

ABSTRACT
Mitophagy is responsible for removal of damaged mitochondria and is therefore a fundamental process in mitochondrial quality control. Both ubiquitin-dependent and receptor-dependent pathways are considered to mediate mitophagy. These distinct mechanisms may be activated in response to distinct mitochondrial stresses. An intriguing question is whether and how crosstalk occurs between the distinct pathways to coordinate mitophagy. We have uncovered a striking piece of evidence to demonstrate that the mitophagy receptor FUNDC1 is a substrate of MARCH5, a mitochondrially localized E3 ubiquitin ligase. In response to hypoxia, MARCH5 degrades redundant FUNDC1 to fine-tune hypoxia-induced mitophagy, whereas ablation of MARCH5 leads to accumulation of FUNDC1 and an exaggerated mitophagic phenotype. Mechanistic studies demonstrate that hypoxic insult enhances the interaction of FUNDC1 with MARCH5, which ubiquitinates FUNDC1 at lysine 119 for subsequent degradation. MARCH5-based ubiquitination and degradation of FUNDC1 circumvents injudicious removal of cellular mitochondria. However, severe hypoxic stress leads to dephosphorylation of FUNDC1, increasing mitophagic flux.
KEYWORDS: degradation, FUNDC1, MARCH5, mitophagy, mitophagy receptor, ubiquitination
Two distinct mitophagy pathways have been elucidated so far in mammalian systems, i.e., the PINK1-PARK2/Parkin pathway and the receptor-mediated pathway. Of these two, PINK1-PARK2 is the most studied, mainly because PARK2 mutations have been linked with mitochondrial dysfunction and Parkinsonism. Upon loss of mitochondrial membrane potential, which serves as signal for loss of mitochondrial function, PINK1 becomes stabilized on the outer membrane of mitochondria. The activated PINK1 phosphorylates ubiquitin and PARK2 to recruit PARK2 and other autophagy receptors such as OPTN and CALCOCO2/NDP52 to mitochondria. Translocated PARK2 ubiquitinates numerous mitochondrial outer-membrane proteins, which is followed by translocation of SQSTM1/p62 to mitochondria and engulfment of the fragmented mitochondrion, or a portion of a mitochondrion, by the autophagy machinery. Conversely, several receptors, such as BNIP3L/Nix, BNIP3, FUNDC1 and PHB2 (prohibitin 2), have been identified to selectively mediate mitophagy. In particular, we have found that FUNDC1 mediates hypoxia-induced mitophagy. In response to hypoxic stress, dephosphorylation of FUNDC1 at Tyr18 occurs due to loss of SRC and CSNK2/CK2 kinase activity and activation of phosphatases including PGAM5. Both biochemical and structural analyses have revealed that dephosphorylated FUNDC1 has a significantly higher affinity for LC3, thus enhancing the FUNDC1-LC3 interaction and recruitment of the autophagy machinery for the formation of mitophagosomes. However, the signaling cascade that links sensing of hypoxia to initiation of mitophagy is poorly understood.
As the hypoxic stress mounts, the signature of mitochondrial membrane proteins such as FUNDC1, TIMM23 and TOMM20 decline. Interestingly, we observed that FUNDC1 is categorically degraded much faster than other mitochondrial proteins (including TIMM23 and TOMM20), and this degradation can be inhibited by MG132 (proteasome inhibitor) and/or chloroquine (autophagic flux inhibitor). These findings led us to the hypothesis that there might be another regulatory mechanism, based on ubiquitination, that governs posttranslational turnover of FUNDC1 and mitophagy.
In an effort to figure out the potential E3 ligases that ubiquitinate FUNDC1, we identified MARCH5, an E3 ligase localized on the mitochondrial outer membrane, which specifically induces FUNDC1 degradation. In contrast to PARK2, ectopic expression of MARCH5 dramatically increases ubiquitination and degradation of FUNDC1. Conversely, the ubiquitination and degradation are suppressed in MARCH5 knockdown cells, and this suppression can be rescued by expression of wild-type MARCH5, but not the C65S,C68S mutant which lacks E3 ligase function.
To explore the molecular mechanism by which MARCH5 mediates FUNDC1 ubiquitination, we individually mutated the 11 lysine (K) sites of FUNDC1 to arginine (R). In vivo and in vitro ubiquitination assays prove that K119 of FUNDC1 is the major site of MARCH5-mediated ubiquitination. Moreover, the K119R mutant is more resistant to degradation than wild-type FUNDC1 even when mitophagy activity is more pronounced. Co-immunoprecipitation assays and glutathione S-transferase (GST) affinity isolation assays confirm the physical interaction between MARCH5 and its substrate FUNDC1. Importantly, the MARCH5-FUNDC1 interaction progressively escalates during hypoxia. Moreover, MARCH5-based FUNDC1 degradation occurred more quickly than dephosphorylation of FUNDC1 at Tyr18, which accounts for the activation of FUNDC1 subsequently leading to induction of mitophagy, demonstrating that the ubiquitin-dependent degradation of FUNDC1 occurs in the initial mitophagy stress before a mature mitophagy phenotype ensues.
We further investigated the biological significance of the MARCH5-FUNDC1 axis in mitophagy regulation. In response to hypoxic stress, knockdown of endogenous MARCH5 induces a remarkable increase in colocalization between GFP-LC3 puncta and mitochondria, but also in degradation of mitochondrial proteins. These data reveal that MARCH5 in intimately involved in hypoxia-induced mitophagy events. Furthermore, MARCH5 and FUNDC1 double RNAi experiments confirm that the substrate FUNDC1 is indispensable for hypoxic mitophagy regulated by MARCH5. Our results demonstrated that the MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy by regulating FUNDC1 ubiquitination and degradation.
MARCH5 has been considered to play roles in mitochondrial homeostasis and cell sensitivity by ubiquitinating different proteins that regulate mitochondrial dynamics. In the mitochondrial fission machinery, MARCH5 ubiquitinates DNM1L/Drp1 or suppresses DNM1L recruitment to mitochondria by mediating the degradation of fission receptors, such as MIEF2/MiD49 and FIS1, and eventually inhibits mitochondrial division (Fig. 1). Intriguingly, FUNDC1 has also been reported to induce mitochondrial fission by increasing mitochondrial recruitment of cytosolic DNM1L. Thus, further in-depth studies are needed to explore whether the MARCH5-FUNDC1 axis participates in the regulation of mitochondrial dynamics, and how mitophagy is linked with the mitochondrial fission machinery.
Figure 1.
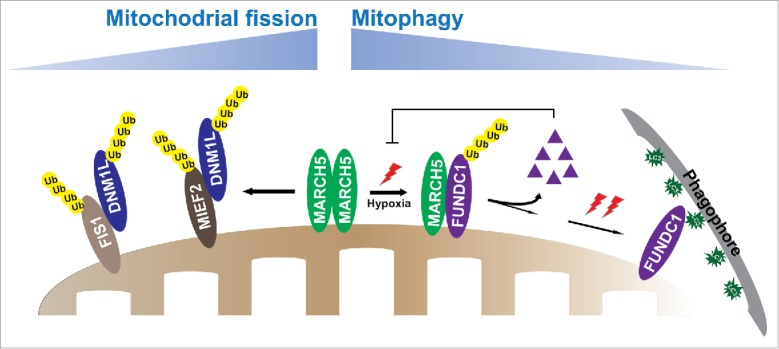
MARCH5 plays a role in mitochondrial morphology and mitophagy. In the mitochondrial dynamic machinery, MARCH5 suppresses mitochondrial fission by ubiquitinating DNM1L or decreasing DNM1L translocation to mitochondria by ubiquitinating and degrading fission receptors FIS1 and MIEF2. In the mitophagy machinery, initial hypoxic stress induces disassembly of MARCH5 oligomers and enhances the interaction of MARCH5 with the mitophagy receptor FUNDC1, which induces ubiquitination and degradation of FUNDC1. Degradation of FUNDC1 conversely inhibits initiation of mitophagy, thus circumventing inappropriate removal of mitochondria. As the exposure time of hypoxia mounts up, FUNDC1 is dephosphorylated and mitophagic flux rapidly increases, thereby recruiting more phagophores to damaged mitochondria for removal.
In conclusion, our data uncovered a novel mechanism, which fine-tunes the regulation of the MARCH5-FUNDC1 axis in the removal of damaged mitochondria. Hypoxia triggers disassembly of MARCH5 oligomers to facilitate access of MARCH5 monomers to the mitophagy receptor FUNDC1 in response to initial mitochondrial stress. MARCH5-based ubiquitination and degradation of FUNDC1 reduces abrupt and excessive initiation of mitochondrial autophagy, thereby avoiding inappropriate removal of cellular mitochondria. However, as the duration of hypoxic stress is prolonged, dephosphorylation of FUNDC1 residues ensues and mitophagic flux builds up exponentially (Fig. 1). Our findings reveal a critical mechanism by which MARCH5 regulates the protein levels of the receptor FUNDC1 to gauge mitochondrial quality.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.