation in unfavorable neuroblastoma.
TFAP2B plays a role in neuronal differentiation of neuroblastoma cells.
Abbreviations
- TFAP2B
transcription factor activating protein 2 beta
- DAC
5-aza-2′-deoxyctidine
- RA
retinoic acid
- INSS
International Neuroblastoma Staging System
- INPC
International Neuroblastoma Pathology Committee
- MAP2
microtubule associated protein 2
- NEFM
neurofilament middle chain
- NSE
neuron specific enolase
- SYP
synaptophysin
- TUBB3
class III beta-tubulin
- EFS
event-free survival
- OS
overall survival
1. Introduction
Neuroblastoma is a pediatric cancer deriving from embryonal precursor cells of the sympathetic nervous system. The clinical and biological behavior of this tumor is remarkably heterogeneous, ranging from fatal tumor progression to spontaneous regression or differentiation into mature ganglioneuroma. A more differentiated histological phenotype of the tumor cells is associated with favorable patient outcome (Cohn et al., 2009). Likewise, low and high risk neuroblastomas exhibit distinct expression patterns of neuronal genes (Fischer et al., 2006; Fredlund et al., 2008; Ohira et al., 2005). Retinoic acid (RA) has been shown to effectively induce differentiation of neuroblastoma cells in experimental systems (Sidell, 1982), and is currently being used in maintenance treatment strategies for high risk neuroblastoma patients to prevent relapse (Matthay et al., 2009). However, the benefit of RA treatment in the clinical setting appears to be limited (Reynolds et al., 2003), and biomarkers that may predict response to RA therapy are lacking to date (Matthay et al., 2009).
Transcription factor activating proteins (TFAP2) are belonging to a sequence‐specific DNA binding transcription factor family, which are encoded by five different but closely related genes, TFAP2‐A, B, C, D and E. TFAP2 family members form homo‐ and heterodimers, and transactivate target genes by binding to GC‐rich consensus elements (Eckert et al., 2005). TFAP2 proteins are RA‐inducible transcriptional activators (Luscher et al., 1989), and have been shown to play important roles during vertebrate development, cell growth and differentiation (Auman et al., 2002; Moser et al., 1997; Zeng et al., 1997; Zhang et al., 1996). In addition, alterations in TFAP2 expression patterns and both oncogenic and tumor suppressive functions have been described in various cancer entities (Fu et al., 2014; Pellikainen and Kosma, 2007). In neuroblastoma, expression of the TFAP2 gene family member TFAP2B, also known as AP‐2β, has been shown to be associated with low risk disease in small tumor cohorts (Fischer et al., 2006; Thorell et al., 2009). In vertebrate embryogenesis, TFAP2B is expressed in migrating neural crest cells and fetal sympathetic neuroblasts, and has been implicated in the development of the sympathetic nervous system (De Preter et al., 2006, 2011, 2008, 2011). These findings prompted us to investigate the role of TFAP2B in neuroblastoma pathogenesis and differentiation.
Here, we examined the expression of TFAP2B in a cohort of 649 primary neuroblastomas using microarrays, and assessed its correlation with prognostic markers and patient outcome. To investigate the mechanisms behind TFAP2B deregulated expression, we determined copy number alterations and DNA methylation patterns of the TFAP2B locus by array‐CGH and methylation array analysis, respectively. The functional relevance of TFAP2B expression was determined by retroviral TFAP2B re‐expression and shRNA‐mediated TFAP2B knockdown in neuroblastoma cell lines. We found that low TFAP2B expression is associated with poor outcome and DNA methylation of the TFAP2B locus in primary neuroblastoma, and that TFAP2B mediates noradrenergic neuronal differentiation of neuroblastoma cells in vitro.
2. Materials and methods
2.1. Patient characteristics
A total of 649 primary neuroblastomas from 9 different countries (Germany, n = 492; USA, n = 59; Italy, n = 23; Japan, n = 20; France, n = 19; Spain, n = 14; Israel, n = 12; Belgium, n = 5; UK, n = 5) were analyzed by expression microarrays as described (Kocak et al., 2013). Stages were classified according to the International Neuroblastoma Staging System (INSS) (Brodeur et al., 1993): stage 1, n = 153; stage 2, n = 113; stage 3, n = 91; stage 4, n = 214; and stage 4S, n = 78. Patient's age at diagnosis ranged from 0 to 25.4 years (median age, 1.1 years). MYCN amplification was observed in 93 tumors, while it was absent in 550 tumors (not determined, n = 6). Loss of chromosome 1p was observed in 141 tumors, while normal chromosome 1p status was found in 411 tumors (not determined, n = 97). Risk estimation of the patients was performed according to NB2004 (low risk, n = 353; intermediate risk, n = 61; high risk, n = 210; unclassified, n = 25). Histological assessment was available for 376 neuroblastomas according to the International Neuroblastoma Pathology Committee (INPC) (Shimada et al., 1999): undifferentiated, n = 22; poorly differentiated, n = 311; and differentiating, n = 43. Prognostic gene expression‐based classification of primary neuroblastomas was performed according to the recently developed prognostic classifier SVM_th10 (favorable, n = 363; unfavorable n = 211; unclassified, n = 75) (Oberthuer et al., 2015).
The neuroblastoma cohort analyzed by aCGH consisted of 199 primary tumors derived from German patients (stage 1, n = 40; stage 2, n = 33; stage 3, n = 38; stage 4, n = 60; and stage 4S, n = 28) (Kocak et al., 2013). Patient's age at diagnosis ranged from 0 to 25.4 years (median age, 1 year). MYCN amplification was observed in 23 tumors, while it was absent in 174 tumors (not determined, n = 2). According to NB2004, 107 patients of the cohort were classified as low risk, 31 as intermediate risk, and 59 as high risk.
The neuroblastoma cohort analyzed by methylation arrays consisted of 105 tumors derived from German patients (stage 1, n = 10; stage 2, n = 9; stage 3, n = 10; stage 4, n = 56; and stage 4S, n = 20). Patient's age at diagnosis ranged from 0 to 24.6 years (median age, 1 year). MYCN amplification was observed in 33 tumors, while it was absent in 72 tumors. According to NB2004, 40 patients of the cohort were classified as low risk, 9 as intermediate risk, and 56 as high risk.
2.2. Analysis of TFAP2B expression patterns and genetic and epigenetic alterations of the TFAP2B locus
Single‐color gene expression profiles from neuroblastoma samples and cell lines were generated using 44K oligonucleotide microarrays as described previously (Kocak et al., 2013; Oberthuer et al., 2010). Array‐based comparative genomic hybridization (aCGH) using 44K or 105K microarrays was used to determine DNA copy number profiles from 199 neuroblastoma tumors as described (Fischer et al., 2010; Kocak et al., 2013; Spitz et al., 2006). All gene expression and aCGH data are available at Gene Expression Omnibus (Accessions: GSE45480, GSE74350 and GSE25771). Whole‐genome DNA methylation profiles of 105 neuroblastoma tumors were generated using the Infinium HumanMethylation450 BeadChip (Illumina) according to the manufacturers' instructions.
2.3. Cell culture
The neuroblastoma cell lines SH‐SY5Y, SK‐N‐DZ, SK‐N‐AS, CHP‐212 and SH‐EP were obtained from American Tissue Culture Collection (ATCC, Rockville, MD, USA), and the neuroblastoma cell lines IMR‐32, LAN‐5, NMB, GI‐ME‐N and NBL‐S were purchased from DSMZ‐German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). KELLY and SK‐N‐BE(2)c cells were provided by Dr. Olaf Witt, German Cancer Research Center (DKFZ), Heidelberg (Germany), and CLB‐GA by Dr. Johannes Schulte, University Children's Hospital, Essen (Germany), and were validated by the DSMZ using STR profiling. Neuroblastoma cells were maintained in media according to the instructions from suppliers supplemented with 10–20% fetal calf serum (FCS) (Invitrogen, Germany). Inducible cell lines were maintained in RPMI‐1640 supplemented with 10% Tet‐free FCS (PAA, Germany), 50 μg/ml G418 and 30–50 μg/ml Hygromycin B. PT67 packaging cells were obtained from Clontech Laboratories and grown in DMEM supplemented with 10% FCS. Cells were maintained in a humidified incubator at 37 °C with 5% CO2 and passaged at 90% confluence using Trypsin‐EDTA. Doxycycline (Sigma–Aldrich, Taufkirchen, Germany) was used at 2 μg/ml and 1 μg/ml to induce transgene expression in SH‐EP cells and IMR‐32 cells, respectively. Neuroblastoma cell lines were shown to be free of mycoplasma by using the Venor® GeM Mycoplasma Detection Kit (Minerva Biolabs, Berlin, Germany) according to the manufacturer's protocol. To assess RA‐mediated differentiation, all‐trans retinoic acid (Sigma–Aldrich) was dissolved in ethanol, and used at 1 μmol/L for 6–18 days in diffused light. Ethanol treated cells were used as negative control. To assess the effects of demethylation, IMR‐32 cells were grown in the presence of 3 μM 5‐aza‐2′‐deoxycytidine (DAC; Sigma–Aldrich) or DMSO (control) for 3–7 days. Media was changed every 24 h containing fresh DAC.
2.4. Cloning of TFAP2B into the pRevTRE vector and generation of TFAP2B inducible neuroblastoma cell lines
A human TFAP2B full open reading frame was obtained by PCR from the pCMV‐Sport6ccdB vector (RZPD) and cloned into the pRevTRE vector (Clontech, Heidelberg, Germany). Neuroblastoma cell lines stably expressing either TFAP2B or GFP under the control of the reverse tetracycline‐controlled transactivator were generated using the RevTet System (Clontech) as described elsewhere (Kocak et al., 2013). Three single clones for SH‐EP pRevTet‐On/pRevTRE‐TFAP2B cells were generated, and all experiments were performed with these clones. Promoter leakage of TFAP2B expression was observed in IMR‐32 pRevTet‐On/pRevTRE‐TFAP2B cells without doxycycline. Therefore, IMR‐32 pRevTet‐On/pRevTRE‐GFP cells were used as control.
2.5. Lentivirus production and infection of neuroblastoma cells
Four short hairpin RNAs targeting the TFAP2B gene were obtained from Sigma–Aldrich (Supplementary Table S1). Replication‐incompetent lentiviruses were produced using the pLKO.1 vector by co‐transfection with Δ8.91 and pHIT‐VSV‐G in HEK 293T cells using TRANS‐IT (Mirus). Infectious lentiviruses were harvested 48 h post‐transfection, centrifuged to remove cell debris, and then filtered through 0.45 μm cellulose acetate filters. Recombinant lentiviruses plus 10 μg/ml of polybrene were used to infect exponentially growing neuroblastoma cells. After transduction, SK‐N‐BE(2)c and SH‐SY5Y cells were selected with 2 μg/ml puromycin and 1 μg/ml puromycin, respectively.
2.6. Western blot hybridization and RT–qPCR
Immunoblots were prepared as described previously (Nowacki et al., 2008). Total protein extracts were isolated either with RIPA or Cell Lysis Buffer (Cell Signaling Technology). RT‐qPCR was carried out two times in technical triplicates as described previously (Fischer et al., 2005). Results of one independent experiment are shown. Primer sequences are given in Supplementary Table S2.
2.7. Analysis of cell cycle distribution, cell growth and senescence
Cell proliferation was assessed using the trypan blue exclusion test. Cells were harvested at days 2, 4, 6 and 8, and evaluated for the number and viability by trypan blue exclusion using a Cedex cell counter (Roche). Cell cycle distribution was assessed by flow cytometry (FACS Canto; BD Biosciences) and analyzed using the DIVA software (BD Biosciences). To examine senescence, cells were fixed and stained using the SA‐ß‐Gal kit (Cell Signaling Technology) according to the manufacturer's instructions.
2.8. Immunohistochemistry, immunofluorescence and microscopy
To assess the tumor cell content in primary neuroblastomas, we performed immunohistochemistry staining using the NB84 monoclonal antibody, which has been established as a useful tool for differential diagnosis of neuroblastoma (Miettinen et al., 1998; Thomas et al., 1991). Sections were fixed in acetone for 5 min and stained with NB84 (Leica Biosystems) using the EnVision™ G|2 System/AP (DAKO); consecutive sections were then stained with rabbit‐anti‐TFAP2B for immunofluorescence (Cell Signaling Technology). For immunofluorescence of cell lines, cells were grown in chamber slides (Nunc Lab‐Tek II Chamber Slide* System, Thermo Scientific), fixed with 4% paraformaldehyde and stained with primary antibody rabbit‐anti‐TFAP2B (1:100; Cell Signaling Technology), rabbit‐anti‐MAP2 (1:200; Santa Cruz) or mouse‐anti‐TUBB3 (1:200, Santa Cruz). TFAP2B and MAP2 signals were detected either by anti‐rabbit IgG (H+L), F(ab′)2 Fragment Alexa Fluor® 488 Conjugate, or by anti‐rabbit IgG (H+L), F(ab′)2 Fragment Alexa Fluor® 555 Conjugate (Cell Signaling Technology). TUBB3 signals were detected by anti‐mouse‐IgG conjugated with TRITC (DAKO). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI), and actin filaments were labeled with rhodamine phalloidin. Fluorescent images were taken with a confocal microscope and FV 1000 software (Olympus Fluoview FV 1000) or widefield microscope Leica DMI6000 B.
2.9. Data analysis and statistics
Analysis of associations between TFAP2B expression levels and prognostic markers or outcome was performed using SPSS version 20.0 (IBM, Mainz, Germany) and R version 2.15.0. Two‐tailed non‐parametric tests (Mann–Whitney U‐test and Kruskal‐Wallis test) were used where appropriate. To assess the prognostic value of TFAP2B expression levels, the neuroblastoma cohort was randomly divided into training and test sets. The optimal cut‐off expression level was calculated by means of maximally selected rank statistics in the training cohort. The optimal cut‐off was defined as the intensity value of TFAP2B that separates the cohort best in two groups in terms of overall‐survival (OS) (Hothorn and Lausen, 2002). The prognostic value of the TFAP2B cut‐off value was then assessed in the validation cohort by Kaplan‐Meier survival analysis using log‐rank test (both for event‐free survival (EFS) and OS), and multivariate Cox regression analysis. The factors age (>18 months versus <18 months), tumor stage (stage 4 versus stages 1–3 and 4S), MYCN (amplified versus normal) and TFAP2B expression were fitted into a stepwise‐backward selection model. The likelihood ratio test p for inclusion was ≤0.05 and for exclusion was ≥0.1. Recurrence, progression and death from disease were considered as events.
To describe the association between CpG methylation and gene expression, methylation cut‐off values for dichotomization of TFAP2B expression and p‐values were estimated by maximally selected log‐rank statistics (Lausen and Schumacher, 1992). The Wilcoxon rank‐sum test was used to test for an association between TFAP2B methylation and established prognostic factors.
Gene Set Enrichment Analysis (GSEA) (www.broadinstitute.org/gsea/; Subramanian et al., 2005) was used to generate enrichment plot of the gene set in the Biocarta cell cycle pathway in TFAP2B and GFP expressing IMR‐32 cells.
Quantitative data of functional analyses are given as means ±S.D. Unpaired two‐tailed student's t‐tests were used where appropriate. The difference was considered to be significant at p < 0.05.
3. Results
3.1. Low TFAP2B expression is associated with unfavorable prognostic markers and poor outcome in neuroblastoma
To investigate whether TFAP2B is associated with prognostic variables in neuroblastoma, we determined TFAP2B expression levels by microarrays in a cohort of 649 primary neuroblastomas representing the entire spectrum of the disease. We observed that low TFAP2B expression was associated with prognostic markers of poor outcome including age of the patient at diagnosis >18 months, stage 4 disease, MYCN amplification, loss of chromosome 1p and high risk disease according to the criteria of the German NB2004 trial (p < 0.001 each, Figure 1a–e). Low TFAP2B expression was also associated with unfavorable gene expression‐based outcome prediction using an accurate multigene expression classifier that we have developed recently (p < 0.001, Figure 1f) (Oberthuer et al., 2015). To assess the impact of TFAP2B expression on neuroblastoma patient outcome, we divided the entire cohort randomly into halves, and determined the optimal prognostic expression threshold in the first set (Supplementary Figure S1). The prognostic value of TFAP2B was then evaluated in the second set. We found that low TFAP2B expression was strongly associated with adverse patient outcome for both event‐free survival (EFS at 5 years, 0.79 ± 0.03 vs 0.45 ± 0.04, p < 0.001; Figure 1g) and overall‐survival (OS at 5 years, 0.91 ± 0.02 vs 0.63 ± 0.04, p < 0.001; Figure 1h). In addition, multivariate Cox regression models based on EFS and OS considering established risk markers (MYCN status, tumor stage and patient age at diagnosis) identified TFAP2B expression as a significant independent prognostic variable for EFS (Table 1). Together, our results demonstrate that high TFAP2B transcript levels are associated with a favorable neuroblastoma phenotype, pointing towards its potential biological relevance.
Figure 1.
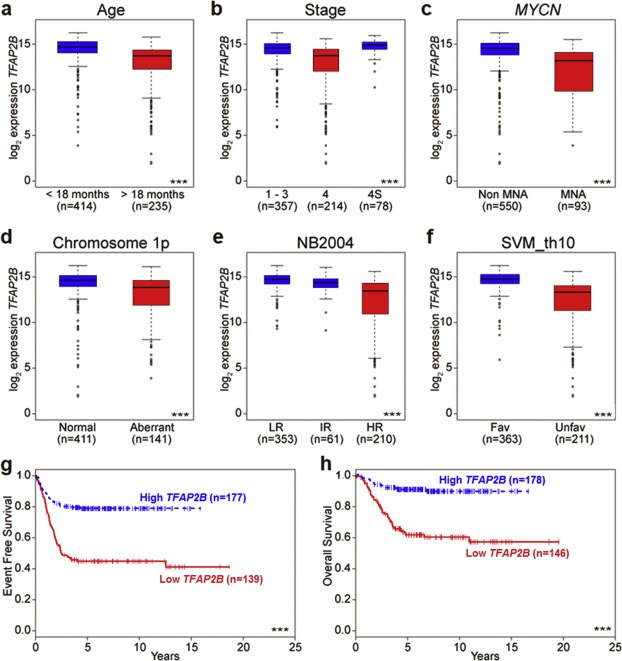
Association of TFAP2B expression levels with prognostic markers and outcome in neuroblastoma. Association of (a) age at diagnosis, (b) tumor stage, (c)MYCN amplification status, (d) chromosome 1p status, (e) risk stratification according to NB2004, and (f) gene expression‐based classification (according to the SVM_th10 classifier) with TFAP2B transcript levels in 649 neuroblastoma samples as determined by microarray gene expression analysis. Boxes, median expression values (horizontal line) and 25th and 75th percentiles; whiskers, distances from the end of the box to the largest and smallest observed values that are <1.5 box lengths from either end of the box; circles, outliers. (g) EFS and (h) OS of neuroblastoma patients in the validation set according to the prognostic TFAP2B expression cut‐off determined in the training set. n = patient number; (***), p < 0.001.
Table Table 1.
Multivariate Cox regression models based on EFS and OS considering single prognostic markers and TFAP2B expression.
| Marker | Patient (N) | Available cases (N) | Hazard ratio | 95% CI | P‐value |
|---|---|---|---|---|---|
| Model based on EFS | 324 | 312 | |||
| Age (≥18 months versus< 18 months) | 2.97 | 1.84; 4.79 | <0.001 | ||
| Stage (4 versus 1–3, 4S) | 1.64 | 1.06; 2.55 | 0.026 | ||
| MYCN (amplified versus normal) | 1.65 | 1.05; 2.59 | 0.035 | ||
| TFAP2B (<19830.07 versus ≥19830.07) | 1.59 | 1.01; 2.52 | 0.042 | ||
| Model based on OS | 324 | 320 | |||
| Age (≥18 months versus < 18 months) | 5.92 | 2.90; 12.07 | <0.001 | ||
| Stage (4 versus 1–3, 4S) | 2.53 | 1.38; 4.63 | 0.002 | ||
| MYCN (amplified versus normal) | 3.72 | 2.23; 6.20 | <0.001 | ||
| TFAP2B (<19830.07 versus ≥19830.07) | n.s. | ||||
HR, Hazard ratio; n.s., not significant.
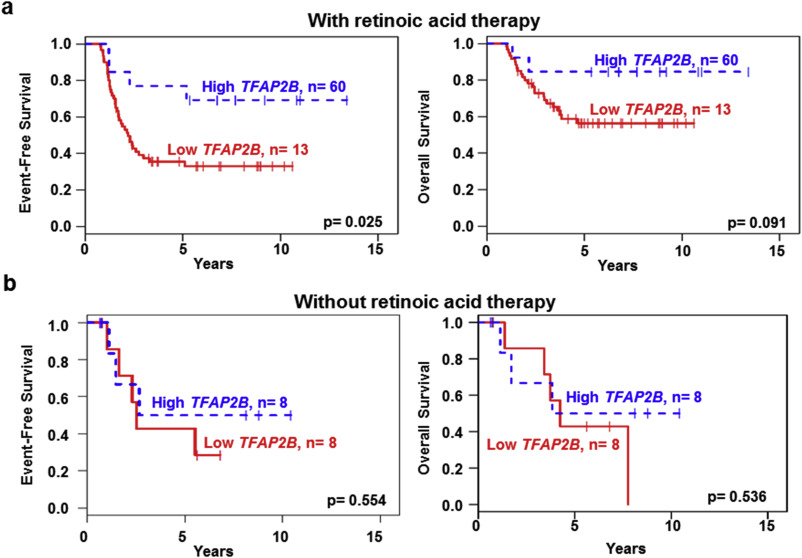
To determine whether TFAP2B expression is also associated with outcome in RA‐treated patients, we selected all German high‐risk patients who had received megatherapy with autologous stem cell transplantation and had been event‐free for at least 3 months after megatherapy (n = 89). In the RA treated subgroup (n = 73), we observed that patients whose tumors had higher TFAP2B expression levels had a significantly better EFS than those with lower TFAP2B expression (5‐year EFS 0.77 ± 0.12 vs 0.36 ± 0.06, p = 0.025; Supplementary Figure S2). Similarly, there was a trend towards a better OS in this cohort (5‐year OS 0.85 ± 0.10 vs 0.56 ± 0.07, p = 0.091). By contrast, TFAP2B expression did not distinguish patients with different outcome in the small subgroup without RA treatment (n = 16; 5‐year EFS, 0.50 ± 0.20 vs 0.43 ± 0.19, p = 0.554; 5‐year OS, 0.50 ± 0.20 vs 0.43 ± 0.19, p = 0.536). While these results have to be validated in larger cohorts, they may indicate that TFAP2B expression might serve as a biomarker to predict RA treatment success.
We also assessed whether TFAP2B expression is associated with the degree of morphological differentiation in primary neuroblastomas. We observed that poorly differentiated tumors had higher TFAP2B expression levels than both undifferentiated and differentiating tumors (Supplementary Figure S3a). On the molecular level, we noticed that TFAP2B expression was significantly correlated with gene expression of the catecholamine biosynthesizing enzymes dopamine beta hydroxylase (DBH) and tyrosine hydroxylase (TH), indicating that TFAP2B might up‐regulate expression of these enzymes in neuroblastoma (Supplementary Figure S3b and S3c).
3.2. Analysis of genetic and epigenetic alterations of the TFAP2B locus
We next aimed at investigating whether genetic or epigenetic alterations of the TFAP2B locus are associated with aberrant TFAP2B expression. First, we examined the TFAP2B methylation status using global methylation profiles of 105 primary neuroblastomas, which were generated by microarrays containing probes for 450K CpG sites. We observed a significant correlation between TFAP2B CpG methylation and low transcript expression levels (Supplementary Table S3), with strongest correlations seen for two CpG sites close to the transcription start site and one CpG site in exon 2 (p < 0.001 each; Figure 2a and 2b). Increased methylation levels of these CpG sites were found to be significantly associated with prognostic variables of poor outcome including MYCN amplification, stage 4, age of the patient at diagnosis >18 months and allocation to the high risk group according to the German NB2004 risk estimation system (p < 0.001 each; Figure 2c and Supplementary Figure S4. Together, these results suggest that low expression of TFAP2B in high risk neuroblastoma might be due to CpG methylation of the TFAP2B locus.
Figure 2.
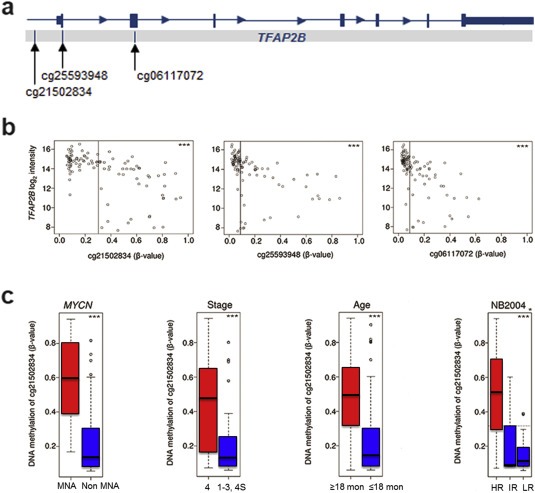
Association of CpG methylation of the TFAP2B locus with prognostic markers and TFAP2B expression in neuroblastoma. (a) TFAP2B genomic structure and localization of CpGs analyzed. (b) Correlation of TFAP2B methylation with TFAP2B expression. Log intensities of probe A_24_P20954 (TFAP2B expression) are plotted against β‐values of probes cg21502834, cg06117072 and cg25593948 representing CpGs in the 5′ region of the TFAP2B locus. (c) Association of TFAP2B methylation (probe cg21502834) with MYCN amplification status, tumor stage, age at diagnosis, and risk group according to NB2004 in 105 neuroblastoma samples as determined by 450K methylation arrays. Boxes, median expression values (horizontal line) and 25th and 75th percentiles; whiskers, distances from the end of the box to the largest and smallest observed values that are <1.5 box lengths from either end of the box; circles, outliers. (*), Wilcoxen test (HR vs IR/LR); (***), p < 0.001; MNA, MYCN‐amplified tumors.
Analysis of the copy number status of TFAP2B in 199 primary neuroblastoma samples using aCGH revealed that the TFAP2B locus is mostly not affected by genomic copy number alterations, and that TFAP2B copy numbers and TFAP2B expression are not correlated (Supplementary Figure S5). We also did not detect any somatic mutations affecting TFAP2B in primary neuroblastoma using whole‐genome (n = 30) and whole‐exome (n = 125) massive parallel sequencing data (unpublished data). Together, our data indicate that neither copy number alterations nor somatic mutations of TFAP2B are a major cause of reduced TFAP2B expression levels in unfavorable neuroblastoma.
3.3. TFAP2B expression is associated with RA responsiveness in neuroblastoma cell lines
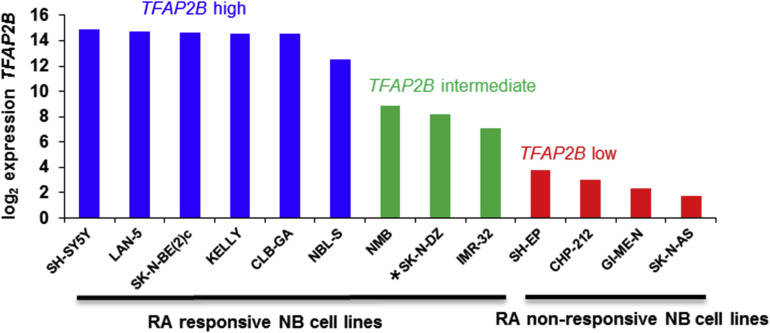
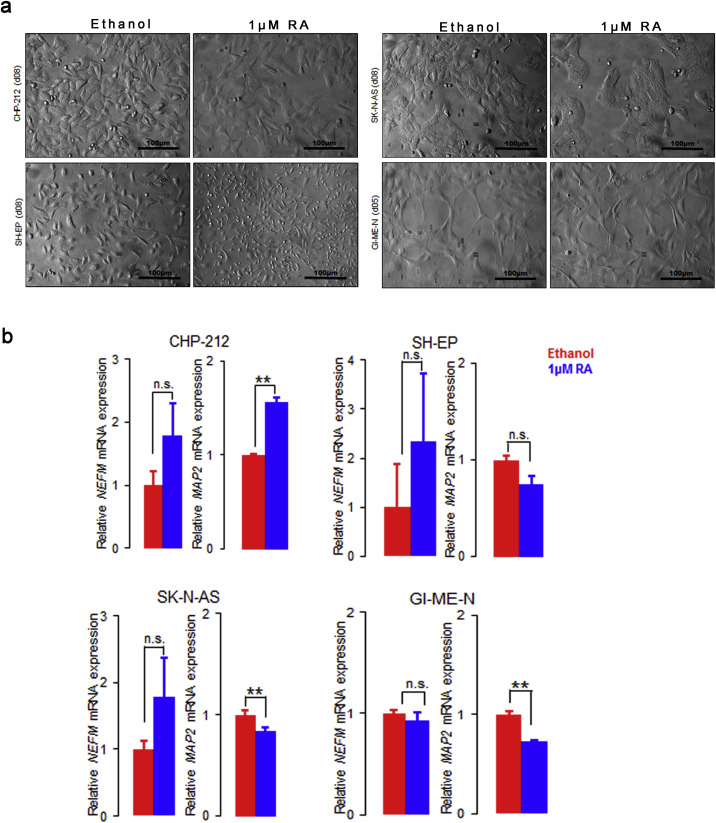
To examine the role of TFAP2B in neuroblastoma pathogenesis, we first screened thirteen neuroblastoma cell lines for endogenous TFAP2B expression both on the RNA and protein level, and categorized them as TFAP2B high (SK‐N‐BE(2)c, SH‐SY5Y, LAN‐5, CLB‐GA, NBL‐S and Kelly), TFAP2B intermediate (IMR‐32, NMB and SK‐N‐DZ) and TFAP2B low (CHP‐212, SH‐EP, GI‐ME‐N and SK‐N‐AS; Figure 3a and Supplementary Figure S6). Next, we asked whether TFAP2B expression of cell lines correlates with their responsiveness to RA. For this purpose, we evaluated morphological signs of differentiation and expression levels of the neuronal marker genes microtubule associated protein 2 (MAP2) and neurofilament middle chain (NEFM) in response to RA treatment. We found that neuronal marker genes were up‐regulated upon RA treatment in all six TFAP2B high cell lines, which was accompanied by induction of neurite‐like processes in five of them (Supplementary Figure S7). Likewise, signs of differentiation were readily detectable in the TFAP2B intermediate cell lines IMR‐32 and NMB (Supplementary Figure S8), while SK‐N‐DZ cells died rapidly upon RA treatment (not shown). By contrast, we did not detect any signs of neuronal differentiation upon RA treatment in TFAP2B low cell lines (Supplementary Figure S9). Contrary to TFAP2B, expression levels of the RA responsive genes RARB and CRABP2 were neither associated with RA sensitivity of the cell lines nor with TFAP2B expression (Supplementary Figure S10).
Figure 3.
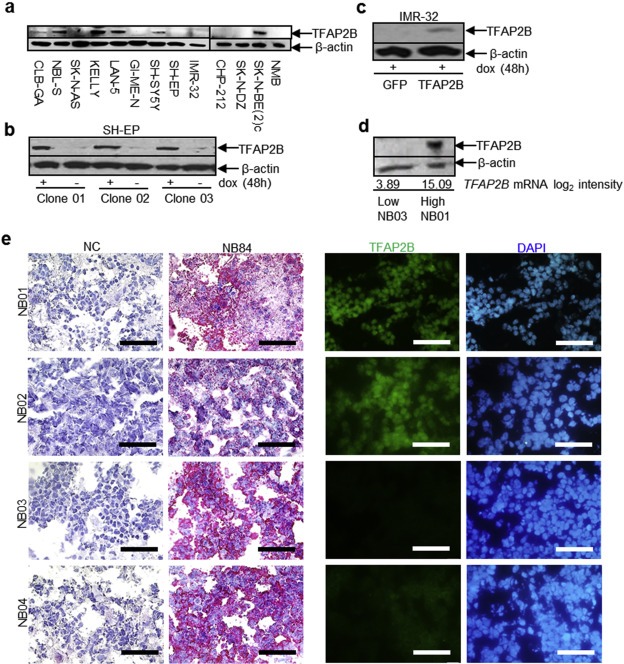
TFAP2B protein level in neuroblastoma cell lines and primary tumors. (a) Endogenous TFAP2B protein level in neuroblastoma cell lines. (b) Inducible expression of TFAP2B in SH‐EP cell clones. (c) Inducible expression of TFAP2B and GFP in IMR‐32 cells. (d) TFAP2B protein level in primary neuroblastoma samples; NB01 with high and NB03 with low TFAP2B mRNA expression according to microarray analysis. (e) Immunohistochemical staining of the neuroblastoma‐specific marker NB84 in four primary neuroblastoma samples (left panel; scale bar = 50 μm), and immunofluorescence staining of TFAP2B in the consecutive sections of the same samples (right panel; scale bar = 50 μm). NC, negative control.
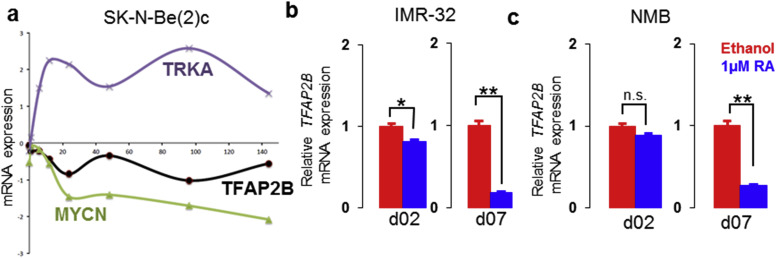
We also evaluated the expression dynamics of TFAP2B after RA treatment in TFAP2B high SK‐N‐BE(2)c cells and TFAP2B intermediate NMB and IMR‐32 cells. In all three cell lines, TFAP2B levels decreased upon RA treatment (Supplementary Figure S11). This finding is in line with TFAP2B expression patterns in primary neuroblastoma, which was highest in poorly differentiated tumors (Supplementary Figure S3a), indicating that high TFAP2B expression may be associated with a specific developmental stage of the progenitor cells, as suggested previously (Bourdeaut et al., 2009). Together with the fact that endogenous TFAP2B expression is closely associated with RA responsiveness of neuroblastoma cell lines, our data suggest that TFAP2B expression might represent rather a precondition to induce RA‐mediated differentiation programs than a downstream target of RA in neuroblastoma.
3.4. TFAP2B re‐expression impairs proliferation and cell cycle progression in neuroblastoma cell lines
To further evaluate the role of TFAP2B in neuroblastoma pathogenesis, TFAP2B expression was reconstituted in two cell lines (IMR‐32 and SH‐EP) using an inducible retroviral gene transfer system. Doxycycline‐induced TFAP2B protein levels in the cell lines were comparable to physiological protein levels observed in primary neuroblastomas with high TFAP2B expression (Figure 3b–e).
In SH‐EP cells, the effect of TFAP2B on neuroblastoma cell growth was explored in three stable clones allowing doxycycline‐inducible TFAP2B expression, whereas polyclonal TFAP2B expressing IMR‐32 cells were compared with polyclonal GFP‐expressing controls. We observed a significant reduction in proliferation after TFAP2B induction in both cell lines (Figure 4 and Supplementary Figure S12a). To determine whether reduced proliferation upon TFAP2B re‐expression might be due to impaired cell cycle progression, the DNA content of TFAP2B‐expressing cells was assessed by flow cytometry. We found a significant increase of the G0/G1 peak at day 6 after TFAP2B induction in both IMR‐32 cells and SH‐EP cells (Figure 4 and Supplementary Figure S12b), indicating that TFAP2B re‐expression impairs cell cycle progression from G1 to S phase. We also detected a significant increase of the sub‐G1 fraction after TFAP2B induction in IMR‐32 cells (Supplementary Figure S13a), indicating that cell death contributes to impaired proliferation in this cell line; a similar effect, however, was not observed in TFAP2B expressing SH‐EP cells (Supplementary Figure S13b).
Figure 4.
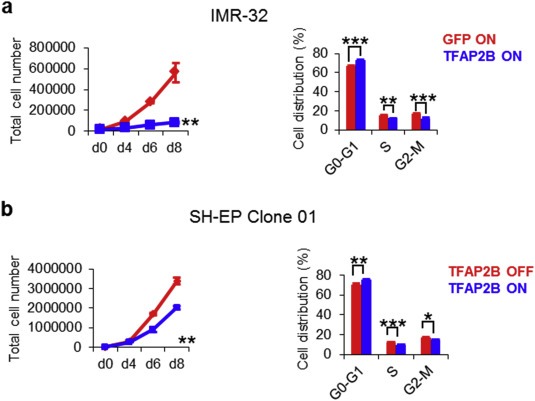
TFAP2B re‐expression inhibits growth of neuroblastoma cells. (a) Proliferation of IMR‐32 and SH‐EP cells upon TFAP2B re‐expression as assessed by trypan blue dye exclusion analysis. (b) Cell cycle analysis of IMR‐32 and SH‐EP cells at day 6 after TFAP2B induction as determined by FACS analysis. Fraction of cells in G0/G1 phase: SH‐EP OFF, 69.05% ± 0.47%; SH‐EP ON, 74.03% ± 0.26%, p < 0.01; IMR‐32 GFP, 65.50% ± 0.84%; IMR‐32 TFAP2B, 70.58% ± 0.59%, p < 0.001. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001).
3.5. TFAP2B induces neuronal differentiation in IMR‐32 cells
Microscopic inspection of TFAP2B‐expressing IMR‐32 cells revealed a high degree of morphological differentiation, as indicated by the development of neurite‐like processes (Figure 5a). To further investigate whether this neuron‐like morphology is associated with induction of neuronal markers, we measured expression levels of class III beta‐tubulin (TUBB3) by immunofluorescence, and MAP2, NEFM, neuron specific enolase (NSE) and synaptophysin (SYP) by RT‐qPCR, all of which were up‐regulated in TFAP2B‐expressing cells as compared to controls (Figure 5b,c). Similarly, expression levels of DBH and TH were induced upon TFAP2B re‐expression (Figure 5d), suggesting that cells differentiate into a noradrenergic phenotype.
Figure 5.
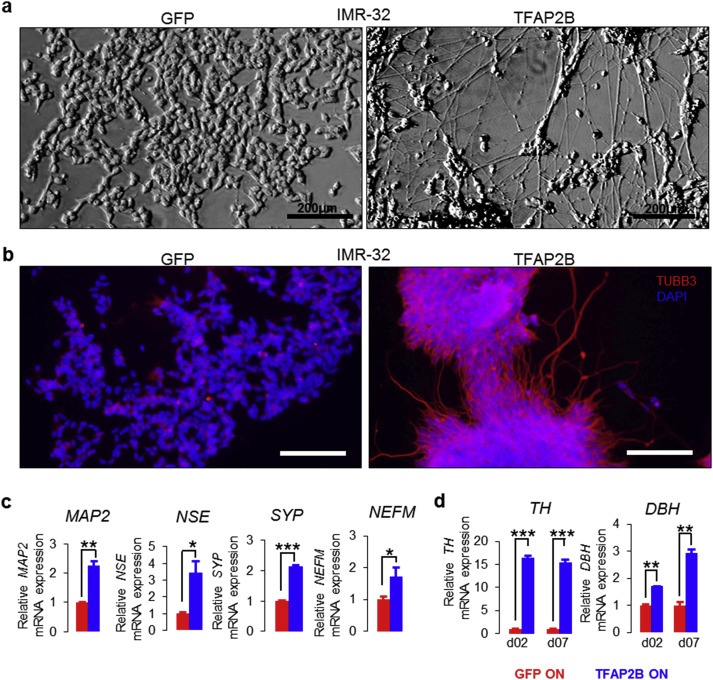
TFAP2B induces neuronal differentiation in IMR‐32 cells. (a) Morphological changes of IMR‐32 cells upon TFAP2B and GFP induction. Bright filed; Scale bar = 200 μm. (b) Immunofluorescence staining of TUBB3 in IMR‐32 after TFAP2B and GFP induction. Scale bar = 75 μm; Red = TUBB3; Blue = DAPI. (c) Analysis of the neuron‐related markers MAP2, NSE, SYP and NEFM in IMR‐32 cells upon TFAP2B re‐expression by RT‐qPCR. (d) Analysis of TH and DBH in IMR‐32 cells after TFAP2B re‐expression by RT‐qPCR. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001).
By contrast, we did not observe morphological differentiation in TFAP2B expressing SH‐EP cells (Supplementary Figure S14a). Consistent with this finding, up‐regulation of the markers TUBB3, MAP2 and DBH was lacking (Supplementary Figure S14b–d). We found, however, that impaired proliferation and G1‐arrest was associated with cellular senescence after TFAP2B induction in SH‐EP cells as assessed by acidic β‐galactosidase (SA‐β‐Gal) activity analysis (Supplementary Figure S15).
Given the fact that methylation of CpG sites in proximity to the TFAP2B transcription start site was strongly correlated with low TFAP2B expression levels in primary neuroblastomas, we wondered whether treatment of IMR‐32 cells with the DNA methyltransferase inhibitor 5‐aza‐2′‐deoxycytidine (DAC) may result in a similar phenotype as observed after transgenic TFAP2B re‐expression. After 3 days of DAC treatment, we indeed detected significantly increased TFAP2B expression levels, as well as induction of MAP2 and NEFM as compared to the controls (Supplementary Figure S16a and S16b). We also observed induction of neurite‐like processes and significant reduction in proliferation upon DAC treatment, which was followed, however, by death of the majority of cells (Supplementary Figure S16c and S16d). Accordingly, we found a significant increase of the sub‐G1 fraction along with a G2/M arrest of DAC treated cells (Supplementary Figure S16e), analogous to previous observations in other cancer entities (Shin et al., 2013). Together, these findings further substantiate that TFAP2B may be silenced by promoter methylation in unfavorable neuroblastoma.
3.6. Microarray‐based gene expression analysis of IMR‐32 cells upon TFAP2B induction
To gain insight into TFAP2B related mechanisms driving growth arrest and differentiation in IMR‐32 cells, we performed microarray‐based gene expression analysis at days 2 and 7 after TFAP2B induction in comparison to GFP vector control cells. Gene set enrichment analysis revealed that, among the entire curated pathway gene set collection, the Biocarta cell cycle pathway gene set was the most significant gene set enriched in TFAP2B samples (normalized enrichment score, 1.59; p = 0.016; Supplementary Figure S17a). Among the genes in this set, we observed that expression of the cell cycle inhibitor gene CDKN2A was significantly induced in TFAP2B expressing cells. By contrast, expression of the cyclin‐dependent kinase genes CDK4 and CDK6 decreased upon TFAP2B re‐expression, supporting the notion of impaired cell cycle progression in TFAP2B expressing cells.
Furthermore, microarray‐based expression analysis validated up‐regulation of the neuronal marker genes investigated in this study (TUBB3, MAP2, NEFM, NSE, SYP, DBH and TH) and the catecholamine biosynthesizing enzyme genes DBH and TH (Supplementary Figure S17b). In addition, we found TFAP2B mediated induction of the neurotrophin receptor gene NTRK1, which has been implicated in neuroblastoma differentiation and regression and is strongly associated with favorable patient outcome (Brodeur, 2003). By contrast, the RE1‐silencing transcription factor REST, a master repressor of neuronal genes (Thiel et al., 2015), was persistently down‐regulated upon TFAP2B induction, indicating that TFAP2B enables differentiation in neuroblastoma cells by repression of REST.
Since TFAP2 transcription factors have been described to act as a negative regulator of Myc‐mediated transactivation (Gaubatz et al., 1995), we also assessed TFAP2B and MYCN expression in primary neuroblastoma and in cell line models. In primary tumors, TFAP2B and MYCN expression levels were inversely correlated (Figure 1c and Supplementary Figure S18a). In line with this observation, MYCN was down‐regulated in IMR‐32 cells upon TFAP2B expression (Supplementary Figure S17b), while TFAP2B was down‐regulated upon MYCN induction in SH‐SY5Y, a non‐MYCN amplified cell line with high endogenous TFAP2B expression (Supplementary Figure S18b and S18c). Together, these data suggest that TFAP2B and MYCN affect each other reciprocally by direct or indirect repression, thereby promoting a favorable and an unfavorable tumor phenotype, respectively.
3.7. Knockdown of TFAP2B abrogates RA‐induced neuronal differentiation
To validate the significance of TFAP2B in neuronal differentiation of neuroblastoma, we evaluated whether silencing of TFAP2B using lentiviral shRNAs in SK‐N‐BE(2)c and SH‐SY5Y neuroblastoma cells may impair RA induced neuronal differentiation. These two cell lines express high endogenous TFAP2B protein levels (Figure 3a) and are known to differentiate in response to RA into neuronal type cells (Supplementary Figure S7) (Jeong et al., 2006; Rebhan et al., 1994). We tested 4 lentiviral constructs expressing short hairpin RNA sequences against human TFAP2B (shTFAP2B), and three of them significantly reduced TFAP2B protein levels in both SH‐SY5Y and SK‐N‐BE(2)c cells (Figure 6a). The effects of RA treatment were assessed in both cell lines after TFAP2B knockdown and in control cells. Upon TFAP2B knockdown, we observed a significant reduction of cells in the G1 phase of the cell cycle in both RA‐treated SH‐SY5Y and SK‐N‐BE(2)c cells (Figure 6b and Supplementary Figure S19). Furthermore, morphological signs of neuronal differentiation were readily detectable in RA‐treated control cells, but absent in neuroblastoma cells lacking TFAP2B expression (Figure 7, Supplementary Figures S20 and S21). In addition, knockdown of TFAP2B severely impaired up‐regulation of the neuronal markers MAP2 (Figure 8a, Supplementary Figures S22 and S23) as well as NEFM and SYP (Figure 8b and 8c). In line with these findings, we found that expression of DBH was significantly diminished in both RA‐treated SH‐SY5Y and SK‐N‐BE(2)c cells after TFAP2B knock‐down in comparison to the controls (Figure 8b and 8c). Similarly, expression of TH was down‐regulated in RA‐treated SK‐N‐BE(2)c cells after TFAP2B knock‐down, while TH expression levels was not measurable by RT‐qPCR in SH‐SY5Y cells in our study. Together, the reverse phenotypes observed in RA‐treated neuroblastoma cells after TFAP2B knockdown and in recombinant TFAP2B expressing neuroblastoma cells suggest that TFAP2B is playing a vital role in retaining RA responsiveness and in mediating differentiation into a noradrenergic neuronal phenotype in neuroblastoma.
Figure 6.
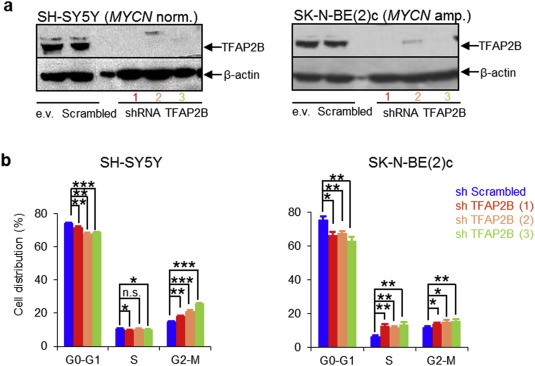
Knockdown of TFAP2B accelerates cell cycle progression upon RA treatment in neuroblastoma cells. (a) Knockdown efficiency of TFAP2B in SH‐SY5Y and SK‐N‐BE(2)c cells as determined by Western blot analysis; e.v., empty vector control. (b) Cell cycle analysis of SH‐SY5Y and SK‐N‐BE(2)c cells after treatment with 1 μM RA for 6 days and 10 days, respectively. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001.
Figure 7.
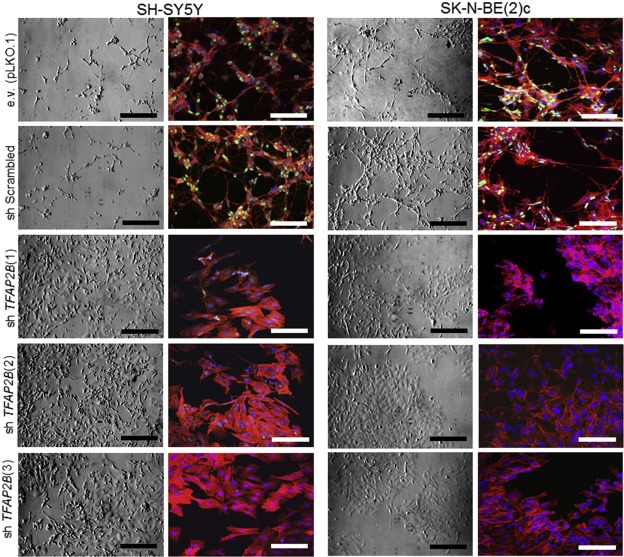
Knockdown of TFAP2B abrogates RA‐induced neuronal differentiation in neuroblastoma cells. Morphological changes in SH‐SY5Y and SK‐N‐BE(2)c cells after treatment with 1 μM RA for 6 and 10 days, respectively. Left panel, bright field; scale bar = 200 μm; right panel, fluorescent; scale bar = 50 μm; Green = TFAP2B; Red = actin filaments; Blue = DAPI.
Figure 8.
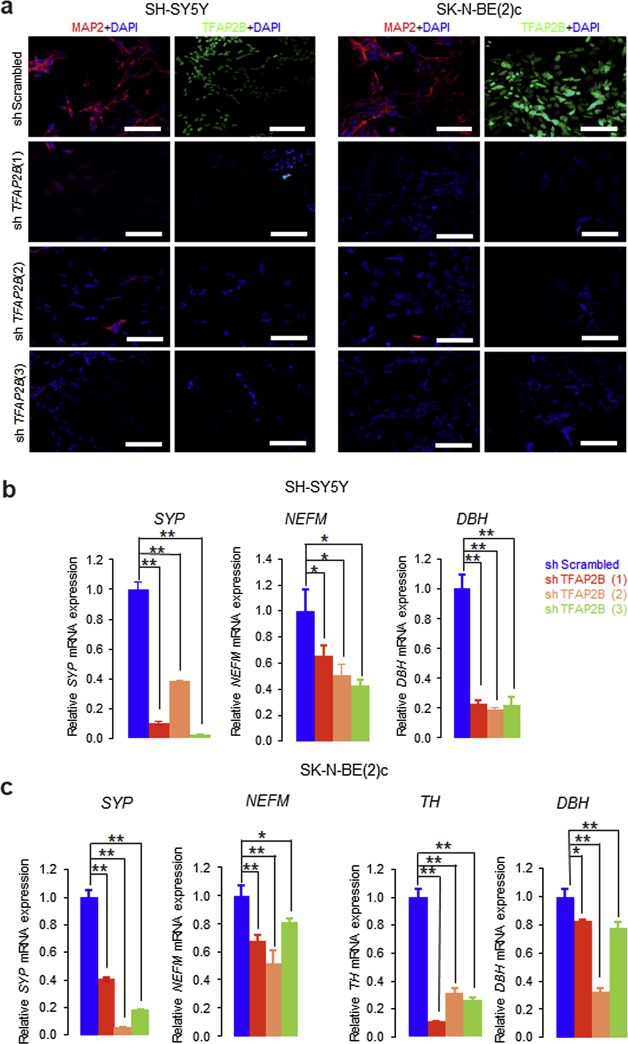
Knockdown of TFAP2B leads to lack of upregulation of RA‐induced neuronal differentiation markers in neuroblastoma cells. (a) Immunofluorescence staining of MAP2 in SH‐SY5Y and SK‐N‐BE(2)c cells expressing shScrambled or shTFAP2B after RA treatment. Scale bar = 50 μm. (b) Analysis of neuronal differentiation markers SYP, NEFM and DBH in SH‐SY5Y and (c)SYP, NEFM, TH and DBH in SK‐N‐BE(2)c cells expressing shScrambled or shTFAP2B after RA treatment by RT‐qPCR. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01.
4. Discussion
Neuroblastoma is a pediatric solid tumor that originates from embryonal neural crest cells committed to the development of the sympathetic nervous system. In most cases, neuroblastoma presents as a tumor consisting of poorly differentiated small round blue cells, which may, however, differentiate into more benign tumor phenotypes (i.e., ganglioneuroblastoma or ganglioneuroma) either spontaneously or following cytotoxic treatment. Since many transcriptional regulators (e.g., PHOX2B, MYCN) involved in developmental differentiation processes are dysregulated in this malignancy (Nakagawara and Ohira, 2004), it has been suggested that the physiologic program of neuroblast differentiation and growth control is disrupted in neuroblastoma (Nakagawara et al., 1993). We here provide further evidence for this hypothesis by showing that the developmental transcription factor TFAP2B is significantly involved in mediating neuroblastoma differentiation.
In our study, we demonstrate that high TFAP2B expression levels are strongly associated with favorable prognostic characteristics and beneficial outcome in neuroblastoma. In neuroblastoma cell line models, re‐expression of TFAP2B led to impaired cell cycle progression associated with noradrenergic neuronal differentiation or senescence. TFAP2B re‐expression in IMR‐32 cells was accompanied by down‐regulation of MYCN and REST, suggesting that direct or indirect repression of transcription factors critically involved in sympathoadrenal development may promote TFAP2B mediated differentiation in neuroblastoma (Marshall et al., 2014). Our observations are in line with previous reports, in which TFAP2B was found to be predominantly expressed in the sympathetic ganglia of developing mouse embryos, pointing towards its role in sympathetic nervous system development (Hong et al., 2008). In addition, knock‐out of Tfap2b in transgenic mice significantly impaired the development of sympathetic ganglia, while forced TFAP2B expression in chick neural crest stem cells favored their differentiation into sympathoadrenergic cells (Hong et al., 2008). In another murine knock‐out model, Tfap2b was found to be essential for the survival of migrating neural crest cells committed to the development of sympathetic neurons (Schmidt et al., 2011). Together, our data suggest that TFAP2B contributes to a favorable phenotype and noradrenergic neuronal differentiation in neuroblastoma, which may reflect its physiological role during sympathetic nervous system development.
We also observed that knockdown of TFAP2B abrogates neuronal differentiation of the tumor cells in the presence of RA. The signaling molecule RA has been shown to induce differentiation of neuroblastoma cells in experimental systems, which has led to the integration of RA‐based maintenance therapies in current treatment protocols to prevent relapse (Matthay et al., 1999). The mechanisms underlying spontaneous and RA‐induced neuroblastoma differentiation are, however, still poorly understood. We here provide evidence that TFAP2B expression may be necessary to enable RA‐mediated differentiation of neuroblastoma cells, indicating that TFAP2B and RA may cooperate in promoting neuronal differentiation in neuroblastoma. Induction of TFAP2 expression upon RA treatment has been previously shown to occur during neuroectodermal differentiation of embryonal carcinoma cells, while it was absent during mesoendodermal differentiation (Philipp et al., 1994). In contrast to these observations, we found that TFAP2B expression was down‐regulated in RA treated neuroblastoma cells, suggesting that TFAP2B expression is rather a precondition to enable RA‐mediated differentiation than a downstream target of RA in neuroblastoma. In line with the latter hypothesis, we found highest TFAP2B levels in poorly differentiated tumors, while it was lower in differentiating and stroma‐rich tumors, as reported previously (Albino et al., 2008; Bourdeaut et al., 2009). Thus, our data are in support of the notion that TFAP2B expression is associated with a specific developmental stage of the neuroblastoma progenitor cells (Bourdeaut et al., 2009), which may render the tumor cells susceptible to signaling molecules inducing neuronal differentiation.
We did not find any evidence of genetic alterations of the TFAP2B locus, but noticed that methylation of CpG sites in the 5′ region of TFAP2B is associated with low TFAP2B expression and unfavorable prognostic markers. We also observed up‐regulation of TFAP2B and neuronal markers in IMR‐32 neuroblastoma cells upon treatment with the DNA methyltransferase inhibitor DAC. These data may suggest that TFAP2B is silenced by DNA methylation of CpG sites at the TFAP2B locus. In neuroblastoma, gene repression by genomic methylation has been shown to be involved in several putative tumor suppressor genes (Abe et al., 2005; Decock et al., 2012; Michalowski et al., 2008). In addition, aberrant DNA methylation of TFAP2 family members and induction of their expression upon DAC treatment have been described previously in cancer (Douglas et al., 2004; Tong et al., 2010). Another recent study reported that CpG sites at the TFAP2B locus are highly methylated in esophageal squamous cell carcinoma but not in normal esophageal mucosa (Takahashi et al., 2013). Together, these data point towards genomic DNA methylation as a mechanism to silence TFAP2B expression in neuroblastoma. Alternatively, other molecular mechanisms, such as upstream transcriptional regulators or microRNAs, may also contribute to regulating expression levels of TFAP2B (Plouhinec et al., 2014).
In conclusion, our results point towards a vital role of TFAP2B in mediating noradrenergic neuronal differentiation in neuroblastoma and in retaining RA responsiveness, and indicate that TFAP2B expression is silenced by aberrant DNA methylation in high risk neuroblastoma. Our data may be used as a starting point to further elucidate the molecular mechanisms of neuroblastoma differentiation, which may eventually lead to the development of biomarker‐based patient stratification systems for differentiation treatment strategies.
Conflict of interest
The authors have no conflict of interest to declare.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary Figure S1 Survival analysis and performance of the prognostic TFAP2B expression. Cut‐off (mRNA intensities <19830.07 vs ≥ 19830.07) in the training set. n = patient number; (***), p < 0.001.
{kind=link}
Supplementary Figure S2 TFAP2B expression as a prognostic marker for RA treatment. (a) EFS and OS of high‐risk neuroblastoma patients who received RA therapy according to TFAP2B expression. n = patient number. (b) EFS and OS of high‐risk neuroblastoma patients who did not receive RA treatment according to TFAP2B expression. n = patient number.
{kind=link}
Supplementary Figure S3 TFAP2B gene expression and differentiation assessment in primary neuroblastomas. (a) Association of TFAP2B expression with differentiation status of primary neuroblastoma tumors. (n.s.), p > 0.05; (**), p < 0.01. (b) Correlation of TFAP2B expression with DBH expression in 649 primary neuroblastomas. 95% confidence interval of r = [0.73; 0.80]; (***), p < 0.001. (c) Correlation of TFAP2B expression with TH expression in 649 primary neuroblastomas. 95% confidence interval of r = [0.51; 0.62]; (***), p < 0.001; n = patient number.
{kind=link}
Supplementary Figure S4 Association of TFAP2B methylation status with prognostic markers. Association of MYCN amplification status, tumor stage, age at diagnosis and risk group (NB2004) in 105 neuroblastoma samples with TFAP2B methylation status as determined by 450K methylome array (probe ID: cg06117072 and cg25593948). Boxes, median expression values (horizontal line) and 25th and 75th percentiles; whiskers, distances from the end of the box to the largest and smallest observed values that are <1.5 box lengths from either end of the box. Wilcoxen test (HR vs IR/LR); (***), p < 0.001; MNA, MYCN‐amplified tumors.
{kind=link}
Supplementary Figure S5 Correlation of TFAP2B copy number with expression in 199 primary neuroblastoma samples.
{kind=link}
Supplementary Figure S6 Microarray expression data showing TFAP2B expression levels in primary neuroblastoma cell lines. Blue, high TFAP2B expression; Green, intermediate TFAP2B expression; Red, low TFAP2B expression.
{kind=link}
Supplementary Figure S7 Responsiveness of TFAP2Bhigh neuroblastoma cells to retinoic acid. (a) Changes in morphology of TFAP2Bhigh neuroblastoma cells treated with 1 μM retinoic acid. Scale bar = 100 μM. (b) Analysis of neuronal markers NEFM and MAP2 in TFAP2Bhigh neuroblastoma cells after treatment with 1 μM retinoic acid by qRT‐PCR. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001; (****), p < 0.0001.
{kind=link}
Supplementary Figure S8 Responsiveness of TFAP2Bintermediate neuroblastoma cells to retinoic acid. (a) Changes in morphology of TFAP2Bintermediate neuroblastoma cells treated with 1 μM retinoic acid. Scale bar = 100 μM. (b) Analysis of neuronal markers NEFM and MAP2 in TFAP2Bintermediate neuroblastoma cells after treatment with 1 μM retinoic acid by qRT‐PCR. Error bars indicate S.D. (*), p < 0.05; (**), p < 0.01; (***), p < 0.001; (****), p < 0.0001. *SK‐N‐DZ cells were not analyzed further as they died rapidly upon RA treatment.
{kind=link}
Supplementary Figure S9 Responsiveness of TFAP2Blow neuroblastoma cells to retinoic acid. (a) Changes in morphology of TFAP2Blow neuroblastoma cells treated with 1 μM retinoic acid. Scale bar = 100 μM. (b) Analysis of neuronal markers NEFM and MAP2 in TFAP2Blow neuroblastoma cells after treatment with 1 μM retinoic acid by qRT‐PCR. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001; (****), p < 0.0001.
{kind=link}
Supplementary Figure S10 Correlation of TFAP2B expression with the expression of RA responsive genes RARB and CRABP2. Microarray expression data showing (a) RARB and (b) CRABP2 expression levels in neuroblastoma cell lines. Blue, high TFAP2B expression; Green, intermediate TFAP2B expression; Red, low TFAP2B expression (c) Correlation of TFAP2B expression with RARB expression in neuroblastoma cell lines. r = [0.15], p = 0.633. (d) Correlation of TFAP2B expression with CRABP2 expression in neuroblastoma cell lines. r = [0], p = 0.999.
{kind=link}
Supplementary Figure S11 Expression of TFAP2B after RA treatment. (a) RNA sequencing analysis of TFAP2B, MYCN and TRKA expression in time series over 144h in TFAP2Bhigh SK‐N‐BE(2)c cells after 10 μM retinoic acid treatment. (b) Analysis of TFAP2B expression in TFAP2Bintermediate IMR‐32 and (c) NMB cells after 1 μM retinoic acid treatment by RT‐qPCR. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01.
{kind=link}
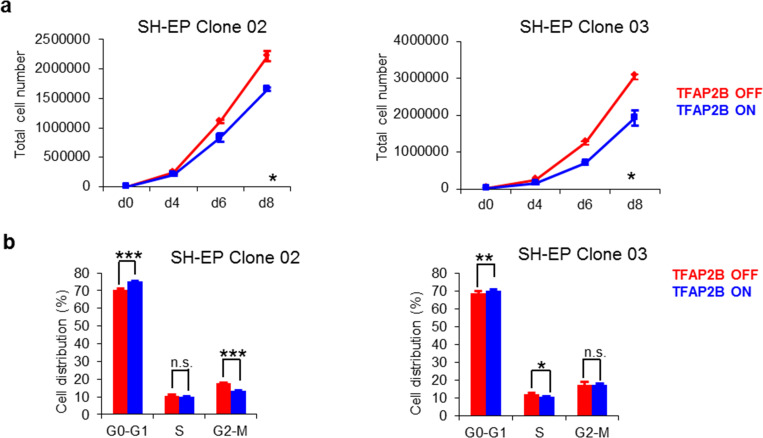
Supplementary Figure S12 TFAP2B re‐expression inhibits growth of neuroblastoma cells. (a) Reduction in proliferation in SH‐EP clones. (b) Increased proportion of SH‐EP cells in G0/G1 phase 6 days after TFAP2B induction. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001.
{kind=link}
Supplementary Figure S13 sub‐G1 phase of cell cycle after TFAP2B induction in neuroblastoma cells. (a) sub‐G1 arrest in IMR‐32 cells after TFAP2B expression. (b) Proportion of SH‐EP cells in sub‐G1 phase at day 6 after TFAP2B induction. Error bars indicate S.D; (n.s.), p > 0.05; (***), p < 0.001.
{kind=link}
Supplementary Figure S14 SH‐EP cells after TFAP2B re‐expression. (a) Bright field microscopy of SH‐EP cells after TFAP2B re‐expression. Scale bar = 200 μM. (b) Immunofluorescence staining of SH‐EP cells for the neuronal marker TUBB3 after TFAP2B induction. Wide field fluorescent; Scale bar = 75 μm; Red = TUBB3; Blue = DAPI. (c) Analysis of the neuronal marker MAP2 in SH‐EP cells after TFAP2B induction at day 2 and 7 by RT‐qPCR. (d) Analysis of the noradrenaline neuronal marker DBH in SH‐EP cells after TFAP2B induction at day 2 and 7 by RT‐qPCR. Error bars indicate S.D; n.s., p > 0.05; (*), p < 0.05; (**), p < 0.01.
{kind=link}
Supplementary Figure S15 Analysis of senescence associated β‐galactosidase activity after TFAP2B re‐expression in SH‐EP cells.
{kind=link}
Supplementary Figure S16 IMR‐32 cells after DAC treatment. (a) Expression of TFAP2B and (b) neuronal markers MAP2 and NEFM in IMR‐32 cells after 3 days of 3 μM DAC treatment as determined by RT‐qPCR. (c) Bright field microscopy of IMR‐32 cells after 3 μM DAC treatment. Scale bar = 100 μM. (d) Growth arrest in IMR‐32 cells after 3 μM DAC treatment. (e) Cell cycle changes in IMR‐32 cells after 3 days of 3 μM DAC or DMSO treatment. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001.
{kind=link}
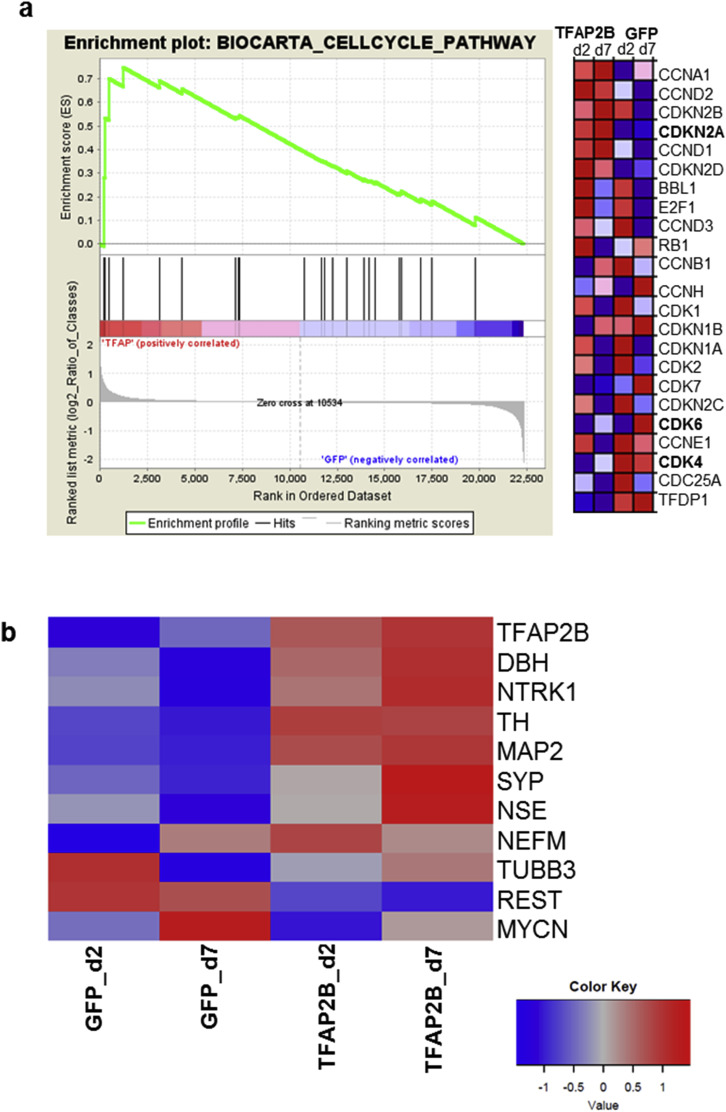
Supplementary Figure S17 TFAP2B induces changes in the expression of genes involved in cell cycle. (a) Gene set enrichment analysis of the Biocarta cell cycle pathway gene set in IMR‐32 cells after 2 and 7 days of TFAP2B induction. Normalized enrichment score, 1.59; p = 0.016. (b) Heat map showing the expression of neuronal genes in IMR‐32 cells after 2 and 7 days of TFAP2B induction.
{kind=link}
Supplementary Figure S18 TFAP2B negatively regulates MYCN in neuroblastoma. (a) Correlation of TFAP2B expression with MYCN expression in 649 primary neuroblastoma. (b) RNA‐sequencing data shows low expression of TFAP2B after 48h of MYCN induction in SH‐SY5Y cells. (c) Normalized TFAP2B expression in SH‐SY5Y cells after MYCN induction as determined by RNA sequencing. (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001.
{kind=link}
Supplementary Figure S19 Proportion of SK‐N‐BE(2)c and SH‐SY5Y cells in sub‐G1 after TFAP2B knock‐down. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01.
{kind=link}
Supplementary Figure S20 TFAP2B knockdown abrogates neuronal differentiation in SH‐SY5Y cells. Induction of differentiation by treatment with 1 μM retinoic acid in SH‐SY5Y cells. Cells with TFAP2B knockdown did not show RA‐induced differentiation. Scale bar = 50 μm. Green = TFAP2B, Red = actin filament, Blue = DAPI. Single channel images are shown in greyscale.
{kind=link}
Supplementary Figure S21 TFAP2B knockdown abrogates neuronal differentiation in SK‐N‐BE(2)c cells. Induction of differentiation by treatment with 1 μM retinoic acid in SK‐N‐BE(2)c cells. Cells with TFAP2B knockdown did not show RA‐induced neuronal differentiation. Scale bar = 50 μm. Green = TFAP2B, Red = actin filament, Blue = DAPI. Single channel images are shown in greyscale.
{kind=link}
Supplementary Figure S22 TFAP2B knockdown inhibits up‐regulation of MAP2 in SH‐SY5Y cells after RA treatment. Immunofluorescence staining for the differentiation marker MAP2 in SH‐SY5Y cells after treatment with retinoic acid for 6 days. Scale bar = 50 μm. Red = MAP2, Blue = DAPI. Single channel images are shown in greyscale.
{kind=link}
Supplementary Figure S23 TFAP2B knockdown inhibits up‐regulation of MAP2 in SK‐N‐BE(2)c neuroblastoma cells after RA treatment. Immunofluorescence staining for the differentiation marker MAP2 in SK‐N‐BE(2)c cells after treatment with retinoic acid for 10 days. Scale bar = 50 μm. Single channel images are shown in greyscale.
{kind=link}
Acknowledgements
The authors thank Andrea Krämer for valuable suggestions and Mirjam Koker for helping us with the lentiviral mediated knockdown experiments. This work was supported by grants from the Cologne Center for Molecular Medicine Cologne (CMMC); the Bundesministerium für Bildung und Forschung (BMBF) through the National Genome Research Network plus (NGFNplus, Grant No. 01GS0895 to MF and Grant No. 01GS0896 to KOH and FW) and through the e:Med initiative (Grant No. 01ZX1303A to MF and Grant No. 01ZX1307D to MF and FW); the Fördergesellschaft Kinderkrebs‐Neuroblastom‐Forschung e.V.; the Competence Network Pediatric Oncology and Hematology (KPOH); the EU (FP6) E.E.T. Pipeline (Grant No. 037260 to DD and FW); the EU (FP7) Asset (Grant No. 259348 to DD and FW); the BMBF MYC‐NET, CancerSys (Grant No. 0316076A to DD and FW). We are thankful to HEC Pakistan and DAAD for 4 years PhD scholarship to FI.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.10.020.
Ikram Fakhera, Ackermann Sandra, Kahlert Yvonne, Volland Ruth, Roels Frederik, Engesser Anne, Hertwig Falk, Kocak Hayriye, Hero Barbara, Dreidax Daniel, Henrich Kai-Oliver, Berthold Frank, Nürnberg Peter, Westermann Frank, Fischer Matthias, (2016), Transcription factor activating protein 2 beta (TFAP2B) mediates noradrenergic neuronal differentiation in neuroblastoma, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.10.020.
References
- Abe, M. , Ohira, M. , Kaneda, A. , Yagi, Y. , Yamamoto, S. , Kitano, Y. , Takato, T. , Nakagawara, A. , Ushijima, T. , 2005. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 65, 828–834. [PubMed] [Google Scholar]
- Albino, D. , Scaruffi, P. , Moretti, S. , Coco, S. , Truini, M. , Di Cristofano, C. , Cavazzana, A. , Stigliani, S. , Bonassi, S. , Tonini, G.P. , 2008. Identification of low intratumoral gene expression heterogeneity in neuroblastic tumors by genome-wide expression analysis and game theory. Cancer. 113, 1412–1422. [DOI] [PubMed] [Google Scholar]
- Auman, H.J. , Nottoli, T. , Lakiza, O. , Winger, Q. , Donaldson, S. , Williams, T. , 2002. Transcription factor AP-2gamma is essential in the extra-embryonic lineages for early postimplantation development. Development. 129, 2733–2747. [DOI] [PubMed] [Google Scholar]
- Bourdeaut, F. , Janoueix-Lerosey, I. , Lucchesi, C. , Paris, R. , Ribeiro, A. , de Pontual, L. , Amiel, J. , Lyonnet, S. , Pierron, G. , Michon, J. , Peuchmaur, M. , Delattre, O. , 2009. Cholinergic switch associated with morphological differentiation in neuroblastoma. J. Pathol. 219, 463–472. [DOI] [PubMed] [Google Scholar]
- Brodeur, G.M. , 2003. Neuroblastoma: biological insights into a clinical enigma. Nature reviews. Cancer. 3, 203–216. [DOI] [PubMed] [Google Scholar]
- Brodeur, G.M. , Pritchard, J. , Berthold, F. , Carlsen, N.L. , Castel, V. , Castelberry, R.P. , De Bernardi, B. , Evans, A.E. , Favrot, M. , Hedborg, F. , 1993. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 11, 1466–1477. [DOI] [PubMed] [Google Scholar]
- Cohn, S.L. , Pearson, A.D. , London, W.B. , Monclair, T. , Ambros, P.F. , Brodeur, G.M. , Faldum, A. , Hero, B. , Iehara, T. , Machin, D. , Mosseri, V. , Simon, T. , Garaventa, A. , Castel, V. , Matthay, K.K. , 2009. The international neuroblastoma risk group (INRG) classification system: an INRG Task Force report. J. Clin. Oncol. 27, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Preter, K. , Vandesompele, J. , Heimann, P. , Yigit, N. , Beckman, S. , Schramm, A. , Eggert, A. , Stallings, R.L. , Benoit, Y. , Renard, M. , De Paepe, A. , Laureys, G. , Pahlman, S. , Speleman, F. , 2006. Human fetal neuroblast and neuroblastoma transcriptome analysis confirms neuroblast origin and highlights neuroblastoma candidate genes. Genome Biol. 7, R84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock, A. , Ongenaert, M. , Hoebeeck, J. , De Preter, K. , Van Peer, G. , Van Criekinge, W. , Ladenstein, R. , Schulte, J.H. , Noguera, R. , Stallings, R.L. , Van Damme, A. , Laureys, G. , Vermeulen, J. , Van Maerken, T. , Speleman, F. , Vandesompele, J. , 2012. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol. 13, R95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas, D.B. , Akiyama, Y. , Carraway, H. , Belinsky, S.A. , Esteller, M. , Gabrielson, E. , Weitzman, S. , Williams, T. , Herman, J.G. , Baylin, S.B. , 2004. Hypermethylation of a small CpGuanine-rich region correlates with loss of activator protein-2alpha expression during progression of breast cancer. Cancer Res. 64, 1611–1620. [DOI] [PubMed] [Google Scholar]
- Eckert, D. , Buhl, S. , Weber, S. , Jager, R. , Schorle, H. , 2005. The AP-2 family of transcription factors. Genome Biol. 6, 246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, M. , Bauer, T. , Oberthur, A. , Hero, B. , Theissen, J. , Ehrich, M. , Spitz, R. , Eils, R. , Westermann, F. , Brors, B. , Konig, R. , Berthold, F. , 2010. Integrated genomic profiling identifies two distinct molecular subtypes with divergent outcome in neuroblastoma with loss of chromosome 11q. Oncogene. 29, 865–875. [DOI] [PubMed] [Google Scholar]
- Fischer, M. , Oberthuer, A. , Brors, B. , Kahlert, Y. , Skowron, M. , Voth, H. , Warnat, P. , Ernestus, K. , Hero, B. , Berthold, F. , 2006. Differential expression of neuronal genes defines subtypes of disseminated neuroblastoma with favorable and unfavorable outcome. Clin. Cancer Res. 12, 5118–5128. [DOI] [PubMed] [Google Scholar]
- Fischer, M. , Skowron, M. , Berthold, F. , 2005. Reliable transcript quantification by real-time reverse transcriptase-polymerase chain reaction in primary neuroblastoma using normalization to averaged expression levels of the control genes HPRT1 and SDHA. J. Mol. Diagn. 7, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredlund, E. , Ringner, M. , Maris, J.M. , Pahlman, S. , 2008. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc. Natl. Acad. Sci. U. S. A. 105, 14094–14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L. , Shi, K. , Wang, J. , Chen, W. , Shi, D. , Tian, Y. , Guo, W. , Yu, W. , Xiao, X. , Kang, T. , Wang, S. , Huang, W. , Deng, W. , 2014. TFAP2B overexpression contributes to tumor growth and a poor prognosis of human lung adenocarcinoma through modulation of ERK and VEGF/PEDF signaling. Mol. Cancer. 13, 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubatz, S. , Imhof, A. , Dosch, R. , Werner, O. , Mitchell, P. , Buettner, R. , Eilers, M. , 1995. Transcriptional activation by Myc is under negative control by the transcription factor AP-2. EMBO J. 14, 1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, S.J. , Huh, Y.H. , Leung, A. , Choi, H.J. , Ding, Y. , Kang, U.J. , Yoo, S.H. , Buettner, R. , Kim, K.S. , 2011. Transcription factor AP-2beta regulates the neurotransmitter phenotype and maturation of chromaffin cells. Mol. Cell Neurosciences. 46, 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, S.J. , Lardaro, T. , Oh, M.S. , Huh, Y. , Ding, Y. , Kang, U.J. , Kirfel, J. , Buettner, R. , Kim, K.S. , 2008. Regulation of the noradrenaline neurotransmitter phenotype by the transcription factor AP-2beta. J. Biol. Chem. 283, 16860–16867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn, T. , Lausen, B. , 2002. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 43, 121–137. [Google Scholar]
- Jeong, H. , Kim, M.S. , Kim, S.W. , Kim, K.S. , Seol, W. , 2006. Regulation of tyrosine hydroxylase gene expression by retinoic acid receptor. J. Neurochem. 98, 386–394. [DOI] [PubMed] [Google Scholar]
- Kocak, H. , Ackermann, S. , Hero, B. , Kahlert, Y. , Oberthuer, A. , Juraeva, D. , Roels, F. , Theissen, J. , Westermann, F. , Deubzer, H. , Ehemann, V. , Brors, B. , Odenthal, M. , Berthold, F. , Fischer, M. , 2013. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 4, e586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lausen, B. , Schumacher, M. , 1992. Maximally selected rank statistics. Biometrics. 48, 73–85. [Google Scholar]
- Luscher, B. , Mitchell, P.J. , Williams, T. , Tjian, R. , 1989. Regulation of transcription factor AP-2 by the morphogen retinoic acid and by second messengers. Genes Development. 3, 1507–1517. [DOI] [PubMed] [Google Scholar]
- Marshall, G.M. , Carter, D.R. , Cheung, B.B. , Liu, T. , Mateos, M.K. , Meyerowitz, J.G. , Weiss, W.A. , 2014. The prenatal origins of cancer. Nature reviews. Cancer. 14, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay, K.K. , Reynolds, C.P. , Seeger, R.C. , Shimada, H. , Adkins, E.S. , Haas-Kogan, D. , Gerbing, R.B. , London, W.B. , Villablanca, J.G. , 2009. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children's oncology group study. J. Clin. Oncol. 27, 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay, K.K. , Villablanca, J.G. , Seeger, R.C. , Stram, D.O. , Harris, R.E. , Ramsay, N.K. , Swift, P. , Shimada, H. , Black, C.T. , Brodeur, G.M. , Gerbing, R.B. , Reynolds, C.P. , 1999. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N. Engl. J. Med. 341, 1165–1173. [DOI] [PubMed] [Google Scholar]
- Michalowski, M.B. , de Fraipont, F. , Plantaz, D. , Michelland, S. , Combaret, V. , Favrot, M.C. , 2008. Methylation of tumor-suppressor genes in neuroblastoma: the RASSF1A gene is almost always methylated in primary tumors. Pediatr. Blood Cancer. 50, 29–32. [DOI] [PubMed] [Google Scholar]
- Miettinen, M. , Chatten, J. , Paetau, A. , Stevenson, A. , 1998. Monoclonal antibody NB84 in the differential diagnosis of neuroblastoma and other small round cell tumors. Am. J. Surg. Pathol. 22, 327–332. [DOI] [PubMed] [Google Scholar]
- Moser, M. , Pscherer, A. , Roth, C. , Becker, J. , Mucher, G. , Zerres, K. , Dixkens, C. , Weis, J. , Guay-Woodford, L. , Buettner, R. , Fassler, R. , 1997. Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Development. 11, 1938–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawara, A. , Arima-Nakagawara, M. , Scavarda, N.J. , Azar, C.G. , Cantor, A.B. , Brodeur, G.M. , 1993. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N. Engl. J. Med. 328, 847–854. [DOI] [PubMed] [Google Scholar]
- Nakagawara, A. , Ohira, M. , 2004. Comprehensive genomics linking between neural development and cancer: neuroblastoma as a model. Cancer Lett. 204, 213–224. [DOI] [PubMed] [Google Scholar]
- Nowacki, S. , Skowron, M. , Oberthuer, A. , Fagin, A. , Voth, H. , Brors, B. , Westermann, F. , Eggert, A. , Hero, B. , Berthold, F. , Fischer, M. , 2008. Expression of the tumour suppressor gene CADM1 is associated with favourable outcome and inhibits cell survival in neuroblastoma. Oncogene. 27, 3329–3338. [DOI] [PubMed] [Google Scholar]
- Oberthuer, A. , Juraeva, D. , Hero, B. , Volland, R. , Sterz, C. , Schmidt, R. , Faldum, A. , Kahlert, Y. , Engesser, A. , Asgharzadeh, S. , Seeger, R. , Ohira, M. , Nakagawara, A. , Scaruffi, P. , Tonini, G.P. , Janoueix-Lerosey, I. , Delattre, O. , Schleiermacher, G. , Vandesompele, J. , Speleman, F. , Noguera, R. , Piqueras, M. , Benard, J. , Valent, A. , Avigad, S. , Yaniv, I. , Grundy, R.G. , Ortmann, M. , Shao, C. , Schwab, M. , Eils, R. , Simon, T. , Theissen, J. , Berthold, F. , Westermann, F. , Brors, B. , Fischer, M. , 2015. Revised risk estimation and treatment stratification of low- and intermediate-risk neuroblastoma patients by integrating clinical and molecular prognostic markers. Clin. Cancer Res. 21, 1904–1915. [DOI] [PubMed] [Google Scholar]
- Oberthuer, A. , Juraeva, D. , Li, L. , Kahlert, Y. , Westermann, F. , Eils, R. , Berthold, F. , Shi, L. , Wolfinger, R.D. , Fischer, M. , Brors, B. , 2010. Comparison of performance of one-color and two-color gene-expression analyses in predicting clinical endpoints of neuroblastoma patients. Pharmacogenomics J. 10, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira, M. , Oba, S. , Nakamura, Y. , Isogai, E. , Kaneko, S. , Nakagawa, A. , Hirata, T. , Kubo, H. , Goto, T. , Yamada, S. , Yoshida, Y. , Fuchioka, M. , Ishii, S. , Nakagawara, A. , 2005. Expression profiling using a tumor-specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell. 7, 337–350. [DOI] [PubMed] [Google Scholar]
- Pellikainen, J.M. , Kosma, V.M. , 2007. Activator protein-2 in carcinogenesis with a special reference to breast cancer–a mini review. Int. J. Cancer. 120, 2061–2067. [DOI] [PubMed] [Google Scholar]
- Philipp, J. , Mitchell, P.J. , Malipiero, U. , Fontana, A. , 1994. Cell type-specific regulation of expression of transcription factor AP-2 in neuroectodermal cells. Developmental Biol. 165, 602–614. [DOI] [PubMed] [Google Scholar]
- Plouhinec, J.L. , Roche, D.D. , Pegoraro, C. , Figueiredo, A.L. , Maczkowiak, F. , Brunet, L.J. , Milet, C. , Vert, J.P. , Pollet, N. , Harland, R.M. , Monsoro-Burq, A.H. , 2014. Pax3 and Zic1 trigger the early neural crest gene regulatory network by the direct activation of multiple key neural crest specifiers. Developmental Biol. 386, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebhan, M. , Vacun, G. , Bayreuther, K. , Rosner, H. , 1994. Altered ganglioside expression by SH-SY5Y cells upon retinoic acid-induced neuronal differentiation. Neuroreport. 5, 941–944. [DOI] [PubMed] [Google Scholar]
- Reynolds, C.P. , Matthay, K.K. , Villablanca, J.G. , Maurer, B.J. , 2003. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 197, 185–192. [DOI] [PubMed] [Google Scholar]
- Schmidt, M. , Huber, L. , Majdazari, A. , Schutz, G. , Williams, T. , Rohrer, H. , 2011. The transcription factors AP-2beta and AP-2alpha are required for survival of sympathetic progenitors and differentiated sympathetic neurons. Developmental Biol. 355, 89–100. [DOI] [PubMed] [Google Scholar]
- Shimada, H. , Ambros, I.M. , Dehner, L.P. , Hata, J. , Joshi, V.V. , Roald, B. , 1999. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer. 86, 349–363. [PubMed] [Google Scholar]
- Shin, D.Y. , Sung Kang, H. , Kim, G.Y. , Kim, W.J. , Yoo, Y.H. , Choi, Y.H. , 2013. Decitabine, a DNA methyltransferases inhibitor, induces cell cycle arrest at G2/M phase through p53-independent pathway in human cancer cells. Biomed. Pharmacother. 67, 305–311. [DOI] [PubMed] [Google Scholar]
- Sidell, N. , 1982. Retinoic acid-induced growth inhibition and morphologic differentiation of human neuroblastoma cells in vitro. J. Natl. Cancer Inst. 68, 589–596. [PubMed] [Google Scholar]
- Spitz, R. , Oberthuer, A. , Zapatka, M. , Brors, B. , Hero, B. , Ernestus, K. , Oestreich, J. , Fischer, M. , Simon, T. , Berthold, F. , 2006. Oligonucleotide array-based comparative genomic hybridization (aCGH) of 90 neuroblastomas reveals aberration patterns closely associated with relapse pattern and outcome. Genes Chromosomes Cancer. 45, 1130–1142. [DOI] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V.K. , Mukherjee, S. , Ebert, B.L. , Gillette, M.A. , Paulovich, A. , Pomeroy, S.L. , Golub, T.R. , Lander, E.S. , Mesirov, J.P. , 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, T. , Matsuda, Y. , Yamashita, S. , Hattori, N. , Kushima, R. , Lee, Y.C. , Igaki, H. , Tachimori, Y. , Nagino, M. , Ushijima, T. , 2013. Estimation of the fraction of cancer cells in a tumor DNA sample using DNA methylation. PloS One. 8, e82302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel, G. , Ekici, M. , Rossler, O.G. , 2015. RE-1 silencing transcription factor (REST): a regulator of neuronal development and neuronal/endocrine function. Cell Tissue Res. 359, 99–109. [DOI] [PubMed] [Google Scholar]
- Thomas, J.O. , Nijjar, J. , Turley, H. , Micklem, K. , Gatter, K.C. , 1991. NB84: a new monoclonal antibody for the recognition of neuroblastoma in routinely processed material. J. Pathol. 163, 69–75. [DOI] [PubMed] [Google Scholar]
- Thorell, K. , Bergman, A. , Caren, H. , Nilsson, S. , Kogner, P. , Martinsson, T. , Abel, F. , 2009. Verification of genes differentially expressed in neuroblastoma tumours: a study of potential tumour suppressor genes. BMC Med. Genomics. 2, 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, W.G. , Wierda, W.G. , Lin, E. , Kuang, S.Q. , Bekele, B.N. , Estrov, Z. , Wei, Y. , Yang, H. , Keating, M.J. , Garcia-Manero, G. , 2010. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics. 5, 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Y.X. , Somasundaram, K. , el-Deiry, W.S. , 1997. AP2 inhibits cancer cell growth and activates p21WAF1/CIP1 expression. Nat. Genet. 15, 78–82. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Hagopian-Donaldson, S. , Serbedzija, G. , Elsemore, J. , Plehn-Dujowich, D. , McMahon, A.P. , Flavell, R.A. , Williams, T. , 1996. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 381, 238–241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary Figure S1 Survival analysis and performance of the prognostic TFAP2B expression. Cut‐off (mRNA intensities <19830.07 vs ≥ 19830.07) in the training set. n = patient number; (***), p < 0.001.
Supplementary Figure S2 TFAP2B expression as a prognostic marker for RA treatment. (a) EFS and OS of high‐risk neuroblastoma patients who received RA therapy according to TFAP2B expression. n = patient number. (b) EFS and OS of high‐risk neuroblastoma patients who did not receive RA treatment according to TFAP2B expression. n = patient number.
Supplementary Figure S3 TFAP2B gene expression and differentiation assessment in primary neuroblastomas. (a) Association of TFAP2B expression with differentiation status of primary neuroblastoma tumors. (n.s.), p > 0.05; (**), p < 0.01. (b) Correlation of TFAP2B expression with DBH expression in 649 primary neuroblastomas. 95% confidence interval of r = [0.73; 0.80]; (***), p < 0.001. (c) Correlation of TFAP2B expression with TH expression in 649 primary neuroblastomas. 95% confidence interval of r = [0.51; 0.62]; (***), p < 0.001; n = patient number.
Supplementary Figure S4 Association of TFAP2B methylation status with prognostic markers. Association of MYCN amplification status, tumor stage, age at diagnosis and risk group (NB2004) in 105 neuroblastoma samples with TFAP2B methylation status as determined by 450K methylome array (probe ID: cg06117072 and cg25593948). Boxes, median expression values (horizontal line) and 25th and 75th percentiles; whiskers, distances from the end of the box to the largest and smallest observed values that are <1.5 box lengths from either end of the box. Wilcoxen test (HR vs IR/LR); (***), p < 0.001; MNA, MYCN‐amplified tumors.
Supplementary Figure S5 Correlation of TFAP2B copy number with expression in 199 primary neuroblastoma samples.
Supplementary Figure S6 Microarray expression data showing TFAP2B expression levels in primary neuroblastoma cell lines. Blue, high TFAP2B expression; Green, intermediate TFAP2B expression; Red, low TFAP2B expression.
Supplementary Figure S7 Responsiveness of TFAP2Bhigh neuroblastoma cells to retinoic acid. (a) Changes in morphology of TFAP2Bhigh neuroblastoma cells treated with 1 μM retinoic acid. Scale bar = 100 μM. (b) Analysis of neuronal markers NEFM and MAP2 in TFAP2Bhigh neuroblastoma cells after treatment with 1 μM retinoic acid by qRT‐PCR. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001; (****), p < 0.0001.
Supplementary Figure S8 Responsiveness of TFAP2Bintermediate neuroblastoma cells to retinoic acid. (a) Changes in morphology of TFAP2Bintermediate neuroblastoma cells treated with 1 μM retinoic acid. Scale bar = 100 μM. (b) Analysis of neuronal markers NEFM and MAP2 in TFAP2Bintermediate neuroblastoma cells after treatment with 1 μM retinoic acid by qRT‐PCR. Error bars indicate S.D. (*), p < 0.05; (**), p < 0.01; (***), p < 0.001; (****), p < 0.0001. *SK‐N‐DZ cells were not analyzed further as they died rapidly upon RA treatment.
Supplementary Figure S9 Responsiveness of TFAP2Blow neuroblastoma cells to retinoic acid. (a) Changes in morphology of TFAP2Blow neuroblastoma cells treated with 1 μM retinoic acid. Scale bar = 100 μM. (b) Analysis of neuronal markers NEFM and MAP2 in TFAP2Blow neuroblastoma cells after treatment with 1 μM retinoic acid by qRT‐PCR. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001; (****), p < 0.0001.
Supplementary Figure S10 Correlation of TFAP2B expression with the expression of RA responsive genes RARB and CRABP2. Microarray expression data showing (a) RARB and (b) CRABP2 expression levels in neuroblastoma cell lines. Blue, high TFAP2B expression; Green, intermediate TFAP2B expression; Red, low TFAP2B expression (c) Correlation of TFAP2B expression with RARB expression in neuroblastoma cell lines. r = [0.15], p = 0.633. (d) Correlation of TFAP2B expression with CRABP2 expression in neuroblastoma cell lines. r = [0], p = 0.999.
Supplementary Figure S11 Expression of TFAP2B after RA treatment. (a) RNA sequencing analysis of TFAP2B, MYCN and TRKA expression in time series over 144h in TFAP2Bhigh SK‐N‐BE(2)c cells after 10 μM retinoic acid treatment. (b) Analysis of TFAP2B expression in TFAP2Bintermediate IMR‐32 and (c) NMB cells after 1 μM retinoic acid treatment by RT‐qPCR. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01.
Supplementary Figure S12 TFAP2B re‐expression inhibits growth of neuroblastoma cells. (a) Reduction in proliferation in SH‐EP clones. (b) Increased proportion of SH‐EP cells in G0/G1 phase 6 days after TFAP2B induction. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001.
Supplementary Figure S13 sub‐G1 phase of cell cycle after TFAP2B induction in neuroblastoma cells. (a) sub‐G1 arrest in IMR‐32 cells after TFAP2B expression. (b) Proportion of SH‐EP cells in sub‐G1 phase at day 6 after TFAP2B induction. Error bars indicate S.D; (n.s.), p > 0.05; (***), p < 0.001.
Supplementary Figure S14 SH‐EP cells after TFAP2B re‐expression. (a) Bright field microscopy of SH‐EP cells after TFAP2B re‐expression. Scale bar = 200 μM. (b) Immunofluorescence staining of SH‐EP cells for the neuronal marker TUBB3 after TFAP2B induction. Wide field fluorescent; Scale bar = 75 μm; Red = TUBB3; Blue = DAPI. (c) Analysis of the neuronal marker MAP2 in SH‐EP cells after TFAP2B induction at day 2 and 7 by RT‐qPCR. (d) Analysis of the noradrenaline neuronal marker DBH in SH‐EP cells after TFAP2B induction at day 2 and 7 by RT‐qPCR. Error bars indicate S.D; n.s., p > 0.05; (*), p < 0.05; (**), p < 0.01.
Supplementary Figure S15 Analysis of senescence associated β‐galactosidase activity after TFAP2B re‐expression in SH‐EP cells.
Supplementary Figure S16 IMR‐32 cells after DAC treatment. (a) Expression of TFAP2B and (b) neuronal markers MAP2 and NEFM in IMR‐32 cells after 3 days of 3 μM DAC treatment as determined by RT‐qPCR. (c) Bright field microscopy of IMR‐32 cells after 3 μM DAC treatment. Scale bar = 100 μM. (d) Growth arrest in IMR‐32 cells after 3 μM DAC treatment. (e) Cell cycle changes in IMR‐32 cells after 3 days of 3 μM DAC or DMSO treatment. Error bars indicate S.D; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001.
Supplementary Figure S17 TFAP2B induces changes in the expression of genes involved in cell cycle. (a) Gene set enrichment analysis of the Biocarta cell cycle pathway gene set in IMR‐32 cells after 2 and 7 days of TFAP2B induction. Normalized enrichment score, 1.59; p = 0.016. (b) Heat map showing the expression of neuronal genes in IMR‐32 cells after 2 and 7 days of TFAP2B induction.
Supplementary Figure S18 TFAP2B negatively regulates MYCN in neuroblastoma. (a) Correlation of TFAP2B expression with MYCN expression in 649 primary neuroblastoma. (b) RNA‐sequencing data shows low expression of TFAP2B after 48h of MYCN induction in SH‐SY5Y cells. (c) Normalized TFAP2B expression in SH‐SY5Y cells after MYCN induction as determined by RNA sequencing. (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01; (***), p < 0.001.
Supplementary Figure S19 Proportion of SK‐N‐BE(2)c and SH‐SY5Y cells in sub‐G1 after TFAP2B knock‐down. Error bars indicate S.D; (n.s.), p > 0.05; (*), p < 0.05; (**), p < 0.01.
Supplementary Figure S20 TFAP2B knockdown abrogates neuronal differentiation in SH‐SY5Y cells. Induction of differentiation by treatment with 1 μM retinoic acid in SH‐SY5Y cells. Cells with TFAP2B knockdown did not show RA‐induced differentiation. Scale bar = 50 μm. Green = TFAP2B, Red = actin filament, Blue = DAPI. Single channel images are shown in greyscale.
Supplementary Figure S21 TFAP2B knockdown abrogates neuronal differentiation in SK‐N‐BE(2)c cells. Induction of differentiation by treatment with 1 μM retinoic acid in SK‐N‐BE(2)c cells. Cells with TFAP2B knockdown did not show RA‐induced neuronal differentiation. Scale bar = 50 μm. Green = TFAP2B, Red = actin filament, Blue = DAPI. Single channel images are shown in greyscale.
Supplementary Figure S22 TFAP2B knockdown inhibits up‐regulation of MAP2 in SH‐SY5Y cells after RA treatment. Immunofluorescence staining for the differentiation marker MAP2 in SH‐SY5Y cells after treatment with retinoic acid for 6 days. Scale bar = 50 μm. Red = MAP2, Blue = DAPI. Single channel images are shown in greyscale.
Supplementary Figure S23 TFAP2B knockdown inhibits up‐regulation of MAP2 in SK‐N‐BE(2)c neuroblastoma cells after RA treatment. Immunofluorescence staining for the differentiation marker MAP2 in SK‐N‐BE(2)c cells after treatment with retinoic acid for 10 days. Scale bar = 50 μm. Single channel images are shown in greyscale.