

Conspectus

The chemistry of hypervalent iodine(III) compounds has gained great interest over the past 30 years. Hypervalent iodine(III) compounds show valuable ionic reactivity due to their high electrophilicity but also express radical reactivity as single electron oxidants for carbon and heteroatom radical generation. Looking at ionic chemistry, these iodine(III) reagents can act as electrophiles to efficiently construct C–CF3, X–CF3 (X = heteroatom), C–Rf (Rf = perfluoroalkyl), X–Rf, C–N3, C–CN, S–CN, and C–X bonds. In some cases, a Lewis or a Bronsted acid is necessary to increase their electrophilicity. In these transformations, the iodine(III) compounds react as formal “CF3+”, “Rf+”, “N3+”, “Ar+”, “CN+”, and “X+” equivalents. On the other hand, one electron reduction of the I(III) reagents opens the door to the radical world, which is the topic of this Account that focuses on radical reactivity of hypervalent iodine(III) compounds such as the Togni reagent, Zhdankin reagent, diaryliodonium salts, aryliodonium ylides, aryl(cyano)iodonium triflates, and aryl(perfluoroalkyl)iodonium triflates. Radical generation starting with I(III) reagents can also occur via thermal or light mediated homolysis of the weak hypervalent bond in such reagents. This reactivity can be used for alkane C–H functionalization. We will address important pioneering work in the area but will mainly focus on studies that have been conducted by our group over the last 5 years.
We entered the field by investigating transition metal free single electron reduction of Togni type reagents using the readily available sodium 2,2,6,6-tetramethylpiperidine-1-oxyl salt (TEMPONa) as an organic one electron reductant for clean generation of the trifluoromethyl radical and perfluoroalkyl radicals. That valuable approach was later successfully also applied to the generation of azidyl and aryl radicals starting with the corresponding benziodoxole (Zhdankin reagent) and iodonium salts. In the presence of alkenes as radical acceptors, vicinal trifluoromethyl-, azido-, and arylaminoxylation products result via a sequence comprising radical addition to the alkene and subsequent TEMPO trapping. Electron-rich arenes also react with I(III) reagents via single electron transfer (SET) to give arene radical cations, which can then engage in arylation reactions. We also recognized that the isonitrile functionality in aryl isonitriles is a highly efficient perfluoroalkyl radical acceptor, and reaction of Rf-benziodoxoles (Togni type reagents) in the presence of a radical initiator provides various perfluoroalkylated N-heterocycles (indoles, phenanthridines, quinolines, etc.). We further found that aryliodonium ylides, previously used as carbene precursors in metal-mediated cyclopropanation reactions, react via SET reduction with TEMPONa to the corresponding aryl radicals. As a drawback of all these transformations, we realized that only one ligand of the iodine(III) reagent gets transferred to the substrate. To further increase atom-economy of such conversions, we identified cyano or perfluoroalkyl iodonium triflate salts as valuable reagents for stereoselective vicinal alkyne difunctionalization, where two ligands from the I(III) reagent are sequentially transferred to an alkyne acceptor.
Finally, we will discuss alkynyl-benziodoxoles as radical acceptors for alkynylation reactions. Similar reactivity was found for the Zhdankin reagent that has been successfully applied to azidation of C-radicals, and also cyanation is possible with a cyano I(III) reagent. To summarize, this Account focuses on the design, development, mechanistic understanding, and synthetic application of hypervalent iodine(III) reagents in radical chemistry.
1. Introduction
Since the early 1990s, chemistry with I(III) compounds has witnessed significant advances in organic chemistry, due to their strong electrophilicity and valuable oxidizing properties, combined with their environment-friendly nature and in many cases commercial availability. The list of useful reactions applying I(III) reagents is meanwhile long.1 They have been used for oxidative I-ligand transfer to transition metals, for electrophilic functionalization, for electrophilic iodonium activation of carbon–carbon π-bonds, for direct C–H functionalization, for conducting iodonium ylide chemistry, as chiral I(III) compounds in enantioselective synthesis, as carbon and heteroatom radical precursors, and as radical acceptors.1 The purpose of this Account is to give an overview on radical chemistry with I(III) reagents focusing on recent work from our group. It covers literature on the use of the I(III) compounds 1–11 up to March 2017 (Figure 1).
Figure 1.

I(III) reagents used in radical chemistry.
The synthesis of I(III) reagents has been reviewed1b,1e,1f and will therefore not be further discussed herein. Compared to regular covalent I–R and I–X bonds, the R–I–X bond in I(III) compounds A, which is termed the “hypervalent bond”, is a three-atom–four-electron bond that is highly polarized, significantly longer, and also weaker than the corresponding I–R and I–X bonds in regular iodides.1b,1e Homolytic cleavage of the weak I–X bond in an I(III) reagent A is achieved thermally or by irradiation to provide the biradical F (Scheme 1). If X• and aryl–I•–R are not covalently linked, X–I homolysis results in the generation of two heteroatom centered radicals. These radicals can react with a substrate R″–H via H-abstraction to generate the C-radical G, which upon further reaction (mostly oxidation and trapping) provides the C–H functionalization product H.2 The iodanyl radical (aryl–I•–R) and also the X• radical can undergo H abstraction.2f,2g,2i−2m However, iodanyl radicals can only express radical chemistry at the I atom if they are longer lived, which is the case if α-fragmentation would lead to a high energy radical R•. For such a system (R = CN), we showed that iodanyl radicals efficiently add to alkynes (see section 7).3
Scheme 1. Generation of Radicals Using I(III) Reagents.

More abundant in radical chemistry of I(III) reagents is the one electron reduction of compounds A. To this end, various single electron transfer (SET) reducing reagents, such as transition metals,4 photocatalysts,5 organic reductants,6 and electron-rich π-systems7 have been used. SET reduction of A first leads to radical B, which fragments to the radical R• and anion C. R• can engage in typical addition reactions to (hetero)arenes, alkenes, and alkynes. The adduct radicals are then oxidized and trapped to afford compounds D and E (for alkene/alkyne additions). If a transition metal is used for SET reduction, oxidation and trapping is often mediated by the oxidized transition metal complex. Alternatively, the SET reduction of iodine(III) by arenes can occur via preformation of a charge-transfer (CT) complex I. SET then provides the radical cation intermediate J, which can be trapped by various carbon or heteroatom type nucleophiles to form after renewed SET oxidation and deprotonation the corresponding arylation products K.7
I(III) reagents were also shown to act as radical acceptors. This is the case for arylalkynyl compounds A where radicals R′• react with A to the iodanyl radical B and the alkynylated product (Scheme 2).8a−8e Similar radical group transfer also occurs with azidyl derivatives A (R = N3) to give alkyl azides,9 and also cyanide transfer to C-radicals with reagents A (R = CN) is possible.8f The following sections give an overview on all these different transformations.
Scheme 2. I(III) Reagents as Radical Acceptors.
2. Radical C–H Functionalization Using I(III) Reagents
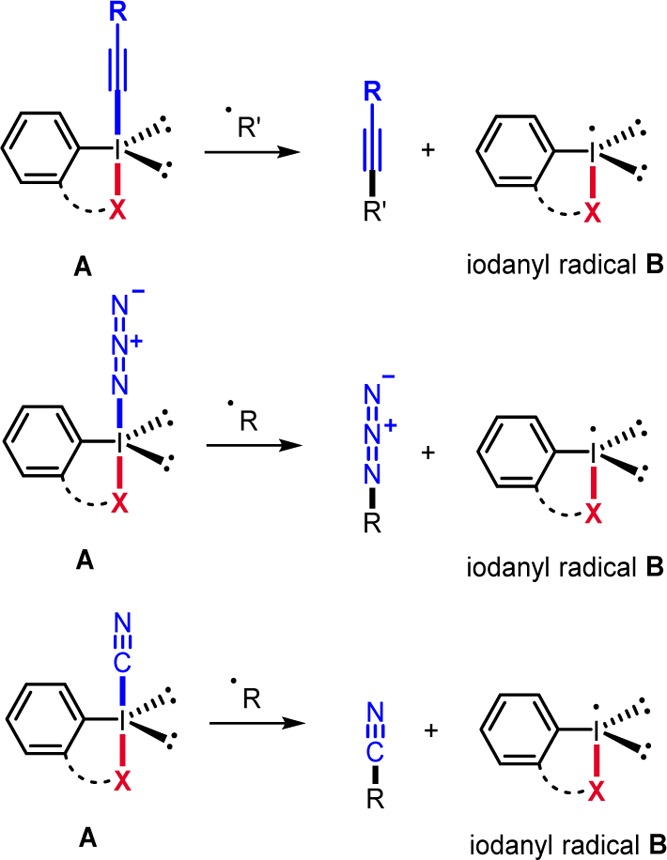
In 1979, Martin reported the benzylic and allylic radical C–H chlorination and bromination with halobenziodoxoles of type 3.2i These reactions likely proceed as chain processes where iodanyl radicals derived from 3 undergo C–H abstraction and the thus generated C-radicals are halogenated by reagents 3 to give the halogenated products along with the corresponding iodanyl radicals thereby sustaining the chain (see Scheme 3, exemplified for halogenation of cyclohexene).2i In analogy, radical C–H iodination by the I(III) reagent 4 was disclosed by Barluenga.2a With I2, 4, and TMSN3 (10 mol %), cyclohexane was transformed to iodocyclohexane. The iodanyl radical B (Scheme 1, X = OCO) derived from 4 abstracts a H atom from cyclohexane to generate the cyclohexyl radical that gets iodinated with I2.
Scheme 3. C–H Halogenation and Oxidative Cross-Coupling.
Antonchik showed that in situ generated PhI(OCOCF3)N3 can be used for transition metal free cross-dehydrogenative coupling of heterocycles with aldehydes2b and alkanes.2c,2d For example, quinoline 12 reacts with PhI(OCOCF3)N3, readily generated from PhI(OCOCF3)2 with NaN3, and cyclohexane to 13 (85%, Scheme 3).2c Homolysis of the weak I–N3 bond generates an N3-radical and B (X = OCOCF3). The N3-radical abstracts a H atom from cyclohexane to give the cyclohexyl radical, which adds to protonated 12 to radical cation 14. Deprotonation and oxidation with B lead to 13, iodobenzene, and trifluoroacetic acid. Note that azido-I(III) reagents derived from PhIO/TMSN3 were previously used by Magnus for radical C–H azidation.2k In these transformations, the C-radical generated by H-transfer is trapped (azidated) by the azido-I(III) reagent (see also section 8).2k Similar reactivity was reported by Kita using PhI(OCOCF3)2/TMSN3 as reagent couple.2l
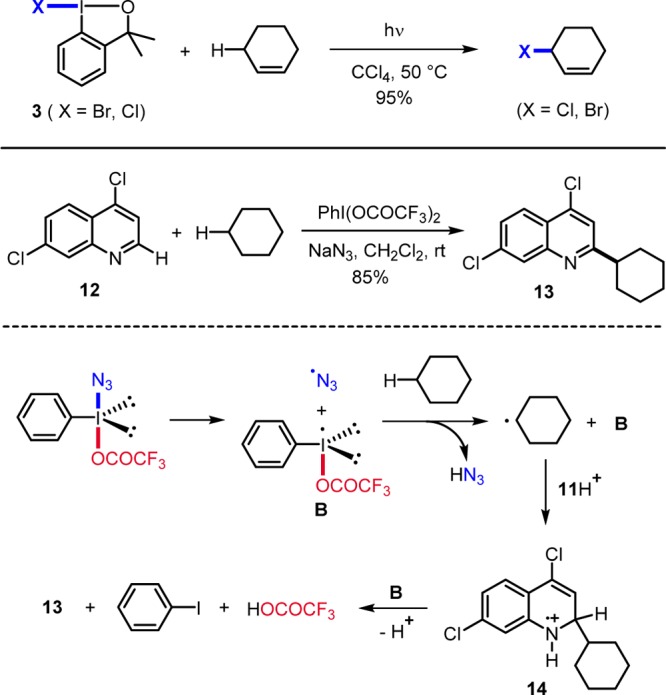
Support for iodanyl radicals acting in H-abstraction reactions was provided by Maruoka.2e Reagents 10 were applied to the oxidation of t-Bu-cyclohexane with t-BuOOH as a cooxidant to give the regioisomeric ketones 15 and 16 (Scheme 4). Reagent 10 first reacts with t-BuOOH to the I(III) intermediate 17. I–O bond homolysis in 17 generates B and t-BuOO•. H-abstraction from t-Bu-cyclohexane by B provides a cyclohexyl radical. As compared to B (R = H) derived from 10a, for the bulkier radical B (R = Ph) derived from 10b, selectivity for C(3)–H abstraction is higher. Cross-coupling of the C-radical with the peroxyl radical10 gives 18. Finally, ionic β-fragmentation affords t-BuOH and the target ketone. Similar reactivity was also noted for alkane C–H functionalization with PhI(OOt-Bu)2.2h Notably, oxidation of benzylic C–H bonds to ketones via iodanyl radicals was already reported by Ochiai in 1992.2j
Scheme 4. C–H Abstraction by Iodanyl Radicals.
3. Trifluoromethylation via SET Reduction of the Togni Reagent
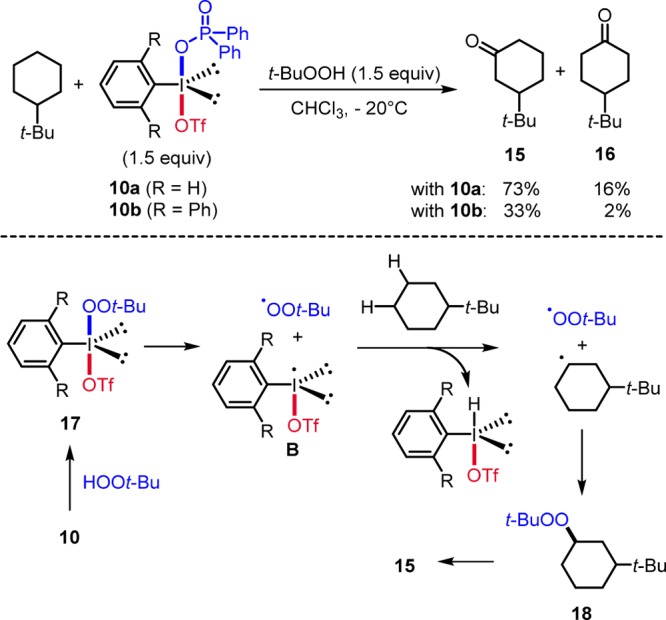
The CF3 group is a valuable substituent for modulation of chemical and physical properties of drug candidates, agrochemicals, and polymers.11 CF3-bearing benziodoxoles 1a, introduced by Togni, have been intensively used for electrophilic trifluoromethylation of carbon and heteroatom based nucleophiles.12 This chemistry has been reviewed12 and we will only address examples on radical transformations focusing on our own contributions. In 2011, Buchwald and Wang described allylic trifluoromethylation of alkenes with reagent 1a (Scheme 5).4a,4b For example, reaction of 19 with 1a using catalytic CuCl provided trifluoromethylated alkene 20. Mechanistically, radical generation occurs by reduction of 1a with CuCl to give the CF3-radical and 21. CF3-radical addition to the alkene leads to the adduct radical that is finally oxidized by the Cu-complex 21. Numerous papers on radical trifluoromethylation with 1a using redox catalysis have appeared since.12,13
Scheme 5. Cu-Catalyzed Allylic Trifluoromethylation.
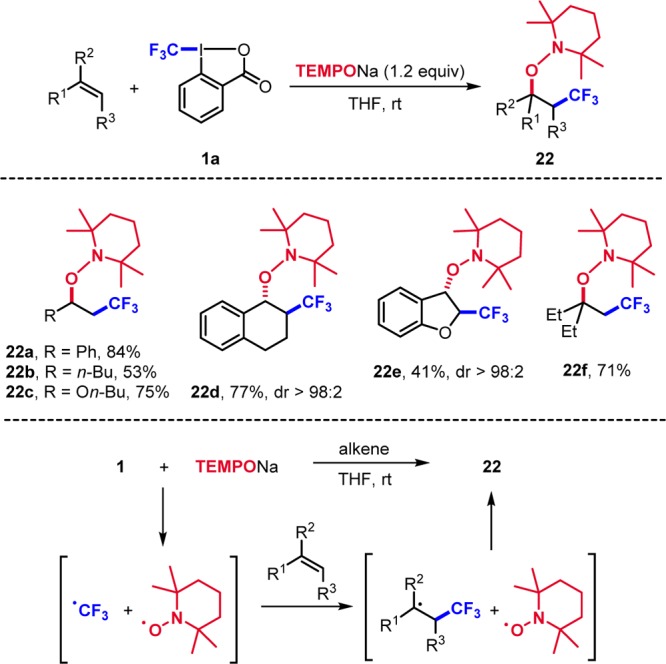
We assumed that the I(III)–CF3 bond in 1a can be reduced with the sodium 2,2,6,6-tetramethylpiperidine-1-oxyl salt (TEMPONa) salt. Indeed, reaction of 1a with TEMPONa cleanly generates the CF3-radical and TEMPO.6 In the presence of a reactive alkene, alkene CF3-radical addition and trapping by TEMPO provide 22 (Scheme 6). Not only activated styrene derivatives (22a) but also nonactivated aliphatic alkenes were successfully converted (22b). Reactions were less efficient with electron-poor alkenes but provided good results with electron-rich vinyl ethers (22c). Trifluoromethylaminoxylation of cyclic alkenes (22d) and of benzofuran (22e) afforded the products with excellent stereo- and regioselectivity. This cascade also worked on β-disubstituted alkenes to give tertiary alkoxyamines (22f).
Scheme 6. Trifluoromethylaminoxylation of Alkenes.
Despite the many contributions toward radical trifluoromethylation, only few studies on the determination of rate constants for CF3-radical addition to alkenes appeared.14 The TEMPONa mediated CF3-radical addition was identified as a tool to estimate such rate constants.15 If the addition of the CF3-radical to the alkene is slow, trapping of the CF3-radical by TEMPO that is concomitantly formed in the initial SET step (see Scheme 6) to form TEMPO–CF3 occurs as a side reaction. The rate constant for trapping of TEMPO by the CF3-radical (ktrap = (8.1 ± 0.3) × 108 M–1 s–1) was determined and used as a radical clock for estimation of all other rate constants (Scheme 7). Product ratio of TEMPO trapping versus trifluoromethylaminoxylation was used to calculate the rate constant kadd for CF3-radical addition according to eq 1.15 CF3-radical addition to styrene and its derivatives is fast and lies in the order of 107 to 108 M–1 s–1. Additions proceed faster for electron-rich styrene derivatives and steric effects play a role.
Scheme 7. Rate Constants for CF3 Radical Addition to Alkenes.

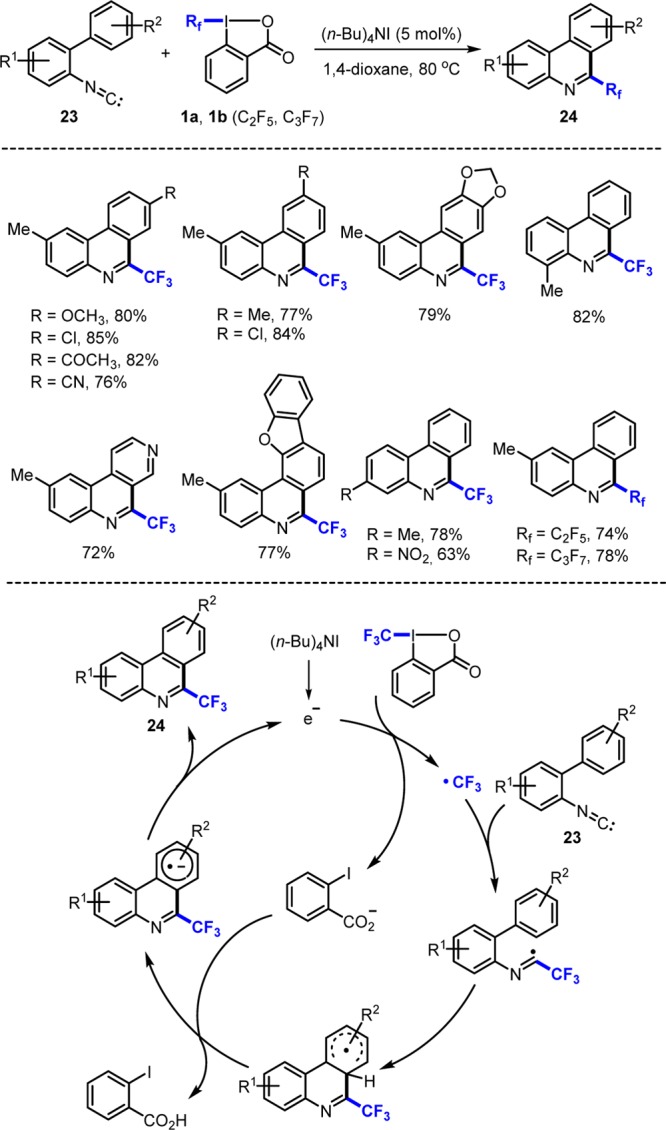
Trifluoromethylated N-heterocycles are prevalent in many drugs or drug candidates.11 Traditionally, the introduction of the CF3 group into an N-heteroarene is achieved by reaction of a halogenated or borylated heteroarene with a trifluoromethylating reagent catalyzed by a transition metal.11 Direct C–H trifluoromethylation of a heteroarene can be achieved by using either radical chemistry or transition metal based processes.11 As an alternative, we chose ortho-isocyanobiaryls 23 as acceptors using reagents 1a and 1b as trifluoromethyl and perfluoroalkyl radical precursors to give 24 (Scheme 8).16,17 The cascade works best in 1,4-dioxane at 70 °C with (n-Bu)4NI as a radical initiator (Scheme 8). Mechanistically, chain initiation occurs by reduction of 1 by the iodide to generate a CF3-radical and ortho-iodobenzoate. CF3-radical addition to the isonitrile generates an imidoyl radical, which undergoes intramolecular base promoted homolytic aromatic substitution to afford 24 formally liberating an electron to close the catalytic cycle.18,19
Scheme 8. Reaction of 2-Isocyanobiaryls with I(III) Reagents.
The aryl isonitrile approach is general for the preparation of perfluoroalkylated heteroarenes.20−23 The concept was also used for preparation of isoquinolines 26 from α-isocyano cinnamic esters 25 and I(III) reagents 1a and 1b (Scheme 9).21 In analogy, indoles 28 were prepared from isonitriles 27,22 and we found that electron-rich isonitriles 29 react with 1a and 1b via two sequential radical additions to indole-3-imines 30.23
Scheme 9. Perfluoroalkylated Heteroarenes from Aryl Isonitriles.

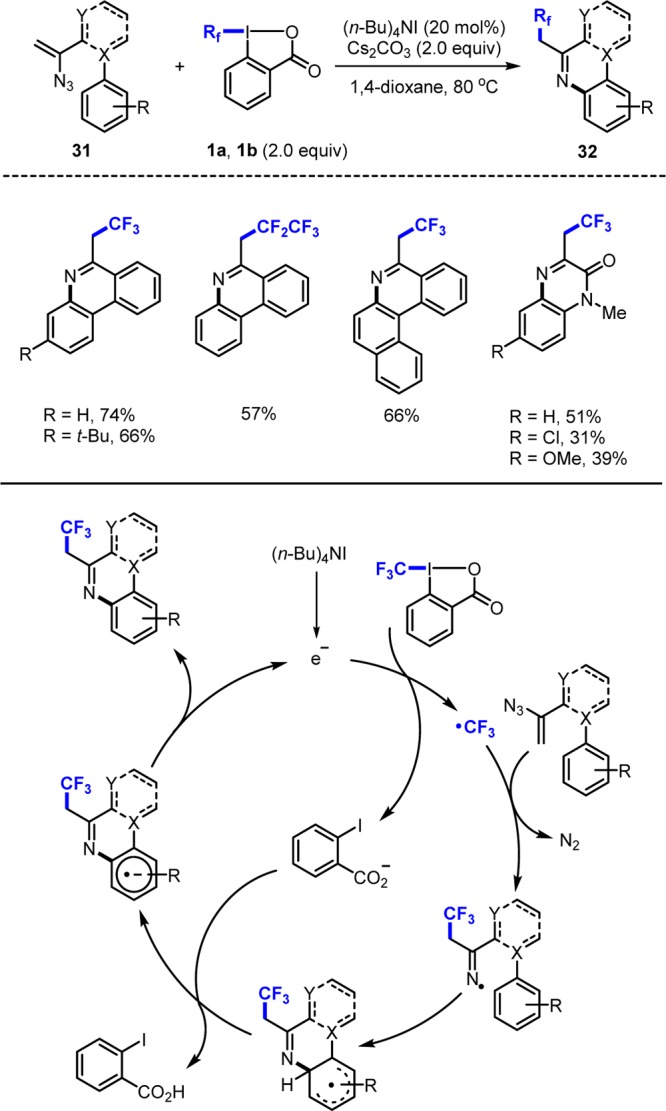
Guided by Chiba who used vinyl azides as CF3-radical acceptors,24 we reacted azides 31 with 1a or 1b (Rf = C2F5) in the presence of (n-Bu)4NI and Cs2CO3 to give the phenanthridines 32 (X = C, Y = CH) (Scheme 10).25 Quinoxalinones 32 (X = N, Y = N) are also accessible by such cascades. The chain is initiated by SET reduction of 1a generating the CF3-radical and the ortho-iodobenzoate anion. Addition of the CF3-radical to the vinyl group followed by N2-fragmentation affords an iminyl radical, which undergoes base promoted homolytic aromatic substitution19 to give the 2,2,2-trifluoroethyl phenanthridines and quinoxalinones 32. In the product forming step, an electron is formally liberated18a closing the catalytic cycle.
Scheme 10. Vinyl Azides as CF3-Radical Acceptors.
4. Alkene Azidation with Azidyl Radicals
N3-radical generation is achieved by homolysis of the I–N3 bond in azido I(III) reagents. Antonchick generated bis(azido)iodobenzene 35in situ by reacting phenyliodine bis(trifluoroacetate) 11 with TMSN3.26 Reagent 35 reacts with acrylamides 33 to α-oxindoles 34 (Scheme 11). Cascades proceed by addition of the N3-radical to 33 to give the radical 36, which cyclizes to the arene to 37. Oxidation by the azidoiodanyl radical generated in the initial homolysis and deprotonation give 34. Amidinyl radicals can also be generated by homolyis of amidinyl I(III) reagents that are easily prepared in situ upon reaction of bezamidines with PhI(OAc)2.27
Scheme 11. Cascades Involving N3-Radicals.
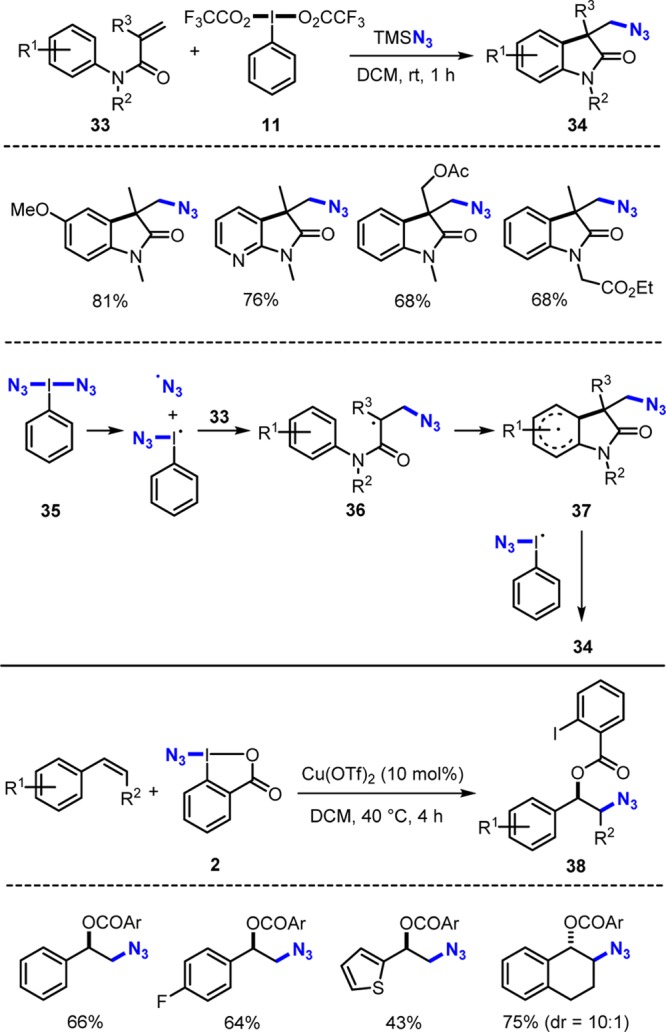
The Zhdankin reagent 2(28) can be SET reduced for clean N3-radical generation. Loh disclosed that styrene derivatives react with 2 under Cu-catalysis to provide the azidooxygenation products 38.9d SET reduction of 2 by a Cu(I)-complex gives the N3-radical that adds to the styrene derivative to provide a benzylic radical and a Cu(II)-complex. Oxygenation to 38 is assisted by the Cu(II)-complex generated in the initial SET step.
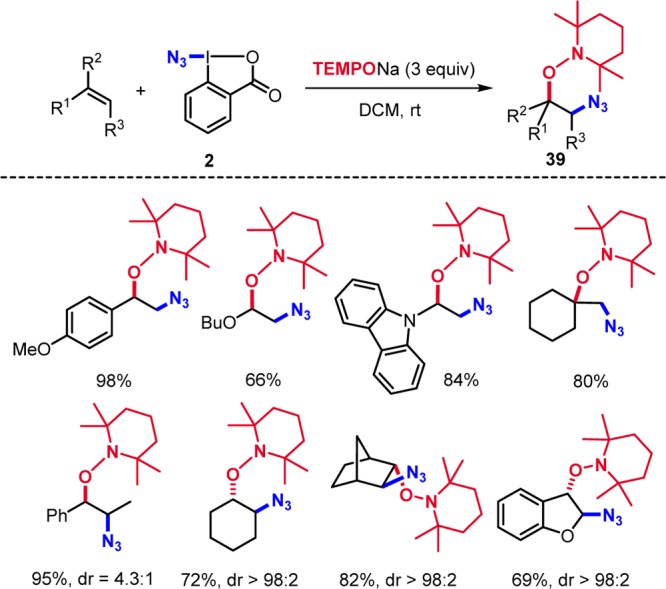
Recognizing the similarity of the Togni and Zhdankin reagents, we explored radical azidooxygenation of alkenes with TEMPONa and found that alkenes react with 2 and TEMPONa to products 39 (Scheme 12).29 Styrene derivatives, enol ethers, enamines, aliphatic alkenes, and benzofurans were substrates for this transformation. The mechanism resembles that for the trifluoromethylaminoxylation discussed in Scheme 6.
Scheme 12. Azidoaminoxylation of Alkenes.
5. Biaryl Formation via Arene Radical Cations
Kita showed that electron-rich arenes are SET oxidized by Lewis acid activated PhI(OCOCF3)2 to arene radical cations that react intra-30a or intermolecularily7a,7b with an arene to give biaryls after one-electron oxidation and deprotonation. For example, reaction of a bibenzyl ether with PhI(OCOCF3)2 and a B-Lewis acid provided 40 (Scheme 13). SET oxidation occurs via formation of a charge transfer complex between the I(III) reagent and the arene with subsequent electron transfer to form an arene radical cation. Intramolecular electrophilic aromatic substitution followed by a renewed SET oxidation and deprotonation lead to 40. Cross dehydrogenative coupling can be achieved using two different arenes with different oxidation potential:7a Reaction of naphthalene and pentamethylbenzene with PhI(OCOCF3)2 provided the cross coupling product 41. More general is the cross coupling between heteroaryl I(III) reagents and electron-rich arenes. As shown for the reaction of I-species 42 with 1,3-dimethoxybenzene to give biaryl 43, the I(III) reagent acts as SET oxidant and aryl donor.7b Likely, C–C bond formation occurs directly from the charge transfer complex. It was also found that arene azidation can be obtained by reaction of intermediately generated arene radical cations with the azide anion.30b Along these lines, cross-dehydrogenative coupling of phthalimide and simple arenes using PhI(OAc)2 was described by the DeBoef group.7c As an example, amidation of para-xylene with phthalimide to give 44 is depicted in Scheme 13. Kita also reported that direct C–H sulfenylation, thiocyanation, and azidation of phenol ethers is possible with PhI(OCOCF3)2 as an oxidant (see Scheme 13).7d,7e These reactions proceed via arene radical cations, as generally discussed in Scheme 1.
Scheme 13. Reaction of Electron-Rich Arenes with I(III) Reagents and Subsequent Trapping with Various Nucleophiles.

6. Alkene Arylaminoxylation
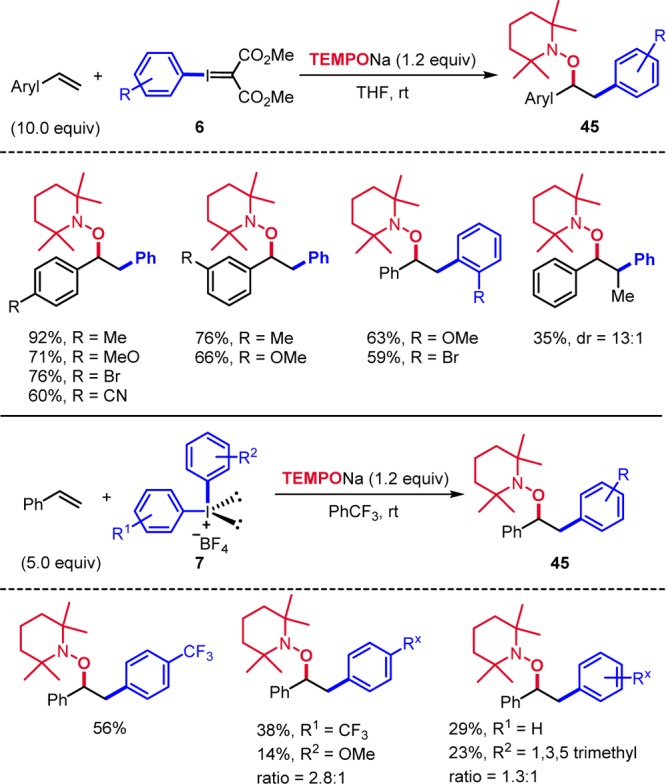
Iodonium salts have been frequently used for aryl radical generation.5a−5d,5g,31 Along these lines, we studied reduction of aryl I(III) reagents by TEMPONa for alkene arylaminoxylation32 and found that the iodanylidene malonate 6, generally used in cyclopropanations,33 can be applied to aryl radical generation (Scheme 14).34 Evaluation of the scope revealed that internal and terminal alkenes are suitable substrates to provide the arylaminoxylated products 45. Mechanistically, these transformations resemble the trifluoromethylaminoxylations discussed in Scheme 6. We also tested diaryliodonium salts as aryl radical precursors,31 which have been applied to electrophilic arylation,35 cross-coupling reactions,36 C–H arylations,37 and for benzyne generation.38 Aryl radical generation was clean upon treatment of the salt 7 with TEMPONa.34 In the presence of a styrene derivative, radical addition and subsequent TEMPO trapping give products 45. Both symmetric and nonsymmetric diaryliodonium tetrafluoroborates were tested showing that SET reduction of electronically different diaryl I(III) reagents occurs with low selectivity. Moreover, steric effects do not play an important role for aryl radical generation (Scheme 14).
Scheme 14. Arylaminoxylation of Styrene Derivatives.
7. Transfer of Two Ligands from Hypervalent I(III) Reagents
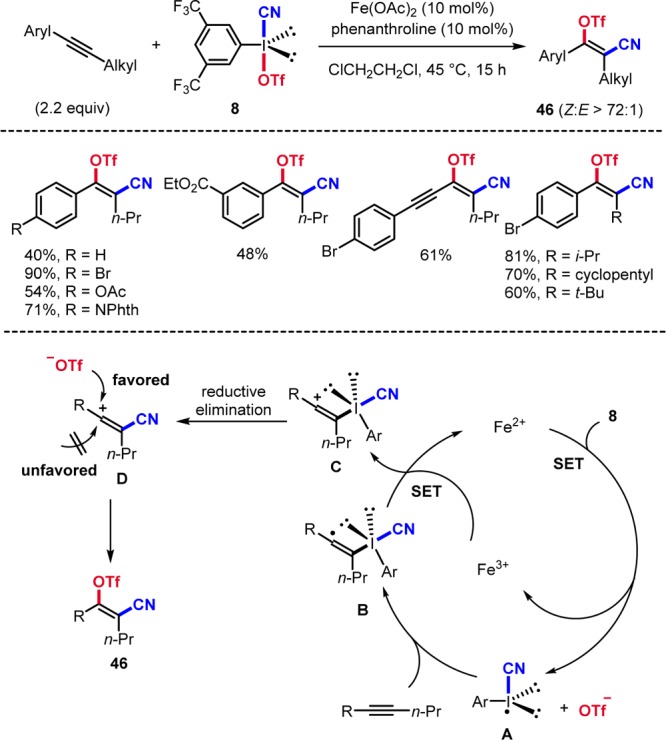
In most transformations, only one ligand of the I(III) reagent gets transferred to the substrate. We were intrigued by the challenge of sequentially transferring two ligands from an I(III) reagent to an alkyne39 and found that aryl(cyano)iodonium triflates 8(40) can be applied to stereo- and regioselective alkyne cyanotriflation.3 This transformation is best conducted with Fe(OAc)2, 1,10-phenanthroline, and triflate 8 (Scheme 15). Cyanotriflation works on alkyl aryl alkynes with complete regiochemistry and excellent Z/E-stereoselectivity to provide 46. Mechanistically, SET-reduction of 8 by the Fe(II)–phen complex generates radical A, which adds to the alkyne to give vinyl radical B. SET oxidation of B by the Fe(III)-complex leads to cation C regenerating the Fe(II)-complex. Reductive elimination at the I(III) center provides the cationic acrylonitrile, which gets trapped by the triflate anion syn to the sterically less shielding cyano group.
Scheme 15. Fe-Catalyzed Alkyne Cyanotriflation.
We also applied aryl(perfluoroalkyl)iodonium triflates 9 for alkyne perfluoroalkyltriflation. Reactions proceed using CuCl initiation and 9 to provide 47 with excellent regio- and stereoselectivity (Scheme 16).41 The perfluoroalkyltriflation works for internal and terminal aryl alkynes. Initiation of the chain occurs by SET reduction of 9 with CuCl to generate the Rf-radical, Cu(II)ClOTf, and iodobenzene. Rf-radical addition to the alkyne leads to a vinyl radical, which is oxidized by 9 in an electron-catalyzed18a process to give a vinylic cation that is stereoselectively trapped by the triflate anion to 47. The high E-stereoselectivity can be explained considering steric shielding and also electrostatic repulsion by the Rf-substituent. Reactions can also be initiated by (n-Bu)4NI supporting that perfluoroalkyltriflation proceeds via a chain process. However, since reactions are more efficiently initiated by the Cu(I)-salt, in some cycles the vinylic radical gets oxidized by Cu(II)ClOTf regenerating the initiator, featuring smart initiation.18b
Scheme 16. Perfluoroalkyltriflation of Various Alkynes.

8. I(III) Reagents As Radical Acceptors
Li and co-workers showed that alkynyl I(III) reagents 5 act as C-radical acceptors.8a They chose carboxylic acids as substrates that are oxidatively decarboxylated to C-radicals using catalytic AgNO3 and K2S2O8 (Scheme 17). Secondary and tertiary carboxylic acids were converted to the alkynylated products 48. Xiao8c and Waser8d conducted similar transformations under photoredox catalysis to convert carboxylic acids with 5 to products 48, and Chen used alkyl trifluoroborates as C-radical precursors in combination with 5 (R = alkynyl) and a photoredox catalyst.8b Li suggested that Ag-catalyzed decarboxylation of the acid leads to a C-radical that adds to reagent 5 to form radical 49. β-Fragmentation provides radical B and the target 48.8a Notably, computational studies by Waser revealed that 49 might not be an intermediate and that the alkynyl transfer might occur by a direct substitution.8f
Scheme 17. C-Radical Alkynylation and Azidation.
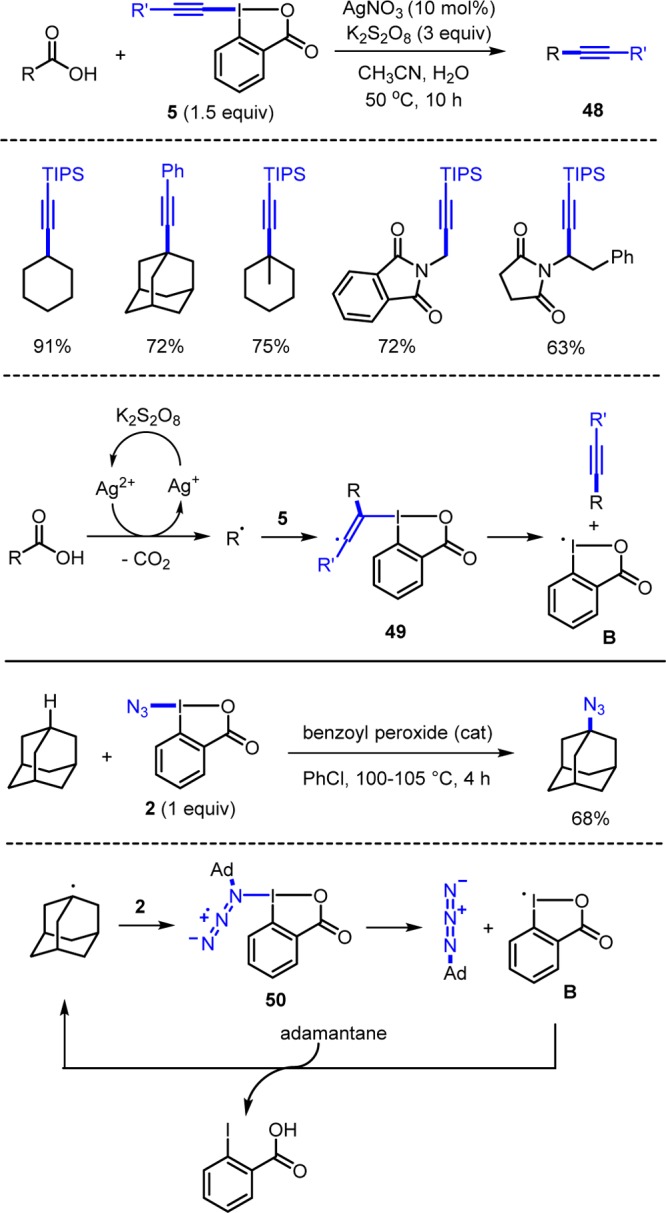
Early studies on radical azidation with azido-I(III) reagents were disclosed by Magnus showing that the PhIO in combination with TMSN3 allows for direct radical C–H azidation.2k In analogy, PhI(OCOCF3)2/TMSN3 can be used for this purpose.2l C-radical azidation can also be achieved with reagent 2 under radical conditions, as first reported by Zhdankin.9b For example, adamantane reacts with 2 in chlorobenzene at 105 °C using dibenzoyl peroxide as an initiator to azidoadamantane. Initiation occurs by thermal decomposition of the peroxide to give a carboxyl radical, which abstracts a H atom from adamantane to generate the adamantyl radical, which adds to 2 leading to adduct 50, which fragments to azidoadamantane and radical B. B abstracts a H atom from adamantane to sustain the chain. Several groups reported successful C-radical azidation with reagent 2 since.9c−9g
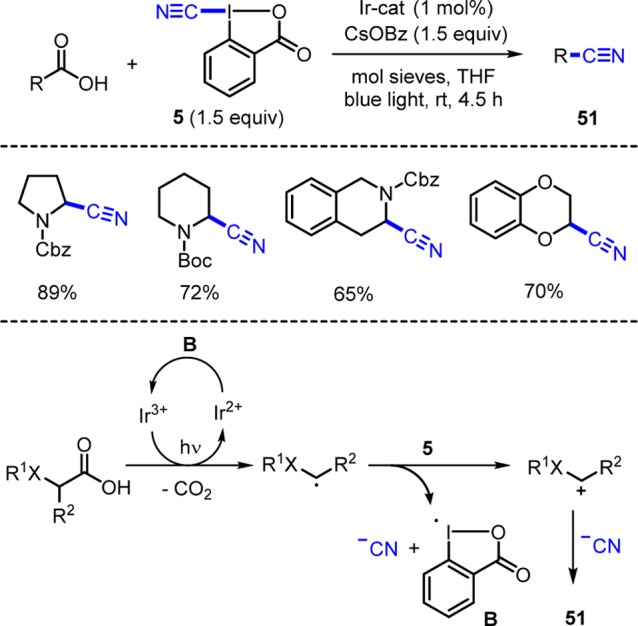
Waser showed that the carboxylic group in acids can be substituted by the cyano group with reagent 5 (R = CN) using photoredox catalysis (Scheme 18).8f Oxidative decarboxylation by a photoexcited Ir(III) complex gives a C-radical that reacts with 5 to the cyano transfer product 51. Calculations reveal that the C-radical generated after oxidative decarboxylation likely reduces 5 to form an iminium cation, an iodanyl radical B, and cyanide. The iminium ion is eventually trapped by the cyanide to 51. Iodanyl radical B is reduced by the Ir(II)-catalyst to regenerate the Ir(III)-complex closing the catalytic cycle. As a byproduct, 2-iodobenzoate is formed.
Scheme 18. Cyanation of Carboxylic Acids.
9. Summary and Perspective
We showed that I(III) compounds are valuable reagents to conduct diverse radical chemistry. The hypervalent bond in these reagents is weak so that thermal homolysis or irradiation leads to reactive intermediates that can be used for direct C–H functionalization. A large body of work deals with single electron reduction of I(III) reagents to provide carbon or heteroatom centered radicals. Radical generation by SET-reduction of I(III) reagents has emerged as a versatile tool for reliable functionalization of π-systems, allowing access to synthetically valuable structures. Notably, often transformations can be conducted in the absence of any transition metal catalyst or toxic reagent. These reactions show high efficiency, and selectivity and often good atom economy, starting materials are easily accessed, and waste handling is not a problem. Further, it is discussed that selected I(III) reagents can also act as radical acceptors for alkynylation, azidation, and cyanation. As can be extracted from the many recent contributions, radical chemistry using I(III) compounds is a timely and active research field. Considering the current ongoing renaissance of radical chemistry, it is obvious that novel interesting applications of I(III) reagents in synthetic radical chemistry will appear in the future.
Acknowledgments
We thank all current and past co-workers who have contributed to our I(III) chemistry. We thank the Alexander von Humboldt Foundation, the Deutsche Forschungsgemeinschaft, and the European Research Council (ERC Advanced Grant Agreement No. 692640) for financial support.
Biographies
Xi Wang was born in Changsha, China, in 1985. He received his B.S. from Hunan University in 2006 and joined the group of Professor Zhangjie Shi at Peking University in 2008. He moved to the laboratory of Professor Jianbo Wang at Peking University in 2010 and obtained his Ph.D. in 2013. Since 2014, he has conducted postdoctoral studies as an Alexander von Humboldt Fellow in the Studer group at the Westfälische Wilhelms-Universität in Münster. His research focuses on trifluoromethylation, cross-coupling reactions involving metal carbenes, and SET reduction of hypervalent iodine(III) reagents.
Armido Studer received his Diploma in 1991 and his Ph.D. in 1995 from ETH Zürich with Prof. Dieter Seebach. He then did postdoctoral studies at the University of Pittsburgh with Prof. Dennis P. Curran. In 1996, he started his independent career at the ETH Zürich. In 2000, he was appointed Associate Professor of Organic Chemistry at the Philipps-University in Marburg, and in 2004 Professor of Organic Chemistry at the Westfälische Wilhelms-University in Münster. In 2006, he received the Novartis Young Investigator Award in Chemistry, in 2007 the Solvias Ligand Contest Award, in 2014 the Research Award of the WWU Münster, and an ERC Advanced Grant in 2016. His research focuses on the development of new synthetic methods in radical chemistry, NHC catalysis, and transition metal catalysis. Polymer chemistry and surface chemistry are other research areas of interest.
The authors declare no competing financial interest.
References
- a Wirth T. Hypervalent Iodine Chemistry in Synthesis: Scope and New Directions. Angew. Chem., Int. Ed. 2005, 44, 3656–3665. 10.1002/anie.200500115. [DOI] [PubMed] [Google Scholar]; b Zhdankin V. V.; Stang P. J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dohi T.; Kita Y. Hypervalent Iodine Reagents as a New Entrance to Organocatalysts. Chem. Commun. 2009, 2073–2085. 10.1039/b821747e. [DOI] [PubMed] [Google Scholar]; d Merritt E. A.; Olofsson B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]; e Yoshimura A.; Zhdankin V. V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]; f Li Y.; Hari P.; Vita M. V.; Waser J. Cyclic Hypervalent Iodine Reagents for Atom-Transfer Reactions: Beyond Trifluoromethylation. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. 10.1002/anie.201509073. [DOI] [PubMed] [Google Scholar]
- a Barluenga J.; Campos-Gómez E.; Rodríguez D.; González-Bobes F.; González J. M. New Iodination Reactions of Saturated Hydrocarbons. Angew. Chem., Int. Ed. 2005, 44, 5851–5854. 10.1002/anie.200501195. [DOI] [PubMed] [Google Scholar]; b Matcha M.; Antonchick A. P. Metal-Free Cross-Dehydrogenative Coupling of Heterocycles with Aldehyde. Angew. Chem., Int. Ed. 2013, 52, 2082–2086. 10.1002/anie.201208851. [DOI] [PubMed] [Google Scholar]; c Antonchick A. P.; Burgmann L. Direct Selective Oxidative Cross-Coupling of Simple Alkanes with Heteroarenes. Angew. Chem., Int. Ed. 2013, 52, 3267–3271. 10.1002/anie.201209584. [DOI] [PubMed] [Google Scholar]; d Narayan R.; Antonchick A. P. Hypervalent Iodine-Mediated Selective Oxidative Functionalization of (Thio)chromones with Alkanes. Chem. - Eur. J. 2014, 20, 4568–4572. 10.1002/chem.201400186. [DOI] [PubMed] [Google Scholar]; e Moteki S. A.; Usui A.; Zhang T.; Solorio Alvarado C. R.; Maruoka K. Site-Selective Oxidation of Unactivated C-H Bonds with Hypervalent Iodine(III) Reagents. Angew. Chem., Int. Ed. 2013, 52, 8657–8660. 10.1002/anie.201304359. [DOI] [PubMed] [Google Scholar]; f Moteki S. A.; Usui A.; Selvakumar S.; Zhang T.; Maruoka K. Metal-Free C-H Bond Activation of Branched Aldehydes with a Hypervalent Iodine(III) Catalyst under Visible-Light Photolysis: Successful Trapping with Electron-Deficient Olefins. Angew. Chem., Int. Ed. 2014, 53, 11060–11064. 10.1002/anie.201406513. [DOI] [PubMed] [Google Scholar]; Early study:; g Ochiai M.; Ito T.; Takahashi H.; Nakanishi A.; Toyonari M.; Sueda T.; Goto S.; Shiro M. Hypervalent (tert-Butylperoxy)iodanes Generate Iodine-Centered Radicals at Room Temperature in Solution: Oxidation and Deprotection of Benzyl and Allyl Ethers, and Evidence for Generation of α-Oxy Carbon Radicals. J. Am. Chem. Soc. 1996, 118, 7716–7730. 10.1021/ja9610287. [DOI] [Google Scholar]; h Zhao Y.; Yim W.-L.; Tan C. K.; Yeung Y.-Y. An Unexpected Oxidation of Unactivated Methylene C–H Using DIB/TBHP Protocol. Org. Lett. 2011, 13, 4308–4311. 10.1021/ol2016466. [DOI] [PubMed] [Google Scholar]; i Amey R. L.; Martin J. C. Synthesis and Reaction of Substituted Arylalkoxyiodinanes: Formation of Stable Bromoarylalkoxy and Aryldialkoxy Heterocyclic Derivatives of Tricoordinate Organoiodine(III). J. Org. Chem. 1979, 44, 1779–1784. 10.1021/jo01325a007. [DOI] [Google Scholar]; j Ochiai M.; Ito T.; Masaki Y.; Shiro M. A Stable Crystalline (Alkylperoxy)iodinane: 1-(tert-Butylperoxy)-1,2-benziodoxol-3(1H)-one. J. Am. Chem. Soc. 1992, 114, 6269–6270. 10.1021/ja00041a069. [DOI] [Google Scholar]; k Magnus P.; Lacour J.; Weber W. Direct N-Alkyl Azidonation of N,N-Dialkylarylamines with the Iodosylbenzene/Trimethylsilylazide Reagent Combination. J. Am. Chem. Soc. 1993, 115, 9347–9348. 10.1021/ja00073a084. [DOI] [Google Scholar]; l Kita Y.; Tohma H.; Takada T.; Mitoh S.; Fujita S.; Gyoten M. A Novel and Direct Alkyl Azidation of p-Alkylanisoles Using Phenyl Iodine(III) Bis(trifluoroacetate) (PIFA) and Trimethylsilyl Azide. Synlett 1994, 1994, 427–428. 10.1055/s-1994-22875. [DOI] [Google Scholar]; m Dohi T.; Takenaga N.; Goto A.; Fujioka H.; Kita Y. Clean and Efficient Benzylic C-H Oxidation in Water Using a Hypervalent Iodine Reagent: Activation of Polymeric Iodosobenzene with KBr in the Presence of Montmorillonite-K10. J. Org. Chem. 2008, 73, 7365–7368. 10.1021/jo8012435. [DOI] [PubMed] [Google Scholar]; n Kang K.; Lee S.; Kim H. Radical Chlorination with Hypervalent Iodine(III) Generated by Ligand Exchange: Revisiting Palladium(II)-Catalyzed Directed C-H Chlorination. Asian J. Org. Chem. 2015, 4, 137–140. 10.1002/ajoc.201402284. [DOI] [Google Scholar]
- Wang X.; Studer A. Regio- and Stereoselective Cyanotriflation of Alkynes Using Aryl(cyano)iodonium Triflates. J. Am. Chem. Soc. 2016, 138, 2977–2980. 10.1021/jacs.6b00869. [DOI] [PubMed] [Google Scholar]
- a Parsons A. T.; Buchwald S. L. Copper-Catalyzed Trifluoromethylation of Unactivated Olefins. Angew. Chem., Int. Ed. 2011, 50, 9120–9123. 10.1002/anie.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang X.; Ye Y.; Zhang S.; Feng J.; Xu Y.; Zhang Y.; Wang J. Copper-Catalyzed C(sp3)-C(sp3) Bond Formation Using a Hypervalent Iodine Reagent: An Efficient Allylic Trifluoromethylation. J. Am. Chem. Soc. 2011, 133, 16410–16413. 10.1021/ja207775a. [DOI] [PubMed] [Google Scholar]; c Mejía E.; Togni A. Rhenium-Catalyzed Trifluoromethylation of Arenes and Heteroarenes by Hypervalent Iodine Reagents. ACS Catal. 2012, 2, 521–527. 10.1021/cs300089y. [DOI] [Google Scholar]; d Wang Y.; Zhang L.; Yang Y.; Zhang P.; Du Z.; Wang C. Alkene Oxyalkylation Enabled by Merging Rhenium Catalysis with Hypervalent Iodine(III) Reagents via Decarboxylation. J. Am. Chem. Soc. 2013, 135, 18048–18051. 10.1021/ja410195j. [DOI] [PubMed] [Google Scholar]
- a Crivello J. V. The Discovery and Development of Onium Salt Cationic Photoinitiators. J. Polym. Sci., Part A: Polym. Chem. 1999, 37, 4241–4254. 10.1002/(SICI)1099-0518(19991201)37:23<4241::AID-POLA1>3.0.CO;2-R. [DOI] [Google Scholar]; b Lalevée J.; Blanchard N.; Tehfe M.-A.; Morlet-Savary F.; Fouassier J. P. Green Bulb Light Source Induced Epoxy Cationic Polymerization under Air Using Tris(2,20-bipyridine)ruthenium(II) and Silyl Radicals. Macromolecules 2010, 43, 10191–10195. 10.1021/ma1023318. [DOI] [Google Scholar]; c Tobisu M.; Furukawa T.; Chatani N. Visible Light-mediated Direct Arylation of Arenes and Heteroarenes Using Diaryliodonium Salts in the Presence and Absence of a Photocatalyst. Chem. Lett. 2013, 42, 1203–1205. 10.1246/cl.130547. [DOI] [Google Scholar]; d Neufeldt S. R.; Sanford M. S. Combining Transition Metal Catalysis with Radical Chemistry: Dramatic Acceleration of Palladium-Catalyzed C-H Arylation with Diaryliodonium Salts. Adv. Synth. Catal. 2012, 354, 3517–3522. 10.1002/adsc.201200738. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mizuta S.; Verhoog S.; Engle K. M.; Khotavivattana T.; O’Duill M.; Wheelhouse K.; Rassias G.; Medebielle M.; Gouverneur V. Catalytic Hydrotrifluoromethylation of Unactivated Alkenes. J. Am. Chem. Soc. 2013, 135, 2505–2508. 10.1021/ja401022x. [DOI] [PubMed] [Google Scholar]; f Tomita R.; Yasu Y.; Koike T.; Akita M. Combining Photoredox-Catalyzed Trifluoromethylation and Oxidation with DMSO: Facile Synthesis of α-Trifluoromethylated Ketones from Aromatic Alkenes. Angew. Chem., Int. Ed. 2014, 53, 7144–7148. 10.1002/anie.201403590. [DOI] [PubMed] [Google Scholar]; g Lalevée J.; Mokbel H.; Fouassier J.-P. Recent Developments of Versatile Photoinitiating Systems for Cationic Ring Opening Polymerization Operating at Any Wavelengths and under Low Light Intensity Sources. Molecules 2015, 20, 7201–7221. 10.3390/molecules20047201. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Jia K.; Zhang F.; Huang H.; Chen Y. Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(sp3)-C(sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc. 2016, 138, 1514–1517. 10.1021/jacs.5b13066. [DOI] [PubMed] [Google Scholar]
- Li Y.; Studer A. Transition-Metal-Free Trifluoromethylaminoxylation of Alkenes. Angew. Chem., Int. Ed. 2012, 51, 8221–8224. 10.1002/anie.201202623. [DOI] [PubMed] [Google Scholar]
- a Dohi T.; Ito M.; Morimoto K.; Iwata M.; Kita Y. Oxidative Cross-Coupling of Arenes Induced by Single-Electron Transfer Leading to Biaryls by Use of Organoiodine(III) Oxidants. Angew. Chem., Int. Ed. 2008, 47, 1301–1304. 10.1002/anie.200704495. [DOI] [PubMed] [Google Scholar]; b Dohi T.; Ito M.; Yamaoka N.; Morimoto K.; Fujioka H.; Kita Y. Unusual ipso Substitution of Diaryliodonium Bromides Initiated by a Single-Electron-Transfer Oxidizing Process. Angew. Chem., Int. Ed. 2010, 49, 3334–3337. 10.1002/anie.200907281. [DOI] [PubMed] [Google Scholar]; c Kantak A. A.; Potavathri S.; Barham R. A.; Romano K. M.; DeBoef B. Metal-Free Intermolecular Oxidative C-N Bond Formation via Tandem C-H and N-H Bond Functionalization. J. Am. Chem. Soc. 2011, 133, 19960–19965. 10.1021/ja2087085. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kita Y.; Takada T.; Mihara S.; Whelan B. A.; Tohma H. Novel and Direct Nucleophilic Sulfenylation and Thiocyanation of Phenol Ethers Using a Hypervalent Iodine(III) Reagent. J. Org. Chem. 1995, 60, 7144–7148. 10.1021/jo00127a018. [DOI] [Google Scholar]; e Kita Y.; Tohma H.; Inagaki M.; Hatanaka K.; Yakura T. A Novel Oxidative Azidation of Aromatic Compounds with Hypervalent Iodine Reagent, Phenyliodine(III) Bis(trifluoroacetate) (PIFA) and Trimethylsilyl Azide. Tetrahedron Lett. 1991, 32, 4321–4324. 10.1016/S0040-4039(00)92160-9. [DOI] [Google Scholar]
- a Liu X.; Wang Z.; Cheng X.; Li C. Silver-Catalyzed Decarboxylative Alkynylation of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc. 2012, 134, 14330–14333. 10.1021/ja306638s. [DOI] [PubMed] [Google Scholar]; b Huang H.; Zhang G.; Gong L.; Zhang S.; Chen Y. Visible-Light-Induced Chemoselective Deboronative Alkynylation under Biomolecule-Compatible Conditions. J. Am. Chem. Soc. 2014, 136, 2280–2283. 10.1021/ja413208y. [DOI] [PubMed] [Google Scholar]; c Zhou Q.-Q.; Guo W.; Ding W.; Wu X.; Chen X.; Lu L.-Q.; Xiao W.-J. Decarboxylative Alkynylation and Carbonylative Alkynylation of Carboxylic Acids Enabled by Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed. 2015, 54, 11196–11199. 10.1002/anie.201504559. [DOI] [PubMed] [Google Scholar]; d Le Vaillant F.; Courant T.; Waser J. Room-Temperature Decarboxylative Alkynylation of Carboxylic Acids Using Photoredox Catalysis and EBX Reagents. Angew. Chem., Int. Ed. 2015, 54, 11200–11204. 10.1002/anie.201505111. [DOI] [PubMed] [Google Scholar]; e Wang H.; Guo L.; Wang S.; Duan X.-H. Decarboxylative Alkynylation of α-Keto Acids and Oxamic Acids in Aqueous Media. Org. Lett. 2015, 17, 3054–3057. 10.1021/acs.orglett.5b01336. [DOI] [PubMed] [Google Scholar]; f Le Vaillant F.; Wodrich M. D.; Waser J. Room Temperature Decarboxylative Cyanation of Carboxylic Acids Using Photoredox Catalysis and Cyanobenziodoxolones: A Divergent Mechanism Compared to Alkynylation. Chem. Sci. 2017, 8, 1790–1800. 10.1039/C6SC04907A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Magnus P.; Lacour J. New Triakylsilyl Enol Ether Chemistry. Direct β-Azido Functionalization of Triisopropylsilyl Enol Ethers. J. Am. Chem. Soc. 1992, 114, 767–769. 10.1021/ja00028a058. [DOI] [Google Scholar]; b Zhdankin V. V.; Krasutsky A. P.; Kuehl C. J.; Simonsen A. J.; Woodward J. K.; Mismash B.; Bolz J. T. Preparation, X-ray Crystal Structure, and Chemistry of Stable Azidoiodinanes–Derivatives of Benziodoxole. J. Am. Chem. Soc. 1996, 118, 5192–5197. 10.1021/ja954119x. [DOI] [Google Scholar]; c Zhou W.; Zhang L.; Jiao N. Direct Transformation of Methyl Arenes to Aryl Nitriles at Room Temperature. Angew. Chem., Int. Ed. 2009, 48, 7094–7097. 10.1002/anie.200903838. [DOI] [PubMed] [Google Scholar]; d Lu M.-Z.; Wang C.-Q.; Loh T.-P. Copper-Catalyzed Vicinal Oxyazidation and Diazidation of Styrenes under Mild Conditions: Access to Alkyl Azides. Org. Lett. 2015, 17, 6110–6113. 10.1021/acs.orglett.5b03130. [DOI] [PubMed] [Google Scholar]; e Sharma A.; Hartwig J. F. Metal-Catalysed Azidation of Tertiary C-H Bonds Suitable for Late-Stage Functionalization. Nature 2015, 517, 600–604. 10.1038/nature14127. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Shinomoto Y.; Yoshimura A.; Shimizu H.; Yamazaki M.; Zhdankin V. V.; Saito A. Tetra-n-butylammonium Iodide Catalyzed C–H Azidation of Aldehydes with Thermally Stable Azidobenziodoxolone. Org. Lett. 2015, 17, 5212–5215. 10.1021/acs.orglett.5b02543. [DOI] [PubMed] [Google Scholar]; g Rabet P. T. G.; Fumagalli G.; Boyd S.; Greaney M. F. Benzylic C-H Azidation Using the Zhdankin Reagent and a Copper Photoredox Catalyst. Org. Lett. 2016, 18, 1646–1649. 10.1021/acs.orglett.6b00512. [DOI] [PubMed] [Google Scholar]
- a Fischer H. The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev. 2001, 101, 3581–3610. 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]; b Studer A. The Persistent Radical Effect in Organic Synthesis. Chem. - Eur. J. 2001, 7, 1159–1164. 10.1002/1521-3765(20010316)7:6<1159::AID-CHEM1159>3.3.CO;2-9. [DOI] [PubMed] [Google Scholar]; c Studer A. Tin-Free Radical Chemistry Using the Persistent Radical Effect: Alkoxyamine Isomerization, Addition Reactions and Polymerizations. Chem. Soc. Rev. 2004, 33, 267–273. 10.1039/b307652k. [DOI] [PubMed] [Google Scholar]
- Reviews:; a Schlosser M. CF3-Bearing Aromatic and Heterocyclic Building Blocks. Angew. Chem., Int. Ed. 2006, 45, 5432–5446. 10.1002/anie.200600449. [DOI] [PubMed] [Google Scholar]; b Furuya T.; Kamlet A. S.; Ritter T. Catalysis for Fluorination and Trifluoromethylation. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tomashenko O. A.; Grushin V. V. Aromatic Trifluoromethylation with Metal Complexes. Chem. Rev. 2011, 111, 4475–4521. 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]; d Besset T.; Schneider C.; Cahard D. Tamed Arene and Heteroarene Trifluoromethylation. Angew. Chem., Int. Ed. 2012, 51, 5048–5050. 10.1002/anie.201201012. [DOI] [PubMed] [Google Scholar]; e Liu X.; Xu C.; Wang M.; Liu Q. Trifluoromethyltrimethylsilane: Nucleophilic Trifluoromethylation and Beyond. Chem. Rev. 2015, 115, 683–730. 10.1021/cr400473a. [DOI] [PubMed] [Google Scholar]
- Charpentier J.; Früh N.; Togni A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 2015, 115, 650–682. 10.1021/cr500223h. [DOI] [PubMed] [Google Scholar]
- Egami H.; Sodeoka M. Trifluoromethylation of Alkenes with Concomitant Introduction of Additional Functional Groups. Angew. Chem., Int. Ed. 2014, 53, 8294–8308. 10.1002/anie.201309260. [DOI] [PubMed] [Google Scholar]
- Avila D. A.; Ingold K. U.; Lusztyk J.; Dolbier W. R. Jr.; Pan H.-Q. Absolute Rate Constants for Radical Additions to Alkenes in Solution. The Synergistic Effect of Perfluorination on the Reactivities of n-Alkyl Radicals. J. Org. Chem. 1996, 61, 2027–2030. 10.1021/jo951782v. [DOI] [PubMed] [Google Scholar]
- Hartmann M.; Li Y.; Studer A. Determination of Rate Constants for Trifluoromethyl Radical Addition to Various Alkenes via a Practical Method. Org. Biomol. Chem. 2016, 14, 206–210. 10.1039/C5OB02210J. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Mück-Lichtenfeld C.; Daniliuc C. G.; Studer A. 6-Trifluoromethyl-Phenanthridines through Radical Trifluoromethylation of Isonitriles. Angew. Chem., Int. Ed. 2013, 52, 10792–10795. 10.1002/anie.201306082. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Studer A. Recent Advances in the Synthesis of Nitrogen Heterocycles via Radical Cascade Reactions Using Isonitriles as Radical Acceptors. Chem. Soc. Rev. 2015, 44, 3505–3521. 10.1039/C5CS00083A. [DOI] [PubMed] [Google Scholar]
- a Studer A.; Curran D. P. The Electron is a Catalyst. Nat. Chem. 2014, 6, 765–773. 10.1038/nchem.2031. [DOI] [PubMed] [Google Scholar]; b Studer A.; Curran D. P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55, 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- Studer A.; Curran D. P. Organocatalysis and C-H Activation Meet Radical- and Electron-Transfer Reactions. Angew. Chem., Int. Ed. 2011, 50, 5018–5022. 10.1002/anie.201101597. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Studer A. 6-Perfluoroalkylated Phenanthridines via Radical Perfluoroalkylation of Isonitriles. Org. Lett. 2014, 16, 3990–3993. 10.1021/ol5018195. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Studer A. 1-Trifluoromethylated Isoquinolines via Radical Trifluoromethylation of Isonitriles. Org. Biomol. Chem. 2014, 12, 9895–9898. 10.1039/C4OB02145B. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Studer A. 2-Trifluoromethylated Indoles via Radical Trifluoromethylation of Isonitriles. Org. Lett. 2014, 16, 1216–1219. 10.1021/ol5001395. [DOI] [PubMed] [Google Scholar]
- Leifert D.; Artiukhin D. G.; Neugebauer J.; Galstyan A.; Strassert C. A.; Studer A. Radical Perfluoroalkylation–Easy Access to 2-Perfluoroalkylindol-3-imines via Electron Catalysis. Chem. Commun. 2016, 52, 5997–6000. 10.1039/C6CC02284G. [DOI] [PubMed] [Google Scholar]
- a Wang Y.-F.; Lonca G. H.; Le Runigo M.; Chiba S. Synthesis of Polyfluoroalkyl Aza-Polycyclic Aromatic Hydrocarbons Enabled by Addition of Perfluoroalkyl Radicals onto Vinyl Azides. Org. Lett. 2014, 16, 4272–4275. 10.1021/ol501997n. [DOI] [PubMed] [Google Scholar]; b Yang T.; Zhu H.; Yu W. Copper-Catalyzed Radical Reactions of 2-Azido-N-arylacrylamides with 1-(Trifluoromethyl)-1,2-benziodoxole and 1-Azidyl-1,2-benziodoxole. Org. Biomol. Chem. 2016, 14, 3376–3384. 10.1039/C6OB00226A. [DOI] [PubMed] [Google Scholar]
- Mackay E. G.; Studer A. Electron-Catalyzed Fluoroalkylation of Vinyl Azides. Chem. - Eur. J. 2016, 22, 13455–13458. 10.1002/chem.201602855. [DOI] [PubMed] [Google Scholar]
- a Matcha K.; Narayan R.; Antonchick A. P. Metal-Free Radical Azidoarylation of Alkenes: Rapid Access to Oxindoles by Cascade C-N and C-C Bond-Forming Reactions. Angew. Chem., Int. Ed. 2013, 52, 7985–7989. 10.1002/anie.201303550. [DOI] [PubMed] [Google Scholar]; b Wu Z.; Xu P.; Zhou N.; Duan Y.; Zhang M.; Zhu C. [3 + 2] Cycloaddition of Azide with Aldehyde Hydrazone through an Aminyl Radical-Polar Crossover Strategy. Chem. Commun. 2017, 53, 1045–1047. 10.1039/C6CC08779E. [DOI] [PubMed] [Google Scholar]
- Huang J.; He Y.; Zhu Q.; Wang Y. Synthesis of Benzimidazoles by PIDA-Promoted Direct C(sp2)-H Imidation of N-Arylamidines. Chem. - Eur. J. 2012, 18, 13964–13967. 10.1002/chem.201202271. [DOI] [PubMed] [Google Scholar]
- a Zhdankin V. V.; Kuehl C. J.; Krasutsky A. P.; Formaneck M. S.; Bolz J. T. Preparation and Chemistry of Stable Azidoiodinanes: 1-Azido-3,3-bis(trifluoromethyl)-3-(1H)-1,2-benziodoxol and 1-Azido-1,2-benziodoxol-3-(1H)-one. Tetrahedron Lett. 1994, 35, 9677–9680. 10.1016/0040-4039(94)88357-2. [DOI] [Google Scholar]; b Akai S.; Okuno T.; Egi M.; Takada T.; Tohma H.; Kita Y. Preparation of Novel Cyclic Hypervalent Iodine(III) Compounds Having Azido, Cyano, and Nitrato Ligands. Heterocycles 1996, 42, 47–51. 10.3987/COM-95-S11. [DOI] [Google Scholar]
- Zhang B.; Studer A. Stereoselective Radical Azidooxygenation of Alkenes. Org. Lett. 2013, 15, 4548–4551. 10.1021/ol402106x. [DOI] [PubMed] [Google Scholar]
- a Takada T.; Arisawa M.; Gyoten M.; Hamada R.; Tohma H.; Kita Y. Oxidative Biaryl Coupling Reaction of Phenol Ether Derivatives Using a Hypervalent Iodine(III) Reagent. J. Org. Chem. 1998, 63, 7698–7706. 10.1021/jo980704f. [DOI] [Google Scholar]; b Kita Y.; Tohma H.; Hatanaka K.; Takada T.; Fujita S.; Mitoh S.; Sakurai H.; Oka S. Hypervalent Iodine-Induced Nucleophilic Substitution of para-Substituted Phenol Ethers. Generation of Cation Radicals as Reactive Intermediates. J. Am. Chem. Soc. 1994, 116, 3684–3691. 10.1021/ja00088a003. [DOI] [Google Scholar]
- a Liu Y.-X.; Xue D.; Wang J.-D.; Zhao C.-J.; Zou Q.-Z.; Wang C.; Xiao J. Room-Temperature Arylation of Arenes and Heteroarenes with Diaryliodonium Salts by Photoredox Catalysis. Synlett 2013, 24, 507–513. 10.1055/s-0032-1318155. [DOI] [Google Scholar]; b Baralle A.; Fensterbank L.; Goddard J.-P.; Ollivier C. Aryl Radical Formation by Copper(I) Photocatalyzed Reduction of Diaryliodonium Salts: NMR Evidence for a CuII/CuI Mechanism. Chem. - Eur. J. 2013, 19, 10809–10813. 10.1002/chem.201301449. [DOI] [PubMed] [Google Scholar]; c Wang R.; Jiang H.; Cheng Y.; Kadi A. A.; Fun H.-K.; Zhang Y.; Yu S. Somophilic Isocyanide Insertion: Synthesis of 6-Arylated and 6-Trifluoromethylated Phenanthridines. Synthesis 2014, 46, 2711–2726. 10.1055/s-0034-1379217. [DOI] [Google Scholar]; d Wen J.; Zhang R.-Y.; Chen S.-Y.; Zhang J.; Yu X.-Q. Direct Arylation of Arene and N-Heteroarenes with Diaryliodonium Salts without the Use of Transition Metal Catalyst. J. Org. Chem. 2012, 77, 766–771. 10.1021/jo202150t. [DOI] [PubMed] [Google Scholar]; e Dohi T.; Ueda S.; Hirai A.; Kojima Y.; Kita Y. Selective Aryl Radical Transfers into N-Heteroaromatics from Diaryliodonoium Salts with Trimethoxybenzene Auxiliary. Heterocycles 2017, 95, 1272–1284. 10.3987/COM-16-S(S)90. [DOI] [Google Scholar]
- Hartmann M.; Li Y.; Studer A. Transition-Metal-Free Oxyarylation of Alkenes with Aryl Diazonium Salts and TEMPONa. J. Am. Chem. Soc. 2012, 134, 16516–16519. 10.1021/ja307638u. [DOI] [PubMed] [Google Scholar]
- a Goudreau S. R.; Marcoux D.; Charette A. B. General Method for the Synthesis of Phenyliodonium Ylides from Malonate Esters: Easy Access to 1,1-Cyclopropane Diesters. J. Org. Chem. 2009, 74, 470–473. 10.1021/jo802208q. [DOI] [PubMed] [Google Scholar]; b Wenz D. R.; Read de Alaniz J. Aza-Piancatelli Rearrangement Initiated by Ring Opening of Donor-Acceptor Cyclopropanes. Org. Lett. 2013, 15, 3250–3253. 10.1021/ol401248p. [DOI] [PubMed] [Google Scholar]; c Ivanov A. S.; Popov I. A.; Boldyrev A. I.; Zhdankin V. V. The I = X (X = O, N, C) Double Bond in Hypervalent Iodine Compounds: Is it Real?. Angew. Chem., Int. Ed. 2014, 53, 9617–9621. 10.1002/anie.201405142. [DOI] [PubMed] [Google Scholar]
- Hartmann M.; Li Y.; Mück-Lichtenfeld C.; Studer A. Generation of Aryl Radicals through Reduction of Hypervalent Iodine(III) Compounds with TEMPONa: Radical Alkene Oxyarylation. Chem. - Eur. J. 2016, 22, 3485–3490. 10.1002/chem.201504852. [DOI] [PubMed] [Google Scholar]
- a Ozanne-Beaudenon A.; Quideau S. Angew. Chem., Int. Ed. 2005, 44, 7065–7069. 10.1002/anie.200501638. [DOI] [PubMed] [Google Scholar]; b Zhang M.-R.; Kumata K.; Suzuki K. A Practical Route for Synthesizing a PET Ligand Containing [18F]Fluorobenzene Using Reaction of Diphenyliodonium Salt with [18F]F–. Tetrahedron Lett. 2007, 48, 8632–8635. 10.1016/j.tetlet.2007.10.025. [DOI] [Google Scholar]; c Jalalian N.; Ishikawa E. E.; Silva L. F. Jr.; Olofsson B. Room Temperature, Metal-Free Synthesis of Diaryl Ethers with Use of Diaryliodonium Salts. Org. Lett. 2011, 13, 1552–1555. 10.1021/ol200265t. [DOI] [PubMed] [Google Scholar]; d Tinnis F.; Stridfeldt E.; Lundberg H.; Adolfsson H.; Olofsson B. Metal-Free N-Arylation of Secondary Amides at Room Temperature. Org. Lett. 2015, 17, 2688–2691. 10.1021/acs.orglett.5b01079. [DOI] [PubMed] [Google Scholar]
- Kang S.-K.; Lee H.-W.; Jang S.-B.; Ho P.-S. Palladium-Catalyzed Cross-Coupling of Organoboron Compounds with Iodonium Salts and Iodanes. J. Org. Chem. 1996, 61, 4720–4724. 10.1021/jo960195m. [DOI] [PubMed] [Google Scholar]
- Kalyani D.; Deprez N. R.; Desai L. V.; Sanford M. S. Oxidative C-H Activation/C-C Bond Forming Reactions: Synthetic Scope and Mechanistic Insights. J. Am. Chem. Soc. 2005, 127, 7330–7331. 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]
- Kitamura T.; Yamane M.; Inoue K.; Todaka M.; Fukatsu N.; Meng Z.; Fujiwara Y. A New and Efficient Hypervalent Iodine-Benzyne Precursor, (Phenyl)[o-(trimethylsilyl)phenyl]iodonium Triflate: Generation, Trapping Reaction, and Nature of Benzyne. J. Am. Chem. Soc. 1999, 121, 11674–11679. 10.1021/ja992324x. [DOI] [Google Scholar]
- a Celik M.; Alp C.; Coskun B.; Gültekin M. S.; Balci M. Synthesis of Diols Using the Hypervalent Iodine(III) Reagent, Phenyliodine(III) Bis(trifluoroacetate). Tetrahedron Lett. 2006, 47, 3659–3663. 10.1016/j.tetlet.2006.03.137. [DOI] [Google Scholar]; b Egami H.; Shimizu R.; Sodeoka M. Oxytrifluoromethylation of Multiple Bonds Using Copper Catalyst under Mild Conditions. Tetrahedron Lett. 2012, 53, 5503–5506. 10.1016/j.tetlet.2012.07.134. [DOI] [Google Scholar]; c Egami H.; Shimizu R.; Usui Y.; Sodeoka M. Oxy-trifluoromethylation of Alkenes and Its Application to the Synthesis of β-Trifluoromethylstyrene Derivatives. J. Fluorine Chem. 2014, 167, 172–178. 10.1016/j.jfluchem.2014.05.009. [DOI] [Google Scholar]; d Janson P. G.; Ghoneim I.; Ilchenko N. O.; Szabó K. J. Electrophilic Trifluoromethylation by Copper-Catalyzed Addition of CF3-Transfer Reagents to Alkenes and Alkynes. Org. Lett. 2012, 14, 2882–2885. 10.1021/ol3011419. [DOI] [PubMed] [Google Scholar]; e Wang Y.; Jiang M.; Liu J.-T. Copper-Catalyzed Regioselective Oxytrifluoromethylation of Allenes Using a CF3-Transfer Reagent. Adv. Synth. Catal. 2014, 356, 2907–2912. 10.1002/adsc.201400320. [DOI] [Google Scholar]; f Hari D. P.; Waser J. Copper-Catalyzed Oxy-Alkynylation of Diazo Compounds with Hypervalent Iodine Reagents. J. Am. Chem. Soc. 2016, 138, 2190–2193. 10.1021/jacs.6b00278. [DOI] [PubMed] [Google Scholar]; g Suero M. G.; Bayle E. D.; Collins B. S. L.; Gaunt M. J. Copper-Catalyzed Electrophilic Carbofunctionalization of Alkynes to Highly Functionalized Tetrasubstituted Alkenes. J. Am. Chem. Soc. 2013, 135, 5332–5335. 10.1021/ja401840j. [DOI] [PubMed] [Google Scholar]
- a Zhdankin V. V.; Crittell C. M.; Stang P. J.; Zefirov N. S. A General Approach to Unsymmetrical Tricoordinate Iodinanes: Single Step Preparation of Mixed Iodosobenzene Sulfonates Phl(X)OSO2R, via Reaction of Iodosobenzene with Me3SiX. Tetrahedron Lett. 1990, 31, 4821–4824. 10.1016/S0040-4039(00)97741-4. [DOI] [Google Scholar]; b Zhdankin V. V.; Scheuller M. C.; Stang P. J. A General Approach to Aryl(cyano)iodonium Triflates-Versatile Iodonium Transfer Reagents. Tetrahedron Lett. 1993, 34, 6853–6856. 10.1016/S0040-4039(00)91812-4. [DOI] [Google Scholar]; c Shu Z.; Ji W.; Wang X.; Zhou Y.; Zhang Y.; Wang J. Iron(II)-Catalyzed Direct Cyanation of Arenes with Aryl(cyano)iodonium Triflates. Angew. Chem., Int. Ed. 2014, 53, 2186–2189. 10.1002/anie.201309791. [DOI] [PubMed] [Google Scholar]; d Zhu D.; Chang D.; Shi L. Transition-Metal-Free Cross-Coupling of Thioethers with Aryl(cyano)iodonium Triflates: A Facile and Efficient Method for the One-Pot Synthesis of Thiocyanates. Chem. Commun. 2015, 51, 7180–7183. 10.1039/C5CC00875A. [DOI] [PubMed] [Google Scholar]
- Wang X.; Studer A.. Regio- and Stereoselective Radical Perfluoroalkyltriflation of Alkynes Using Phenyl(perfluoroalkyl)iodonium Triflates. Manuscript in preparation, 2017. [DOI] [PMC free article] [PubMed]