

Abstract
Postzygotic mutations in somatic cells lead to genome mosaicism and can be the cause of cancer, possibly other human diseases and aging. Somatic mutations are difficult to detect in bulk tissue samples. Here, we review the available assays for measuring somatic mutations, with a focus on recent single-cell, whole genome sequencing methods.
Impact statement
Somatic mutations cause cancer, possibly other diseases and aging. Yet, very little is known about the frequency of such mutations in vivo, their distribution across the genome, and their possible functional consequences other than cancer. Even in cancer, we do not know the heterogeneity of mutations within a tumor and if seemingly normal cells in its surroundings already have elevated mutation frequencies. Here, we review a new, whole genome amplification system that allows accurate quantification and characterization of single-cell mutational landscapes in human cells and tissues in relation to disease.
Keywords: Aging, cancer, genomic features, heterogeneity, somatic mutations, whole genome amplification
Introduction
Mutations in the genome of somatic cells of multicellular organisms are the inevitable consequence of errors during DNA repair or replication. They can vary from base substitution mutations to genome structural variations and large chromosomal changes. Due to their random nature, most mutations have no or little phenotypic effects. Very rarely, mutations have beneficial effects, something that has been coopted by evolution to give rise to adaptation and speciation through natural selection. Somatic mutations are often pathogenic. For example, cancer is now generally accepted to be a genetic disease caused by accidental mutations that inactivate tumor suppressor genes, such as TP53, or activate oncogenes, such as those belonging to the Ras family.1
Because mutations are both inevitable and irreversible (in contrast to DNA damage, DNA mutations cannot be repaired), they accumulate with age in the cells that make up tissues and organs. This is likely one of the reasons why cancer occurs more often in older people who have had more opportunities for mutations to accumulate. Also people who are exposed to DNA-damaging agents, such as radiation, are generally at a higher risk for cancer, most likely because of an elevated load of somatic mutations. Mutations may also causally contribute to other chronic diseases, including neurodegenerative diseases and even to the biological process of aging.2
Due to the relatively simple nature of the genetic material, DNA mutations are in principle amenable to reliable detection using current sequencing technology. However, even with the most advanced sequencing technology, direct testing for somatic mutations is not straightforward due to the very low abundance of such mutations, both spontaneous mutations and mutations induced by exposure to an agent. Here, we will discuss available assays for somatic mutations with a special focus on single-cell, next-generation sequencing assays and their application in (1) studying cancer and aging, (2) the identification of pro-mutagenic hazards, and (3) quantitative mutagenicity risk assessment of human populations.
Current methods for mutation analysis
For a long time, the only types of mutations readily detectable in human or animal primary cells were chromosomal alterations, such as chromosomal aneuploidy, using cytogenetics.3 More recently, cytogenetic methods gained accuracy and could be applied on a much larger scale due to the development of fluorescence in situ hybridization (FISH). Using these methods it has, for example, been shown that lymphocytes with chromosomal aberrations increase with age in the blood of both humans and mice.4,5 Interphase FISH can even be applied on non-dividing cells in tissues such as brain6 for the detection of aneuploidy, i.e. gain or loss of entire chromosomes. Aneuploidy levels, even in postmitotic tissue, appeared to be remarkably high.7,8 In mice, we found that in the cerebral cortex, the frequency of aneuploid cells can rise to a level as high as 5%.8 Chromosomal aneuploidy is a hallmark of pathological conditions and a causal factor of birth defects and cancer.9
However, large chromosomal aberrations are merely the tip of an underlying iceberg of different types of mutations. Indeed, mutations that are much more frequent on a per genome basis include base substitutions, deletions, and genome rearrangements. Methods have become available over the last decade to detect such mutations. For example, methods using endogenous selectable marker genes, such as hypoxanthine-guanine phosphoribosyltransferase (HGPRT), were used to assess mutation frequencies in blood cells.10 Interestingly, application of these methods showed that similar to the aforementioned chromosomal aberrations and aneuploidy also the frequency of these generally much smaller mutations was found to increase with age, both in humans and rodents.11,12
Somewhat later, the development of transgenic mice harboring reporter genes that can be recovered in E. coli to study mutations that had occurred in the animal, for the first time allowed mutagenicity testing in any possible target organ in an animal.13,14 These mouse models remain in use as substitutes for the expensive, long-term rodent bioassays to predict carcinogenicity of environmental compounds.15 The use of these animals also revealed that spontaneous mutations accumulate with age in essentially all organs and tissues,16–19 albeit at greatly different rates (Figure 1).
Figure 1.

Somatic mutations accumulate during aging in four types of tissues in mouse. A transgenic mouse model harboring chromosomally integrated plasmids containing the lacZ reporter gene were excised and transferred into E. coli to select for mutants that inactivate the lacZ-encoded beta-galactosidase. Using this model, mutation frequency (y-axis) was estimated as a function of the age of the animals. Each determination point is the average of at least five individual mice. Data were from Dollé et al.16,17 (A color version of this figure is available in the online journal.)
Reporter assays are quite sensitive and specific20 and quickly became the method of choice in mutagenicity studies. However, reporter genes, the size of which does not exceed about 3000 bp, are heavily methylated and not transcribed. Hence, they are unlikely to be fully representative of the somatic genome with all its complex, tissue-specific features. In addition, similar relatively simple transgenic reporter systems are not readily available for human cells or cell lines.
To address these limitations, assays should be able to comprehensively characterize the total complement of mutations in individual cells across the genome in primary cells and tissues. In theory, this can be done by next-generation sequencing, which should allow the detection of a wide range of somatic mutations. However, since somatic mutations in normal tissues are unique for each individual cell (except for those cells that are derived from the same ancestor cell in which the mutation occurred), whole genome sequencing will simply provide the germline mutational landscape. When sequencing at very high depth, one could occasionally find a sequencing read with a true mutation. However, those true mutations would drown in the sequencing errors, which are as high as 0.1–1%.21 To study the very low abundant mutations in normal somatic tissues, it is necessary to analyze single cells or clones derived from single cells. To some extent, tumors can serve as surrogates for single cells. Tumors, as clonal expansions of single cells, can provide information about the somatic mutations present in these cells prior to tumorigenesis. Using data from The Cancer Genome Atlas (TCGA) to systematically study the frequency and spectrum of somatic mutations in thousands of cancer patients and different tumor types as a function of the age of the patient, we found that the number of identified somatic mutations increases exponentially with age.22 However, since mutations can also arise after neoplastic transformation, during tumor progression, it is difficult to draw definite conclusions other than that mutation frequency increases with age. Others have demonstrated aging-specific signature mutations in human tumors.23
More recently, whole genome sequencing of clonal organoid cultures derived from mouse or human primary multipotent cells revealed hundreds of base substitution mutations per genome increasing with age.24,25 However, clonal amplification through organoid technology requires extensive cell culture and essentially limits analysis to stem or progenitor cells. Single-cell technology allows direct analysis of all types of cells, including postmitotic cells, such as neurons and muscle fibers.
Single-cell methods to analyze somatic mutations
Analyzing mutations in single cells by next-generation sequencing requires whole genome amplification (WGA), which suffers from artifacts. We recently developed a new protocol, i.e. Single-Cell Multiple Displacement Amplification (SCMDA), with a single-cell variant caller (SCcaller), to accurately identify somatic mutations across the genome from a single cell after whole genome amplification.26 The procedure was validated by directly comparing mutation frequency and spectrum between amplified single cells and unamplified clones derived from cells in the same population of early passage, human primary fibroblasts. We also sequenced SCMDA-amplified single cells and non-amplified clones derived from the same clone, reasoning that there should be significant overlap between the single cells and their kindred clone. The entire procedure is schematically depicted in Figure 2.
Figure 2.

Schematic representation of validating mutation detection using single-cell sequencing after whole genome amplification. Mutations identified using whole genome sequencing of single human, primary fibroblasts after amplification and in unamplified clones from single fibroblasts taken from the same population were compared. After Dong et al.26 (A color version of this figure is available in the online journal.)
The number of somatic mutations in human primary fibroblasts was about 1000, in the same range as the numbers in unamplified clones (Figure 3). While slightly lower than the numbers observed in the aforementioned clonal organoid cultures, it should be noted that those are representative of stem cells. There is evidence that stem cells have lower somatic mutation frequencies than normal cells,27,28 but we have thus far not directly compared the two types of cells using our SCMDA technology.
Figure 3.
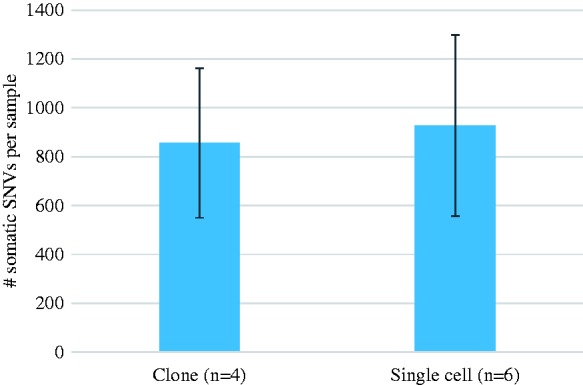
Somatic mutation frequency in human primary fibroblasts as determined by whole genome sequencing of unamplified DNA from clones and from SCMDA-amplified single cells from the same population. Error bars indicate S.D. After Dong et al.26 (A color version of this figure is available in the online journal.)
Interestingly, these somatic mutation frequencies are much higher than the germline mutation frequency. In humans, the germline mutation frequency has been determined by whole genome sequencing of parents and children and calling de novo mutations in the offspring. This resulted in a germline mutation frequency of 1.2 ×10−8,29 confirming earlier indirect estimates.30,31 When directly comparing the somatic mutation frequencies observed by us in primary human and mouse fibroblasts with the germline mutation frequencies obtained as described above, by sequencing parents and children in both mice and humans, we observed an almost two orders of magnitude higher somatic mutation frequency.32
An interesting question that arises from the observation of such a high level of base substitution mutations in somatic cells is why we do not suffer from a much higher frequency of cancers than we do. Indeed, assuming that a typical organ such as liver, contains 1012 cells and that there are 6 × 109 base pairs per human genome, at a mutation load per cell of about 1000, there should be multiple mutational hits per nucleotide. The most likely explanation as to why this does not result in cancer much more frequently is that many mutant cells may be eliminated, for example, through apoptosis or via the immune system. Indeed, overall mutation load of tumors correlates with neoantigen load.33 Furthermore, mutations alone are insufficient in causing cancer and require a permissive microenvironment.34
In addition to base substitutions also other types of mutations, such as small insertions and deletions (INDELS), copy number variations (CNVs) and genome structural variations (SVs) are in principle detectable in single cells using variants of the same single-cell procedures as described above. This would allow the complete characterization of the somatic mutational landscape in human or animal primary cells.
Functional consequences of somatic mutations other than cancer
Cancer is caused by genetic mutations in normal cells, which are subsequently selected in cycles of progression, ultimately resulting in a malignant tumor. This universally accepted model of cancer initiation and progression was first proposed by Fearon and Vogelstein for colorectal cancer.35 It explains why cancer is so adaptable and, through sequential mutations, capable of gaining favorable attributes, such as the capacity to invading tissues, suppressing immune responses and becoming resistant to therapies. Each mutation is followed by clonal outgrowth in which a particularly advantageous mutation is selected. In this way, initially rare somatic mutations can eventually become predominant through selection.
However, it is now also clear that somatic mutations can lead to genome mosaicism and contribute to diseases other than cancer, for example, type I neurofibromatosis.36 Most of these cases, which may amount to 6–20% of single gene disorders,37 are due to combined germline and somatic mosaicism. However, in a rare case of sporadic Alzheimer’s disease, the cause was a de novo, somatic mutation in the presenilin-1 gene.38 Somatic mutations have also been found in atherosclerotic plaques,39 although in this case, a causal role of the mutations in atherogenesis and vascular disease development is uncertain. Clearly, somatic mutations early during development, which can give rise to clonal enrichment, are more likely to have a phenotypic effect than mutations occurring later. In many cases, the critical somatic mutation is a second mutation turning a recessive germline mutation in one allele into a dominant phenotype after somatic inactivation of the other allele. Postzygotic mutations are now considered as a possible cause of human disease.40,41
A question that would come up at this stage is whether somatic mutations, over time, can ever reach a level that is high enough to exert adverse effects that reduce cell fitness. This possibility has been considered in the 1950s as a possible cause of aging.2,42,43 The results described in the previous section indicate that the numbers of somatic mutations in human primary cells can be as high as about 1000 per genome. And these are only base substitutions. Adding up INDELs, CNVs, SVs, and aneuploidies would yield levels of genome instability that could have functional consequences after the significant age-related increases in somatic mutation frequencies indicated by work from the past. Application of methods such as SCMDA will soon reveal the entire landscape of somatic mutations in aging tissues and organs of humans and experimental animals.44 This brings us to the question as to how a stochastic process of somatic mutation accumulation can cause loss of cell fitness.
Of the approximately 1000 base substitutions per cell, which we observed in the early passage primary fibroblasts isolated from the skin of a very young individual,26 approximately 10 proved to be non-synonymous mutations in protein-coding sequences (Lei Zhang, Xiao Dong, unpublished). Cells from aged individuals may contain at least twice that number of potentially deleterious mutations. Together with the small number of loss-of-function mutations (LOFs) typically present in the germline of human individuals,45 these de novo somatic mutations could impact on cell function. Of note, de novo mutations in most somatic cells are difficult if not impossible to eliminate through selection.
In addition to mutations directly affecting proteins, the far majority of base substitutions affect parts of the genome that are not coding for proteins. While most of those mutations will have no effect at all, a fair number of them are bound to affect the substantial part of the genome that is involved in gene regulation, which can be as high as 11%.46 This potential target for mutagenesis is far larger than the about 1.5% protein-coding part of the genome. Hence, at a mutation load of about 1000 base substitutions in a typical cell, gene regulatory regions are more likely to be functionally affected than protein-coding regions.
Hence, while the jury is still out on whether somatic mutations have any functional impact other than cancer, the availability of single-cell assays for accurate identification of all possible de novo sequence variants in the somatic genome now allows for the first time to quantitatively analyze genome instability in normal cells directly without the need for surrogate markers. This will likely lead to new advances in different areas of application, some of which will be discussed below.
Applications of single-cell mutation analysis
Heterogeneity in tumor and normal tissues
The most obvious application of the new single-cell assays for measuring genome instability is cellular heterogeneity in normal and diseased tissues. Elsewhere, the application of single-cell genomics in aging research has been extensively discussed.47 Here, we focus on cellular heterogeneity within and surrounding tumors. As mentioned above, because they are clonal lineages, tumors lend themselves well for measuring somatic mutations, and information is now available for mutations in whole exomes or whole genomes of many thousands of human tumors. However, access to the high level of intra-tumor heterogeneity in mutation frequency, spectrum and distribution across the genome48 requires single-cell assays. Moreover, very little is known about seemingly normal cells surrounding the tumor or elsewhere in the body. Such cells could be primed to develop into tumors due to an overall high mutation load or the presence of specific mutations in high-risk genes. Thus far, hidden due to their low abundance, all these mutations can now be analyzed by methods such as SCMDA. Direct assessment of mutations in normal, pre-cancerous and cancerous cells using single-cell technology will fundamentally change the way research is conducted, i.e. away from studying bulk and clonal tissues towards a single-cell approach in studying heterogeneity within the tumor and its surrounding regions. Once sequencing costs will come down further, this shift in molecular cancer research will likely lead to a corresponding shift in clinical practice, most notably in diagnosing cancer patients not only based on the tumor but also on adjacent and even distant, normal tissue. This will impact treatment by tailoring it better to the individual.
Testing agents for mutagenic effects
A second potential application of single-cell genomics is in testing chemicals and other agents for hazards associated with mutagenesis, most notably cancer. Several decades after its introduction, the Ames bacterial mutagenesis assay49,50 is still widely used to test whether a chemical can cause mutations and, therefore, would be a carcinogen. Current genetic toxicity test batteries consist of Ames bacterial mutagenesis, mutagenesis at reporter loci or chromosomal aberrations in mouse or human cells and the in vivo rodent bone marrow micronucleus assay.51 However, the field is still confronted with major challenges to the interpretation of genotoxicity test results in the context of human health risk assessment.51,52 Single-cell mutation analysis would allow the use of primary human cells, such as hepatocytes, as well as cell lines and cells and tissues from mice or rats to study mutagenic effects of various agents. This should greatly increase the predictivity of genotoxicity test as compared with current assays.
Environmental and inherited, individual risk assessment
In addition to hazard identification, i.e. how likely is it that an agent is a human mutagen and therefore a carcinogen, mutagenicity assays could be applied in assessing individual disease risk, either due to environmental exposure or inherited susceptibility. As discussed above, cancer is a genetic disease caused by genetic instability. Individual risk for cancer, and for other diseases in which somatic mutations may play a causal role, is therefore determined by inherited and acquired factors to increase or decrease genome stability. Environmental exposure may interact with genetic predisposition to either increase or decrease risk. The best example is smoking, which is the main cause of lung cancer, albeit the majority of even heavy smokers never get lung cancer. It is generally assumed that inherited factors can explain this individual variation in risk.
Single-cell mutation analysis can now be used to test if individuals genetically predisposed to cancer show higher background levels of somatic mutations in their normal somatic cells, e.g. from blood or tissue biopsies. For this purpose, the mutation load of a representative number of single cells would then be compared with the whole genome sequence of the individual's bulk DNA, which serves as the control for possible polymorphic variation with the reference genome. Similarly, single-cell genomics can also assess how many mutations have been produced in an individual by exposure to radiation or mutagenic chemicals. This would provide a direct measure of internal exposure to a mutagen. This is important for the quantification of risk in individuals and in populations and allows the recognition of specific risk factors, such as occupation, lifestyle or social status. Internal exposure is often determined by measuring signs of the agents to which individuals or populations might have been exposed, such as a mutagenic chemical or its metabolites, the products of the interaction of the agent with cellular macromolecules (protein and/or DNA) in body fluids and tissues. However, mutations are generally considered as the true end point that determines cancer risk and, therefore, the end point of choice in individual risk assessment. The only mutations that can currently be analyzed in human bodily fluids or biopsies are chromosomal aberrations, sister chromatid exchanges, increased frequency of micronuclei and inactivating mutations at the HPRT locus. The single-cell assays described above could be readily used to measure all possible mutations across the genome in any cell or tissue sample.
For example, after the nuclear power plant accident at the Fukushima nuclear power station in 2011, individual exposure was measured using dosimeters. However, this does not address the problem that there might be Fukushima residents with greater than average sensitivity to radiation-induced mutations because of their genetic background. Single-cell analysis would readily uncover such an increased risk.
The future of single-cell genomics
Single-cell genomics has now been developed to a level where it can accurately assess somatic mutations in human primary cells and tissues. As we have seen, this opens up multiple applications in basic science and translational medicine. However, there are two major obstacles that need to be overcome before single-cell mutation analysis can be applied on a large scale. First, current cost of sequencing genomes, which have come down to about $1000, still essentially constrains the analysis of tens to hundreds of single cells from an individual. However, progress in next-generation sequencing continues, with the emergence of new systems and improved instruments.53 Hence, it seems realistic to assume that within several years we will enter the era of a $100 genome. By that time, single-cell genomics will likely beginning to come into its own having acquired a niche in studying cellular heterogeneity in human individuals.
Acknowledgements
We thank an anonymous reviewer for the suggestion to briefly discuss the observed high somatic mutation frequency in the context of cancer risk. Research in the Vijg lab is supported by grants from US National Institutes of Health (AG017242, CA180126, AG047200, AG038072) and the Glenn Foundation for Medical Research.
Authors’ contributions
JV wrote the article based on experimental results obtained by XD and LZ. The figures were made by XD and LZ.
Declaration of conflicting interests
JV, XD and LZ are three of the founders of SingulOmics Corp.
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science 2013; 339: 1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vijg J. Aging of the genome, New York: Oxford, 2007. [Google Scholar]
- 3.Ferguson-Smith MA. History and evolution of cytogenetics. Mol Cytogenet 2015; 8: 19–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramsey MJ, Moore DH, 2nd, Briner JF, Lee DA, Olsen L, Senft JR, Tucker JD. The effects of age and lifestyle factors on the accumulation of cytogenetic damage as measured by chromosome painting. Mutat Res 1995; 338: 95–106. [DOI] [PubMed] [Google Scholar]
- 5.Tucker JD, Spruill MD, Ramsey MJ, Director AD, Nath J. Frequency of spontaneous chromosome aberrations in mice: effects of age. Mutat Res 1999; 425: 135–41. [DOI] [PubMed] [Google Scholar]
- 6.Faggioli F, Vijg J, Montagna C. Four-color FISH for the detection of low-level aneuploidy in interphase cells. Methods Mol Biol 2014; 1136: 291–305. [DOI] [PubMed] [Google Scholar]
- 7.Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci U S A 2001; 98: 13361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faggioli F, Wang T, Vijg J, Montagna C. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum Mol Genet 2012; 21: 5246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faggioli F, Vijg J, Montagna C. Chromosomal aneuploidy in the aging brain. Mech Ageing Dev 2011; 132: 429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albertini RJ, Castle KL, Borcherding WR. T-cell cloning to detect the mutant 6-thioguanine-resistant lymphocytes present in human peripheral blood. Proc Natl Acad Sci U S A 1982; 79: 6617–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones IM, Thomas CB, Tucker B, Thompson CL, Pleshanov P, Vorobtsova I, et al. Impact of age and environment on somatic mutation at the hprt gene of T lymphocytes in humans. Mutat Res 1995; 338: 129–39. [DOI] [PubMed] [Google Scholar]
- 12.Grist SA, McCarron M, Kutlaca A, Turner DR, Morley AA. In vivo human somatic mutation: frequency and spectrum with age. Mutat Res 1992; 266: 189–96. [DOI] [PubMed] [Google Scholar]
- 13.Gossen JA, de Leeuw WJ, Tan CH, Zwarthoff EC, Berends F, Lohman PH, et al. Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc Natl Acad Sci U S A 1989; 86: 7971–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vijg J, van Steeg H. Transgenic assays for mutations and cancer: current status and future perspectives. Mutat Res 1998; 400: 337–54. [DOI] [PubMed] [Google Scholar]
- 15.Long AS, Lemieux CL, Arlt VM, White PA. Tissue-specific in vivo genetic toxicity of nine polycyclic aromatic hydrocarbons assessed using the MutaMouse transgenic rodent assay. Toxicol Appl Pharmacol 2016; 290: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dollé ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet 1997; 17: 431–4. [DOI] [PubMed] [Google Scholar]
- 17.Dollé ME, Snyder WK, Gossen JA, Lohman PH, Vijg J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci U S A 2000; 97: 8403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dollé ME, Snyder WK, Dunson DB, Vijg J. Mutational fingerprints of aging. Nucleic Acids Res 2002; 30: 545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ono T, Ikehata H, Nakamura S, Saito Y, Hosoi Y, Takai Y, Yamada S, Onodera J, Yamamoto K. Age-associated increase of spontaneous mutant frequency and molecular nature of mutation in newborn and old lacZ-transgenic mouse. Mutat Res 2000; 447: 165–77. [DOI] [PubMed] [Google Scholar]
- 20.Wahnschaffe U, Bitsch A, Kielhorn J, Mangelsdorf I. Mutagenicity testing with transgenic mice. Part II: comparison with the mouse spot test. J Carcinog 2005; 4: 4.–4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E, Heisler LE, Beck TA, Simpson JT, Tonon L, Sertier AS, Patch AM, Jager N, Ginsbach P, Drews R, Paramasivam N, Kabbe R, Chotewutmontri S, Diessl N, Previti C, Schmidt S, Brors B, Feuerbach L, Heinold M, Grobner S, Korshunov A, Tarpey PS, Butler AP, Hinton J, Jones D, Menzies A, Raine K, Shepherd R, Stebbings L, Teague JW, Ribeca P, Giner FC, Beltran S, Raineri E, Dabad M, Heath SC, Gut M, Denroche RE, Harding NJ, Yamaguchi TN, Fujimoto A, Nakagawa H, Quesada V, Valdes-Mas R, Nakken S, Vodak D, Bower L, Lynch AG, Anderson CL, Waddell N, Pearson JV, Grimmond SM, Peto M, Spellman P, He M, Kandoth C, Lee S, Zhang J, Letourneau L, Ma S, Seth S, Torrents D, Xi L, Wheeler DA, Lopez-Otin C, Campo E, Campbell PJ, Boutros PC, Puente XS, Gerhard DS, Pfister SM, McPherson JD, Hudson TJ, Schlesner M, Lichter P, Eils R, Jones DT, Gut IG. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun 2015;6:10001. [DOI] [PMC free article] [PubMed]
- 22.Milholland B, Auton A, Suh Y, Vijg J. Age-related somatic mutations in the cancer genome. Oncotarget 2015; 6: 24627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome I, Consortium IBC, Consortium IM-S, PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature 2013;500:415–21.
- 24.Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EE, Verstegen MM, van der Laan LJ, de Jonge J, Jzermans JN, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, van Boxtel R. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016;538:260–4. [DOI] [PMC free article] [PubMed]
- 25.Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, Tarpey PS, Roerink S, Blokker J, Maddison M, Mudie L, Robinson B, Nik-Zainal S, Campbell P, Goldman N, van de Wetering M, Cuppen E, Clevers H, Stratton MR. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 2014;513:422–5. [DOI] [PMC free article] [PubMed]
- 26.Dong X, Zhang L, Milholland B, Lee M, Maslov AY, Wang T, Vijg J. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods 2017; 14: 491–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cervantes RB, Stringer JR, Shao C, Tischfield JA, Stambrook PJ. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc Natl Acad Sci U S A 2002; 99: 3586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rouhani FJ, Nik-Zainal S, Wuster A, Li Y, Conte N, Koike-Yusa H, Kumasaka N, Vallier L, Yusa K, Bradley A. Mutational history of a human cell lineage from somatic to induced pluripotent stem cells. PLoS Genet 2016;12:e1005932. [DOI] [PMC free article] [PubMed]
- 29.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K. Rate of de novo mutations and the importance of father's age to disease risk. Nature 2012;488:471–5. [DOI] [PMC free article] [PubMed]
- 30.Keightley PD. Rates and fitness consequences of new mutations in humans. Genetics 2012; 190: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondrashov A. Genetics: the rate of human mutation. Nature 2012; 488: 467–8. [DOI] [PubMed] [Google Scholar]
- 32.Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J. Differences between germline and somatic mutation rates in humans and mice. Nat Commun 2017; 8: 15183–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348: 69–74. [DOI] [PubMed] [Google Scholar]
- 34.Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 2011; 17: 320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–67. [DOI] [PubMed] [Google Scholar]
- 36.Erickson RP. Somatic gene mutation and human disease other than cancer. Mutat Res 2003; 543: 125–36. [DOI] [PubMed] [Google Scholar]
- 37.Erickson RP. Somatic gene mutation and human disease other than cancer: an update. Mutat Res 2010; 705: 96–106. [DOI] [PubMed] [Google Scholar]
- 38.Beck JA, Poulter M, Campbell TA, Uphill JB, Adamson G, Geddes JF, Revesz T, Davis MB, Wood NW, Collinge J, Tabrizi SJ. Somatic and germline mosaicism in sporadic early-onset Alzheimer's disease. Hum Mol Genet 2004;13:1219–24. [DOI] [PubMed]
- 39.Andreassi MG. Coronary atherosclerosis and somatic mutations: an overview of the contributive factors for oxidative DNA damage. Mutat Res 2003; 543: 67–86. [DOI] [PubMed] [Google Scholar]
- 40.Lupski JR. Genetics. Genome mosaicism – one human, multiple genomes. Science 2013; 341: 358–9. [DOI] [PubMed] [Google Scholar]
- 41.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science 2013; 341: 1237758–1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Failla G. The aging process and carcinogenesis. Ann N Y Acad Sci 1958; 71: 1124–35. [DOI] [PubMed] [Google Scholar]
- 43.Szilard L. On the nature of the aging process. Proc Natl Acad Sci U S A 1959; 45: 30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gundry M, Vijg J. Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants. Mutat Res 2012; 729: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, O'Dushlaine C, Van Hout CV, Staples J, Gonzaga-Jauregui C, Metpally R, Pendergrass SA, Giovanni MA, Kirchner HL, Balasubramanian S, Abul-Husn NS, Hartzel DN, Lavage DR, Kost KA, Packer JS, Lopez AE, Penn J, Mukherjee S, Gosalia N, Kanagaraj M, Li AH, Mitnaul LJ, Adams LJ, Person TN, Praveen K, Marcketta A, Lebo MS, Austin-Tse CA, Mason-Suares HM, Bruse S, Mellis S, Phillips R, Stahl N, Murphy A, Economides A, Skelding KA, Still CD, Elmore JR, Borecki IB, Yancopoulos GD, Davis FD, Faucett WA, Gottesman O, Ritchie MD, Shuldiner AR, Reid JG, Ledbetter DH, Baras A, Carey DJ. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354. [DOI] [PubMed]
- 46.Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, Dunham I, Elnitski LL, Farnham PJ, Feingold EA, Gerstein M, Giddings MC, Gilbert DM, Gingeras TR, Green ED, Guigo R, Hubbard T, Kent J, Lieb JD, Myers RM, Pazin MJ, Ren B, Stamatoyannopoulos JA, Weng Z, White KP, Hardison RC. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A 2014;111:6131–8. [DOI] [PMC free article] [PubMed]
- 47.Vijg J, Dong X, Milholland B, Zhang L. Genome instability: a conserved mechanism of ageing? Essays Biochem. Epub ahead of print 26 May 2017. doi:10.1042/EBC20160082. [DOI] [PMC free article] [PubMed]
- 48.Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol 2010; 5: 51–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ames BN, Lee FD, Durston WE. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc Natl Acad Sci U S A 1973; 70: 782–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCann J, Choi E, Yamasaki E, Ames BN. Detection of carcinogens as mutagens in the Salmonella/microsome test: assay of 300 chemicals. Proc Natl Acad Sci U S A 1975; 72: 5135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahadevan B, Snyder RD, Waters MD, Benz RD, Kemper RA, Tice RR, Richard AM. Genetic toxicology in the 21st century: reflections and future directions. Environ Mol Mutagen 2011; 52: 339–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hernandez LG, Slob W, van Steeg H, van Benthem J. Can carcinogenic potency be predicted from in vivo genotoxicity data?: a meta-analysis of historical data. Environ Mol Mutagen 2011; 52: 518–28. [DOI] [PubMed] [Google Scholar]
- 53.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016; 17: 333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]