

Abstract
Neuropsychiatric symptom development has become more prevalent with 270,000 blast exposures occurring in the past 10 years in the United States. How blast injury leads to neuropsychiatric symptomology is currently unknown. Preclinical models of blast-induced traumatic brain injury have been used to demonstrate blood-brain barrier disruption, degenerative pathophysiology, and behavioral deficits. Vascular injury is a primary effect of neurotrauma that can trigger secondary injury cascades and neurodegeneration. Here we present data from a novel scaled and clinically relevant mouse blast model that was specifically developed to assess the outcome of vascular injury. We look at the biochemical effects and behavioral changes associated with blast injury in young-adult male BALB/c mice. We report that blast exposure causes focal vascular injury in the Somatosensory Barrel Field cortex, which leads to perivascular astrocyte reactivity, as well as acute aberrant behavior. Biochemical analysis revealed that mild blast exposure also invokes tauopathy, neuroinflammation, and oxidative stress. Overall, we propose our model to be used to evaluate focal blood-brain barrier disruption and to discover novel therapies for human neuropsychiatric symptoms.
Keywords: Traumatic Brain Injury, Blood-Brain Barrier Disruption, Astrocyte Reactivity, Tauopathy, Neuropsychiatric Behavior
1. Introduction
Neurotrauma causes serious adverse symptoms in soldiers and has been linked to behavioral deficits (1). Recent evidence suggests that blood-brain barrier (BBB) disruption exacerbates the initial injury and increases inflammation following neurotrauma (2). BBB disruption can lead to robust oxidative stress (3), and contribute to persistent inflammation (4). What is unknown is the relationship between BBB disruption and subsequent gliosis and tauopathy.
Gliosis following TBI can disrupt important signaling cascades (5), and may account for the progression of neurodegenerative pathology. Herein, we show evidence from our clinically relevant mouse blast model that TBI causes focal BBB disruption, oxidative stress, and neuroinflammation in the somatosensory barrel field cortex (S1BF). The S1BF is important because it is linked to temporal behavioral responses and can become disrupted after injury (6). Furthermore, it has been shown to be a region susceptible to gliosis following TBI (7). In this paper, we show convincing evidence that vascular disruption and the subsequent biochemical events may contribute to a perivascular distribution of reactive astrocytes, as well as acute aberrant behavior.
An important component of this cascade is tauopathy. We have recently shown that tau conformational change occurs in a perivascular distribution (8). Kayed et al. recently reported tau oligomer formation, neuronal cell death and cognitive deficits in rodents after TBI (9). In this paper, we also show tau oligomer formation correlated with focal BBB disruption in the S1BF cortex. Furthermore, acute behavioral deficits were observed when mice were subjected to the elevated plus maze (EPM). Elucidating a link between vascular disruption, tauopathy, and neuropsychiatric disorders requires further investigation to discover potential treatment options.
We present Friedlander blast wave curves generated by our model in order to discuss possible mechanical causes of the observed injury changes. The impulse of the Friedlander wave is important for understanding depth of penetration into tissue (10). Our model is unique in that we can produce a high peak overpressure over a very short duration compared to other blast injury models (10). Other rodent models of blast injury have linked tauopathy with aberrant behavior (11), and vascular injury with oxidative stress and neuroinflammation (12). Going forward we will use the clinically relevant table-top mouse model in order to further investigate the molecular underpinnings of neurotrauma related neurodegeneration with a focus on developing innovative treatment solutions.
2. Results
2.1. Mouse Blast Model and Experimental Design
We designed a shock tube model for mice (Figure 1A and 1B) by scaling down our validated rat model (10). The new model has a smaller diameter (4 cm) and a longer driven (15 cm) to driver section (9 cm) ratio to allow conformity of the wave upon exit. We subjected mice to a single mild blast (~ 20 psi, 0.5 ms; Figure 1C) and euthanized at 24 and 72 hours. The brains were subsequently assessed for acute vascular injury, glial reactivity, and tauopathy. Prior to sacrifice the mice were assessed for aberrant behavior. The experimental design is portrayed in Figure 1D.
Figure 1. Mouse Blast Model, Freidlander Curve and Experimental Design.
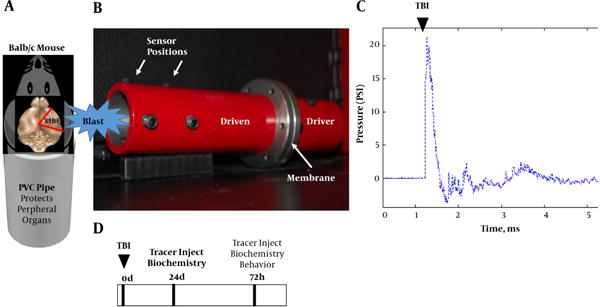
A, schematic shows mouse placed into PVC pipe with the blast wave impacting the right side of the head (S1BF cortex); B, photograph of mouse shock tube model; C, classic Freidlander curve produced by the blast wave with a peak overpressure of ~ 20 psi for a duration of 0.5 ms; D, experimental design.
2.2. Focal Vascular Disruption and Astrogliosis Observed After Blast
We peripherally injected 3 kDa texas red dye (TRD) to image vascular disruption after blast exposure, and subsequently perfused with indocyanine green (ICG) to label blood vessels. Moreover, TRD and ICG are only circulated for a relatively small amount of time compared to more typical Evan’s blue and give a more accurate acute depiction of clinical BBB physiology (13). We observed TRD distribution (Red) in the third and lateral ventricles, but also in the ipsilateral S1BF cortex, outlined by the white box, for all five mice at 72 hours post-blast (Figure 2A). No significant changes were seen at 24 hours (data not shown). We also found the 50 kDa ICG to distribute in a similar pattern to TRD after blast injury (Figure 2A, bottom). TRD distribution in the S1BF cortex seemed highest at 72 hours post-blast (Figure 2B). Astrocyte immune-reactivity was also observed in the S1BF cortex of slides adjacent to Figure 2B (Figure 2C). These data suggest that blast causes BBB disruption and may indicate the release of pro-inflammatory factors.
Figure 2. Blast Induced Blood-Brain Barrier Disruption in the S1BF Cortex.
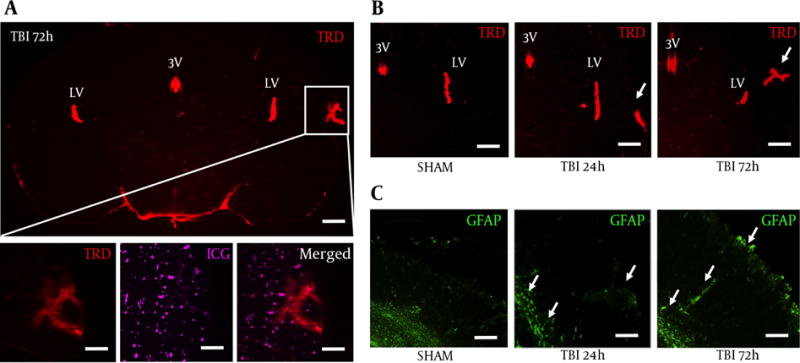
A, a coronal brain slice imaged at 1.25× shows Texas Red conjugated dextran 3 kDa distribution within the S1BR region (outlined by white box) at 72 hours post-blast; scale bars = 500 μm; B, Bottom panels are from white box above imaged at 6.3× to show vascular disruption: TRD distribution (left), ICG distribution (middle), merged (right); scale bars = 100μm. Brain slices imaged at 3.2x show increased TRD distribution in the S1BF region at 72 hour post-blast compared to SHAM and 24 hours post-blast animals; Scale bars = 200 μm. Arrows demarcate vascular disruption within the ipsilateral S1BF region; C, adjacent brain slices imaged at 3.2× show increased GFAP fluorescence in the S1BF region at 72 hour post-blast compared to SHAM and 24 hours post-blast animals; scale bars = 200 μm. Arrows demarcate astrocyte reactivity within the ipsilateral S1BF region and near grey/white matter junctions; LV = lateral ventricle; 3V = third ventricle; TRD = texas red dextran; ICG = indocyanine green; GFAP = glial fibrillary acidic protein.
2.3. Blood-Brain Barrier Disruption Observed in the S1BF Cortex After Blast
TRD distribution was observed in a diffuse pattern within the S1BF cortex of TBI mice at 72 hours post-blast (Figure 3A). Interestingly, under high magnification, we observed 3 kDa TRD (Figure 3B) to distribute in a similar pattern to 50 kDa ICG-albumin (Figure 3C) as outlined by the white box in Figure 3A. TRD distribution was observed in a punctate pattern within blood vessels and brain ventricles of SHAM mice (Figure 3D). The white box outlines punctate TRD (Figure 3E) and ICG (Figure 3F) within intact brain vessels of SHAM mice.
Figure 3. Vascular Injury Observed in the Ipsilateral S1BF Cortex After Blast.
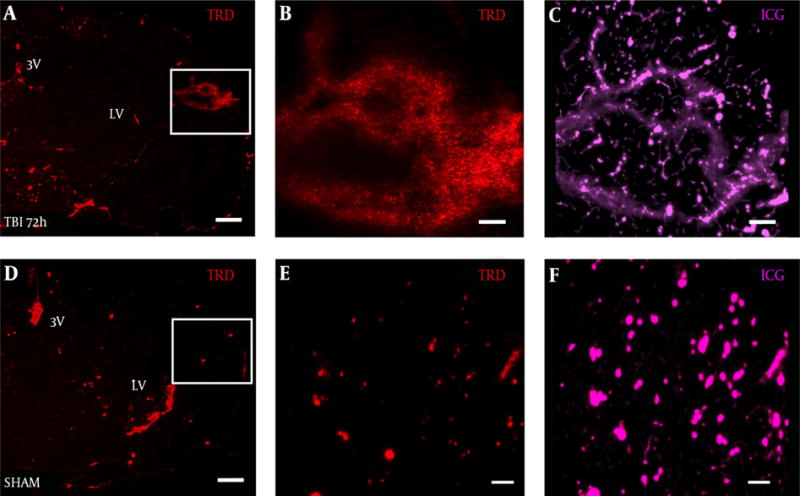
Brain slice from a A, TBI mouse shows TRD distribution in the S1BF region compared to D, SHAM mouse brain slice; panels A, D both imaged at 2.5×, scale bar = 200 μm. White box in panel A, outlines rightward panels showing vascular disruption with B, TRD distributed in a similar pattern to C, ICG-albumin; White box in panel D, outlines rightward panels showing intact blood vessels E, with TRD and F, ICG-albumin; panels B, C, E, F, all imaged at 12.6×, scale bars = 50 μm. TRD = Texas red dextran; ICG = indocyanine green.
2.4. Robust Astrocyte Reactivity Observed in the S1BF Cortex After Blast
In a slide adjacent to Figure 3A, robust astrocyte reactivity was observed in the S1BF cortex of blast mice at 72 hours (Figure 4A). The white box in Figure 4A outlines the S1BF region showing robust GFAP staining after blast (Figure 4B). The white box in Figure 4B outlines the swelling of an astrocytic process after blast (Figure 4C). In a slide adjacent to Figure 3D, weak GFAP staining was observed in the S1BF cortex of SHAM mice (Figure 4D). The white box in Figure 4D outlines the S1BF region showing weak GFAP staining in SHAM mice (Figure 4E). The white box in Figure 4E outlines the weak GFAP staining of astrocytic processes in SHAM mice (Figure 4F).
Figure 4. Robust Astrocyte Reactivity Observed in the Ipsilateral S1BF Cortex After Blast.
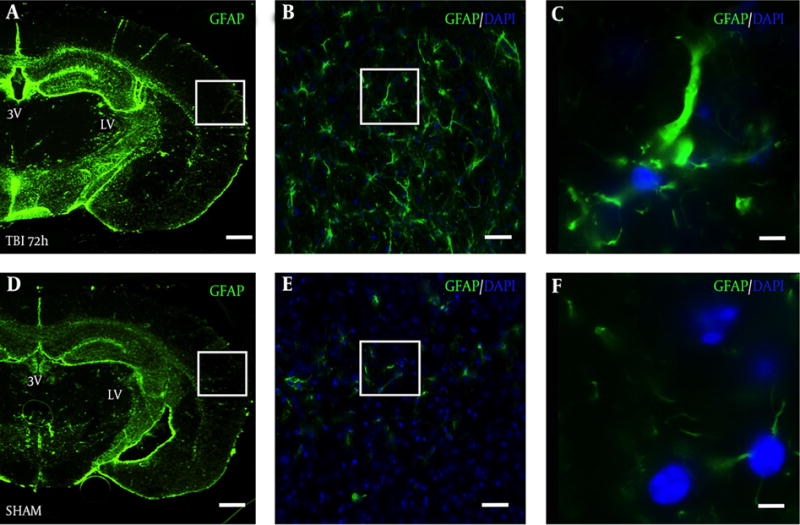
Brain slice from a TBI mouse adjacent to the TRD slices from Figure 3A shows robust GFAP staining in the A, S1BF region compared to an adjacent brain slice from Figure 3D from D, a SHAM mouse; A and D, both imaged at 2.5×, scale bar = 200 μm; White box in panel A outlines panel B showing astrocyte reactivity with GFAP at 20×, B, scale bar = 30 μm; white box in panel B outlines panel C showing robust astrocytic processes at 63×, C, scale bar =10 μm; White box in panel D outlines panel E showing weak GFAP staining at 20x, E, scale bar = 30 μm; white box in panel E outlines panel F showing weak GFAP staining at 63×, F, scale bar =10 μm. GFAP = glial fibrillary acidic protein; DAPI = 4′,6-diamidino-2-phenylindole (nuclear stain).
2.5. Blast Exposure Increased GFAP Expression in the S1BF Cortex
A significant difference in GFAP mRNA abundance was observed in the ipsilateral S1BF region after blast (F (2, 12) = 5.73; P < 0.05). We revealed a significant increase in GFAP mRNA abundance at 24 hours post-blast when compared to control animals (q = 3.36; P < 0.05) (Figure 5A). One-way ANOVA with Dunnetts post-hoc values represented as mean ± sem. A significant difference in protein expression of GFAP was observed in the ipsilateral S1BF region after blast (F (2, 12) = 4.42; P < 0.05). We measured a significant increase in GFAP protein expression at 72 hours post-blast when compared to control animals (q = 2.68; P < 0.05) (Figure 5B). One-way ANOVA with Dunnetts post-hoc values represented as mean ± sem. The limited GFAP expression at 24 hours post-blast, but substantially increased gene expression is consistent with previous data from our rat model (10), where peak protein expression was also observed at 72 hours post-blast. Under high magnification, we also observed reactive astrocytes along ICG-labeled blood vessels within the S1BF region of mice at 72 hours after blast exposure (Figure 5C).
Figure 5. Increased GFAP Expression Measured Following Blast Injury.
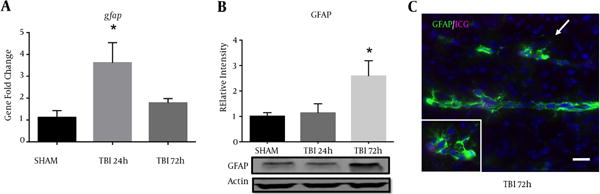
A, a significant increase in GFAP mRNA abundance was measured in the ipsilateral S1BF region at 24 hours post-blast (*P < 0.05 vs SHAM); B, one-way ANOVA, Dunnetts post-hoc. Mean ± sem. n = 5. A significant increase in GFAP protein expression was also observed in the ipsilateral S1BF region at 72 hours post-blast (*P < 0.05). One-way ANOVA, Dunnetts post-hoc. Mean ± sem. n = 5. Image from a TBI brain slice shows blood vessels (marked by residual ICG-albumin after washout) surrounded by robust astrocyte reactivity (GFAP); imaged at 20x, scale bar = 30 μm. Arrow demarcates inset; imaged at 63x.
2.6. Markers of Oxidative Stress and Neuroinflammation Increased After Blast
Oxidative stress is an important component of the inflammasome, which contributes to neurodegeneration following TBI (14). We observed an increase in NOX4 fluorescence within the ipsilateral S1BF cortex after blast exposure (Figure 6A). We measured a significant increase in NOX4 total cell fluorescence in mice at 72 hours post-blast compared to controls (t = 4.07; P < 0.001) (Figure 6B). We also observed an increase in fluorescence of pro-inflammatory marker iNOS in the ipsilateral S1BF cortex after blast (Figure 6C). We measured a significant increase in iNOS total cell fluorescence in mice at 72 hours post-blast compared to controls (t = 4.18; P < 0.001) (Figure 6D). Standard t-test with two-tailed critical values represented as mean ± sem.
Figure 6. Increased NOX4 and iNOS Cell Fluorescence Observed After Blast.
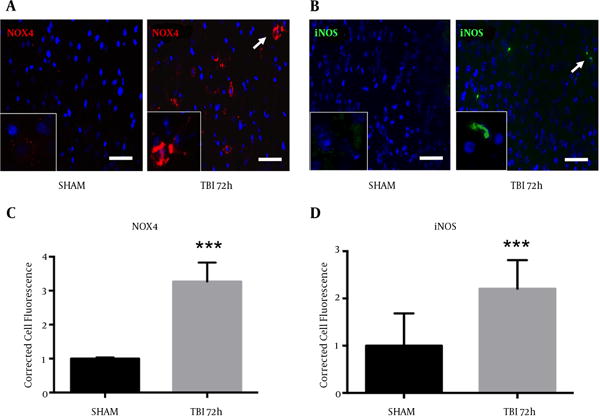
A, NOX4 fluorescence; B, and iNOS fluorescence; C, were both elevated in the ipsilateral S1BF region. A significant increase in NOX4 total cell fluorescence was measured at 72 hours post-blast (***P < 0.001 vs SHAM). A significant increase in iNOS total cell fluorescence was also measured at 72 hours post-blast (***P < 0.001 vs SHAM) (D). Two-tailed Standard t-test. Mean ± sem. n = 5.
2.7. Blast Increased mRNA Abundance of Pro-Inflammatory Markers
Markers of neuroinflammation are commonly observed after TBI (12, 15). Recently, our group has also shown an upregulation of pro-inflammatory markers in a rat model of blast injury (16). A significant difference in iNOS mRNA abundance was observed in the ipsilateral S1BF region after blast (F (2, 12) = 5.60; P < 0.05). We revealed a significant increase in iNOS mRNA abundance at 24 hours post-blast when compared to control animals (q = 3.21; P < 0.05) (Figure 7A). A significant difference in TNFα mRNA abundance was also observed in the ipsilateral S1BF region after blast (F (2, 12) = 4.53; P < 0.05). We revealed a significant increase in TNFα mRNA abundance at 24 hours post-blast when compared to control animals (q = 2.91; P < 0.05) (Figure 7B). One-way ANOVA with Dunnetts post-hoc values represented as mean ± sem.
Figure 7. Pro-Inflammatory Gene Abundance Elevated After Blast Injury.
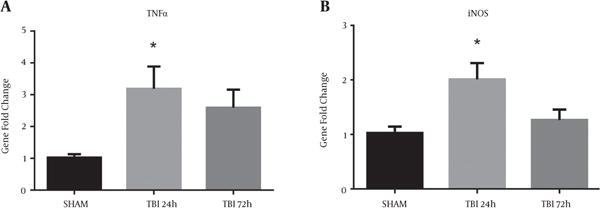
A, A significant increase in iNOS mRNA abundance was measured in the ipsilateral S1BF region at 24 hours post-blast (*P < 0.05 vs SHAM); B, A significant increase in TNFα mRNA abundance was also observed at 24 hours post-blast (*P < 0.05 vs SHAM). One-way ANOVA, Dunnetts post-hoc. Mean ± sem. n = 5.
2.8. Oligomeric Tau Observed in the Ipsilateral S1BF Cortex After Blast
A novel, oligomer-specific, tau antibody was produced to better understand the role of tau pathology throughout disease progression (17). We discovered the oligomer-specific antibody (T22) within the ipsilateral S1BF cortex after blast. We revealed an increase in T22 fluorescence at 72 hours post-blast (Figure 8A). We measured a significant increase in T22 total cell fluorescence at 72 hours post-blast compared to sham controls (t = 5.59; P < 0.001) (Figure 8B). Standard t-test with two-tailed critical values represented as mean ± sem.
Figure 8. Oligimeric Tau Observed in the S1BF Region After Blast.
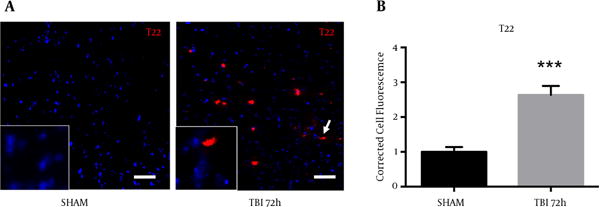
A, blast exposure revealed an elevated fluorescence of T22 (oligomeric tau); B, A significant increase in T22 total cell fluorescence was measured at 72 hours post-blast (*P < 0.001 vs SHAM). Two-tailed Standard t-test. Mean ± sem. n = 5.
2.9. Aberrant Behavior Observed in Mice After Blast Exposure
Behavioral testing, including the EPM, can activate the S1BF region in rodents (18). Moreover, injury to the S1BF region in rats was shown to produce an anxiety-like behavior as measured in the EPM (19). As such, we sought to measure impulsive-like behavior and disinhibition in mice after exposure to our blast model. We revealed a significant increase in percent time spent in the open arm of the EPM in mice at 72 hours post-blast compared to SHAM animals (t = 2.61; P < 0.05) (Figure 9A). We found no difference in total movement between 72 hour and SHAM animals (t = 0.29; P > 0.05) (Figure 9B). Standard t-test with two-tailed critical values represented as mean ± sem.
Figure 9. Aberrant Behavior Was Observed in Mice After Blast Injury.
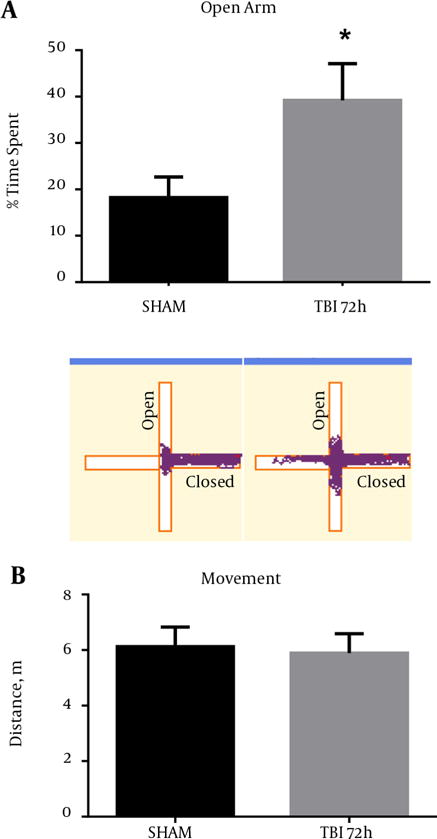
A, A significant increase in the time spent in the open arms of the EPM was observed at 72 hours post-blast (*P < 0.05 vs SHAM) (). Track plots from Anymaze™ are shown below to provide a clear visual representation of the mouse’s behavior during the EPM trials; B, no significant difference was observed in total movement at 72 hours post-blast (P > 0.05). Two-tailed Standard t-test. Mean ± sem. n = 10.
3. Discussion
TBI has been associated with astrogliosis in subcortical regions (20). It has been proposed that this robust astrogliosis may be due to microvascular damage and acute disruption of the BBB (21). BBB disruption has been well reported in more severe TBI models, but its role in mild TBI is poorly understood (22). Herein we show that BBB disruption occurs at 72 hours following mild blast TBI. Interestingly, the mRNA increase occurred at 24 hours. The subsequent 72-hour increase in GFAP protein expression that coincides with the BBB disruption may potentially be due to the initial inflammatory cytokine burst triggered post-injury. The vessels with increased permeability showed robust GFAP fluorescence, suggesting astrogliosis. A similar astrogliosis distribution was also observed in another rodent model of TBI (23). The Friedlander wave generated by our model produces a short duration impulse (area under the curve). This is important for two reasons: 1) with a freely mobile head the mouse’s skull and brain undergoes extensive acceleration/deceleration from the propulsion of the wave and 2) the wave may directly penetrate through the skull causing injury at density gradients such as the cerebrovasculature. Further work is warranted in collaboration with our physics colleagues to determine the true extent of wave penetration through the skull.
The characteristic finding of chronic traumatic encephalopathy is neurofibrillary tau tangles (24, 25). Our group previously reported tau hyperphosphorylation in a perivascular distribution following TBI in non-transgenic rats (26). Ojo et al. recently found reduced blood flow, glial reactivity, and oligomeric tau formation in tau transgenic mice after repetitive TBI (27). In this paper, we show tau oligomer formation adjacent to regions of astrogliosis and BBB disruption in non-transgenic mice after blast injury. This finding is important in that tau oligomers and tau hyperphosphorylation for human TBI specimens have been shown to be perivascular in distribution (28, 29).
Recent evidence suggests that pathologic tau plays an active role in disease progression (30). Secondary injury cascades are activated in conjunction with these tau changes. We have previously shown that endoplasmic reticulum stress and oxidative stress contribute to long-term tau hyperphosphorylation changes and are activated simultaneously following neurotrauma (8, 31). Furthermore, the interaction between endoplasmic reticulum stress, oxidative stress, and neuroinflammation is a topic of growing importance in the field of neurotrauma (16).
Secondary injury cascades and tauopathy may contribute to aberrant behavioral changes (32). One of the most well characterized behavioral changes in relation to TBI is impulsivity. Impulsivity is a primary presenting symptom in warfighters exposed to blast TBI (11). In this paper, we observed a significant increase in open arm exploratory behavior, without an increase in total distance traveled on the EPM. Going forward it will be critical to elucidate the molecular underpinnings responsible for this aberrant behavior. The mouse blast model provides the opportunity to investigate causative mechanisms for aberrant behavior with genetic manipulation.
Modeling cognitive behavior in rodents relies on functional performance. In this paper, we observed robust histopathologic changes in perivascular regions of the S1BF. It has also been reported that TBI can contribute to nigrostriatal damage (33). Cognitive deficits have also been observed in a model of diffuse axonal injury (34), and blast injury (11). Herein, we utilized the EPM to evaluate aberrant behaviors associated with blast injury. In future investigations, we plan to investigate functional deficits and cortical connectivity after blast injury. It is interesting to note the injury to the S1BF cortex, and the subsequent behavioral deficits observed. The aberrant behavior we observed following neurotrauma may be the result of dysfunctional cortical circuitry and warrants further investigation.
We have previously shown behavioral changes in rats at acute time points after single TBI (33) and repeat TBI (16). In addition, Kondo et al. also observed impulsive-like behavior in mice after severe rotational acceleration TBI (35). These results suggest that the acute injury we observed in the S1BF region may be associated with impulsive-like behavior. It is also interesting to point out that this behavior seems to be acute. The cortical circuitry should be further examined between the orbital frontal cortex and S1BF region to determine the underpinnings of the impulsive-like behavior. The ultimate goal is to find therapeutic targets that can be successfully targeted clinically. These interventions have broad clinical utility for warfighters, athletes, and those suffering concussions from falls or motor vehicle collisions. Such pharmaceutical options must have well-known efficacy in targeting secondary injury cascades, but more importantly be able to provide symptomatic improvement that is maintained over time.
One of the most important considerations will be the ability to detect subtle improvements with advanced imaging. Advanced imaging modalities are now being used to evaluate the positive benefit of future therapeutics longitudinally (36). Diffusion tensor imaging has also become a valuable tool for the detection of brain structure abnormalities after blast injury (37, 38) In order to continue to improve these imaging modalities; it is imperative that the key secondary injury pathways are well characterized in pre-clinical models. In order for this to occur, it will be necessary to determine the number and interval-between injuries that causes neurodegeneration (26). Furthermore, characterization of the interplay between secondary injury cascades must be well elucidated, which was the primary focus of this paper.
4. Conclusions
Blast TBI continues to be a significant problem with limited diagnostic or treatment solutions. In this paper, we present compelling evidence that blood brain barrier disruption and tauopathy may contribute to behavioral deficits following blast TBI in BALB/c mice. The vascular injury was complemented with astrocyte reactivity, oxidative stress, and neuroinflammation. The focal injury observed in the S1BF region may have led to the aberrant behavior observed with the EPM. We will continue to investigate the physiological pathology of neurotrauma to better diagnose injury progression and ultimately develop key therapeutic agents to help treat patients.
5. Experimental Procedure
5.1. Animals
Thirty (30) male BALB/c mice (Charles River) at 2 – 3 months of age were used in this study. The West Virginia University animal care and use committee approved all procedures involving mice. Mice were acclimated for 1 week prior to use and housed under 12 hour light/dark conditions with food and water available ad libitum. Animal experiments were performed according to the principles of the guide for the care and use of laboratory animals.
5.2. Chemicals and Reagents
Texas red conjugated dextran (3 kDa) (TRD) was purchased from Molecular Probes-Life Technologies (Carlsbad, CA). Indocyanine Green (ICG) was purchased from Sigma Aldrich (St. Louis, MO). Bovine Serum Albumin was purchased from Santa Cruz (Dallas, TX). All chemicals and reagents used were of analytical grade and were used as supplied.
5.3. Blast Exposure
Blast exposure was induced as previously described (10). Briefly, a 0.003″ polyester membrane was fastened between two steel tubes (Driver and Driven sections; See Figure 1A). Nitrogen gas filled the Driver section until the membrane ruptured and expelled a mild blast wave through the Driven section and out of the tube exit (~ 20 psi, 0.5 ms duration; See Figure 1B). The animal’s head was placed perpendicular to the tube exit (~ 2cm), with the right side being exposed to the mild blast wave. Following blast exposure, mice were returned to a holding cage equipped with a homeothermic heating pad. Once basic reflexes were restored, mice were returned to their home cage. No mortality was observed after this blast intensity.
5.4. Tissue Preparation
Mice (n = 15) used for biochemical analysis were euthanized and their brains were rapidly removed in an ice-cold HaltTM protease/phosphatase inhibitor cocktail mix (Thermo Scientific). The ipsilateral S1BF was dissected out, halved, and submerged in isopentane (−65°C), and stored at −80°C for later measurement of protein expression and mRNA abundance. Mice (n = 15) used for vascular permeability assessment and immunohistochemistry (IHC) were first anesthetized with ketamine/xylazine (100 and 8 mg/kg respectively). To examine vascular permeability we injected 3 kDa TRD (5 μg/g) into the femoral vein at 24 and 72 hours after blast exposure. After 10 minutes of TRD circulation, we transcardially perfused 50 kd ICG/albumin (5 and 11 μg/g respectively) at rate of 5 mL/min for 2 minutes to washout TRD and label blood vessels. Following perfusion, brains were removed and submerged in isopentane (−65°C) and stored at −80°C for IHC preparation and fluorescence imaging.
5.5. Quantitative Real-Time Polymerase Chain Reaction
S1BF tissue for gene analysis was homogenized in TRIzol® reagent (Thermo Scientific) and confirmed for quality (1.8 - 2.1 absorbance ratio). Reverse transcription was conducted and real-time PCR analyses were performed on cDNA using the oligonucleotide primer for GFAP (Mm01253033_m1), iNOS (Mm00435175_m1), TNFa (Mm00443258_m1), and 18s rRNA (Hs99999901_s1; endogenous control) (Thermo Scientific). Changes in mRNA abundance were determined by the ΔΔCt method with a threshold cycle value of 0.2 normalized to 18s rRNA.
5.6. Immunoblotting
S1BF tissue for protein analysis was sonicated in 1% SDS and Western blot analysis was performed as previously described (39). A rabbit anti-GFAP monoclonal antibody (1:1000), and a rabbit anti-β-actin monoclonal antibody (1:10,000) (Cell Signaling) were used with a secondary antibody IRDye® 800CW (goat anti-rabbit) (LI-COR Biosciences). Images were collected using the odyssey classic infrared imaging system (LI-COR Biosciences). Images were converted to grey scale, and detected bands were quantified using image studio lite software (LI-COR Biosciences). Bands were normalized to β-actin values to measure relative intensity.
5.7. Immunohistochemistry
Whole brains from mice were mounted on a Leica CM3050S cryostat (Leica Microsystems) set to −20°C. Coronal sections were sliced at a thickness of 20 μm and mounted onto glass slides for all IHC staining as previously described (8). Three immediate adjacent sections of 20 μm were cut per each slide. Briefly, brain slices were circumscribed, and incubated overnight with primary antibodies: GFAP (Cell Signaling); Oligimeric tau (T22; kindly gifted by Dr. Rakez Kayed); NADPH-oxidase 4 (NOX4; Santa Cruz); and inducible nitric oxide synthase (iNOS; Santa Cruz). The next day, an Alexa Flour® secondary antibody (Invitrogen) was applied to slides for 3 hours, and coverslip mounted with Vectashield® 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain (vector).
All Images were acquired from the ipsilateral S1BF region (10 slides per animal). Antibody-stained fluorescence images were acquired using a confocal microscope (Z1 Axio Observer; Zeiss, Oberkochen, Germany). TRD and ICG fluorescence images were acquired using a stereomicroscope (Olympus MVX10; Olympus, Center Valley, PA) equipped with a 0.5 NA 2 × objective and a monochromatic cooled CCD scientific camera (Retiga 4000R, QIMaging, Surrey, BC, Canada). Texas red fluorescence was imaged using a DsRed sputter filter (excitation/band λ 545/25 nm, emission/band λ 605/70 nm and dichromatic mirror at λ 565 nm) (Chroma Technologies, Bellows Falls, VT).
For antibody-stained fluorescence quantification, 10 distinct cells with clear morphology were randomly selected per slide, outlined, and measured with ImageJ software (NIH) by an observer blinded to experimental group. Density was adjusted per mean area to give total cell fluorescence normalized to background.
5.8. Behavior on Elevated Plus Maze
Behavior was assessed in mice using the elevated plus maze (EPM). Increased time spent in the open arms was considered a sign of impulsive-like behavior in the rodent (16, 40, 41). The EPM was set at a height of 60 cm above the floor. The two open arms intersected perpendicular to the two closed arms. Each arm was 58 cm × 5 cm. The closed arms were encased by black siding 15 cm tall. Each mouse was placed in the middle of the EPM facing an open arm and tracking was performed for 10 minutes using AnyMaze™ software (Stoelting, Wood Dale, IL), which pinpointed the location of the mouse’s head and body continuously throughout the testing trial.
5.9. Statistical Analysis
Data comparing three groups were analyzed using a one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc tests. Data comparing two groups were analyzed using a Standard t-test with two-tailed critical values corresponding to P < 0.05. Statistical analyses were performed using Graph Pad software (San Diego, CA). All data are reported as the average standard error of the mean (sem). Sample sizes were determined using a power analysis with an α of 0.05, a β of 0.2, and a sample effect of 0.4 for behavioral data and 0.3 for all other data (DSS Research Power Analysis). P < 0.05 was considered significant for all data.
Acknowledgments
Paul Lockman provided funding and support for this project with an NIH grant (RO1 CA166067-01 A1). Aric Logs-don and Brandon Lucke-Wold received funding and support from the American Foundation for pharmaceutical education pre-doctoral fellowship. Brandon Lucke-Wold also received support from Sigma Xi grants in aid of research, the American medical association foundation seed grant, American Association of Pharmaceutical Scientists Pre-doctoral Fellowship, and the neurosurgery research and education foundation medical student summer research fellowship. We would like to acknowledge the generous gift of the T22 antibody from Dr. Rakez Kayed. We would also like to thank Matt Robson PhD, and his father James Robson for help creating the model.
Footnotes
Footnotes
Authors’ Contribution: Aric F. Logsdon conceived the study, designed and performed the experiments, analyzed the data, and wrote the manuscript. Brandon P. Lucke-Wold conceived the study, designed and performed the experiments, analyzed the data, and helped write the manuscript. Afroz S. Mohammad helped design and perform experiments. Chris E. Adkins helped design and perform experiments, analyze data, and manuscript preparation. Ryan C. Turner designed the blast model and helped write the manuscript. Xinlan Li helped run experiments and analyze data. Charles L. Rosen provided invaluable clinical knowledge, in addition to experimental design and manuscript preparation. Jason D. Huber provided invaluable scientific knowledge, in addition to helping with experimental design and manuscript preparation. Paul R. Lockman provided funding for the project and also helped to design the study, analyze the data, and manuscript preparation.
Conflict of Interests: The authors have no conflicts of interest to disclose.
References
- 1.Meabon JS, Huber BR, Cross DJ, Richards TL, Minoshima S, Pagulayan KF, et al. Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci Transl Med. 2016;8(321):321ra6. doi: 10.1126/scitranslmed.aaa9585. [DOI] [PubMed] [Google Scholar]
- 2.Gama Sosa MA, De Gasperi R, Janssen PL, Yuk FJ, Anazodo PC, Pricop PE, et al. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol Commun. 2014(2):67. doi: 10.1186/2051-5960-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toklu H, Tümer N. Frontiers in Neuroengineering. CRC Press; 2015. Oxidative Stress, Brain Edema, Blood–Brain Barrier Permeability, and Autonomic Dysfunction from Traumatic Brain Injury. [PubMed] [Google Scholar]
- 4.Yuan F, Xu ZM, Lu LY, Nie H, Ding J, Ying WH, et al. SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-kappaB p65 acetylation and activation. J Neurochem. 2016;136(3):581–93. doi: 10.1111/jnc.13423. [DOI] [PubMed] [Google Scholar]
- 5.Prochnow N. Relevance of gap junctions and large pore channels in traumatic brain injury. Front Physiol. 2014;5:31. doi: 10.3389/fphys.2014.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eldawlatly S, Oweiss KG. Temporal precision in population-but not individual neuron-dynamics reveals rapid experience-dependent plasticity in the rat barrel cortex. Front Comput Neurosci. 2014;8:155. doi: 10.3389/fncom.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miremami JD, Talauliker PM, Harrison JL, Lifshitz J. Neuropathology in sensory, but not motor, brainstem nuclei of the rat whisker circuit after diffuse brain injury. Somatosens Mot Res. 2014;31(3):127–35. doi: 10.3109/08990220.2014.897602. [DOI] [PubMed] [Google Scholar]
- 8.Lucke-Wold BP, Turner RC, Logsdon AF, Nguyen L, Bailes JE, Lee JM, et al. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg. 2016;124(3):687–702. doi: 10.3171/2015.3.JNS141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerson J, Castillo-Carranza DL, Sengupta U, Bodani R, Prough DS, DeWitt DS, et al. Tau Oligomers Derived from Traumatic Brain Injury Cause Cognitive Impairment and Accelerate Onset of Pathology in Htau Mice. J Neurotrauma. 2016;33(22):2034–43. doi: 10.1089/neu.2015.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner RC, Naser ZJ, Logsdon AF, DiPasquale KH, Jackson GJ, Robson MJ, et al. Modeling clinically relevant blast parameters based on scaling principles produces functional & histological deficits in rats. Exp Neurol. 2013;248:520–9. doi: 10.1016/j.expneurol.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic Traumatic Encephalopathy in Blast-Exposed Military Veterans and a Blast Neurotrauma Mouse Model. Sci Translat Med. 2012;4(134):134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2015;51(3):966–79. doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adkins CE, Mohammad AS, Terrell-Hall TB, Dolan EL, Shah N, Sechrest E, et al. Characterization of passive permeability at the blood-tumor barrier in five preclinical models of brain metastases of breast cancer. Clin Exp Metastasis. 2016;33(4):373–83. doi: 10.1007/s10585-016-9784-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman LC, Ting JP. The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem. 2016;136(Suppl 1):29–38. doi: 10.1111/jnc.13217. [DOI] [PubMed] [Google Scholar]
- 15.Abdul-Muneer PM, Long M, Conte AA, Santhakumar V, Pfister BJ. High Ca2+ Influx During Traumatic Brain Injury Leads to Caspase-1-Dependent Neuroinflammation and Cell Death. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-9949-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Logsdon AF, Lucke-Wold BP, Nguyen L, Matsumoto RR, Turner RC, Rosen CL, et al. Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res. 2016;1643:140–51. doi: 10.1016/j.brainres.2016.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012;26(5):1946–59. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen NK, Sartori SB, Herzog H, Tasan R, Sperk G, Singewald N. Effect of neuropeptide Y Y2 receptor deletion on emotional stress-induced neuronal activation in mice. Synapse. 2009;63(3):236–46. doi: 10.1002/syn.20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnstone VP, Wright DK, Wong K, O’Brien TJ, Rajan R, Shultz SR. Experimental Traumatic Brain Injury Results in Long-Term Recovery of Functional Responsiveness in Sensory Cortex but Persisting Structural Changes and Sensorimotor, Cognitive, and Emotional Deficits. J Neurotrauma. 2015;32(17):1333–46. doi: 10.1089/neu.2014.3785. [DOI] [PubMed] [Google Scholar]
- 20.Carron SF, Yan EB, Alwis DS, Rajan R. Differential susceptibility of cortical and subcortical inhibitory neurons and astrocytes in the long term following diffuse traumatic brain injury. J Comp Neurol. 2016;524(17):3530–60. doi: 10.1002/cne.24014. [DOI] [PubMed] [Google Scholar]
- 21.Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma. 2014;31(13):1180–93. doi: 10.1089/neu.2013.3080. [DOI] [PubMed] [Google Scholar]
- 22.Cunningham TL, Cartagena CM, Lu XC, Konopko M, Dave JR, Tortella FC, et al. Correlations between blood-brain barrier disruption and neuroinflammation in an experimental model of penetrating ballistic-like brain injury. J Neurotrauma. 2014;31(5):505–14. doi: 10.1089/neu.2013.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plantman S, Ng KC, Lu J, Davidsson J, Risling M. Characterization of a novel rat model of penetrating traumatic brain injury. J Neurotrauma. 2012;29(6):1219–32. doi: 10.1089/neu.2011.2182. [DOI] [PubMed] [Google Scholar]
- 24.Lucke-Wold BP, Turner RC, Logsdon AF, Bailes JE, Huber JD, Rosen CL. Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma. 2014;31(13):1129–38. doi: 10.1089/neu.2013.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131(1):75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner RC, Lucke-Wold BP, Logsdon AF, Robson MJ, Dashnaw ML, Huang JH, et al. The Quest to Model Chronic Traumatic Encephalopathy: A Multiple Model and Injury Paradigm Experience. Front Neurol. 2015;6:222. doi: 10.3389/fneur.2015.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ojo JO, Mouzon B, Algamal M, Leary P, Lynch C, Abdullah L, et al. Chronic Repetitive Mild Traumatic Brain Injury Results in Reduced Cerebral Blood Flow, Axonal Injury, Gliosis, and Increased T-Tau and Tau Oligomers. J Neuropathol Exp Neurol. 2016;75(7):636–55. doi: 10.1093/jnen/nlw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKee AC, Daneshvar DH, Alvarez VE, Stein TD. The neuropathology of sport. Acta Neuropathol. 2014;127(1):29–51. doi: 10.1007/s00401-013-1230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC. Characterization of Early Pathological Tau Conformations and Phosphorylation in Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol. 2016;75(1):19–34. doi: 10.1093/jnen/nlv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A. 2014;111(41):E4376–85. doi: 10.1073/pnas.1411649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lucke-Wold BP, Naser ZJ, Logsdon AF, Turner RC, Smith KE, Robson MJ, et al. Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Transl Res. 2015;166(6):509–528 e1. doi: 10.1016/j.trsl.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, et al. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M, Bachstetter AD, Cass WA, Lifshitz J, Bing G. Pioglitazone Attenuates Neuroinflammation and Promotes Dopaminergic Neuronal Survival in the Nigrostriatal System of Rats after Diffuse Brain Injury. J Neurotrauma. 2017;34(2):414–22. doi: 10.1089/neu.2015.4361. [DOI] [PubMed] [Google Scholar]
- 34.Morawska MM, Buchele F, Moreira CG, Imbach LL, Noain D, Baumann CR. Sleep Modulation Alleviates Axonal Damage and Cognitive Decline after Rodent Traumatic Brain Injury. J Neurosci. 2016;36(12):3422–9. doi: 10.1523/JNEUROSCI.3274-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523(7561):431–6. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenmenger LB, Huo EJ, Hoffman JM, Minoshima S, Matesan MC, Lewis DH, et al. Advances in PET Imaging of Degenerative, Cerebrovascular, and Traumatic Causes of Dementia. Semin Nucl Med. 2016;46(1):57–87. doi: 10.1053/j.semnuclmed.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Budde MD, Shah A, McCrea M, Cullinan WE, Pintar FA, Stemper BD. Primary blast traumatic brain injury in the rat: relating diffusion tensor imaging and behavior. Front Neurol. 2013;4:154. doi: 10.3389/fneur.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamnaksh A, Budde MD, Kovesdi E, Long JB, Frank JA, Agoston DV. Diffusion tensor imaging reveals acute subcortical changes after mild blast-induced traumatic brain injury. Sci Rep. 2014;4:4809. doi: 10.1038/srep04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, et al. Bryostatin-1 Restores Blood Brain Barrier Integrity following Blast-Induced Traumatic Brain Injury. Mol Neurobiol. 2015;52(3):1119–34. doi: 10.1007/s12035-014-8902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mosienko V, Bert B, Beis D, Matthes S, Fink H, Bader M, et al. Exaggerated aggression and decreased anxiety in mice deficient in brain serotonin. Transl Psychiatry. 2012;2:ee122. doi: 10.1038/tp.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson EM, Traver KL, Hoffman SW, Harrison CR, Herman JP. Environmental enrichment protects against functional deficits caused by traumatic brain injury. Front Behav Neurosci. 2013;7:44. doi: 10.3389/fn-beh.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]