

Broadening of inner molecular orbitals upon adsorption can predict chemoselectivity of metal catalysts.
Abstract
The mechanistic understanding and control over transformations of multi-unsaturated hydrocarbons on transition metal surfaces remains one of the major challenges of hydrogenation catalysis. To reveal the microscopic origins of hydrogenation chemoselectivity, we performed a comprehensive theoretical investigation on the reactivity of two α,β-unsaturated carbonyls—isophorone and acrolein—on seven (111) metal surfaces: Pd, Pt, Rh, Ir, Cu, Ag, and Au. In doing so, we uncover a general mechanism that goes beyond the celebrated frontier molecular orbital theory, rationalizing the C═C bond activation in isophorone and acrolein as a result of significant surface-induced broadening of high-energy inner molecular orbitals. By extending our calculations to hydrogen-precovered surface and higher adsorbate surface coverage, we further confirm the validity of the “inner orbital broadening mechanism” under realistic catalytic conditions. The proposed mechanism is fully supported by our experimental reaction studies for isophorone and acrolein over Pd nanoparticles terminated with (111) facets. Although the position of the frontier molecular orbitals in these molecules, which are commonly considered to be responsible for chemical interactions, suggests preferential hydrogenation of the C═O double bond, experiments show that hydrogenation occurs at the C═C bond on Pd catalysts. The extent of broadening of inner molecular orbitals might be used as a guiding principle to predict the chemoselectivity for a wide class of catalytic reactions at metal surfaces.
INTRODUCTION
Interaction of unsaturated hydrocarbons with transition metal surfaces has a pivotal importance for numerous applications of heterogeneous catalysis related to fine chemical and pharmaceutical industries (1–6). Particularly important is the surface chemistry of multi-unsaturated hydrocarbons, such as α,β-unsaturated ketones and aldehydes, and their derivatives because they represent a broad class of valuable intermediates for practically important processes (7–9). One of the most useful target products of this type of conversions is unsaturated alcohols, which are produced by heterogeneous chemoselective hydrogenation of the C═O bond in unsaturated ketones and aldehydes (10). However, hydrogenation of the C═C bond in these components is experimentally observed to be much easier than that of the C═O bond, which obstructs the selective hydrogenation of the carbonyl bond while keeping the C═C double bond intact (10–12). In recent model studies on acrolein hydrogenation over Pd(111), some of us spectroscopically followed the evolution of surface species formed during the reaction. The first reaction product appearing on the initially clean Pd surface was identified to be an oxopropyl species that results from half-hydrogenation of the C═C bond. C═O bond hydrogenation in acrolein does not occur on the clean Pd(111) surface but can only be induced by formation of a densely packed overlayer of spectator species (6).
Although numerous experimental studies on both single-crystal surfaces and powdered materials provide a clear evidence that α,β-unsaturated ketones and aldehydes are preferentially hydrogenated at the C═C bond over metal catalysts, particularly for the simplest molecules containing no protecting groups such as acrolein, calculations fail to explain these observations and instead predict hydrogenation of the C═O bond on the clean Pd and Pt surfaces (13–16). The origin of this discrepancy remains largely unexplored.
To obtain atomistic- and electronic-level insights into the surface chemistry and factors governing chemoselectivity for this important class of reactions, we perform here a comprehensive theoretical study on the activation of C═C and C═O bonds in two multi-unsaturated hydrocarbons on seven transition metal surfaces. Our calculations reveal a mechanism that goes beyond the widely accepted frontier molecular orbital (FMO) theory, which states that the frontier orbitals, that is, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), are mainly responsible for chemical reactions. The FMO theory proved itself to be a useful tool for understanding the reactivity of small molecules on metal surfaces, for example, 1-butene on the Pt(111) surface. As shown in fig. S1, the HOMO of 1-butene, which is spatially located on the C═C double bond, is strongly perturbed upon interaction with the catalyst. On the basis of the FMO theory, 1-butene is activated preferentially at the C═C bond, which agrees with experimental findings that 1-butene is hydrogenated to butane in the presence of hydrogen (17). However, this theory largely fails in predicting the reactivity of more complex molecules, such as α,β-unsaturated carbonyls. Here, we systematically study 14 molecule/surface combinations to show that chemoselectivity on metal surfaces can be mostly determined by the inner-lying molecular orbitals. These predictions are confirmed by our experimental studies on partial hydrogenation of isophorone and acrolein on Pd. Significant surface-induced broadening of the HOMO−1 orbital is found for both systems, which is shown to decisively contribute to the activation of the C═C bond, and unambiguously explains the preferential hydrogenation of the C═C bond in these molecules found in experimental studies. We have also extended our calculations to more realistic conditions and demonstrated that the “inner orbital broadening mechanism” still holds for hydrogen-precovered surfaces and high surface coverage. Hence, our study provides an important generalization of the FMO concept as applied to catalytic reactions on metal surfaces.
RESULTS
We first carried out experimental reaction studies for two α,β-unsaturated carbonyls—isophorone and acrolein—over Pd nanoparticles mostly terminated by (111) facets. Figure 1 shows the evolution of two possible reaction products—trimethylcyclohexanone and isophorol—upon hydrogenation of isophorone. In these experiments, isophorone and hydrogen were dosed via molecular beams (MBs) onto a model supported catalysts consisting of ~7-nm-sized Pd nanoparticles supported on a thin Fe3O4/Pt(111) oxide film (for details of the experimental procedure, see Materials and Methods). As a result, the evolution of trimethylcyclohexanone resulting from hydrogenation of the C═C bond was observed at a high reaction rate (upper line). In contrast, formation of neither isophorol nor trimethylcyclohexanol was detected, implying that the C═O bond was not hydrogenated under these reaction conditions, whereas the chemical transformation of the C═C bond is facile. Similar observations have also been obtained for acrolein (Fig. 1, right), which are consistent with the large body of existing experimental observations (6, 12, 18).
Fig. 1. Experimental observations of hydrogenation products.
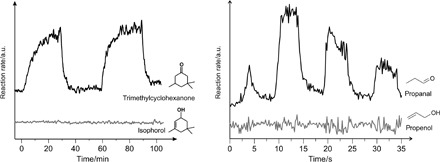
The evolution of two possible reaction products upon hydrogenation of isophorone (left) and acrolein (right) over Pd nanoparticles supported on the model Fe3O4/Pt(111) oxide film. Experimental details are given in the text. a.u., arbitrary units.
The experimental observed selectivity can be elucidated by the computed reaction barriers for isophorone over the Pd(111) surface. We determined the structure of the transition state using the linear/quadratic synchronous transit method (19). At low coverage, we found that the energy barriers are 0.61 and 1.08 eV for the first and second hydrogenation step, respectively, at the C═C bond, being significantly smaller than those when hydrogen atoms attack the C═O bond (0.99 and 1.40 eV, respectively). Similarly, the desorption energy of the C═C bond hydrogenation product (1.14 eV) is also smaller than that of the C═O hydrogenation product (1.57 eV). The theoretical explanation of the increased chemical reactivity of the C═C bond of adsorbed isophorone, as compared to the C═O bond, is in an excellent agreement with the experimentally measured reaction rates of isophorone partial hydrogenation along different reaction pathways (Fig. 1, left; for details of the calculations, see fig. S2).
Motivated by the experimental evidence and the calculated kinetic barriers, we performed comprehensive density functional theory (DFT) calculations on the reactivity of the isophorone and acrolein molecules over different metal surfaces. Note that both experiments and our calculations indicate two alternative pathways in hydrogenation of unsaturated hydrocarbons. As mentioned above, hydrogenation of the C═O bond, with high selectivity and activity, is an essential step in the industrial synthesis of fine chemicals. However, the commonly used metal catalysts, such as Pt, Pd, Ru, and Rh, show no or low selectivity toward the production of unsaturated alcohols. On the basis of our DFT calculations, Fig. 2 summarizes the two alternative reaction pathways for isophorone on different transition metal surfaces. One of them involves the hydrogenation of the C═C double bond over reactive Pd(111), Pt(111), Rh(111), and Ir(111) surfaces to form saturated ketone with high activity. The other leads to the hydrogenation of the C═O bond to form unsaturated alcohol when using Cu(111), Ag(111), and Au(111) as catalysts, albeit with very low activity. To understand the microscopic origin of selectivity, we start by discussing the orbitals of the isolated gas-phase isophorone molecule corresponding to the frontier HOMO and the inner HOMO−1 orbitals. As shown in Fig. 2B, the HOMO and HOMO−1 of an isolated isophorone molecule are spatially located on the C═O and C═C double bonds, respectively. The LUMO is rather homogeneously distributed over the entire conjugated system. The difference in energy between HOMO and HOMO−1 is larger than 1 eV. As such, isophorone is typically assumed to be activated preferentially at the C═O double bond according to the FMO theory.
Fig. 2. Two alternative reaction pathways for isophorone on different metal surfaces.
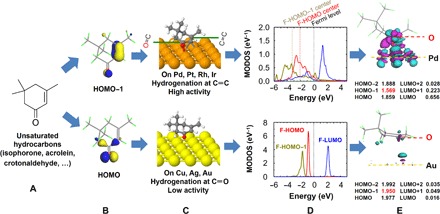
(A and B) Structure and molecular orbitals for the isolated isophorone molecule. (C) Structures of isophorone adsorbed on transition metal surfaces. (D) Molecular orbital density of states (MODOS) projected on the free isophorone HOMO−1, HOMO, and LUMO orbitals for isophorone on the Pd(111) surface (top) and Au(111) surface (bottom). The zero of energy corresponds to the Fermi level, and the d-band center of the clean Pd(111) surface is located at −2.11 eV. (E) Side view of the electron density difference upon isophorone adsorption on Pd(111) and Au(111) at their equilibrium adsorption structures. The values of the isosurface 0.04 and 0.015 Å−3 were used for the former and the latter. Cyan and purple indicate electron depletion and accumulation, respectively. The computed projected occupation of selected molecular orbitals near the Fermi level is also shown in this plot.
To obtain more insights into the adsorption geometry and the reactivity of the adsorbed isophorone, we performed electronic structure calculations using the DFT + vdWsurf method (20), with the Perdew-Burke-Ernzerhof (PBE) functional (21). The PBE + vdWsurf method is an accurate and efficient approach that allows quantitative treatment of both weakly and strongly adsorbed molecules on metal surfaces (for more details on the methodology and computational settings, see Materials and Methods) (20, 22–25). To obtain the most stable adsorption structure, we started by exploring the potential energy surface for isophorone on the Pd(111) surface in a 4 × 4 unit cell. We relaxed a single isophorone at eight adsorption sites of the metal substrate (fig. S3). The oxygen atom was taken as reference in the molecule because all stable configurations correspond to this atom, which forms covalent bonds with Pd atoms at the Pd(111) surface. The most stable adsorption structure is shown in the upper panel in Fig. 2C, from which isophorone adsorbs in a flat-lying adsorption geometry on the Pd(111) surface, with the C═C and C═O bonds oriented parallel to the substrate surface plane. The computed results agree well with our experimental observations by infrared reflection absorption spectroscopy (IRAS) and near-edge x-ray absorption fine structure (NEXAFS). As shown in Fig. 3, the IRAS experiments revealed that there are no features detected for the C═O or C═C stretching modes, suggesting that the two bonds are parallel to the substrate according to the metal-surface selection rule (26). In addition, an independent NEXAFS experiment, which was carried out with a different polarization of the x-ray beam with respect to the Pd(111) surface, indicated that the tilt of the adsorbed isophorone is less than 13° ± 6° at 0.2 monolayer coverage (22).
Fig. 3. IR and NEXAFS spectra of isophorone on Pd(III).
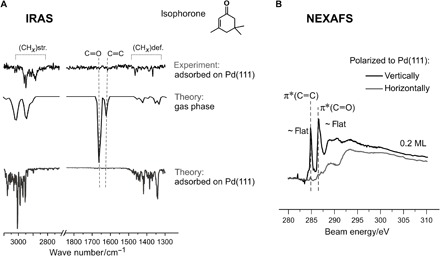
(A) Experimental and theoretical IR spectra of gas-phase and surface-adsorbed isophorone on Pd(111). The experimental spectra were measured at 120 K, and the calculated anharmonic IR spectra for isophorone on Pd(111) were obtained on the basis of the relaxed structures using the PBE + vdWsurf method. str., stretching; def., deformation. (B) NEXAFS experimental spectra of the isophorone/Pd(111) system obtained at different polarization of the x-ray beam with respect to the Pd(111) surface at 120 K. ML, monolayer.
Having obtained the adsorption geometry that correctly reproduces the experimental observations from IRAS and NEXAFS for the isophorone molecule, we performed a detailed analysis of its electronic structure and the degree of the electronic perturbations by the interaction with the metal surface. To quantify the extent of the electron density redistribution, we projected the density of states of the full adsorption system onto selected molecular orbitals of the free molecule (Fig. 2D) (27). The most important finding is that the extent of broadening of molecular orbitals, and not their average energetic positions with respect to the Pd(111) Fermi level, largely determines the activation of the corresponding bonds for hydrogenation. Figure 2D shows that both the former HOMO (F-HOMO) and F-HOMO−1 orbitals are strongly broadened because of the coupling to the metal d bands, but the width of the F-HOMO−1 (−6 to 3 eV) is larger than that of the F-HOMO (−6 to 1 eV). Consequently, a notably larger amount of charge transfer is found from the F-HOMO−1 than from the F-HOMO (0.43 versus 0.14 electrons; Fig. 2E), further confirming that the C═C bond is chemically more activated by the catalyst. Our results indicate that the F-HOMO−2 and F-LUMO+1 of isophorone also participate in the charge-transfer process, showing that the interaction of isophorone with the catalyst is originated from the collective delocalized mechanism induced by the Pd(111) surface. Note that the average position of the F-HOMO−1 center with respect to the F-HOMO remains the same as in gas-phase isophorone, which indicates that the position of the molecular orbital center relative to the Fermi level alone cannot explain the selectivity in the hydrogenation of isophorone.
To make a clear analysis of the origin of the selectivity, we explicitly consider the details of the broadening of the molecular orbitals of isophorone upon adsorption on metal surfaces. In the case of isophorone/Pd(111), we find that a smaller broadening of the F-HOMO compared to F-HOMO−1 is due to the charge compensation by the F-LUMO orbital of the adsorbate. As shown in Fig. 2D, because of the interaction with Pd atoms, the F-HOMO of isophorone donates electrons to the metal and becomes substantially broadened. Meanwhile, the F-LUMO is shifted below the Fermi level and partially filled, indicating a donation/back-donation type of bonding (28, 29). Note that the back-donation process would first compensate for depopulation of the F-HOMO and that this leads to the F-HOMO−1 being more chemically activated than the F-HOMO. The here-presented mechanism requires a complex interplay of the electronic interactions between the adsorbate molecules and the metal surface and has not been observed so far for small molecular adsorbates.
The proposed orbital broadening mechanism, that is, that the extent of the broadening of molecular orbitals that determines the chemical reactivity of the C═C and C═O bonds, is found to be a general phenomenon that holds for a variety of molecules and metallic surfaces. To demonstrate this point, besides the Pd(111) surface, we further carried out extensive calculations for isophorone and acrolein—the smallest α,β-unsaturated aldehyde—on the (111) surfaces of Pt, Rh, Ir, Cu, Ag, and Au. In analogy to the isophorone/Pd(111) studied here, the HOMO−1 orbitals of both adsorbates are strongly broadened upon interaction with the Pd(111), Pt(111), Rh(111), and Ir(111) surfaces, which is accompanied by a large amount of charge transfer from the inner orbital to the substrate (Fig. 4 and figs. S4 and S5). In the scope of our model, strong broadening of the HOMO−1 orbital localized at the C═C bond results in activation and preferential hydrogenation of this bond.
Fig. 4. Structure and electronic properties of acrolein.
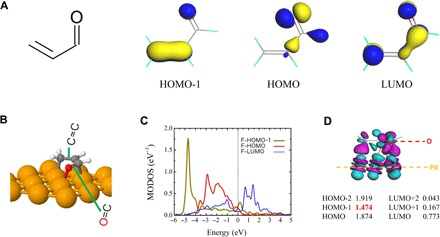
(A) Structure and molecular orbitals for the isolated acrolein molecule. (B) Structure of acrolein adsorbed on the Pd(111) surface. (C) MODOS projected on the free acrolein HOMO−1, HOMO, and LUMO orbitals for the adsorption system. The zero of energy corresponds to the Fermi level. (D) Side view of the electron density difference upon acrolein adsorption on Pd(111) at its equilibrium adsorption structure, using the value of the isosurface 0.04 Å−3. Cyan and purple indicate electron depletion and accumulation, respectively. The computed projected occupation of selected molecular orbitals near the Fermi level is also shown in this plot.
Although isophorone and acrolein have similar broadening of inner orbitals on reactive surfaces, they perform very differently upon interaction with coinage metal surfaces, that is, Cu(111), Ag(111), and Au(111). For isophorone, high selectivity for C═O bond hydrogenation is typically observed over noble metals, such as Ag and Au, but with relatively low activity [for a recent review, see the study of Mäki-Arvela et al. (12)]. Generally, the molecules are weakly interacting with these noble metals and behave similarly to their gas-phase counterparts (Fig. 2C, bottom). Weaker interacting systems, such as noble metals, show much lesser extent or no broadening effect on the inner molecular orbitals (Fig. 2D, bottom, and fig. S4). Consequently, the selectivity toward hydrogenation of the C═C bond of isophorone is expected to decrease on these metals, whereas the relative amount of C═O hydrogenation events should increase, resulting in a total increase in the selectivity toward hydrogenation of the carbonyl group. Conversely, the C═C bond in acrolein is found to be much closer to coinage metals than for isophorone, leading to apparent broadening of the inner orbital for the adsorbed acrolein (fig. S5). Consistent with the study of Brandt et al. (30), our results suggest the activation of the C═C bond when acrolein adsorbs at the coinage metal surfaces.
The proposed inner orbital broadening mechanism was discussed so far under idealized conditions in calculations. However, the same conclusions remain for more realistic conditions of hydrogen-precovered surface and higher surface coverage. For hydrogen-precovered Pd(111) surface, our calculations show that the coadsorption of hydrogen leads to a negligible variation, less than 0.05 Å, in the structure of the adsorbed isophorone (fig. S6). This finding is supported by IRAS experiments, which show that the C═C and C═O bonds of isophorone are lying flat at low coverage, no matter whether hydrogen atoms are coadsorbed or not on Pd(111). IRAS measurements indicate that the C═C and C═O vibrations have the same frequency for the clean and hydrogen-precovered surface. Because the molecular structures and thereby the molecular orbital distributions are not affected by hydrogen atoms, we thus conclude that our proposed mechanism still holds when hydrogen atoms are present on the metal surface.
In addition to hydrogen preadsorption, high surface coverage is an important condition that is relevant to practical applications. The relaxed adsorption structure for isophorone on Pd(111) in a smaller (3 × 2) unit cell is shown in fig. S7A. In the case of high coverage, the C═C bond in isophorone is significantly tilted and hinders its interaction with Pd atoms. This is in excellent agreement with the observations and conclusion from our NEXAFS measurements. Note that the reorientation of the adsorbed isophorone, in turn, can lead to completely different electronic structure. As evidenced in fig. S7B, the F-HOMO of the adsorbed isophorone is considerably broadened at high coverage, whereas the F-HOMO−1 inner orbital localized at the C═C bond remains largely unperturbed. In the scope of our model, the activation of the C═C bond is thus prohibited, which agrees with experiments that saturated ketone would not be produced at higher surface coverage. These observations suggest that the inner orbital broadening mechanism would hold for a range of coverages.
DISCUSSION
In summary, we reported a combined theoretical and experimental study on the interaction of the α,β-unsaturated carbonyls isophorone and acrolein with a range of transition metal surfaces. We found that the extent of broadening of the inner orbital HOMO−1, located at the C═C bond of isophorone and acrolein, is significantly greater than that of the frontier HOMO orbital at the C═O bond, suggesting preferential activation of C═C bond partial hydrogenation. This observation directly correlates with the reactivity behavior in partial hydrogenation of these molecules: Although facile hydrogenation of the C═C double bond was detected experimentally, no products resulting from hydrogenation of the carbonyl bond could be observed. By going beyond the widely accepted FMO theory, we explain the preferential hydrogenation of the C═C bond in small α,β-unsaturated carbonyls observed experimentally and resolve the long-standing controversy in the previous theoretical studies predicting preferential hydrogenation of the C═O bond. These results provide detailed atomistic-level insights into the origin of selective hydrogenation of multi-unsaturated hydrocarbon compounds and hold a great potential to significantly contribute to the rational design of chemoselective catalysts. Moreover, the related effects are expected to play a key role in controlling activity and selectivity of a broad variety of technically important chemical processes involving (multi-)unsaturated hydrocarbon compounds, such as selective partial oxidation of alkenes or hydrogenation of multi-unsaturated carbonyl compounds.
MATERIALS AND METHODS
Experimental
MB experiments were performed at the Fritz-Haber-Institut (Berlin) in an ultrahigh vacuum (UHV) apparatus described in detail previously (31). An effusive doubly differentially pumped multichannel array source operated at room temperature was used to continuously supply the H2. The beam fluxes for H2 of 5 × 1015 molecules cm−2 s−1 were used in these experiments. Another effusive doubly differentially pumped multichannel array source was used to dose isophorone (Acros Organics; purity, 98%) at a flux of 2 × 1012 molecules cm−2 s−1 with the 30-min long pulses. The diameter of the beams exceeded the sample diameter. An automated quadrupole mass spectrometer (QMS) system (ABB Extrel) continuously monitored the partial pressure of the reactant [isophorone, 138 atomic mass unit (amu) with the main fragment detected at 82 amu] and products (trimethylcyclohexanone, main fragment at 83 amu; isophorol, expected fragments at 125, 107, 91, and 84 amu). The QMS data were corrected for the natural abundance of 13C.
The IRAS data were acquired on Pd(111) by using a vacuum-adapted Fourier transform infrared spectrometer (Bruker IFS 66v/S) with a spectral resolution of 2 cm−1. A mid-infrared polarizer was used to select the p component of the IR light. d5-isophorone [Quotient Bioresearch (Radiochemicals) Limited; purity, 90%] and trimethylcyclohexanone (Sigma-Aldrich; purity, 98%) were supplied via an effusive doubly differentially pumped MB onto the sample surface to exactly control the exposure. Acquisition of the spectra was performed at 120 K.
The thin (~100 Å) Fe3O4 film was grown on a Pt(111) single crystal by repeated cycles of Fe (>99,99%, Goodfellow) physical vapor deposition and subsequent oxidation [see the works by Schalow et al. (32, 33) for details]. The cleanliness and quality of the oxide films were checked by low-energy electron diffraction. Pd particles (>99.9%, Goodfellow) were grown by physical vapor deposition using a commercial evaporator (Focus, EFM 3,; flux calibrated by a quartz microbalance) with the sample temperature at 115 K. During Pd evaporation, the sample was biased to +800 V to avoid creation of defects by accelerated metal ions. The final Pd coverage was 2.7 × 1015 atoms cm−2. The resulting surfaces were stabilized via several cycles of oxygen (8 × 10−7 mbar for 1000 s) and CO exposures (8 × 10−7 mbar for 3000 s) at 500 K (33), followed by flash annealing in UHV to 485 K.
NEXAFS experiments were performed at the undulator beamline UE52-PGM at the BESSY II synchrotron facility in Berlin. The Pd(111) single crystal was cleaned by repeating sputtering-annealing-oxidizing cycles. Isophorone was deposited onto the Pd(111) at 120 K by placing the crystal in front of the gas doser. The actual coverage of isophorone was determined by the position of the C1s peak in x-ray photoelectron spectroscopy (34). Following deposition of isophorone onto the Pd(111) crystal, the sample was transferred to a separate analysis chamber for NEXAFS spectra collection. The analysis chamber was equipped with a channeltron detector with a retarding field of 150 V for partial electron-yield NEXAFS measurements. The energy of the incident x-ray beam was scanned from 250 to 350 eV with a resolution of 0.1 eV in the range of 280 to 300 eV and 0.5 eV elsewhere. Spectra were taken at incident beam angles of 70° and 80° with respect to the sample normal, each with s- and p-polarization. The spectra were normalized by their C K-edge. The orientations of the molecular orbitals were calculated from the ratio of the corresponding peak areas in the spectra measured with p- and s-polarized light (35).
Theoretical calculations
The DFT calculations used the numeric atom–centered basis set all-electron code FHI-aims (36, 37), together with the PBE exchange-correlation functional (21). We used the PBE + vdWsurf method (20) to account for the van der Waals (vdW) interactions and collective response effects. The PBE + vdWsurf method extends pairwise vdW approaches to modeling of adsorbates on surfaces by a synergetic combination of the PBE + vdW method (38) for intermolecular interactions with the Lifshitz-Zaremba-Kohn theory (39, 40) for the nonlocal Coulomb screening within the bulk. We used the scaled zeroth-order regular approximation (41) to treat relativistic effects for Pd atoms. The “tight” settings, including the “tier2” standard basis set in the FHI-aims code, were used for H, C, and O, and “tier1” was used for Pd. All atomic positions were relaxed until the maximal force on each atom was smaller than 0.01 eV Å−1. For all computations, the convergence criteria of 10−5 electrons per unit volume for the charge density and 10−4 eV for the total energy of the system were used. Adopting these settings, the numerical accuracies in determining the binding energy and equilibrium distance are better than 0.01 eV and 0.01 Å, respectively. In slab calculations, we used a 3 × 3 × 1 Monkhorst-Pack mesh for the sampling of the Brillouin zone of the (4 × 4) surface.
Supplementary Material
Acknowledgments
We thank M. Oehzelt for the experimental support on the NEXAFS experiments. Funding: A.T. acknowledges support from the European Research Council (ERC) (Consolidator Grant BeStMo). W.L. acknowledges support from the NSF of China (21403113) and Jiangsu Province (BK20150035). S.S. acknowledges support from the Fonds der Chemischen Industrie and ERC (ERC Starting Grant ENREMOS). Author contributions: A.T., S.S., and W.L. designed the research. W.L., Y.J., and A.T. performed the calculations and analyzed and discussed the results. K.-H.D., C.P.O., W.R., A.S., and S.S. conducted the experiments. W.L., S.S., and A.T. wrote the paper. All authors commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700939/DC1
Supplementary Text
fig. S1. Structure and electronic properties of 1-butene.
fig. S2. Hydrogenation pathways of isophorone on Pd(111).
fig. S3. Adsorption structures of isophorone on Pd(111).
fig. S4. MODOS of isophorone on metal surfaces.
fig. S5. MODOS of acrolein on metal surfaces.
fig. S6. Coadsorption structure of isophorone on Pd(111).
fig. S7. Isophorone on Pd(111) at high coverage.
REFERENCES AND NOTES
- 1.Zaera F., An organometallic guide to the chemistry of hydrocarbon moieties on transition metal surfaces. Chem. Rev. 95, 2651–2693 (1995). [Google Scholar]
- 2.Gallezot P., Richard D., Selective hydrogenation of α,β-unsaturated aldehydes. Catal. Rev. 40, 81–126 (1998). [Google Scholar]
- 3.Marshall S. T., O’Brien M., Oetter B., Corpuz A., Richards R. M., Schwartz D. K., Medlin J. W., Controlled selectivity for palladium catalysts using self-assembled monolayers. Nat. Mater. 9, 853–858 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Chudasama V., Fitzmaurice R. J., Caddick S., Hydroacylation of α,β-unsaturated esters via aerobic C–H activation. Nat. Chem. 2, 592–596 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Liu W., Tkatchenko A., Scheffler M., Modeling adsorption and reactions of organic molecules at metal surfaces. Acc. Chem. Res. 47, 3369–3377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dostert K.-H., O’Brien C. P., Ivars-Barceló F., Schauermann S., Freund H.-J., Spectators control selectivity in surface chemistry: Acrolein partial hydrogenation over Pd. J. Am. Chem. Soc. 137, 13496–13502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pirnot M. T., Rankic D. A., Martin D. B. C., MacMillan D. W. C., Photoredox activation for the direct β-arylation of ketones and aldehydes. Science 339, 1593–1596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Y., Qian H., Drake B. A., Jin R., Atomically precise Au25(SR)18 nanoparticles as catalysts for the selective hydrogenation of α,β-unsaturated ketones and aldehydes. Angew. Chem. Int. Ed. 122, 1317–1320 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Fleischer S., Zhou S., Junge K., Beller M., General and highly efficient iron-catalyzed hydrogenation of aldehydes, ketones, and α,β-unsaturated aldehydes. Angew. Chem. Int. Ed. 52, 5120–5124 (2013). [DOI] [PubMed] [Google Scholar]
- 10.M. Baerns, Basic Principles in Applied Catalysis, vol. 75 of Springer Series in Chemical Physics (Springer-Verlag, 2004).
- 11.P. N. Rylander, Hydrogenation Methods (Academic Press, 1985). [Google Scholar]
- 12.Mäki-Arvela P., Hájek J., Salmi T., Murzin D. Y., Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts. Appl. Catal. A 292, 1–49 (2005). [Google Scholar]
- 13.Loffreda D., Delbecq F., Vigné F., Sautet P., Catalytic hydrogenation of unsaturated aldehydes on Pt(111): Understanding the selectivity from first-principles calculations. Angew. Chem. Int. Ed. 44, 5279–5282 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Tuokko S., Pihko P. M., Honkala K., First principles calculations for hydrogenation of acrolein on Pd and Pt: Chemoselectivity depends on steric effects on the surface. Angew. Chem. Int. Ed. 55, 1670–1674 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Loffreda D., Delbecq F., Vigné F., Sautet P., Chemo-regioselectivity in heterogeneous catalysis: Competitive routes for C═O and C═C hydrogenations from a theoretical approach. J. Am. Chem. Soc. 128, 1316–1323 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Yang B., Wang D., Gong X.-Q., Hu P., Acrolein hydrogenation on Pt(211) and Au(211) surfaces: A density functional theory study. Phys. Chem. Chem. Phys. 13, 21146–21152 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Yoon C., Yang M. X., Somorjai G. A., Reactions of 1-butene and cis-2-butene on platinum surfaces: Structure sensitivity of cis-2-butene isomerization. J. Catal. 176, 35–41 (1998). [Google Scholar]
- 18.Ponec V., On the role of promoters in hydrogenations on metals; α,β-unsaturated aldehydes and ketones. Appl. Catal. A 149, 27–48 (1997). [Google Scholar]
- 19.Delley B., An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92, 508–517 (1990). [Google Scholar]
- 20.Ruiz V. G., Liu W., Zojer E., Scheffler M., Tkatchenko A., Density-functional theory with screened van der Waals interactions for the modeling of hybrid inorganic-organic systems. Phys. Rev. Lett. 108, 146103 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Dostert K.-H., O’Brien C. P., Liu W., Riedel W., Savara A. A., Tkatchenko A., Schauermann S., Freund H.-J., Adsorption of isophorone and trimethyl-cyclohexanone on Pd(111): A combination of infrared reflection absorption spectroscopy and density functional theory studies. Surf. Sci. 650, 149–160 (2016). [Google Scholar]
- 23.Liu W., Carrasco J., Santra B., Michaelides A., Scheffler M., Tkatchenko A., Benzene adsorbed on metals: Concerted effect of covalency and van der Waals bonding. Phys. Rev. B 86, 245405 (2012). [Google Scholar]
- 24.Egger D. A., Ruiz V. G., Saidi W. A., Bučko T., Tkatchenko A., Zojer E., Understanding structure and bonding of multilayered metal-organic nanostructures. J. Phys. Chem. C 117, 3055–3061 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Ruitenbeek J., Metal/molecule interfaces: Dispersion forces unveiled. Nat. Mater. 11, 834–835 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann F. M., Infrared reflection-absorption spectroscopy of adsorbed molecules. Surf. Sci. Rep. 3, 109–192 (1983). [Google Scholar]
- 27.Rangger G. M., Romaner L., Heimel G., Zojer E., Understanding the properties of interfaces between organic self-assembled monolayers and noble metals—A theoretical perspective. Surf. Interface Anal. 40, 371–378 (2008). [Google Scholar]
- 28.Dewar M. J. S., A review of the pi-complex theory. Bull. Soc. Chim. Fr. 18, C71–C79 (1951). [Google Scholar]
- 29.Chatt J., Duncanson L. A., Olefin co-ordination compounds. Part III. Infra-red spectra and structure: Attempted preparation of acetylene complexes. J. Chem. Soc., 2939–2947 (1953). [Google Scholar]
- 30.Brandt K., Chiu M. E., Watson D. J., Tikhov M. S., Lambert R. M., Chemoselective catalytic hydrogenation of acrolein on Ag(111): Effect of molecular orientation on reaction selectivity. J. Am. Chem. Soc. 131, 17286–17290 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Libuda J., Meusel I., Hartmann J., Freund H.-J., A molecular beam/surface spectroscopy apparatus for the study of reactions on complex model catalysts. Rev. Sci. Instrum. 71, 4395 (2000). [Google Scholar]
- 32.Schalow T., Laurin M., Brandt B., Schauermann S., Guimond S., Kuhlenbeck H., Starr D. E., Shaikhutdinov S. K., Libuda J., Freund H.-J., Oxygen storage at the metal/oxide interface of catalyst nanoparticles. Angew. Chem. Int. Ed. 44, 7601–7605 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Schalow T., Brandt B., Starr D. E., Laurin M., Schauermann S., Shaikhutdinov S. K., Libuda J., Freund H.-J., Oxygen-induced restructuring of a Pd/Fe3O4 model catalyst. Catal. Lett. 107, 189–196 (2006). [Google Scholar]
- 34.Beaumont S. K., Kyriakou G., Watson D. J., Vaughan O. P. H., Papageorgiou A. C., Lambert R. M., Influence of adsorption geometry in the heterogeneous enantioselective catalytic hydrogenation of a prototypical enone. J. Phys. Chem. C 114, 15075–15077 (2010). [Google Scholar]
- 35.J. Stöhr, NEXAFS Spectroscopy (Springer-Verlag, 1992). [Google Scholar]
- 36.Blum V., Gehrke R., Hanke F., Havu P., Havu V., Ren X., Reuter K., Scheffler M., Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 180, 2175–2196 (2009). [Google Scholar]
- 37.Havu V., Blum V., Havu P., Scheffler M., Efficient O(N) integration for all-electron electronic structure calculation using numeric basis functions. J. Comput. Phys. 228, 8367–8379 (2009). [Google Scholar]
- 38.Tkatchenko A., Scheffler M., Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Lifshitz E. M., The theory of molecular attractive forces between solids. Sov. Phys. JETP 2, 73–83 (1956). [Google Scholar]
- 40.Zaremba E., Kohn W., Van der Waals interaction between an atom and a solid surface. Phys. Rev. B 13, 2270–2285 (1976). [Google Scholar]
- 41.van Lenthe E., Baerends E.-J., Snijders J. G., Relativistic total energy using regular approximations. J. Chem. Phys. 101, 9783 (1994). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700939/DC1
Supplementary Text
fig. S1. Structure and electronic properties of 1-butene.
fig. S2. Hydrogenation pathways of isophorone on Pd(111).
fig. S3. Adsorption structures of isophorone on Pd(111).
fig. S4. MODOS of isophorone on metal surfaces.
fig. S5. MODOS of acrolein on metal surfaces.
fig. S6. Coadsorption structure of isophorone on Pd(111).
fig. S7. Isophorone on Pd(111) at high coverage.