

ABSTRACT
During mycobacterial infection, macroautophagy/autophagy, a process modulated by cytokines, is essential for mounting successful host responses. Autophagy collaborates with human immune responses against Mycobacterium tuberculosis (Mt) in association with specific IFNG secreted against the pathogen. However, IFNG alone is not sufficient to the complete bacterial eradication, and other cytokines might be required. Actually, induction of Th1 and Th17 immune responses are required for protection against Mt. Accordingly, we showed that IL17A and IFNG expression in lymphocytes from tuberculosis patients correlates with disease severity. Here we investigate the role of IFNG and IL17A during autophagy in monocytes infected with Mt H37Rv or the mutant MtΔRD1. Patients with active disease were classified as high responder (HR) or low responder (LR) according to their T cell responses against Mt. IL17A augmented autophagy in infected monocytes from HR patients through a mechanism that activated MAPK1/ERK2-MAPK3/ERK1 but, during infection of monocytes from LR patients, IL17A had no effect on the autophagic response. In contrast, addition of IFNG to infected monocytes, increased autophagy by activating MAPK14/p38 α both in HR and LR patients. Interestingly, proteins codified in the RD1 region did not interfere with IFNG and IL17A autophagy induction. Therefore, in severe tuberculosis patients' monocytes, IL17A was unable to augment autophagy because of a defect in the MAPK1/3 signaling pathway. In contrast, both IFNG and IL17A increased autophagy levels in patients with strong immunity to Mt, promoting mycobacterial killing. Our findings might contribute to recognize new targets for the development of novel therapeutic tools to fight the pathogen.
KEYWORDS: autophagy, cytokines, IFNG, IL17A, immune response, Mycobacterium tuberculosis, patients, tuberculosis
Introduction
Tuberculosis is considered to be the leading cause of death from an infectious microorganism. In fact, Mycobacterium tuberculosis causes nearly 10.4 million new cases and 1.8 million deaths per year.1 Currently, the major objectives to fight this pathogen are to develop new diagnosis methods, novel treatments, and more successful vaccines. The immune response elicited after M. tuberculosis infection is critically dependent on CD4+ T cells. In particular, Th1 cells play an important role in granuloma formation and clearance of M. tuberculosis infection.2-4 Indeed, reduced IFNG production is a marker of severe disease.5 In spite of that, IFNG alone is not sufficient to the complete eradication of the bacteria, indicating that other cytokines might be required for pathogen removal. It is proposed that CD4+ T cells producing IL17 and IL22 might contribute to adaptive immunity to M. tuberculosis.6 Certainly, IL17A is rapidly induced by γδ T cells during infection7 and IL17A secretion by CD4+T lymphocytes is required to eliminate primary infections and for the establishment of an effective memory response.8-11 Furthermore, IL17A has a crucial role in granuloma formation12 and mediates the induction of CXCL13.13 Nevertheless, excessive IL17 levels could be detrimental, exacerbating inflammation and increasing neutrophil recruitment and tissue damage.14 In line with this, we have recently demonstrated that the ratio of antigen-expanded CD4+ IFNG+ IL17+ lymphocytes, in peripheral blood and pleural fluid from tuberculosis patients, is directly correlated with clinical parameters associated with disease severity.15 Despite the great strides made in the characterization of the acquired cellular response in tuberculosis patients, it remains to be elucidated what exactly constitutes a protective response.16 Moreover, how M. tuberculosis is able to evade host immune surveillance and persist, particularly inside macrophages, remains to be understood.
Autophagy, a fundamental homeostatic mechanism, plays a role in innate and adaptive immunity against intracellular pathogens, including M. tuberculosis.17 Moreover, enhanced autophagy mediates elimination of intracellular M. tuberculosis through lytic and antimicrobial properties unique to autolysosomes.18 The role of autophagy as a defense mechanism makes it possible to speculate that vaccines that induce an autophagic response might be more successful in preventing the acquisition of tuberculosis or reactivation of latency. In addition, considering the ability of M. tuberculosis to modulate autophagy process, whole genome microarray analysis reveals regions like RD1, which encodes potential antigenic determinants that could increase the immunogenicity of a vaccine and could affect the autophagy process.19 Furthermore, the importance of host autophagy in orchestrating successful antimicrobial responses to mycobacteria during chemotherapy has been reported.20
On the other hand, autophagy can be modulated by cytokines and other immunological signals.21,22 Accordingly, TNF induces autophagy in Ewing sarcoma cells.21 Besides, in macrophages and other cells IFNG augments the autophagy process.17,23 In contrast, Harris et al. report that Th2 cytokines (IL4 and IL13) abrogate autophagy and autophagy-mediated killing of intracellular mycobacteria in murine macrophages and human U937 or THP-1 cells.24 Furthermore, IL10 also inhibits autophagy induction in murine macrophages.25 Therefore, critical cytokines modulate both positively and negatively the autophagy response affecting survival of mycobacteria. Moreover, it has been demonstrated that activation of RAW cells with IFNG induces the autophagic process.17 Accordingly, we have recently reported that autophagy collaborates with human immune responses against M. tuberculosis in close association with specific IFNG secreted against the pathogen.26 Additionally, it has been suggested that there is a mechanism through which CD4+ Th lymphocyte polarization might differentially affect the immune control of intracellular pathogens, like M. tuberculosis.24 However, the ability of IL17 produced in response to M. tuberculosis to modulate the autophagy process has not been studied.
Given that IL17 plays a pivotal role in the immune response to mycobacteria, in this study we investigated the role of this important cytokine during the process of autophagy in human monocytes from patients with active disease infected with M. tuberculosis strains. Moreover, we also analyzed the role of IFNG during infection of human monocytes with the M. tuberculosis H37Rv pathogenic strain and the nonpathogenic RD1-deleted- M. tuberculosis strain (MtΔRD1). Our results indicate that IL17A increases autophagy against live M. tuberculosis strains in monocytes from tuberculosis patients that display strong immunity against the bacteria through a mechanism that activates MAPK1/3. During infection of monocytes from patients with severe disease and weak immunity to the pathogen, IL17A did not modify the levels of autophagy and no MAPK1/3 phosphorylation was detected. In contrast, addition of IFNG to infected monocytes from tuberculosis patients with either strong or weak immunity to M. tuberculosis augmented autophagy through a mechanism that involves MAPK14 activation.
Together, our present findings suggest that IL17A, a cytokine that would be required to eliminate primary infection and for the establishment of an effective memory response,8-11 participates in the immune response of the human host against M. tuberculosis through the activation of the autophagy process in correlation with the severity of the disease.
Results
Autophagy rate in human PBMC infected with M. tuberculosis strains
To compare the rate of autophagy of human monocytes infected by the 2 M. tuberculosis strains (Mt H37Rv or MtΔRD1) used in this study, we determined the autophagic flux in monocytes from TB patients in the absence or presence of Bafilomycin A1 (BafA1), which inhibits the vacuolar-type H+-translocatingATPase and prevents fusion between autophagosomes and lysosomes, leading to inhibition of MAP1LC3A,B-II/LC3A,B-II degradation.27 We observed that, in adherent cells, infection with Mt H37Rv in the presence of BafA1 did not modify LC3 puncta levels, indicating that LC3-II degradation due to lysosome fusion to the autophagosome is impaired during this strain infection, which blocks the autophagic pathway (Fig. 1A). In contrast, MtΔRD1 significantly increased LC3 puncta levels in the presence of BafA1, strongly suggesting that this strain induced autophagosome formation and that the autophagic pathway was functional (Fig. 1B). Similar results were observed after stimulation with sonicated Mt-Ag (Similar results were observed after stimulation with sonicated M. tuberculosis antigen (Mt-Ag)).26 (Fig. S1).
Figure 1.
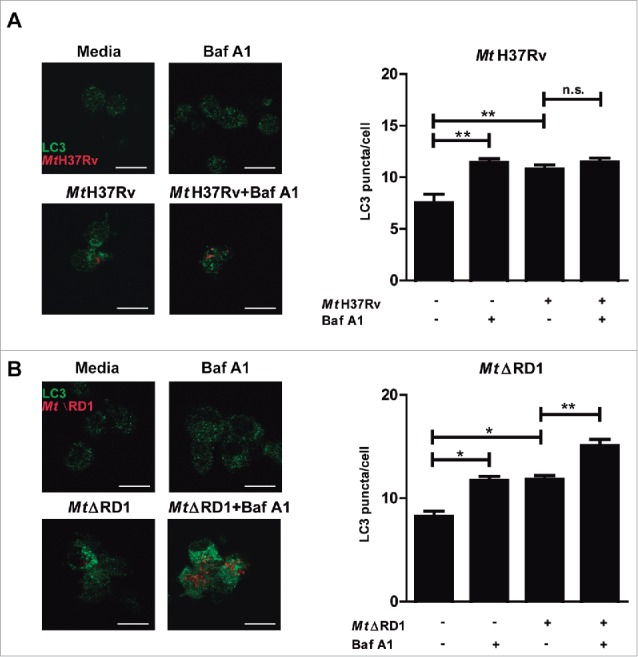
Modulation of autophagy of monocytes infected with Mt H37Rv or MtΔRD1. PBMC from TB patients were incubated at 2 × 106 cells/ml for 16 h without stimulus to allow monocyte adherence. Cells were then infected with (A) Mt H37Rv or (B) MtΔRD1 (MOI: 20). After 2 h of infection, the culture medium was replaced and cells were cultured for 24 h. BafA1 (1 µg/ml) was added for the last 2 h of culture and then LC3 was determined by immunofluorescence. Bars represent the mean values of LC3 puncta per cell ± SEM. *P < 0.05, **P < 0.01. P values were calculated using one-way ANOVA with the post hoc Tukey multiple comparisons test.
Taken together, these findings demonstrate a differential impact of both strains on autophagy flux during infection of human monocytes, probably caused by the intracellular activity of the proteins encoded in the RD1 region of the Mt genome.
IFNG upregulates autophagy in monocytes from TB patients infected with M. tuberculosis strains
In human and murine models it has been demonstrated that autophagy is a defense mechanism that inhibits mycobacterial survival.17 However, very limited information exists about the autophagic process in patients with active tuberculosis disease. Recently we investigated the potential role of autophagy during the human immune response against M. tuberculosis antigens. To this end, we studied M. tuberculosis-induced autophagy in 2 groups of patients with active tuberculosis classified on their T cell responses to the bacterium: High responder (HR) tuberculosis patients displayed significant T cell proliferation and IFNG production against Mt-Ag, while low responder (LR) tuberculosis patients displayed weak or no T cell responses to the antigen.28 Our previous data show that Mt-Ag induces the highest levels of autophagy in healthy donors (HD) and the lowest levels in LR tuberculosis patients, in direct association with the amounts of IFNG secreted by each individual.26 Besides, by addition of recombinant IFNG, the percentages of CD14+ LC3A,B-II+ cells detected were significantly increased in the 3 groups of individuals, even in LR patients26 (Fig. S2). In the present work, we further investigated the role of IFNG by studying autophagy in monocytes from HD, HR and LR tuberculosis patients upon infection with M. tuberculosis strains. Then, in contrast to those previous studies with Mt-Ag, similar levels of LC3 puncta were observed in adherent cells from HD and in both groups of TB patients infected with either the Mt H37Rv or MtΔRD1 strains (Fig. 2A and B). However, and in line with our previous data with Mt-Ag, addition of exogenous IFNG significantly augmented the levels of autophagy in the 3 groups of individuals under study (Fig. 2). Together, the present results indicate that live M. tuberculosis augment LC3 puncta levels in TB patients independently of their pathogenicity and, as expected, addition of recombinant IFNG significantly increases the autophagy response.
Figure 2.
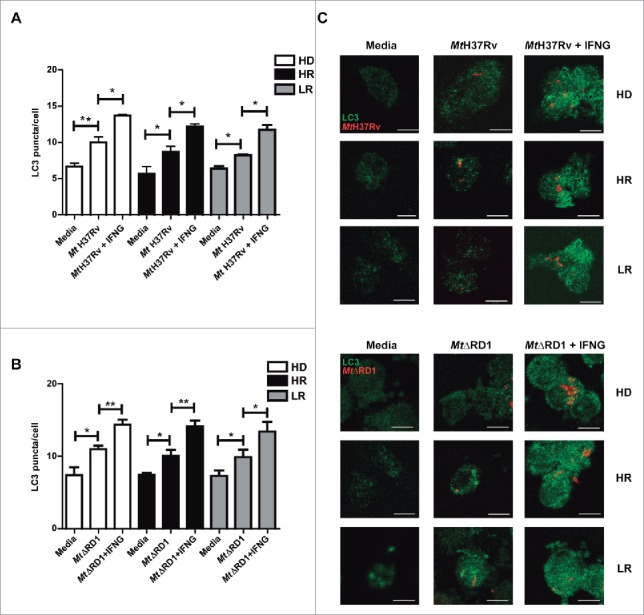
Modulation of autophagy by IFNG in monocytes infected with Mt H37Rv or MtΔRD1. PBMC from HD, High responder (HR) and Low responder (LR) TB patients were incubated at 2 × 106 cells/ml for 16 h without stimulus to allow monocyte adherence. Cells were then incubated uninfected in media or infected with (A) Mt H37Rv or (B) MtΔRD1 (MOI: 20). After 2 h of infection, the culture medium was replaced and cells were cultured with or without recombinant IFNG (1.8 ng/ml) for 24 h. Autophagy levels were evaluated by immunofluorescence against LC3 on monocytes. Bars represent the mean values of LC3 puncta per cell ± SEM. (C) Representative images of a HD, HR and LR TB patient are shown. Scale bars: 5 μm. *P < 0.05, **P < 0.01. P values were calculated using one-way ANOVA with the post hoc Tukey multiple comparisons test.
To analyze the molecular mechanism by which IFNG increased autophagy in monocytes from tuberculosis patients and considering that it has been demonstrated that IFNG activates autophagy via MAPK14/p38 α (mitogen-activated protein kinase14),29 we next investigated the phosphorylation of MAPK14 in monocytes from HR and LR tuberculosis patients in the presence or absence of IFNG. To this end, monocytes were obtained and stimulated with Mt-Ag ± recombinant IFNG during 24 h. Afterwards, phosphorylated and total MAPK14 expression was determined. As shown in Fig. 3, phosphorylation of MAPK14 was marked increased in extracts from adherent cells from HR tuberculosis patients while, in LR individuals, no differences in MAPK14 phosphorylation were detected in Mt-Ag stimulated cells as compared with control cells (cultured with media). Interestingly, significant expression of activated MAPK14 was measured in extracts from HR and LR tuberculosis patients upon addition of exogenous IFNG (Fig. 3). These results indicate a direct association between the levels of autophagy induced in tuberculosis patients' monocytes stimulated with Mt-Ag and the activation of MAPK14 by IFNG, suggesting that this cytokine activates autophagy via the MAPK14 signaling pathway, as previously reported.29
Figure 3.
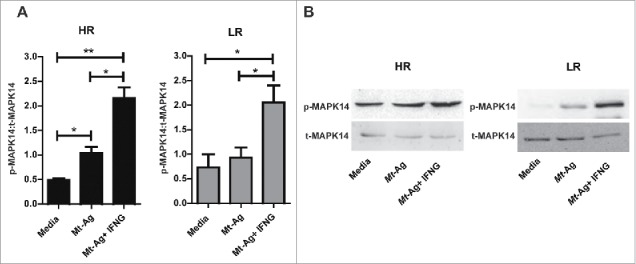
IFNG promotes MAPK14 phosphorylation in monocytes from HR and LR TB patients. Adherent cells from HR and LR TB patients were stimulated with sonicated M. tuberculosis (Mt- Ag, 10 µg/ml) with or without recombinant IFNG (1.8 ng/ml) for 24 h. Phosphorylated and total MAPK14 (p-MAPK14 and t-MAPK14, respectively) expressions were then measured by western blot. (A) Densitometry of the blots was performed, and the ratios of p-MAPK14 to t-MAPK14 protein expression was expressed as arbitrary units ± SEM. (B) Results from a representative HR and a LR TB patient are shown. *P < 0.05. P values were calculated using one-way ANOVA with the post hoc Tukey multiple comparisons test.
Role of IL17A during autophagy of monocytes from TB patients infected with M. tuberculosis strains
IFNG plays a crucial role in immunity to tuberculosis but although it is necessary, it is not sufficient for protection against tuberculosis.30 Our present and previous data demonstrate that autophagy against M. tuberculosis can be increased by IFNG.26 We next investigated the role of IL17A, another cytokine that participates in the immune response of the host against tuberculosis infection. Besides, it has been reported that during the chronic phase of tuberculosis, a balance between Th1 and Th17 responses is required to control bacterial growth and limit immunopathology.31 Some reports have analyzed the role of IL17A on the autophagy process, with diverse results in different cell types.32-35 However, to our knowledge, there are no reports showing the role of IL17A on autophagy during human active tuberculosis. Therefore, we analyzed whether IL17A might influence the autophagy process in monocytes from patients with active tuberculosis infected with M. tuberculosis strains. Figure 4A shows that addition of exogenous IL17A positively regulates the percentages of CD14+ LC3A,B-II+ cells in HD and HR tuberculosis patients upon Mt-Ag stimulation, but we observed no effect on the levels of autophagy in cells from LR tuberculosis patients. When adherent cells from the 3 groups of individuals were infected either with the pathogenic Mt H37Rv or nonpathogenic MtΔRD1 strains, similar levels of autophagy were detected in HD and tuberculosis patients (Fig. 4B, C and D). However, and in sharp contrast to our findings with IFNG (Fig. 2), exogenous IL17A did not increase either LC3 puncta or percentages of CD14+ LC3A,B-II+ cells in LR tuberculosis patients (Fig. 4). Importantly, treatment of adherent cells with IFNG or IL17A alone had no effect on the levels of autophagy in tuberculosis patients as detected by confocal microscopy (Fig. S3). Notably, we observed that both cytokines analyzed, IFNG and IL17A, induced autophagosome formation and that autophagy flux was functional (Fig. S4). Thus, these data indicate that both IFNG and IL17A positively modulate the autophagy process in M. tuberculosis infected cells from HD and HR tuberculosis patients, but only IFNG augmented autophagy in LR patients with the weakest response to the pathogen (Fig. 2 and Fig. S5).
Figure 4.

Modulation of autophagy by IL17A in monocytes infected with Mt H37Rv or MtΔRD1 from healthy donors and TB patients. PBMC from HD, HR TB and LR TB were incubated at 2 × 106 cells/ml for 16 h without stimulus to allow monocyte adherence. (A) Cells were then stimulated with or without sonicated M. tuberculosis antigen (Mt- Ag, 10 µg/ml) with or without recombinant IL17A (10 ng/ml) for 24 h. Autophagy levels were evaluated by flow cytometry against intracellular saponin-resistant LC3A,B-II on CD14+ cells. Bars represent the mean values of the percentage of CD14+ LC3A,B-II+ cells ± SEM. (B and C) Cells were incubated uninfected in media or infected with (B) Mt H37Rv or (C) MtΔRD1 (MOI: 20). After 2 h of infection, the culture medium was replaced and cells were cultured ± recombinant IL17A (10 ng/ml) for 24 h. Autophagy levels were evaluated by immunofluorescence against LC3 on monocytes. Bars represent the mean values of LC3 puncta per cell ± SEM. (D) Representative images of a HD, HR and LR TB patients are shown. Scale bars: 5 μm. *P < 0.05, **P < 0.01, ***P < 0.001. P values were calculated using one-way ANOVA with the post hoc Tukey multiple comparisons test.
Monocyte bactericidal activity is modulated by IFNG and IL17A in patients with active tuberculosis
It has been demonstrated that macrophage bactericidal activity is potentiated by IFNG stimulation36 and that IL17F significantly decreases the intracellular counts of Mycobacterium terrae in RAW macrophages.33 Thus, we next treated adherent cells from HR patients with the Mt H37Rv pathogenic strain or the MtΔRD1 mutant strain in the presence or absence of IFNG or IL17A, and bacterial survival in the monocytes was calculated on the basis of colony-forming units (CFU). As shown in Fig. 5A, an increase in bactericidal activity against the pathogen was observed in infected cells that had been treated with IFNG or IL17A. To confirm that the effect of both cytokines was related to autophagy induction, we blocked the autophagy process with the inhibitor 3-methyladenine (3-MA), which blocks autophagosome formation via the inhibition of class III phosphatidylinositol 3-kinase. As expected, we observed no differences between the CFU in cells infected with Mt HR37v and monocytes infected with the pathogen in the presence of the blocking reagent (Fig. 5B). However, the increase in the bactericidal effect of IFNG and IL17A on adherent cells of HR patients was only detected in the absence of the autophagy inhibitor, confirming our findings on the role of IFNG and IL17A on the modulation of autophagy in monocytes infected with live M. tuberculosis strains and its potential as a mycobactericidial mechanism.
Figure 5.
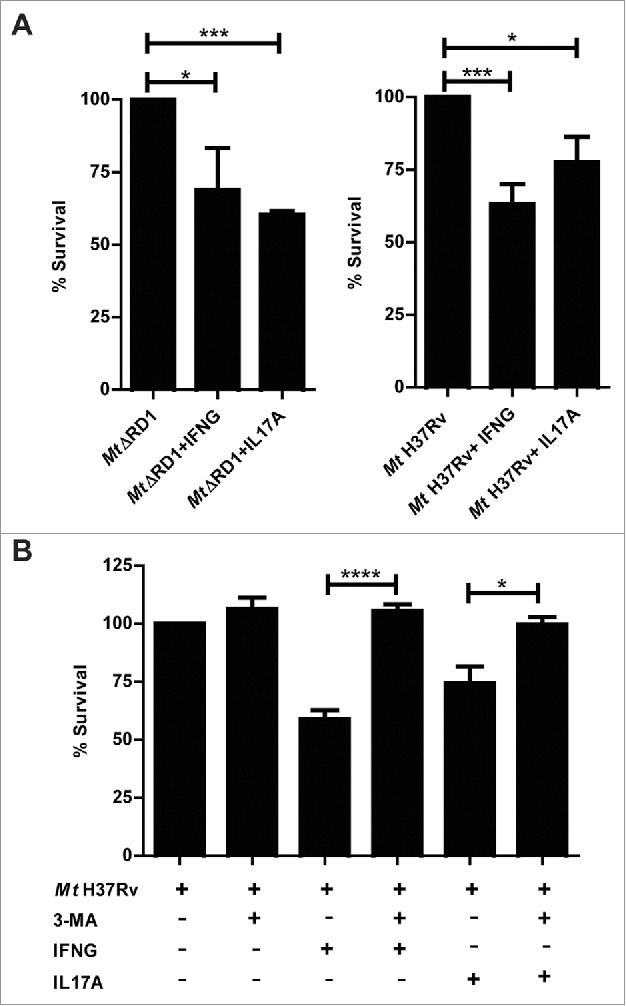
Intracellular survival of Mt H37Rv and MtΔRD1 in cytokine-treated monocytes from TB patients. PBMC from HR TB patients were incubated at 2 × 106 cells/ml for 16 h without stimulus to allow monocyte adherence. Cells were infected with MtΔRD1 or Mt H37Rv (MOI: 20). (A) After 2 h of infection, the culture medium was replaced and cells were cultured ± IFNG (1.8 ng/ml) or IL17A (10 ng/ml) for 24 h. In a separate set of experiments (B) cells were incubated with or without IFNG (1.8 ng/ml) or IL17A (10 ng/ml) for 24 h and 10 mM 3-methyladenine (3-MA) was added where indicated. Cells were washed and lysed for mycobacterial colony-forming units (CFU) determination. Data are presented as means of bacterial viability (CFU expressed as percentage of the control) ± SEM.*P < 0.05, **P < 0.01 and ***P < 0.001. P values were calculated using one-way ANOVA with the post hoc Tukey multiple comparisons test.
Signaling through IL17A regulates the modulation of autophagy in monocytes from tuberculosis patients
It has been reported that il17ra−/− mice are defective in exerting long-term control of M. tuberculosis infection.37 Moreover, it is known that intracellular signaling induced by IL17A strongly correlates with the surface expression of IL17RA (interleukin 17 receptor A) and, in contrast to most cytokine receptors, high levels of IL17RA are required for effective responses.38 Then, considering our present findings showing that IFNG upregulates the levels of autophagy in infected monocytes from tuberculosis patients, even in LR patients, and that IL17A augmented the percentage of CD14+ LC3A,B-II+ cells in HR patients but had no effect in the autophagy process on patients with weak response to M. tuberculosis, we wondered if there were differences in the levels of the expression of IL17RA in LR tuberculosis patients as compared with HR patients. As shown in Fig. 6A and B, analysis by flow cytometry of CD14+ monocytes, indicated that no differences were detected between HR and LR tuberculosis patients neither in the surface expression nor in the percentages of CD14+ IL17RA+ monocytes respectively. In addition, these results demonstrate for the first time that monocytes from patients with active tuberculosis express IL17RA.
Figure 6.
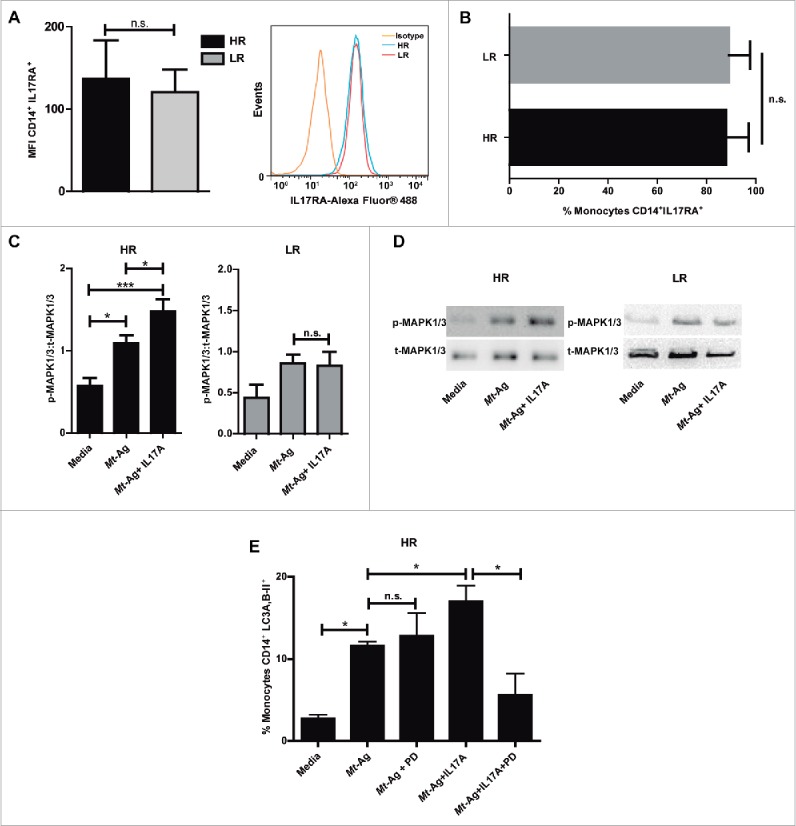
IL17 receptor signaling promotes autophagy through MAPK1/3 in high-responder TB patients. (A and B) IL17RA levels were evaluated by flow cytometry in CD14+ monocytes from HR TB and LR TB patients. (A) Bars represent the MFI (mean fluorescence intensity) ± SEM and a representative histogram of flow cytometry is shown. (B) Bars represents the percentage of CD14+ IL17RA+ cells ± SEM (C and D) Adherent cells from HR TB and LR TB were stimulated with sonicated M. tuberculosis antigen (Mt- Ag, 10 µg/ml) ± recombinant IL17A (10 ng/ml) for 24 h. Phosphorylated and total MAPK1/3 expressions were then measured by western blot. (C) Densitometry of the images was performed, and the ratios of p-MAPK1/3 to t-MAPK1/3 protein expression were expressed as arbitrary units. (D) Results from a representative HR and a LR TB patient are shown. (E) PBMC from HR TB were incubated with or without an inhibitor of activation of MAPK1/3 (PD98059 [PD], 50 μM) for 1 h and then stimulated with sonicated M. tuberculosis (Mt- Ag, 10 µg/ml) ± recombinant IL17A (10 ng/ml) for 24 h. Autophagy levels were evaluated by flow cytometry against intracellular saponin-resistant LC3A,B-II on CD14+ cells. Bars represent the mean values of the percentage of CD14+ LC3A,B-II+ cells ± SEM. *P < 0.05 and **P < 0.01. P values were calculated by the Mann-Whitney test for unpaired samples (A, B) and one-way ANOVA with the post hoc Tukey multiple comparisons test (D, E).
Therefore, we next analyzed whether IL17 signaling would be impaired in LR tuberculosis patients. Treatment of MKN28 cells with IL17A activates MAPK1/3.39 Moreover IL17A activates the phosphorylation of MAPK1/3 triggered by BCG (Bacillus Calmette-Guerin).40 Furthermore, autophagy against Listeria monocytogenes is dependent selectively on the MAPK1/3 pathway.41 Thus, we evaluated the phosphorylation of MAPK1/3 after Mt-Ag stimulation of HR and LR adherent cells. For this purpose, PBMC from tuberculosis patients were stimulated with Mt-Ag in the presence or absence of recombinant IL17A for 24 h. Afterwards, phosphorylated and total extracellular MAPK1/3 expression was measured by western blot. Figure 6C and D showed a clear increase in the levels of activated MAPK1/3 in HR tuberculosis patients. Moreover, these levels were significantly augmented by IL17A. In contrast, in LR tuberculosis patients, no differences in the phosphorylation of MAPK1/3 were detected upon stimulation of monocytes with Mt-Ag in the presence of IL17A (Fig. 6C and D), indicating an impaired IL17 signaling in patients with weak response to M. tuberculosis.
Finally, we treated PBMC from HR tuberculosis patients with or without PD98059, an specific inhibitor of activation of MAPK1/3, for 1 h. Then the cells were stimulated with Mt-Ag in the presence or absence of IL17A and autophagy levels were determined. Figure 6E clearly showed that inhibition of MAPK1/3 significantly diminished the percentage of CD14+ LC3A,B-II+ cells from HR tuberculosis patients, suggesting that MAPK1/3 participates in the signaling pathway of IL17A on the autophagy process against M. tuberculosis.
Taken together our current results suggest the participation of IFNG and IL17A in the activation of the autophagy process in patients with active disease as a mechanism to fight M. tuberculosis infection.
Discussion
Autophagy has been shown to be involved in host defense, cell survival or death and innate and adaptive immunity.42 Moreover, recent studies have revealed a potential role for autophagy in vaccine development for preventing acquisition or reactivation of latent tuberculosis, through enhanced immune activation.43,44 Furthermore, antituberculosis antibiotics positively activate autophagy,45 indicating a probable way to improve chemotherapy against M. tuberculosis. Thus, either as a potential successful vaccine or a complementary immunotherapy, efforts are needed to further elucidate the basic mechanisms of autophagy in immunity against mycobacteria, which will allow to achieve protective and therapeutic benefits in human tuberculosis.46 Therefore, considering that different studies have demonstrated the function of some cytokines in the activation and inhibition of autophagy,24,47,48,49 and that certain cytokines are crucial in the defense of the host against M. tuberculosis, here we investigated the effect of IFNG and IL17A on the modulation of autophagy in infected monocytes from patients with active tuberculosis.
We have previously demonstrated that autophagy in Mt-Ag-stimulated monocytes is regulated by Th1 responses against the pathogen.19 In the present report, we further analyzed the role of IFNG on autophagy during infection of human monocytes with M. tuberculosis strains. Thus, in HR and LR patients, we observed that IFNG increased the levels of autophagy in monocytes infected with the pathogenic strain M. tuberculosis H37Rv and with the nonpathogenic strain MtΔRD1 (Fig. 2). Apart from the well-established activity of IFNG on autophagy in animal models,17,24,50,51 it has been previously demonstrated that addition of recombinant IFNG alone increases the LC3-II levels in human monocytes.47 However, it is important to mention that those studies used 10 ng/ml of recombinant IFNG, whereas a considerable lower concentration of the cytokine was used in our study (1.8 ng/ml). Thus, this critical cytokine modulates positively the autophagic response affecting mycobacterial survival in tuberculosis patients regardless of their immune status.
Previously we have demonstrated that Mt-induced IFNG production by human T cells correlates with phosphorylation of the MAPKs, MAPK1/3 and MAPK14.52 Given that upon Mt-Ag stimulation we have detected a positive correlation between IFNG and LC3-II levels,26 we next investigated the phosphorylation of MAPK14 in monocytes from tuberculosis patients. IFNG increased phosphorylation of MAPK14 in both groups of tuberculosis patients, although marked higher levels of MAPK14 phosphorylation were observed in HR patients. These data are in close relationship with the levels of autophagy detected in patients with active disease upon stimulation with Mt-Ag (Fig. S2 and ref.26). It has been reported that both phosphatidylinositol 3-kinase (PIK3C3) and MAPK/p38 are required for IFNG induced activation of autophagy in macrophages in an IRGM-independent manner.29,36 Moreover, IFNG mediated autophagy via MAPK14, contributes to the ability of macrophages to kill intracellular bacteria.36 Therefore, given that autophagy participates in the defense against M. tuberculosis in association with specific IFNG secreted to the pathogen,26 the present findings extend those results on the participation of MAPK14 signaling pathway in this mechanism.
The role of IL17 on the autophagy process against M. tuberculosis had not been yet investigated in monocytes from patients with active tuberculosis. Here, we observed that addition of exogenous IL17A significantly increased CD14+ LC3A,B-II+ cells infected with mycobacteria in HD and HR patients. Interestingly, in contrast to our results with IFNG, IL17A was unable to modulate the autophagy process in tuberculosis patients with weak immune response to the pathogen (LR). The fact that IL17A did not regulate autophagy in severe LR tuberculosis patients highlights the important role of IL17 during the disease, participating in the increase of autophagy in individuals with strong immunity to M. tuberculosis. It is important to note that the antibody used for flow cytometry detects the LC3A isoform besides the LC3B isoform. Therefore, we cannot rule out the contribution of LC3A isoform to the autophagy process in our experiments. However, the results of both techniques (flow cytometry and confocal microscopy) tested are conclusive as to the induction of autophagy by IL17A.
Our data are consistent with previous observations that IL17A stimulates autophagy in B lymphocytes32 and that IL17A and IL17F promote autophagy in RAW 264.7 murine macrophages.33 Other studies have found that IL17 inhibits autophagy in lung epithelial cells34 and in hepatocellular carcinoma,35 reinforcing the notion that the role of IL17 on autophagy is cell-type- and cell-context-dependent. Our present work has extended the knowledge about the effect of IL17A on the immune response of patients with tuberculosis, emphasizing the induction of autophagy activity by IL17A in human monocytes infected with M. tuberculosis. We focused this work on the role of IL17A since we previously found that IL17A and IFNG expression in lymphocytes from tuberculosis patients correlate with the severity of the disease15 and that there is an association of the IL17A rs2275913 single nucleotide polymorphism with tuberculosis severity in Argentina.53 Nevertheless, it would be interesting to analyze the effect of other IL17 family members, especially IL17F, given that this cytokine is also a Th17 cytokine, shares a 55% of sequence homology with IL17A and the same receptor. The sequences of IL17B, IL17C, and IL17E differ substantially from those of IL17A and IL17F and bind distinct receptors, suggesting that these other IL17 family members may form a distinct subclass.54,55
Induction of autophagy overcomes the inhibition of phagosome-lysosome fusion by M. tuberculosis,56,57 promoting the maturation and acidification of mycobacteria containing compartments,17,24 and the incorporation of different components with enhanced antimycobacterial properties.18,58,59 We observed that IFNG and IL17A significantly increased the bactericidal activity of monocytes from infected HR tuberculosis patients, and that this effect was mostly related to autophagy. In RAW 264.7 macrophages, autophagy contributes to bactericidal activity at an early stage of IFNG stimulation against Listeria monocytogenes and intravacuolar Salmonella typhimurium.36 Moreover, our data are in agreement with the growing evidence that shows that IFNG-induced autophagy plays a protective role during M. tuberculosis infection.10,33 Besides, Orosz et al., have recently demonstrated that IL17A and IL17F induce autophagy in murine macrophages, contributing to the elimination of Mycobacterium terrae.30 Nevertheless, the effects of IL17 family members on autophagy had not been yet investigated in human monocytes and macrophages. Importantly, we observed an antibacterial activity triggered by IL17A that is autophagy-dependent, in contrast with the slight decrease in M. terrae survival detected in murine macrophages.30 Altogether, important cytokines that participate in the defense against M. tuberculosis activate the autophagic response affecting mycobacterial survival.
Recently, Kimmey et al. have used a genetic approach to elucidate the role for multiple ATG genes and the requirement for autophagy in resistance to M. tuberculosis infection in vivo,60 concluding that autophagic capacity in myeloid cells does not correlate with the outcome of M. tuberculosis infection. Nevertheless, other authors have demonstrated that excessive IL1 activation by autophagy-deficient macrophages leads to Th17 polarization as a likely contributor to neutrophil-associated effects in vivo.61 Besides, the studies by Kimmey and coworkers cover only the early stages of M. tuberculosis infection in a mouse model, whereas observations beyond the early 80 d are required in this model that inherently controls mycobacteria better than humans. It is important to point out that our experiments were performed entirely in human cells from patients with tuberculosis, and are in agreement with other reports, which demonstrate that activation of autophagy promotes mycobacteria elimination.17,24,61
The role of MAPK1/3 on the autophagy process has been described before.36 Furthermore, it has been previously reported that IL17 induced by BCG triggers the MAPK1/3 signaling cascade involving the phosphorylation of MAPK1/3.35 Here we investigated for the first time the influence of this signaling pathway induced by IL17A on the autophagy process of monocytes from tuberculosis patients. In HR tuberculosis patients, phosphorylation of MAPK1/3 was increased by IL17A, whereas no differences in the phosphorylation of these MAPK's were detected in LR tuberculosis patients. Moreover, our results clearly indicated that IL17A-induced autophagy was impaired in the presence of a MAPK1/3 inhibitor, confirming that MAPK1/3 participate in the role of IL17A on the positive modulation of autophagy in HR tuberculosis patients.
The so-called genomic region of difference (RD1) of M. tuberculosis is required for the full virulence of the bacteria and the loss of RD1 contributes to the attenuation of Mycobacterium bovis BCG.36,62 Then, considering the importance of the RD1 region in the immunopathology of tuberculosis, we performed our studies infecting cells from tuberculosis patients with the M. tuberculosis H37Rv strain and the nonpathogenic MtΔRD1 strain. Similar to our results after stimulation with Mt-Ag,19 MtΔRD1 significantly increased LC3 levels in the presence of BafA1. However, infection of cells with the Mt H37Rv strain in the presence of BafA1 did not modify LC3B-II levels, indicating that this strain blocks the autophagic pathway. These findings are in line with previous reports from Romagnoli et al. where the authors demonstrate in human primary dendritic cells that M. tuberculosis H37Rv impairs autophagy at the step of autophagosome-lysosome fusion, whereas neither the M. tuberculosis H37Ra nor BCG strains obstructed autophagosome maturation.63 Previously, Petruccioli et al. have demonstrated that M. tuberculosis inhibits autophagy in human primary macrophages, and that specific T cells can restore functional autophagic flux through cell-cell contact.64 Maintaining the whole cell populations together, we have used monocytes instead of macrophages to preserve the most physiological way to investigate the role of cytokines on autophagy. This model allows us to study the differences that already carry the cellular populations of HD and both groups of patients with tuberculosis. In line with our results, Yang et al. show that Mycobacterium leprae upregulates IRGM expression in both monocytes and monocyte-derived macrophages.65
Recent investigations have shown that LC3, previously considered as an exclusively autophagic marker, is also implicated in LC3-associated phagocytosis (LAP), although the LAP result differs depending on its cellular context. Importantly, most studies that describe LAP use murine cell lines or mouse models.66-69 Accordingly, Romao et al. described that the frequency of LC3-positive phagosomes seems to differ between species, with 40% and 80% of the total phagosomes in mouse but only 5% to 10% in human cells being LC3-associated.70 Because the present work was performed with human cells, it would be unlikely that LAP would alter our results.
Th1-Th17 lymphocytes are the main source of IL17A produced by PBMC from tuberculosis patients, in direct correlation with disease severity.15 Although previous findings point out that when IL17A levels are elevated and sustained in time, this cytokine might be harmful for the host, generating tissue damage and reducing IFNG levels, our present data on the role of IL17A in active tuberculosis let us to hypothesize a dual role for this cytokine, where moderate levels of IL17A would be required at early stages of M. tuberculosis infection to induce autophagy and recruit neutrophils and IFNG-secreting cells to activate macrophages and eliminate the pathogen.8-10 This could explain partially why in patients with weak immune response to M. tuberculosis, and poor immunological and clinical conditions, IL17A is elevated15 but unable to activate autophagy.
Thus, in the present work we demonstrated that 2 important cytokines that participate in the host immune defense during tuberculosis,31,9 IFNG and IL17A, activate autophagy in infected monocytes from patients with active disease, contributing to the elimination of the pathogen and promoting antimycobacterial immune defenses despite the ability of the bacteria to evade immune responses. Additionally, we extended our studies on T cells from HR and LR tuberculosis patients to infected monocytes from these individuals. We observed that monocytes from LR individuals were unable to activate autophagy through IL17A at least in part because of a defect in the MAPK1/3 signaling pathway. In contrast, both IFNG and IL17A increased the levels of autophagy in patients with strong immunity to M. tuberculosis. Therefore, our findings provide new insights about the role of autophagy in pathogen-host interactions during active tuberculosis, contributing to identify new targets and design novel therapeutic tools to fight this pathogen.
Materials and methods
Subjects
HIV-uninfected patients with tuberculosis were diagnosed at the “Dr. F Muniz” Hospital (Buenos Aires, Argentina), based on clinical and radiological data, together with the identification of acid-fast bacilli in sputum. All patients had received less than one wk of antituberculosis therapy, and were classified as high-responder (HR) or low responder patients (LR) as previously reported,28 based on in vitro lymphocyte responses to M. tuberculosis antigen. Briefly, HR patients are individuals displaying significant proliferative responses, IFNG production and an increased signaling lymphocyte activation molecule (SLAMF1) expression against the antigen; whereas LR patients exhibit low proliferative responses, IFNG production and SLAM expression. LR patients had more severe pulmonary disease, lower leukocyte counts, and a more prolonged illness, compared with HR individuals. In our patient population, no differences regarding age distribution, sex, ethnicity, or frequency of extra pulmonary forms of TB were found between HR and LR tuberculosis patients. However, significant differences were detected regarding X-ray radiography severity, leukocyte count, and time of disease evolution (days previous to hospital admission, during which, the patient displays clinical symptoms) (Table S1).15 As controls, we enrolled Bacillus Calmette-Guerin (BCG)-vaccinated healthy adults lacking a history of tuberculosis (Table S2). Individuals with latent infection were excluded following assessment using the QuantiFERON TB Gold In-tube test (Cellestis Ltd., T0590–0301 and 0594–0201). All participants provided a written, informed consent for the collection of samples and subsequent analysis. The protocols conducted in this work were approved by the Ethics Committee of the Muñiz Hospital.
Antigen
In vitro stimulation of cells was performed with a cell lysate from the virulent Mycobacterium tuberculosis strain H37Rv, prepared by probe sonication (Mt-Ag) (BEI Resources, NIAID, NIH: Mycobacterium tuberculosis, Strain H37Rv, Whole Cell Lysate, NR-14822).
Bacterial growth conditions
M. tuberculosis H37Rv (Mt H37Rv) and M. tuberculosis mc26230 (delta RD1 delta panCD) (MtΔRD1) were grown in Middlebrook 7H9 broth (BD, 271310) or on 7H10 agar (BD, 283810) with 0.5% Tween 20 (Biopack, 2003.08), 0.2% glycerol, and albumin-dextrose-catalase-oleic acid supplement (BD BBL™, 212351), and pantothenic acid for MtΔRD1. Cultures were harvested at exponential growing phase at 37°C. To disaggregate clumps, mycobacteria were sonicated at 2.5 W output for 4 min (Elma d-7700 Singentrans sonic), then centrifuged for 10 min at 300 x g and the supernatant diluted in phosphate-buffered saline (PBS; 1.37 mM NaCl, 27 mM KCl, 2 mM KH2PO4, 80 mM Na2H2PO4 anhydrous in water). In some cases, mycobacteria were incubated with Rhodamine (Sigma, R6626) for 30 min and then washed 2 times with PBS for 10 min at 300 x g. Finally, the OD at 600 nm was determined. Bacterial growth of Mt H37Rv in any experiment involving the pathogenic strain was performed in BSL3 security cabinets at the Malbran Institute, Buenos Aires, Argentina.
Human cell culture and infection
Peripheral blood mononuclear cells (PBMC) were isolated by centrifugation over Ficoll-Hypaque (GE Healthcare, 17–1440–03) and cultured (2 × 106 cells/mL) in flat-bottom 24- or 48-well plates with RPMI 1640 (Gibco, 22400–071) supplemented with L-glutamine (2 mM; Sigma, G5792), penicillin-streptomycin, and 10% fetal bovine serum (Gibco, 10437028) for 16 h without stimulus to allow monocyte adherence. Cells were then either stimulated with sonicated Mt (Mt-Ag, BEI Resources, NIH, NR-14822; 10 μg/ml) or infected with a RD1 deleted-Mt-strain (MtΔRD1) or with the Mt H37Rv (Mt H37Rv) pathogenic strain (MOI 20) for 2 h. Then, cells were washed twice with warm RPMI and cultured in complete media ± IFNG (1.8 ng/ml, eBioscience 14–8319) or IL17A (10 ng/ml, eBioscience, 14–8179) for 24 h. To determine the effect of different treatments on the autophagic flux, the vacuolar-type HC-ATPase inhibitor BafA1 (100 nM; Fermentek, 88899–55–2) was added for the last 2 h of culture before LC3 determination by immunofluorescence. In some experiments, cells were cultured with or without the MAPK1/3 inhibitor (PD98059, 50 μM; Calbiochem, 513000) for 1 h and stimulated with Mt-Ag for 24 h, after which autophagy levels were determined. PD98059 specifically inhibits activation of MAPK1/3 but does not affect activation of other related dual-specificity protein kinases or that of 18 other serine/threonine protein kinases.
Colony-forming unit assay
PBMC were infected with MtΔRD1 or Mt H37Rv for 2 h, washed with RPMI1640 (Gibco, 22400–071), and incubated for 24 h in the presence or absence of cytokines. Afterward, cells were washed 2 times with warm PBS and lysed with 0.05% Triton X-100 (Sigma, X100) in PBS. Serial dilution of adherent cells lysates was made, and 40-µl aliquots were inoculated (in duplicate) on Middlebrook 7H10 agar plates supplemented with oleic acid-albumin-dextrose-catalase or pantothenic acid in the case of MtΔRD1. Plates were incubated for 3 wk, and colonies were counted from dilutions yielding 10 to 100 visible colonies.
Flow cytometry
PBMC from tuberculosis patients and healthy donors were stimulated with Mt-Ag or infected with MtΔRD1 or Mt H37Rv as described above. Cells were then stained with specific fluorophore-marked antibodies against CD14 (Biolegend, 325608) and IL17RA (Alexa Fluor® 647, BG/hIL17RA; Biolegend, 340903). Intracellular staining of endogenous saponin-resistant LC3 was done as described by Eng KE et al.71 Briefly, PBMC were washed with PBS and then permeabilized with PBS containing 0.05% saponin (Sigma, 47036). In this protocol, the cells are not fixed, therefore LC3-I is washed out of the cell because, unlike LC3-II, it is not anchored to the autophagosome.9 Cells were then incubated with mouse anti-human LC3A,B-II antibody (MBL International, M152–3) for 20 min, rinsed with PBS, incubated with anti-mouse secondary antibody conjugated to phycoerythrin (eBioscience, 12–4012) for 20 min and rinsed twice more with PBS. Afterwards, cells were stained with an anti-CD14 antibody (Biolegend, 325608) to detect the monocyte population. Negative control samples were incubated with irrelevant isotype matched monoclonal antibody (eBioscience, 12–4717). Samples were analyzed on a FACSAria II flow cytometer (BD Biosciences, San José, CA, USA).
Western blot
Total cell protein extracts were prepared from adherent cells from PBMCs stimulated with Mt-Ag in presence or absence of IL17A or IFNG for 24 h. Western blotting was performed by standard methods. Each nitrocellulose membrane was blotted, stripped, and reblotted with rabbit monoclonal antibodies to phospho-MAPK1/3 (p-MAPK1/3 Tyr-204, Santa Cruz Biotechnology, sc-7383), total MAPK1/3 (Santa Cruz Biotechnology, sc-154), phospho-MAPK14 (p-MAPK14 Thr-180/Tyr-182, R&D Systems, MAB8691), and total MAPK14 (Santa Cruz Biotechnology, sc-535 g). Bound antibodies were revealed with horseradish peroxidase-conjugated anti-rabbit antibody (Bio-Rad, 170–6515) or anti-mouse antibody (BioRad, 170–6516) using ECL PLUS (Amersham Biosciences). Images were obtained with an Intelligent Dark Box (Fujifilm LAS1000, Tokio, Japan) and analyzed with ImageJ Analysis software. The intensity of each band was expressed in arbitrary units.
Confocal microscopy
Cells were cultured and stimulated or infected on coverslips for 24 h. After incubation under the different experimental conditions, cells were washed to remove nonadherent cells. Adherent cells were then fixed with cold methanol for 20 sec, then washed and subsequently permeabilized and blocked with blocking buffer (PBS containing 0.5% saponin (Santa Cruz Biotechnology, sc-280079A) and 1% bovine serum albumin (Santa Cruz Biotechnology, sc-2323A) for 15 min. The buffer was then removed and the anti-LC3B primary antibody (Cell Signaling Technology, 2775) was added. Anti-LC3B detects endogenous levels of total LC3B protein. Because it only appears to weakly cross-react with LC3A and LC3C at transfected levels, it is very doubtful that it cross-reacts with LC3A or LC3C to any appreciable extent at endogenous levels (unpublished data provided by the manufacturer). Cells were then incubated for 16 h at 4°C. Afterwards, cells were washed with blocking buffer and incubated with the secondary antibody (Alexa Fluor® 488 goat anti-rabbit IgG [HCL]; Invitrogen, A11008) for 2 h at room temperature. Finally, nuclei were stained with DAPI. The coverslips were mounted with PBS-glycerol (Sigma-Aldrich, G2025) and fixed cells were imaged using a Zeiss Spectral LSM 510 confocal microscope (Zeiss, Jena, Germany) using objective 63, numerical aperture (NA) 1.42. The number of LC3 puncta per cell was then quantified using ImageJ image analysis software.
Image processing
All the images were processed using ImageJ software (Wayne Rasband, National Institutes of Health). After the image binarization using a defined threshold, the puncta number was quantified using the Particle Analyzer plugin. Brightness and contrast was adjusted in all images belonging to the same individual, when needed.
Statistical analysis
Analysis of variance (ANOVA) and the post hoc Tukey multiple comparisons test were used as indicated in the figure legends. Wilcoxon rank sum test was used to analyze differences between unpaired samples. P values of < 0.05 were considered statistically significant.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- Ag
antigen
- BCG
Bacillus Calmette-Guerin
- CFU
colony-forming units
- HD
healthy donors
- HR
high responder (tuberculosis patient)
- IFNG
interferon gamma
- IL
interleukin
- IL17RA
interleukin 17 receptor A
- LR
low responder (tuberculosis patient)
- MAPK
mitogen-activated protein kinase
- MAPK1/ERK2
mitogen-activated protein kinase 1
- MAPK3/ERK1
mitogen-activated protein kinase 3
- MAPK14/p38 α
mitogen-activated protein kinase 14
- MAP1LC3/LC3
microtubule associated protein 1 light chain 3
- Mt
Mycobacterium tuberculosis
- MtΔRD1
RD1-deleted M. tuberculosis strain
- PBMC
peripheral blood mononuclear cells
- TB
tuberculosis
- Th
T helper
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are thankful to Dr. William R. Jacobs, Jr, from the Albert Einstein College of Medicine, for providing MtΔRD1 strain. We are grateful to Dr. Mariana Piuri, Liliana Rondón and Estafania Urdaniz for constant support. We are also thankful to Dr. Beatriz Lopez and Noemi Yokobori for their constant support and technical assistance.
Funding
This work received financial support from Agencia Nacional de Promoción Científica y Tecnológica (PICT 0240 and PICT 1762 to V. E. G.); the University of Buenos Aires (20020130100236BA to V.E.G.); and Consejo Nacional de Investigaciones Científicas y Tecnológicas (PIP2012–2014 to V.E.G.). N.L.T., N.O.A., M.I.C and V.E.G. are members of the Researcher Career of CONICET (Argentina).
References
- [1].WHO WHO. Global tuberculosis report 2016 2016.
- [2].Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med 1993; 178:2243-7; PMID:8245795; https://doi.org/ 10.1084/jem.178.6.2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to mycobacterium tuberculosis infection. J Exp Med 1993; 178:2249-54; PMID:7504064; https://doi.org/ 10.1084/jem.178.6.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, Levin M. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 1996; 335:1941-9; PMID:8960473; https://doi.org/ 10.1056/NEJM199612263352602 [DOI] [PubMed] [Google Scholar]
- [5].Gong JH, Zhang M, Modlin RL, Linsley PS, Iyer D, Lin Y, Barnes PF. Interleukin-10 downregulates mycobacterium tuberculosis-induced Th1 responses and CTLA-4 expression. Infect Immun 1996; 64:913-8; PMID:8641800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, Black G, Hassan HY, Wilkinson RJ, Walzl G, Gelderbloem SJ, et al.. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol 2008; 180:1962-70; PMID:18209095; https://doi.org/ 10.4049/jimmunol.180.3.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during mycobacterium tuberculosis infection. J Immunol 2006; 177:4662-9; PMID:16982905; https://doi.org/ 10.4049/jimmunol.177.7.4662 [DOI] [PubMed] [Google Scholar]
- [8].Etna MP, Giacomini E, Severa M, Coccia EM. Pro- and anti-inflammatory cytokines in tuberculosis: A two-edged sword in TB pathogenesis. Semin Immunol 2014; 26:543-51; PMID:25453229; https://doi.org/ 10.1016/j.smim.2014.09.011 [DOI] [PubMed] [Google Scholar]
- [9].Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, et al.. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during mycobacterium tuberculosis challenge. Nat Immunol 2007; 8:369-77; PMID:17351619; https://doi.org/ 10.1038/ni1449 [DOI] [PubMed] [Google Scholar]
- [10].Torrado E, Robinson RT, Cooper AM. Cellular response to mycobacteria: Balancing protection and pathology. Trends Immunol 2011; 32:66-72; PMID:21216195; https://doi.org/ 10.1016/j.it.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Khader SA, Cooper AM. IL-23 and IL-17 in tuberculosis. Cytokine 2008; 41:79-83; PMID:18218322; https://doi.org/ 10.1016/j.cyto.2007.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Okamoto Yoshida Y, Umemura M, Yahagi A, O'Brien RL, Ikuta K, Kishihara K, Hara H, Nakae S, Iwakura Y, Matsuzaki G. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol 2010; 184:4414-22; PMID:20212094; https://doi.org/ 10.4049/jimmunol.0903332 [DOI] [PubMed] [Google Scholar]
- [13].Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, Reinhart TA, Kolls J, Randall TD, Connell TD, et al.. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunol 2013; 6:972-84; PMID:23299616; https://doi.org/ 10.1038/mi.2012.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, Porcelli SA, Almo SC, Nathenson SG, Jacobs WR Jr. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U S A 2010; 107:13402-7; PMID:20624978; https://doi.org/ 10.1073/pnas.1007394107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jurado JO, Pasquinelli V, Alvarez IB, Pena D, Rovetta AI, Tateosian NL, Romeo HE, Musella RM, Palmero D, Chuluyán HE, et al.. IL-17 and IFN-gamma expression in lymphocytes from patients with active tuberculosis correlates with the severity of the disease. J Leukoc Biol 2012; 91:991-1002; PMID:22416258; https://doi.org/ 10.1189/jlb.1211619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cooper AM. Editorial: Be careful what you ask for: Is the presence of IL-17 indicative of immunity? J Leukoc Biol 2010; 88:221-3; PMID:20679070; https://doi.org/ 10.1189/jlb.0310146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753-66; PMID:15607973; https://doi.org/ 10.1016/j.cell.2004.11.038 [DOI] [PubMed] [Google Scholar]
- [18].Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, 4th Virgin HW, Kyei GB, Johansen T, Vergne I, et al.. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 2010; 32:329-41; PMID:20206555; https://doi.org/ 10.1016/j.immuni.2010.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sambandamurthy VK, Jacobs WR Jr. Live attenuated mutants of mycobacterium tuberculosis as candidate vaccines against tuberculosis. Microbes Infect 2005; 7:955-61; PMID:15914065; https://doi.org/ 10.1016/j.micinf.2005.04.001 [DOI] [PubMed] [Google Scholar]
- [20].Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, Lee SH, Cha GH, Kim JM, Lee ZW, et al.. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2012; 11:457-68; PMID:22607799; https://doi.org/ 10.1016/j.chom.2012.03.008 [DOI] [PubMed] [Google Scholar]
- [21].Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G, Codogno P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 2006; 281:30373-82; PMID:16857678; https://doi.org/ 10.1074/jbc.M602097200 [DOI] [PubMed] [Google Scholar]
- [22].Goletti D, Petruccioli E, Romagnoli A, Piacentini M, Fimia GM. Autophagy in Mycobacterium tuberculosis infection: A passepartout to flush the intruder out? Cytokine Growth Factor Rev 2013; 24:335-43; PMID:23395260; https://doi.org/ 10.1016/j.cytogfr.2013.01.002 [DOI] [PubMed] [Google Scholar]
- [23].Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, et al.. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280:20722-9; PMID:15778222; https://doi.org/ 10.1074/jbc.M413934200 [DOI] [PubMed] [Google Scholar]
- [24].Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular mycobacterium tuberculosis. Immunity 2007; 27:505-17; PMID:17892853; https://doi.org/ 10.1016/j.immuni.2007.09.003 [DOI] [PubMed] [Google Scholar]
- [25].Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, Park CG, Chun T. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol 2011; 48:720-7; PMID:21095008; https://doi.org/ 10.1016/j.molimm.2010.10.020 [DOI] [PubMed] [Google Scholar]
- [26].Rovetta AI, Pena D, Hernandez Del Pino RE, Recalde GM, Pellegrini J, Bigi F, Musella RM, Palmero DJ, Gutierrez M, Colombo MI, et al.. IFNG-mediated immune responses enhance autophagy against mycobacterium tuberculosis antigens in patients with active tuberculosis. Autophagy 2014; 10:2109-21; PMID:25426782; https://doi.org/ 10.4161/15548627.2014.981791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Man N, Chen Y, Zheng F, Zhou W, Wen LP. Induction of genuine autophagy by cationic lipids in mammalian cells. Autophagy 2010; 6:289-54; PMID:20383065; https://doi.org/ 10.4161/auto.6.4.11612 [DOI] [PubMed] [Google Scholar]
- [28].Pasquinelli V, Quiroga MF, Martinez GJ, Zorrilla LC, Musella RM, Bracco MM, Belmonte L, Malbrán A, Fainboim L, Sieling PA, et al.. Expression of signaling lymphocytic activation molecule-associated protein interrupts IFN-gamma production in human tuberculosis. J Immunol 2004; 172:1177-85; PMID:14707094; https://doi.org/ 10.4049/jimmunol.172.2.1177 [DOI] [PubMed] [Google Scholar]
- [29].Matsuzawa T, Kim BH, Shenoy AR, Kamitani S, Miyake M, Macmicking JD. IFN-gamma elicits macrophage autophagy via the p38 MAPK signaling pathway. J Immunol 2012; 189:813-8; PMID:22675202; https://doi.org/ 10.4049/jimmunol.1102041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smith SG, Lalor MK, Gorak-Stolinska P, Blitz R, Beveridge NE, Worth A, McShane H, Dockrell HM. Mycobacterium tuberculosis PPD-induced immune biomarkers measurable in vitro following BCG vaccination of UK adolescents by multiplex bead array and intracellular cytokine staining. BMC Immunol 2010; 11:35; PMID:20609237; https://doi.org/ 10.1186/1471-2172-11-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Torrado E, Cooper AM. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev 2010; 21:455-62; PMID:21075039; https://doi.org/ 10.1016/j.cytogfr.2010.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yuan J, Yu M, Li HH, Long Q, Liang W, Wen S, Wang M, Guo HP, Cheng X, Liao YH. Autophagy contributes to IL-17-induced plasma cell differentiation in experimental autoimmune myocarditis. Int Immunopharmacol 2014; 18:98-105; PMID:24269624; https://doi.org/ 10.1016/j.intimp.2013.11.008 [DOI] [PubMed] [Google Scholar]
- [33].Orosz L, Papanicolaou EG, Seprenyi G, Megyeri K. IL-17A and IL-17F induce autophagy in RAW 264.7 macrophages. Biomed Pharmacother 2016; 77:129-34; PMID:26796276; https://doi.org/ 10.1016/j.biopha.2015.12.020 [DOI] [PubMed] [Google Scholar]
- [34].Liu H, Mi S, Li Z, Hua F, Hu ZW. Interleukin 17A inhibits autophagy through activation of PIK3CA to interrupt the GSK3B-mediated degradation of BCL2 in lung epithelial cells. Autophagy 2013; 9:730-42; PMID:23514933; https://doi.org/ 10.4161/auto.24039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhou Y, Wu PW, Yuan XW, Li J, Shi XL. Interleukin-17A inhibits cell autophagy under starvation and promotes cell migration via TAB2/TAB3-p38 mitogen-activated protein kinase pathways in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci 2016; 20:250-63; PMID:26875893 [PubMed] [Google Scholar]
- [36].Matsuzawa T, Fujiwara E, Washi Y. Autophagy activation by interferon-gamma via the p38 mitogen-activated protein kinase signalling pathway is involved in macrophage bactericidal activity. Immunology 2014; 141:61-9; PMID:24032631; https://doi.org/ 10.1111/imm.12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Freches D, Korf H, Denis O, Havaux X, Huygen K, Romano M. Mice genetically inactivated in interleukin-17A receptor are defective in long-term control of mycobacterium tuberculosis infection. Immunology 2013; 140:220-31; PMID:23721367; https://doi.org/ 10.1111/imm.12130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem 2006; 281:24138-48; PMID:16798734; https://doi.org/ 10.1074/jbc.M604597200 [DOI] [PubMed] [Google Scholar]
- [39].Sebkova L, Pellicano A, Monteleone G, Grazioli B, Guarnieri G, Imeneo M, Pallone F, Luzza F. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in helicobacter pylori-infected human gastric epithelial cells. Infect Immun 2004; 72:5019-26; PMID:15321994; https://doi.org/ 10.1128/IAI.72.9.5019-5026.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fang JW, Li JC, Au KY, Yim HC, Lau AS. Interleukin-17A differentially modulates BCG induction of cytokine production in human blood macrophages. J Leukoc Biol 2011; 90:333-41; PMID:21521755; https://doi.org/ 10.1189/jlb.0510311 [DOI] [PubMed] [Google Scholar]
- [41].Anand PK, Tait SW, Lamkanfi M, Amer AO, Nunez G, Pages G, Pouysségur J, McGargill MA, Green DR, Kanneganti TD. TLR2 and RIP2 pathways mediate autophagy of listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J Biol Chem 2011; 286:42981-91; PMID:22033934; https://doi.org/ 10.1074/jbc.M111.310599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xu Y, Eissa NT. Autophagy in innate and adaptive immunity. Proc Am Thorac Soc 2010; 7:22-8; PMID:20160145; https://doi.org/ 10.1513/pats.200909-103JS [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Buffen K, Oosting M, Quintin J, Ng A, Kleinnijenhuis J, Kumar V, van de Vosse E, Wijmenga C, van Crevel R, Oosterwijk E, et al.. Autophagy controls BCG-induced trained immunity and the response to intravesical BCG therapy for bladder cancer. PLoS Pathog 2014; 10:e1004485; PMID:25356988; https://doi.org/ 10.1371/journal.ppat.1004485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ni Cheallaigh C, Keane J, Lavelle EC, Hope JC, Harris J. Autophagy in the immune response to tuberculosis: Clinical perspectives. Clin Exp Immunol 2011; 164:291-300; PMID:21438870; https://doi.org/ 10.1111/j.1365-2249.2011.04381.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, Lee SH, Cha GH, Kim JM, Lee ZW, et al.. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2013; 11:457-68; PMID:22607799; https://doi.org/ 10.1016/j.chom.2012.03.008 [DOI] [PubMed] [Google Scholar]
- [46].Jo EK. Autophagy as an innate defense against mycobacteria. Pathog Dis 2013; 67:108-18; PMID:23620156; https://doi.org/ 10.1111/2049-632X.12023 [DOI] [PubMed] [Google Scholar]
- [47].Duan L, Yi M, Chen J, Li S, Chen W. Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3. Biochem Biophys Res Commun 2016; 473:1229-34; PMID:27079235; https://doi.org/ 10.1016/j.bbrc.2016.04.045 [DOI] [PubMed] [Google Scholar]
- [48].Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during mycobacterium tuberculosis infection. Immunity 2011; 35:1023-34; PMID:22195750; https://doi.org/ 10.1016/j.immuni.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al.. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; https://doi.org/ 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- [50].Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, et al.. Ubiquilin 1 Promotes IFN-gamma-Induced xenophagy of mycobacterium tuberculosis. PLoS Pathog 2015; 11:e1005076; PMID:26225865; https://doi.org/ 10.1371/journal.ppat.1005076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang D, Bao L. Mycobacterium tuberculosis EspB protein suppresses interferon-gamma-induced autophagy in murine macrophages. J Microbiol Immunol Infect 2014; 49(6):859-865. [DOI] [PubMed] [Google Scholar]
- [52].Pasquinelli V, Rovetta AI, Alvarez IB, Jurado JO, Musella RM, Palmero DJ, Malbrán A, Samten B, Barnes PF, García VE. Phosphorylation of mitogen-activated protein kinases contributes to interferon gamma production in response to mycobacterium tuberculosis. J Infect Dis 2013; 207:340-50; PMID:23125442; https://doi.org/ 10.1093/infdis/jis672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rolandelli A, Hernandez Del Pino RE, Pellegrini JM, Tateosian NL, Amiano NO, de la Barrera S, Casco N, Gutiérrez M, Palmero DJ, García VE. The IL-17A rs2275913 single nucleotide polymorphism is associated with protection to tuberculosis but related to higher disease severity in Argentina. Sci Rep 2017; 7:40666; PMID:28098168; https://doi.org/ 10.1038/srep40666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008; 28:454-67; PMID:18400188; https://doi.org/ 10.1016/j.immuni.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity 2011; 34:149-62; PMID:21349428; https://doi.org/ 10.1016/j.immuni.2011.02.012 [DOI] [PubMed] [Google Scholar]
- [56].Vergne I, Fratti RA, Hill PJ, Chua J, Belisle J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: Mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell 2004; 15:751-60; PMID:14617817; https://doi.org/ 10.1091/mbc.E03-05-0307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hart PD, Young MR, Gordon AH, Sullivan KH. Inhibition of phagosome-lysosome fusion in macrophages by certain mycobacteria can be explained by inhibition of lysosomal movements observed after phagocytosis. J Exp M 1987; 166:933-46; PMID:3309128; https://doi.org/ 10.1084/jem.166.4.933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, Lee ZW, Lee SH, Kim JM, Jo EK. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009; 6:231-43; PMID:19748465; https://doi.org/ 10.1016/j.chom.2009.08.004 [DOI] [PubMed] [Google Scholar]
- [59].Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, et al.. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med 2011; 3:104ra2; PMID:21998409; https://doi.org/ 10.1126/scitranslmed.3003045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015; 528:565-9; PMID:26649827; https://doi.org/ 10.1038/nature16451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Deretic V. Autophagy in leukocytes and other cells: Mechanisms, subsystem organization, selectivity, and links to innate immunity. J Leukoc Biol 2016; 100:969-78; PMID:27493243; https://doi.org/ 10.1189/jlb.4MR0216-079R [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 2002; 46:709-17; PMID:12410828; https://doi.org/ 10.1046/j.1365-2958.2002.03237.x [DOI] [PubMed] [Google Scholar]
- [63].Romagnoli A, Etna MP, Giacomini E, Pardini M, Remoli ME, Corazzari M, Falasca L, Goletti D, Gafa V, Simeone R, et al.. ESX-1 dependent impairment of autophagic flux by mycobacterium tuberculosis in human dendritic cells. Autophagy 2012; 8:1357-70; PMID:22885411; https://doi.org/ 10.4161/auto.20881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Petruccioli E, Romagnoli A, Corazzari M, Coccia EM, Butera O, Delogu G, Piacentini M, Girardi E, Fimia GM, Goletti D, et al.. Specific T cells restore the autophagic flux inhibited by Mycobacterium tuberculosis in human primary macrophages. J Infect Dis 2012; 205:1425-35; PMID:22457295; https://doi.org/ 10.1093/infdis/jis226 [DOI] [PubMed] [Google Scholar]
- [65].Yang D, Chen J, Zhang L, Cha Z, Han S, Shi W, Ding R, Ma L, Xiao H, Shi C, et al.. Mycobacterium leprae upregulates IRGM expression in monocytes and monocyte-derived macrophages. Inflammation 2014; 37:1028-34; PMID:24469081; https://doi.org/ 10.1007/s10753-014-9825-1 [DOI] [PubMed] [Google Scholar]
- [66].Akoumianaki T, Kyrmizi I, Valsecchi I, Gresnigt MS, Samonis G, Drakos E, Boumpas D, Muszkieta L, Prevost MC, Kontoyiannis DP, et al.. Aspergillus cell wall melanin blocks LC3-Associated phagocytosis to promote pathogenicity. Cell Host Microbe 2016; 19:79-90; PMID:26749442; https://doi.org/ 10.1016/j.chom.2015.12.002 [DOI] [PubMed] [Google Scholar]
- [67].Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 2011; 108:17396-401; PMID:21969579; https://doi.org/ 10.1073/pnas.1113421108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, et al.. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for rubicon, NOX2 and autophagy proteins. Nat Cell Biol 2015; 17:893-906; PMID:26098576; https://doi.org/ 10.1038/ncb3192 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [69].Li X, Prescott M, Adler B, Boyce JD, Devenish RJ. Beclin 1 is required for starvation-enhanced, but not rapamycin-enhanced, LC3-associated phagocytosis of burkholderia pseudomallei in RAW 264.7 cells. Infect Immun 2013; 81:271-7; PMID:23115045; https://doi.org/ 10.1128/IAI.00834-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Romao S, Munz C. LC3-associated phagocytosis. Autophagy 2014; 10:526-8; PMID:24413059; https://doi.org/ 10.4161/auto.27606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Eng KE, Panas MD, Karlsson Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy 2010; 6:634-41; PMID:20458170; https://doi.org/ 10.4161/auto.6.5.12112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.