

Abstract
Liquid biopsies, i.e. the analysis of circulating tumor cells (CTCs) or circulating tumor DNA (ctDNA), are evolving into promising clinical tools. Indeed, a plethora of liquid biopsy technologies to deduce non‐invasively characteristics of the tumor genome from the peripheral blood have been developed over the last few years. For example, liquid biopsies have been used to assess the tumor burden, to monitor the evolution of tumor genomes, to unravel mechanisms of resistance, to establish the tumor heterogeneity, and for the identification of prognostic and predictive markers. In this review we focus on methods to establish genome‐wide profiles of somatic copy number alterations (SCNAs) from plasma DNA and show how they provide novel insights into the biology of cancer and their impact on the management of patients.
Keywords: Circulating tumor cells (CTCs), Circulating tumor DNA (ctDNA), Plasma DNA, Whole‐genome sequencing, Somatic copy number alterations
Highlights
The analysis of circulating tumor cells (CTCs) or circulating tumor DNA (ctDNA) are evolving into promising clinical tools.
Novel methods allow establishing non‐invasively genome‐wide profiles of somatic copy number alterations (SCNAs) from ctDNA.
Analyzing the evolution of SCNAs is important as it may alter the status of oncogenes (OGs) or tumor suppressor genes (TGs).
1. Introduction
Cancer cells harbor a plethora of somatic alterations in their tumor genomes, such as base substitutions, insertions or deletions of small DNA segments (indels), copy number changes of large chromosomal regions, structural rearrangements, such as translocations, and epigenetic changes which alter chromatin structure and gene expression (Stratton et al., 2009; Vogelstein et al., 2013). Furthermore, cancer genomes are often unstable and accumulate new changes depending on exerted selection pressures. Therefore, tools allowing the monitoring of tumor genomes with easy means should be of great importance.
To this end, considerable progress has recently been achieved with the analyses of circulating tumor cells (CTCs) and plasma DNA. The cell‐free fraction of blood consists in patients with cancer of circulating tumor DNA (ctDNA) and DNA fragments released from normal cells in variable proportions (Crowley et al., 2013, 2014, 2013, 2015, 2011). The variable allele fraction of mutant DNA fragments may reflect tumor dynamics (Diehl et al., 2005) and has an impact on the selection of methods for their subsequent analyses (Belic et al., 2015; Heitzer et al., 2015).
A specific biomarker of disease burden are mutations which have previously been identified in the primary tumor, and several studies have focused on the detection of such specific and predetermined mutations in corresponding peripheral blood from the same patient (Diehl et al., 2005, 2008, 2008, 2010, 2010, 1996, 2009). Tumor‐specific structural chromosomal rearrangements, i.e. breakpoints, identified by whole‐genome sequencing of primary tumors, which are then subsequently used to monitor in plasma of the respective patients minimal residual disease (MRD) by high‐sensitive PCR approaches have a particularly high specificity (Leary et al., 2010; McBride et al., 2010; Olsson et al., 2015). Various approaches for the non‐invasive identification of somatic mutations in blood at high resolution have been developed (Bettegowda et al., 2014; Dawson et al., 2013; Forshew et al., 2012; Misale et al., 2012; Murtaza et al., 2013; Newman et al., 2014; Thierry et al., 2014). Mutation analyses assisted in the elucidation of resistance mechanisms (Diaz et al., 2012, 2012, 2014, 2013, 2015), for the assessment of tumor heterogeneity (Reinert et al., 2015), and establishment of methylation patterns (Chan et al., 2013a; Sun et al., 2015). Furthermore, we and others explored approaches for analyses of somatic copy number alterations (SCNAs) at a genome‐wide scale (Chan et al., 2013b; Heitzer et al., 2013b; Heitzer et al., 2013d; Leary et al., 2012; Murtaza et al., 2013).
Indeed, among the outstanding characteristics of cancer genomes are their frequent SCNAs and often extensive aneuploidies (Stratton et al., 2009; Vogelstein et al., 2013). Their identification and characterization are of utmost importance for basic research, understanding disease mechanisms, and tumor classification. Furthermore, they may contribute to the identification of prognostic and predictive tumor markers, which is of significance for personalized medicine, or “biologically personalized therapeutics” (Cherny et al., 2014). Here, we review methods and strategies for copy number detection and interpretation as well as their advantages and limitations.
1.1. Relevance and biology of somatic copy number alterations in cancer
90% of solid tumors and 50% of blood‐related cancers are aneuploid and have SCNAs (Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer; http://cgap.nci.nih.gov/Chromosomes/Mitelman; (Beroukhim et al., 2010)). SCNAs alter a larger percentage of the genome than any other somatic genetic alterations (Beroukhim et al., 2010; Stratton et al., 2009; Vogelstein et al., 2013). SCNAs comprise losses (i.e. deletions), gains (e.g. duplications), and high‐level amplifications (Figure 1). In principle, gains and losses are copy number changes of any length and amplitude. However, a “gain” is usually a relatively moderate copy number increase (e.g. trisomy or tetrasomy), whereas an “amplification” is a high level gain with sometimes up to several hundred copies (Stratton et al., 2009), frequently occurring of only a restricted size. There is no exact definition for an upper size limit of a focal amplification and previous studies have analyzed focal SCNAs with a size of up to 85 Mb (Beroukhim et al., 2010) or shorter than the chromosome arm (Zack et al., 2013). Similarly, homozygous deletions are usually also only observed for relatively small regions and accordingly a correlation between amplitude and size for both high‐level amplifications and homozygous deletions has been reported (Beroukhim et al., 2010).
Figure 1.
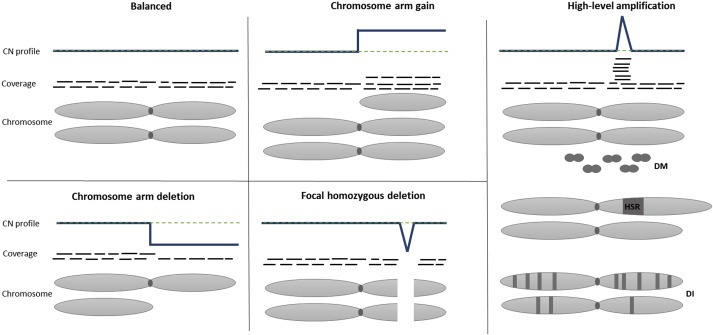
Copy number alterations detectable with plasma‐Seq. Schematic representation of frequently occurring SCNAs in tumor cells (depicted as chromosomes), sequence reads (coverage; illustrated as short black lines), and resulting copy number profile (CN profile; the green dotted line represents the balanced status, the blue line the copy number profile) for a balanced chromosome, chromosome arm gain; high‐level amplification (may be caused by double minutes (DM), homogenously staining regions (HSR), or amplifications where the amplified region is scattered throughout the genome as small insertions (distributed insertions, DI)), chromosome arm deletion, and smaller focal deletion, e.g. homozygous deletion. In contrast, structural rearrangements, which do not result in copy number changes, e.g. balanced translocations, are not detected by plasma‐Seq. Due to the shallow sequencing read depth the number of reads spanning breakpoints is too low for a reliable detection.
SCNAs may affect activation of oncogenes (OGs) or inactivation of tumor suppressor genes (TSGs) (Beroukhim et al., 2010; Stratton et al., 2009; Vogelstein et al., 2013). The loss of TSGs and gain of OGs may propel further karyotype changes, including whole or partial chromosome gains and losses. This may result in clonal aneuploidy karyotype patterns, which are frequently characteristic of a specific cancer (Davoli et al., 2013; Santaguida and Amon, 2015).
When applying novel parameters for predicting TSGs and OGs, TSGs were found to be enriched in recurring focal deletions, whereas OGs were enriched in amplifications and depleted from focal deletions. Hence, recurrent focal deletions and amplifications both may represent “Cancer Gene Islands”, which are characterized by particular densities of TSGs and OGs (Solimini et al., 2012) (Figure 2a).
Figure 2.
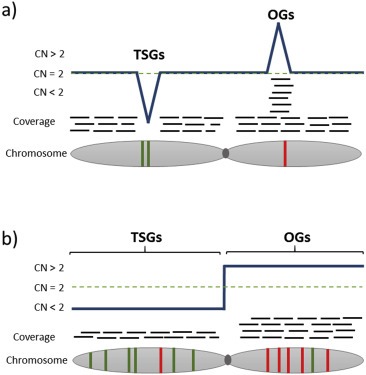
Models for the selection of SCNAs. (a) The “cancer gene island model” (Solimini et al., 2012) for focal SCNAs suggests that focal deletions are mainly enriched for tumor suppressor genes (TSGs), whereas focal amplifications are mainly enriched for oncogenes (OGs) to exert a negative or positive phenotypic effect on tumorigenesis. (b) The “gene dosage balance model” (Davoli et al., 2013) for larger SCNAs predicts that patterns of aneuploidy are determined by cumulative gene dosage: deletions frequently harbor TSGs and only rarely OGs and the reverse pattern is present in gained regions.
However, gaining or losing large chromosomal regions or even whole chromosomes affects not specifically TSGs and OGs, but results frequently yet not universally in expression changes of large numbers of genes. Indeed, recent large‐scale integrated analyses of copy number and gene expression have found that SCNAs comprise a major mechanism driving carcinogenesis in epithelial cancers, such as breast and prostate carcinoma (Curtis et al., 2012; Grasso et al., 2012; Taylor et al., 2010). A study of 2000 primary breast tumors has reported that the expression landscape was dominated by cis‐ and trans‐acting acquired SCNAs and the patterns of salient SCNAs even allowed the definition of new breast‐cancer subtypes (Curtis et al., 2012). In prostate cancer genome‐wide comprehensive analyses have revealed that outlying expression coincided with copy number events (Grasso et al., 2012; Taylor et al., 2010). Hence, the phenotypes of aneuploidy cells are caused by gene imbalances and by simultaneous changes in the gene dosage of many genes (Santaguida and Amon, 2015).
Indeed, the distribution and potency of TSGs and OGs on chromosomes was shown to influence the frequency of whole‐chromosome arm SCNAs in cancer. Applying a newly developed score that measures positive and negative growth and survival potential that wild‐type OGs or TSGs normally impart to the respective chromosome arm it was calculated how SCNAs might impact cancer evolution by altering this balance (Davoli et al., 2013).
Importantly, Davoli et al., (2013) suggested the existence of two classes for both TSGs and OGs. One TSG class is haploinsufficient (Haploinsufficiency: loss of one copy of a gene causes in a diploid organism a phenotype) and contributes to sporadic cancer, whereas the other class is haplosufficient without significant contribution to cancer. In case of the first class TSGs cancer may occur by loss of one functional allele, which may produce a selectable phenotype, whereas the latter class refers to the familial TSGs and the two‐hit model of tumorigenesis (Knudson, 2001). Estimates suggest ∼30% haploinsufficiency overall among human genes (Davoli et al., 2013). In fact, the vast majority, if not all, of sporadic TSGs are likely to be haploinsufficient, and these genes cannot be identified by mutation analyses but rather by systematic mapping of recurrently deleted chromosomal regions (Davoli et al., 2013). Hence, a “gene dosage balance model” for the prediction of aneuploidy patterns was developed (Figure 2b).
Likewise, one OG class are oncogenes which are frequently activated by mutations (e.g. KRAS), whereas another OG class shows triplosensitivity (Triplosensitivity: an additional copy of a gene causes a phenotype). The latter OG class must not show specific mutational signatures and often correlates with amplifications (e.g. CCND1) (Davoli et al., 2013).
This model supports a view that chromosomal arm and whole‐chromosome SCNAs are selected for by the relative densities and potencies of positively and negatively acting cancer drivers on a particular chromosome (Davoli et al., 2013). Hence, cumulative haploinsufficiency for deletions and cumulative triplosensitivity for amplifications are important contributors to tumorigenesis. Hence, systematic mapping of SCNAs will reveal the distribution of such TSGs and OGs on chromosomes and will allow explaining of copy number changes on chromosomal regions through a process of cumulative haploinsufficiency and triplosensitivity (Davoli et al., 2013) (Figure 2b).
This is also likely a reason why many cancer cells have an increased ploidy, because polyploidy attenuates aneuploidy‐associated phenotypes. Polyploidy helps in tolerating gains and losses of large chromosomal regions and may facilitate utilization of potentially beneficial traits occurring as consequence of altered dosages of specific cancer driver genes (Santaguida and Amon, 2015).
Furthermore, tumor genomes are characterized by various forms of instability and a common instability is chromosomal instability (CIN) (Geigl et al., 2008), where the chromosomal composition of tumor cells constantly changes. The temporal ordering of SCNAs is greatly unknown and genome‐wide copy number screens of plasma during a disease course may contribute to an improved understanding. In addition, SCNA mapping may have clinical relevance, as increased SCNAs have been linked to poor prognosis in several cancer types (Carter et al., 2006; Zack et al., 2013).
1.2. Whole‐genome copy number analyses approaches
Hence, there is growing evidence that aneuploidy contributes to promote tumorigenesis. However, our current knowledge how certain karyotypes may promote tumorigenesis is limited, due to lack of data how aneuploidy may change and its plasticity during tumor evolution. To this end, liquid biopsies offer novel opportunities to address this issue.
A genome‐wide screen for copy number changes has the advantage that it is an untargeted approach, which does not require any prior knowledge about characteristics of the primary tumor genome or its metastatic deposits.
To this end, one of the first studies employed Affymetrix Genome‐Wide Human SNP 6.0 arrays for the analysis of plasma DNA from patients with breast cancer and this approach allowed mainly the detection of high‐level amplifications (Shaw et al., 2012).
In our first plasma DNA study we generated random DNA libraries by converting the plasma DNA fragments into PCR‐amplifiable OmniPlex Library molecules flanked by universal priming sites for whole‐genome amplification (WGA) and subjected the WGA products to array CGH on a 60 K microarray platform (i.e. 55,077 oligonucleotides). Subsequently, we calculated for each of these oligonucleotides whether the ratio values were decreased, balanced, or increased (Heitzer et al., 2013b). In a subset of 32 patients (34.4%) with stage IV colorectal carcinoma we were able to establish genome‐wide tumor‐specific copy number alterations directly from plasma DNA (Heitzer et al., 2013b). The copy number changes, which we deduced from plasma, displayed a high correlation to those observed in the respective primary tumors and also to those in CTCs, which were collected at the same time (Heitzer et al., 2013, 2013).
As massively parallel sequencing of plasma DNA from the maternal circulation was emerging to a clinical tool for the routine detection of fetal aneuploidy (Chiu et al., 2008; Chiu and Lo, 2012; Fan et al., 2008) the essentially same approach, i.e. next‐generation sequencing from plasma, was used for the detection of chromosomal alterations in the circulation of 3 patients with hepatocellular carcinoma and 1 patient with both breast and ovarian cancer (Chan et al., 2013b) and from 10 patients with colorectal and breast cancer (Leary et al., 2012). This approach is based on sequencing and enumerating genomic DNA tags, similar to the previously described digital karyotyping (Wang et al., 2002). The genome is divided into windows or bins of a certain size, and a GC‐corrected read count for each of these windows is then used to determine if the respective region is increased or decreased. In order to establish z‐scores a statistical means for the presence of an SCNA the read counts can be compared to a reference group (Chan et al., 2013b; Heitzer et al., 2013d; Leary et al., 2012). Due to the relative high coverage (i.e. >10× (Leary et al., 2012) and >15× (Chan et al., 2013b)), these plasma sequencing analyses were time‐consuming and the costs were prohibitive for routine clinical implementation. As it had been previously shown that whole‐genome sequencing with a shallow sequencing depth of about 0.1× is sufficient for a robust and reliable analysis of copy number changes from single cells (Navin et al., 2011), we developed a whole‐genome plasma sequencing approach employing also such a shallow sequencing depth. Using a benchtop high‐throughput sequencing instrument (i.e. the Illumina MiSeq) we performed whole‐genome sequencing from plasma DNA and measured copy number from sequence read depth. We refer to this approach as plasma‐Seq (Heitzer et al., 2013d). In addition, we sequence a panel of high‐interest genes with high coverage for mutation analyses. Up to date, we have used plasma‐Seq to analyze blood from patients with prostate (Heitzer et al., 2013d), colon (Mohan et al., 2014), and breast (Heidary et al., 2014) cancer, an example is outlined in the next section and in Figure 3. A distinct advantage of this approach is the speed, as it allows generation of results in “real time” (<48 h). Furthermore the costs are low, making it an affordable tool, which is an important prerequisite for implementing this technique in the clinic.
Figure 3.
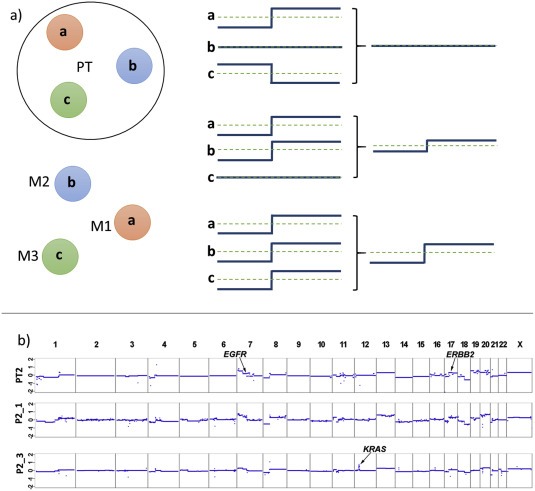
Scenarios and outcomes observable with plasma‐Seq. (a) Plasma‐Seq copy number profiles represent an average of all cells releasing DNA into the circulation, e.g. from different clones within a tumor lesion (PT) or from various tumor lesions, i.e. metastatic deposits (M1–M3) (designated as a, b, and c). If all lesions have the same SCNAs they are visible with large amplitude (bottom), the amplitude decreases, if the SCNA is present in a subset of lesions only (center). If the SCNA pattern varies in different lesions they may even not be visible at all (top). (b) Copy number analyses of a primary tumor (PT2) and two plasma analyses (P2_1, 27 months after diagnosis of the primary tumor; P2_3, 6 months after P2_1) from the same patient. The X‐ and Y‐axes indicate the chromosome and the log2‐ratios, respectively. The locations of the EGFR, ERBB2 and KRAS genes are indicted in PT2 or P2_3. The profiles were taken from one of our previous studies (Mohan et al., 2014).
A similar approach has been applied by others for SCNAs analysis in blood from patients with prostate cancer (Xia et al., 2015). Furthermore, whole‐exome sequencing data has also been used to assess SCNAs in plasma (Murtaza et al., 2013).
Assessment of SCNAs can be performed with CTCs as well. All available single‐cell strategies depend on initial whole‐genome amplification (WGA), of which several commonly used single‐cell WGA techniques exist (Fiegler et al., 2007; Garvin et al., 2015; Geigl et al., 2009; Geigl and Speicher, 2007). After WGA, SCNA analyses by array‐CGH of single CTCs (Heitzer et al., 2013a; Mohlendick et al., 2013; Neves et al., 2014; Polzer et al., 2014; Steinert et al., 2014), several CTCs (Magbanua et al., 2013), or SNP arrays (Chiu et al., 2014) have been reported. Due to the aforementioned observation that SCNA determination from single cells requires only sparse sequence coverage (Baslan et al., 2012, 2015, 2011), others and we used this approach to establish SCNA profiles from CTCs (Heidary et al., 2014; Ni et al., 2013).
1.3. Comparison of SCNA profiling by ctDNA and CTCs
Both ctDNA and CTCs are powerful resources to deduce SCNA profiles from peripheral blood. However, differences in acquisition of the material, the need for WGA, and purity of tumor DNA exist.
Although CTC capture methods have been improved, CTCs remain difficult to isolate, as they constitute only few cells in the background of a million mononuclear cells (Alix‐Panabieres and Pantel, 2014; Pantel and Speicher, 2015). In contrast, acquiring samples for ctDNA analyses implies just a simple blood draw. Furthermore, plasma DNA analysis does not require WGA. The dependence on initial WGA for single CTC analysis is problematic, as yields of amplified DNA may vary among CTCs, WGA introduces amplification bias, and the commonly used single‐cell WGA techniques differ in their suitability for SCNA analysis (Garvin et al., 2015).
However, CTC analyses provide highly specific approaches, as they represent a pure tumor cell population and yield information on a cellular level, whereas ctDNA reflects an average of all tumor cells releasing DNA into the circulation as discussed in the next section. Furthermore, ctDNA is derived from apoptotic and necrotic cells, whereas tumor DNA can be obtained from intact viable CTC, which may justify the increased effort to capture CTCs (Pantel and Speicher, 2015). Moreover, it is possible to analyze the DNA of CTCs with a particular phenotype, for example CTCs expressing stem cell markers or immune checkpoint molecules like PDL1 (Mazel et al., 2015).
1.4. Stability assessment of aneuploid karyotypes by liquid biopsies
SCNAs in plasma are averages from various tumor sites, which release tumor DNA into the circulation and which may reflect either different clones within a heterogeneous primary tumor or represent different metastatic deposits (Figure 3a). As such they may blur copy number alterations occurring in only a subset of cells; however, they reliably reflect the most prominent copy number alterations and allow conclusions about tumor biology. For example, in one of our previous studies we analyzed patients with colorectal cancer receiving anti‐EGFR therapy (Mohan et al., 2014). In one of these patients (i.e. #2) we analyzed a first plasma sample after a disease course of 2 years and 3 months and observed that the copy number alterations in the plasma sample were almost identical to those of the primary tumor (Figure 3b). This stability of the SCNA pattern – despite the long time interval and despite intensive treatment in between – confirms the existence of selected, stable aneuploidy karyotypes that have evolved to support maximal proliferation and in which the stresses caused by aneuploidy karyotypes are suppressed (Santaguida and Amon, 2015). Furthermore, this example illustrates that aneuploidy cannot be equated with CIN (Geigl et al., 2008). However, within 6 months of treatment with panitumumab, this stable aneuploid karyotype developed a novel change, i.e. a KRAS amplification (Figure 3b; (Mohan et al., 2014)), which is an established resistance marker against anti‐EGFR therapy (Misale et al., 2012; Valtorta et al., 2013).
Hence, plasma‐Seq may reveal highly stable aneuploid karyotypes, which withstand selection pressures over long periods of time, or new small changes, such as novel focal amplifications, which may have a great impact on the disease course. Importantly, plasma‐Seq may also reveal the emergence of tremendous clonal shifts or even the occurrence of new clones (Ulz et al.; manuscript submitted). Thus, the cancer genome may also be characterized by continuously changing karyotypes, indicating that aneuploidy has suboptimal impact on cellular fitness and these important features can now be unraveled by appropriate liquid biopsy approaches.
1.5. Limitations to confirm liquid biopsy findings with tissue biopsies
All plasma DNA analyses have some important limitations in common. A vital question is the confirmation of plasma results by another approach. An option could be to biopsy a metastasis at the same time of the blood collection. However, metastases may be inaccessible and in many instances this should be only applicable if there is a medical reason to biopsy or remove metastases, otherwise there are important ethical issues to consider. Furthermore, a biopsy in metastatic disease is just a random, relatively small sample of the entire tumor events, which might not be representative and might not be a major source of ctDNA into the circulation. Hence, beside rigorously testing and verifying the liquid biopsy methods, a relatively easy confirmation may be achieved by comparing ctDNA results with those obtained from CTCs collected at the same time (Heitzer et al., 2013, 2013, 2013). Indeed, recent studies have suggested that SCNAs of CTCs are often highly similar and shared between CTCs, the primary, and the metastatic tumor cells (Heitzer et al., 2013a; Ni et al., 2013).
Furthermore, one should bear in mind that plasma copy number analyses reveal only relative copy number changes, which neither allow to establish the ploidy level of tumors cells, nor the exact prediction of the absolute copy number due to the dilution effects with DNA from normal cells.
Reliable copy number analyses require a relatively high allele frequency of ctDNA estimated to be at least 5–10% of plasma DNA (Belic et al., 2015; Carreira et al., 2014; Heitzer et al., 2013d). Considering that ctDNA is detected with lesser frequency in localized than in metastasized tumor disease (Bettegowda et al., 2014), this approach may be less suited for the detection of MRD or for screening of at‐risk populations. However, to this end there is another very interesting aspect from the aforementioned non‐invasive prenatal testing for fetal aneuploidies using maternal plasma DNA. As this approach has rapidly evolved to a frequently used test, there is by now an enormous data set of plasma DNA sequencing data from healthy, relatively young individuals where whole‐genome plasma sequencing was performed not within the context of cancer. Nevertheless, this prenatal testing resulted in the incidental detection of occult maternal malignancies in 10 of 125,426 (Bianchi et al., 2015) and 3 of 4000 (Amant et al., 2015) noninvasive prenatal testing cases. Hence, it may be interesting to screen an older population (e.g. above 60 years of age) to test how many occult tumors may be detected. The disadvantage that the ctDNA content must be relatively high is compensated by the fact that copy number screening is untargeted and that it does not depend on the previous identification of highly specific somatic mutations. Furthermore, using appropriate prescreening tools (Belic et al., 2015), the costs for such a population screening would be in a moderate range.
2. Conclusions
At present, our knowledge how certain karyotypes contribute to tumorigenesis is very limited. The identification of cancer driver genes, either oncogenes or tumor suppressor genes, which contribute to tumorigenesis often by haplo‐ or triploinsufficiency if their copy number is changed, is incomplete.
Analyses of ctDNA offer a new and exciting possibility to study the role of aneuploidy in tumorigenesis, in particular in advanced stage cancer. As aneuploidy is a hallmark of cancer but rare in normal cells, therapeutics targeting the aneuploidy state may provide ideal therapeutic properties.
Of course, mutation analyses should accompany SCNAs studies, as they complement each other. Expression analyses and other markers may get added. Furthermore, methylation changes of plasma DNA, which may even allow the identification of the cell of origin (Sun et al., 2015), could broaden our knowledge. Hence, future studies should comprehensively cover as many parameters as possible, so that liquid biopsies evolve to an indispensable clinical tool which improves the management of patients.
Conflicts of interest
The authors do not declare conflicts of interest.
Acknowledgments
We are grateful to Maria Langer‐Winter for critical reading and editing of this manuscript. Funding was provided by the Austrian Science Fund (FWF) (grant#: P20338, P23284 and W 1226‐B18, DKplus Metabolic and Cardiovascular Disease), and Cancer‐ID, a project funded by the Innovative Medicines Initiative Joint Undertaking (IMI JU) under grant agreement no. 115749.
Heitzer Ellen, Ulz Peter, Geigl Jochen B., Speicher Michael R., (2016), Non-invasive detection of genome-wide somatic copy number alterations by liquid biopsies, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.12.004.
This is a contribution to the special issue edited by Klaus Pantel and Catherine Alix‐Panabieres, Liquid Biopsies.
Contributor Information
Ellen Heitzer, Email: ellen.heitzer@medunigraz.at.
Peter Ulz, Email: peter.ulz@medunigraz.at.
Jochen B. Geigl, Email: jochen.geigl@medunigraz.at
Michael R. Speicher, Email: michael.speicher@medunigraz.at
References
- Alix-Panabieres, C. , Pantel, K. , 2014. Challenges in circulating tumour cell research. Nat. Rev. Cancer. 14, 623–631. [DOI] [PubMed] [Google Scholar]
- Amant, F. , Verheecke, M. , Wlodarska, I. , Dehaspe, L. , Brady, P. , Brison, N. , Van Den Bogaert, K. , Dierickx, D. , Vandecaveye, V. , Tousseyn, T. , Moerman, P. , Vanderstichele, A. , Vergote, I. , Neven, P. , Berteloot, P. , Putseys, K. , Danneels, L. , Vandenberghe, P. , Legius, E. , Vermeesch, J.R. , 2015. Presymptomatic identification of cancers in pregnant women during noninvasive prenatal testing. JAMA Oncol. 1, 814–819. [DOI] [PubMed] [Google Scholar]
- Baslan, T. , Kendall, J. , Rodgers, L. , Cox, H. , Riggs, M. , Stepansky, A. , Troge, J. , Ravi, K. , Esposito, D. , Lakshmi, B. , Wigler, M. , Navin, N. , Hicks, J. , 2012. Genome-wide copy number analysis of single cells. Nat. Protoc. 7, 1024–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baslan, T. , Kendall, J. , Ward, B. , Cox, H. , Leotta, A. , Rodgers, L. , Riggs, M. , D'Italia, S. , Sun, G. , Yong, M. , Miskimen, K. , Gilmore, H. , Saborowski, M. , Dimitrova, N. , Krasnitz, A. , Harris, L. , Wigler, M. , Hicks, J. , 2015. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 25, 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belic, J. , Koch, M. , Ulz, P. , Auer, M. , Gerhalter, T. , Mohan, S. , Fischereder, K. , Petru, E. , Bauernhofer, T. , Geigl, J.B. , Speicher, M.R. , Heitzer, E. , 2015. Rapid identification of plasma DNA samples with increased ctDNA levels by a modified FAST-SeqS approach. Clin. Chem. 61, 838–849. [DOI] [PubMed] [Google Scholar]
- Beroukhim, R. , Mermel, C.H. , Porter, D. , Wei, G. , Raychaudhuri, S. , Donovan, J. , Barretina, J. , Boehm, J.S. , Dobson, J. , Urashima, M. , Mc Henry, K.T. , Pinchback, R.M. , Ligon, A.H. , Cho, Y.J. , Haery, L. , Greulich, H. , Reich, M. , Winckler, W. , Lawrence, M.S. , Weir, B.A. , Tanaka, K.E. , Chiang, D.Y. , Bass, A.J. , Loo, A. , Hoffman, C. , Prensner, J. , Liefeld, T. , Gao, Q. , Yecies, D. , Signoretti, S. , Maher, E. , Kaye, F.J. , Sasaki, H. , Tepper, J.E. , Fletcher, J.A. , Tabernero, J. , Baselga, J. , Tsao, M.S. , Demichelis, F. , Rubin, M.A. , Janne, P.A. , Daly, M.J. , Nucera, C. , Levine, R.L. , Ebert, B.L. , Gabriel, S. , Rustgi, A.K. , Antonescu, C.R. , Ladanyi, M. , Letai, A. , Garraway, L.A. , Loda, M. , Beer, D.G. , True, L.D. , Okamoto, A. , Pomeroy, S.L. , Singer, S. , Golub, T.R. , Lander, E.S. , Getz, G. , Sellers, W.R. , Meyerson, M. , 2010. The landscape of somatic copy-number alteration across human cancers. Nature. 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettegowda, C. , Sausen, M. , Leary, R.J. , Kinde, I. , Wang, Y. , Agrawal, N. , Bartlett, B.R. , Wang, H. , Luber, B. , Alani, R.M. , Antonarakis, E.S. , Azad, N.S. , Bardelli, A. , Brem, H. , Cameron, J.L. , Lee, C.C. , Fecher, L.A. , Gallia, G.L. , Gibbs, P. , Le, D. , Giuntoli, R.L. , Goggins, M. , Hogarty, M.D. , Holdhoff, M. , Hong, S.M. , Jiao, Y. , Juhl, H.H. , Kim, J.J. , Siravegna, G. , Laheru, D.A. , Lauricella, C. , Lim, M. , Lipson, E.J. , Marie, S.K. , Netto, G.J. , Oliner, K.S. , Olivi, A. , Olsson, L. , Riggins, G.J. , Sartore-Bianchi, A. , Schmidt, K. , Shih, l.M. , Oba-Shinjo, S.M. , Siena, S. , Theodorescu, D. , Tie, J. , Harkins, T.T. , Veronese, S. , Wang, T.L. , Weingart, J.D. , Wolfgang, C.L. , Wood, L.D. , Xing, D. , Hruban, R.H. , Wu, J. , Allen, P.J. , Schmidt, C.M. , Choti, M.A. , Velculescu, V.E. , Kinzler, K.W. , Vogelstein, B. , Papadopoulos, N. , Diaz, L.A. , 2014. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, D.W. , Chudova, D. , Sehnert, A.J. , Bhatt, S. , Murray, K. , Prosen, T.L. , Garber, J.E. , Wilkins-Haug, L. , Vora, N.L. , Warsof, S. , Goldberg, J. , Ziainia, T. , Halks-Miller, M. , 2015. Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA: J. Am. Med. Assoc. 314, 162–169. [DOI] [PubMed] [Google Scholar]
- Carreira, S. , Romanel, A. , Goodall, J. , Grist, E. , Ferraldeschi, R. , Miranda, S. , Prandi, D. , Lorente, D. , Frenel, J.S. , Pezaro, C. , Omlin, A. , Rodrigues, D.N. , Flohr, P. , Tunariu, N. , de Bono, J.S. , Demichelis, F. , Attard, G. , 2014. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 6, 254ra125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, S.L. , Eklund, A.C. , Kohane, I.S. , Harris, L.N. , Szallasi, Z. , 2006. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048. [DOI] [PubMed] [Google Scholar]
- Chan, K.C. , Jiang, P. , Chan, C.W. , Sun, K. , Wong, J. , Hui, E.P. , Chan, S.L. , Chan, W.C. , Hui, D.S. , Ng, S.S. , Chan, H.L. , Wong, C.S. , Ma, B.B. , Chan, A.T. , Lai, P.B. , Sun, H. , Chiu, R.W. , Lo, Y.M. , 2013. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. U. S. A. 110, 18761–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, K.C. , Jiang, P. , Zheng, Y.W. , Liao, G.J. , Sun, H. , Wong, J. , Siu, S.S. , Chan, W.C. , Chan, S.L. , Chan, A.T. , Lai, P.B. , Chiu, R.W. , Lo, Y.M. , 2013. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin. Chem. 59, 211–224. [DOI] [PubMed] [Google Scholar]
- Cherny, N.I. , de Vries, E.G. , Emanuel, L. , Fallowfield, L. , Francis, P.A. , Gabizon, A. , Piccart, M.J. , Sidransky, D. , Soussan-Gutman, L. , Tziraki, C. , 2014. Words matter: distinguishing “personalized medicine” and “biologically personalized therapeutics”. J. Natl. Cancer Inst. 106, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, R.W. , Lo, Y.M. , 2012. Clinical applications of maternal plasma fetal DNA analysis: translating the fruits of 15 years of research. Clin. Chem. Lab. Med.: CCLM/FESCC. 0, 1–8. [DOI] [PubMed] [Google Scholar]
- Chiu, R.W. , Chan, K.C. , Gao, Y. , Lau, V.Y. , Zheng, W. , Leung, T.Y. , Foo, C.H. , Xie, B. , Tsui, N.B. , Lun, F.M. , Zee, B.C. , Lau, T.K. , Cantor, C.R. , Lo, Y.M. , 2008. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc. Natl. Acad. Sci. U. S. A. 105, 20458–20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, C.G. , Nakamura, Y. , Chong, K.K. , Huang, S.K. , Kawas, N.P. , Triche, T. , Elashoff, D. , Kiyohara, E. , Irie, R.F. , Morton, D.L. , Hoon, D.S. , 2014. Genome-wide characterization of circulating tumor cells identifies novel prognostic genomic alterations in systemic melanoma metastasis. Clin. Chem. 60, 873–885. [DOI] [PubMed] [Google Scholar]
- Crowley, E. , Di Nicolantonio, F. , Loupakis, F. , Bardelli, A. , 2013. Liquid biopsy: monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 10, 472–484. [DOI] [PubMed] [Google Scholar]
- Curtis, C. , Shah, S.P. , Chin, S.F. , Turashvili, G. , Rueda, O.M. , Dunning, M.J. , Speed, D. , Lynch, A.G. , Samarajiwa, S. , Yuan, Y. , Graf, S. , Ha, G. , Haffari, G. , Bashashati, A. , Russell, R. , McKinney, S. , Group, M. , Langerod, A. , Green, A. , Provenzano, E. , Wishart, G. , Pinder, S. , Watson, P. , Markowetz, F. , Murphy, L. , Ellis, I. , Purushotham, A. , Borresen-Dale, A.L. , Brenton, J.D. , Tavare, S. , Caldas, C. , Aparicio, S. , 2012. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli, T. , Xu, A.W. , Mengwasser, K.E. , Sack, L.M. , Yoon, J.C. , Park, P.J. , Elledge, S.J. , 2013. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 155, 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, S.J. , Tsui, D.W. , Murtaza, M. , Biggs, H. , Rueda, O.M. , Chin, S.F. , Dunning, M.J. , Gale, D. , Forshew, T. , Mahler-Araujo, B. , Rajan, S. , Humphray, S. , Becq, J. , Halsall, D. , Wallis, M. , Bentley, D. , Caldas, C. , Rosenfeld, N. , 2013. Analysis of circulating tumor DNA to monitor metastatic breast cancer. New Engl. J. Med. 368, 1199–1209. [DOI] [PubMed] [Google Scholar]
- Diaz, L.A. , Bardelli, A. , 2014. Liquid biopsies: genotyping circulating tumor DNA. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 32, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, L.A. , Williams, R.T. , Wu, J. , Kinde, I. , Hecht, J.R. , Berlin, J. , Allen, B. , Bozic, I. , Reiter, J.G. , Nowak, M.A. , Kinzler, K.W. , Oliner, K.S. , Vogelstein, B. , 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl, F. , Li, M. , Dressman, D. , He, Y. , Shen, D. , Szabo, S. , Diaz, L.A. , Goodman, S.N. , David, K.A. , Juhl, H. , Kinzler, K.W. , Vogelstein, B. , 2005. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. U. S. A. 102, 16368–16373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl, F. , Schmidt, K. , Choti, M.A. , Romans, K. , Goodman, S. , Li, M. , Thornton, K. , Agrawal, N. , Sokoll, L. , Szabo, S.A. , Kinzler, K.W. , Vogelstein, B. , Diaz, L.A. , 2008. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl, F. , Schmidt, K. , Durkee, K.H. , Moore, K.J. , Goodman, S.N. , Shuber, A.P. , Kinzler, K.W. , Vogelstein, B. , 2008. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology. 135, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H.C. , Blumenfeld, Y.J. , Chitkara, U. , Hudgins, L. , Quake, S.R. , 2008. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. U. S. A. 105, 16266–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiegler, H. , Geigl, J.B. , Langer, S. , Rigler, D. , Porter, K. , Unger, K. , Carter, N.P. , Speicher, M.R. , 2007. High resolution array-CGH analysis of single cells. Nucleic Acids Res. 35, e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshew, T. , Murtaza, M. , Parkinson, C. , Gale, D. , Tsui, D.W. , Kaper, F. , Dawson, S.J. , Piskorz, A.M. , Jimenez-Linan, M. , Bentley, D. , Hadfield, J. , May, A.P. , Caldas, C. , Brenton, J.D. , Rosenfeld, N. , 2012. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 4, 136ra168 [DOI] [PubMed] [Google Scholar]
- Garvin, T. , Aboukhalil, R. , Kendall, J. , Baslan, T. , Atwal, G.S. , Hicks, J. , Wigler, M. , Schatz, M.C. , 2015. Interactive analysis and assessment of single-cell copy-number variations. Nat. Methods. 12, 1058–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geigl, J.B. , Speicher, M.R. , 2007. Single-cell isolation from cell suspensions and whole genome amplification from single cells to provide templates for CGH analysis. Nat. Protoc. 2, 3173–3184. [DOI] [PubMed] [Google Scholar]
- Geigl, J.B. , Obenauf, A.C. , Schwarzbraun, T. , Speicher, M.R. , 2008. Defining ‘chromosomal instability’. Trends Genet.: TIG. 24, 64–69. [DOI] [PubMed] [Google Scholar]
- Geigl, J.B. , Obenauf, A.C. , Waldispuehl-Geigl, J. , Hoffmann, E.M. , Auer, M. , Hormann, M. , Fischer, M. , Trajanoski, Z. , Schenk, M.A. , Baumbusch, L.O. , Speicher, M.R. , 2009. Identification of small gains and losses in single cells after whole genome amplification on tiling oligo arrays. Nucleic Acids Res. 37, e105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso, C.S. , Wu, Y.M. , Robinson, D.R. , Cao, X. , Dhanasekaran, S.M. , Khan, A.P. , Quist, M.J. , Jing, X. , Lonigro, R.J. , Brenner, J.C. , Asangani, I.A. , Ateeq, B. , Chun, S.Y. , Siddiqui, J. , Sam, L. , Anstett, M. , Mehra, R. , Prensner, J.R. , Palanisamy, N. , Ryslik, G.A. , Vandin, F. , Raphael, B.J. , Kunju, L.P. , Rhodes, D.R. , Pienta, K.J. , Chinnaiyan, A.M. , Tomlins, S.A. , 2012. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 487, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidary, M. , Auer, M. , Ulz, P. , Heitzer, E. , Petru, E. , Gasch, C. , Riethdorf, S. , Mauermann, O. , Lafer, I. , Pristauz, G. , Lax, S. , Pantel, K. , Geigl, J.B. , Speicher, M.R. , 2014. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res.: BCR. 16, 421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer, E. , Auer, M. , Gasch, C. , Pichler, M. , Ulz, P. , Hoffmann, E.M. , Lax, S. , Waldispuehl-Geigl, J. , Mauermann, O. , Lackner, C. , Hofler, G. , Eisner, F. , Sill, H. , Samonigg, H. , Pantel, K. , Riethdorf, S. , Bauernhofer, T. , Geigl, J.B. , Speicher, M.R. , 2013. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 73, 2965–2975. [DOI] [PubMed] [Google Scholar]
- Heitzer, E. , Auer, M. , Hoffmann, E.M. , Pichler, M. , Gasch, C. , Ulz, P. , Lax, S. , Waldispuehl-Geigl, J. , Mauermann, O. , Mohan, S. , Pristauz, G. , Lackner, C. , Hofler, G. , Eisner, F. , Petru, E. , Sill, H. , Samonigg, H. , Pantel, K. , Riethdorf, S. , Bauernhofer, T. , Geigl, J.B. , Speicher, M.R. , 2013. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int. J. Cancer. 133, 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer, E. , Auer, M. , Ulz, P. , Geigl, J.B. , Speicher, M.R. , 2013. Circulating tumor cells and DNA as liquid biopsies. Genome Med. 5, 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer, E. , Ulz, P. , Belic, J. , Gutschi, S. , Quehenberger, F. , Fischereder, K. , Benezeder, T. , Auer, M. , Pischler, C. , Mannweiler, S. , Pichler, M. , Eisner, F. , Haeusler, M. , Riethdorf, S. , Pantel, K. , Samonigg, H. , Hoefler, G. , Augustin, H. , Geigl, J.B. , Speicher, M.R. , 2013. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 5, 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer, E. , Ulz, P. , Geigl, J.B. , 2015. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 61, 112–123. [DOI] [PubMed] [Google Scholar]
- Knudson, A.G. , 2001. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer. 1, 157–162. [DOI] [PubMed] [Google Scholar]
- Leary, R.J. , Kinde, I. , Diehl, F. , Schmidt, K. , Clouser, C. , Duncan, C. , Antipova, A. , Lee, C. , McKernan, K. , De La Vega, F.M. , Kinzler, K.W. , Vogelstein, B. , Diaz, L.A. , Velculescu, V.E. , 2010. Development of personalized tumor biomarkers using massively parallel sequencing. Sci. Transl. Med. 2, 20ra14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary, R.J. , Sausen, M. , Kinde, I. , Papadopoulos, N. , Carpten, J.D. , Craig, D. , O'Shaughnessy, J. , Kinzler, K.W. , Parmigiani, G. , Vogelstein, B. , Diaz, L.A. , Velculescu, V.E. , 2012. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 4, 162ra154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magbanua, M.J. , Sosa, E.V. , Roy, R. , Eisenbud, L.E. , Scott, J.H. , Olshen, A. , Pinkel, D. , Rugo, H.S. , Park, J.W. , 2013. Genomic profiling of isolated circulating tumor cells from metastatic breast cancer patients. Cancer Res. 73, 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazel, M. , Jacot, W. , Pantel, K. , Bartkowiak, K. , Topart, D. , Cayrefourcq, L. , Rossille, D. , Maudelonde, T. , Fest, T. , Alix-Panabieres, C. , 2015. Frequent expression of PD-L1 on circulating breast cancer cells. Mol. Oncol. 9, 1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride, D.J. , Orpana, A.K. , Sotiriou, C. , Joensuu, H. , Stephens, P.J. , Mudie, L.J. , Hamalainen, E. , Stebbings, L.A. , Andersson, L.C. , Flanagan, A.M. , Durbecq, V. , Ignatiadis, M. , Kallioniemi, O. , Heckman, C.A. , Alitalo, K. , Edgren, H. , Futreal, P.A. , Stratton, M.R. , Campbell, P.J. , 2010. Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes Chromosomes Cancer. 49, 1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale, S. , Yaeger, R. , Hobor, S. , Scala, E. , Janakiraman, M. , Liska, D. , Valtorta, E. , Schiavo, R. , Buscarino, M. , Siravegna, G. , Bencardino, K. , Cercek, A. , Chen, C.T. , Veronese, S. , Zanon, C. , Sartore-Bianchi, A. , Gambacorta, M. , Gallicchio, M. , Vakiani, E. , Boscaro, V. , Medico, E. , Weiser, M. , Siena, S. , Di Nicolantonio, F. , Solit, D. , Bardelli, A. , 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 486, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale, S. , Arena, S. , Lamba, S. , Siravegna, G. , Lallo, A. , Hobor, S. , Russo, M. , Buscarino, M. , Lazzari, L. , Sartore-Bianchi, A. , Bencardino, K. , Amatu, A. , Lauricella, C. , Valtorta, E. , Siena, S. , Di Nicolantonio, F. , Bardelli, A. , 2014. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci. Transl. Med. 6, 224ra226 [DOI] [PubMed] [Google Scholar]
- Mohan, S. , Heitzer, E. , Ulz, P. , Lafer, I. , Lax, S. , Auer, M. , Pichler, M. , Gerger, A. , Eisner, F. , Hoefler, G. , Bauernhofer, T. , Geigl, J.B. , Speicher, M.R. , 2014. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 10, e1004271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohlendick, B. , Bartenhagen, C. , Behrens, B. , Honisch, E. , Raba, K. , Knoefel, W.T. , Stoecklein, N.H. , 2013. A robust method to analyze copy number alterations of less than 100 kb in single cells using oligonucleotide array CGH. PloS One. 8, e67031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaza, M. , Dawson, S.J. , Tsui, D.W. , Gale, D. , Forshew, T. , Piskorz, A.M. , Parkinson, C. , Chin, S.F. , Kingsbury, Z. , Wong, A.S. , Marass, F. , Humphray, S. , Hadfield, J. , Bentley, D. , Chin, T.M. , Brenton, J.D. , Caldas, C. , Rosenfeld, N. , 2013. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 497, 108–112. [DOI] [PubMed] [Google Scholar]
- Navin, N. , Kendall, J. , Troge, J. , Andrews, P. , Rodgers, L. , McIndoo, J. , Cook, K. , Stepansky, A. , Levy, D. , Esposito, D. , Muthuswamy, L. , Krasnitz, A. , McCombie, W.R. , Hicks, J. , Wigler, M. , 2011. Tumour evolution inferred by single-cell sequencing. Nature. 472, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawroz, H. , Koch, W. , Anker, P. , Stroun, M. , Sidransky, D. , 1996. Microsatellite alterations in serum DNA of head and neck cancer patients. Nat. Med. 2, 1035–1037. [DOI] [PubMed] [Google Scholar]
- Neves, R.P. , Raba, K. , Schmidt, O. , Honisch, E. , Meier-Stiegen, F. , Behrens, B. , Mohlendick, B. , Fehm, T. , Neubauer, H. , Klein, C.A. , Polzer, B. , Sproll, C. , Fischer, J.C. , Niederacher, D. , Stoecklein, N.H. , 2014. Genomic high-resolution profiling of single CKpos/CD45neg flow-sorting purified circulating tumor cells from patients with metastatic breast cancer. Clin. Chem. 60, 1290–1297. [DOI] [PubMed] [Google Scholar]
- Newman, A.M. , Bratman, S.V. , To, J. , Wynne, J.F. , Eclov, N.C. , Modlin, L.A. , Liu, C.L. , Neal, J.W. , Wakelee, H.A. , Merritt, R.E. , Shrager, J.B. , Loo, B.W. , Alizadeh, A.A. , Diehn, M. , 2014. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, X. , Zhuo, M. , Su, Z. , Duan, J. , Gao, Y. , Wang, Z. , Zong, C. , Bai, H. , Chapman, A.R. , Zhao, J. , Xu, L. , An, T. , Ma, Q. , Wang, Y. , Wu, M. , Sun, Y. , Wang, S. , Li, Z. , Yang, X. , Yong, J. , Su, X.D. , Lu, Y. , Bai, F. , Xie, X.S. , Wang, J. , 2013. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. U. S. A. 110, 21083–21088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson, E. , Winter, C. , George, A. , Chen, Y. , Howlin, J. , Tang, M.H. , Dahlgren, M. , Schulz, R. , Grabau, D. , van Westen, D. , Ferno, M. , Ingvar, C. , Rose, C. , Bendahl, P.O. , Ryden, L. , Borg, A. , Gruvberger-Saal, S.K. , Jernstrom, H. , Saal, L.H. , 2015. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 7, 1034–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantel, K. , Speicher, M.R. , 2015. The biology of circulating tumor cells. Oncogene. 10.1038/onc.2015.192 [DOI] [PubMed] [Google Scholar]
- Polzer, B. , Medoro, G. , Pasch, S. , Fontana, F. , Zorzino, L. , Pestka, A. , Andergassen, U. , Meier-Stiegen, F. , Czyz, Z.T. , Alberter, B. , Treitschke, S. , Schamberger, T. , Sergio, M. , Bregola, G. , Doffini, A. , Gianni, S. , Calanca, A. , Signorini, G. , Bolognesi, C. , Hartmann, A. , Fasching, P.A. , Sandri, M.T. , Rack, B. , Fehm, T. , Giorgini, G. , Manaresi, N. , Klein, C.A. , 2014. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 6, 1371–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert, T. , Scholer, L.V. , Thomsen, R. , Tobiasen, H. , Vang, S. , Nordentoft, I. , Lamy, P. , Kannerup, A.S. , Mortensen, F.V. , Stribolt, K. , Hamilton-Dutoit, S. , Nielsen, H.J. , Laurberg, S. , Pallisgaard, N. , Pedersen, J.S. , Orntoft, T.F. , Andersen, C.L. , 2015. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 10.1136/gutjnl-2014-308859 [DOI] [PubMed] [Google Scholar]
- Santaguida, S. , Amon, A. , 2015. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 16, 473–485. [DOI] [PubMed] [Google Scholar]
- Schwarzenbach, H. , Hoon, D.S. , Pantel, K. , 2011. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer. 11, 426–437. [DOI] [PubMed] [Google Scholar]
- Shaw, J.A. , Page, K. , Blighe, K. , Hava, N. , Guttery, D. , Ward, B. , Brown, J. , Ruangpratheep, C. , Stebbing, J. , Payne, R. , Palmieri, C. , Cleator, S. , Walker, R.A. , Coombes, R.C. , 2012. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 22, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siravegna, G. , Mussolin, B. , Buscarino, M. , Corti, G. , Cassingena, A. , Crisafulli, G. , Ponzetti, A. , Cremolini, C. , Amatu, A. , Lauricella, C. , Lamba, S. , Hobor, S. , Avallone, A. , Valtorta, E. , Rospo, G. , Medico, E. , Motta, V. , Antoniotti, C. , Tatangelo, F. , Bellosillo, B. , Veronese, S. , Budillon, A. , Montagut, C. , Racca, P. , Marsoni, S. , Falcone, A. , Corcoran, R.B. , Di Nicolantonio, F. , Loupakis, F. , Siena, S. , Sartore-Bianchi, A. , Bardelli, A. , 2015. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 21, 795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini, N.L. , Xu, Q. , Mermel, C.H. , Liang, A.C. , Schlabach, M.R. , Luo, J. , Burrows, A.E. , Anselmo, A.N. , Bredemeyer, A.L. , Li, M.Z. , Beroukhim, R. , Meyerson, M. , Elledge, S.J. , 2012. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 337, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert, G. , Scholch, S. , Niemietz, T. , Iwata, N. , Garcia, S.A. , Behrens, B. , Voigt, A. , Kloor, M. , Benner, A. , Bork, U. , Rahbari, N.N. , Buchler, M.W. , Stoecklein, N.H. , Weitz, J. , Koch, M. , 2014. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 74, 1694–1704. [DOI] [PubMed] [Google Scholar]
- Stratton, M.R. , Campbell, P.J. , Futreal, P.A. , 2009. The cancer genome. Nature. 458, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, K. , Jiang, P. , Chan, K.C. , Wong, J. , Cheng, Y.K. , Liang, R.H. , Chan, W.K. , Ma, E.S. , Chan, S.L. , Cheng, S.H. , Chan, R.W. , Tong, Y.K. , Ng, S.S. , Wong, R.S. , Hui, D.S. , Leung, T.N. , Leung, T.Y. , Lai, P.B. , Chiu, R.W. , Lo, Y.M. , 2015. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. U. S. A. 112, E5503–E5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, B.S. , Schultz, N. , Hieronymus, H. , Gopalan, A. , Xiao, Y. , Carver, B.S. , Arora, V.K. , Kaushik, P. , Cerami, E. , Reva, B. , Antipin, Y. , Mitsiades, N. , Landers, T. , Dolgalev, I. , Major, J.E. , Wilson, M. , Socci, N.D. , Lash, A.E. , Heguy, A. , Eastham, J.A. , Scher, H.I. , Reuter, V.E. , Scardino, P.T. , Sander, C. , Sawyers, C.L. , Gerald, W.L. , 2010. Integrative genomic profiling of human prostate cancer. Cancer Cell. 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry, A.R. , Mouliere, F. , El Messaoudi, S. , Mollevi, C. , Lopez-Crapez, E. , Rolet, F. , Gillet, B. , Gongora, C. , Dechelotte, P. , Robert, B. , Del Rio, M. , Lamy, P.J. , Bibeau, F. , Nouaille, M. , Loriot, V. , Jarrousse, A.S. , Molina, F. , Mathonnet, M. , Pezet, D. , Ychou, M. , 2014. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 20, 430–435. [DOI] [PubMed] [Google Scholar]
- Valtorta, E. , Misale, S. , Sartore-Bianchi, A. , Nagtegaal, I.D. , Paraf, F. , Lauricella, C. , Dimartino, V. , Hobor, S. , Jacobs, B. , Ercolani, C. , Lamba, S. , Scala, E. , Veronese, S. , Laurent-Puig, P. , Siena, S. , Tejpar, S. , Mottolese, M. , Punt, C.J. , Gambacorta, M. , Bardelli, A. , Di Nicolantonio, F. , 2013. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cancer. 133, 1259–1265. [DOI] [PubMed] [Google Scholar]
- Vogelstein, B. , Papadopoulos, N. , Velculescu, V.E. , Zhou, S. , Diaz, L.A. , Kinzler, K.W. , 2013. Cancer genome landscapes. Science. 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T.L. , Maierhofer, C. , Speicher, M.R. , Lengauer, C. , Vogelstein, B. , Kinzler, K.W. , Velculescu, V.E. , 2002. Digital karyotyping. Proc. Natl. Acad. Sci. U. S. A. 99, 16156–16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, S. , Kohli, M. , Du, M. , Dittmar, R.L. , Lee, A. , Nandy, D. , Yuan, T. , Guo, Y. , Wang, Y. , Tschannen, M.R. , Worthey, E. , Jacob, H. , See, W. , Kilari, D. , Wang, X. , Hovey, R.L. , Huang, C.C. , Wang, L. , 2015. Plasma genetic and genomic abnormalities predict treatment response and clinical outcome in advanced prostate cancer. Oncotarget. 6, 16411–16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung, T.K. , Chan, K.C. , Mok, T.S. , Tong, J. , To, K.F. , Lo, Y.M. , 2009. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin. Cancer Res. 15, 2076–2084. [DOI] [PubMed] [Google Scholar]
- Zack, T.I. , Schumacher, S.E. , Carter, S.L. , Cherniack, A.D. , Saksena, G. , Tabak, B. , Lawrence, M.S. , Zhang, C.Z. , Wala, J. , Mermel, C.H. , Sougnez, C. , Gabriel, S.B. , Hernandez, B. , Shen, H. , Laird, P.W. , Getz, G. , Meyerson, M. , Beroukhim, R. , 2013. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]