

Abstract
Caveolin-1 is a target for academic and pharmaceutical research due to its many cellular roles and associated diseases. We report peptide WL47 (1), a small, high-affinity, selective disrupter of cavolin-1 oligomers. Developed and optimized though screening and analysis of synthetic peptide libraries, ligand 1 has 7500-fold improved affinity compared to its T20 parent ligand, and an 80% decrease in sequence length. Ligand 1 will permit targeted study of caveolin-1 function.
TOC image
INTRODUCTION
The family of caveolin membrane proteins contribute to the cell’s structural, signaling, and transportation processes. With such diverse roles, caveolins are also associated with myriad diseases, including cancer, cardiovascular disease, muscular dystrophies, atherosclerosis, diabetes, Alzheimer’s disease, and HIV.1–5 Although the caveolins represent excellent candidate targets for drug discovery, progress has been hampered by a lack of techniques available to probe and control caveolin structure and functions with specificity and precision. Caveolins are typically studied in cells and organisms through ablation via knock-outs, mutagenesis, deletions, or nonspecific cholesterol depletion using compounds such as nystatin or filipin.6–11 Here we report a molecular tool to disrupt oligomerization of the most widely distributed and commonly studied caveolin, caveolin-1 (CAV). This CAV ligand could facilitate future research by providing a mechanism for perturbing CAV function, and provides a potential lead compound for a new class of therapeutics.
As a monotonic membrane protein, CAV, a 22 kDa protein, penetrates only one leaflet of the lipid bilayer, and both the N- and C-termini remain on the cytoplasmic side.12 Multiple copies of CAV oligomerize to form high molecular weight complexes that bend the membrane inward to form invaginations, termed “caveolae,” of 50–100 nm in diameter.13,14 The cholesterol- and sphingolipid-rich membrane of these caveolae regions are a sub-type of lipid raft.15 While these invaginations can mediate endocytosis in a manner similar to clathrin-coated pits, current research emphasizes their role in signaling pathways. CAV’s binding partners include cAMP-dependent protein kinase A (PKA), endothelial nitric oxide synthase (eNOS), insulin receptors, and the HIV coat protein gp41.16,17 CAV modulates signaling pathways by binding, and thus sequestering, both enzymes and receptors engaged in cell signaling. The complex with oligomeric CAV can stabilize such enzymes or receptors in either their active or inactive conformations. CAV also mediates cholesterol trafficking by binding and transporting cholesterol to the cell membrane. The oligomeric state of CAV also influences early cellular response to mechanical stress.18,19
Previous research in the Weiss laboratory used the known interaction between CAV and gp41 (2) as a starting point for generating a ligand for CAV.20 The FDA-approved drug T20 (Fuzeon) (3), a 36 amino acid peptide derived from gp41, blocks HIV viral fusion with CD4+ T-cells.21,22
The T20 sequence was mutated extensively in a phage-displayed library for screening and selections targeting CAV residues (1-104), a truncated form of CAV that can be solubly expressed in a heterologous system, in contrast to wild type CAV which cannot. From this library, 36-mer sequences were isolated with dissociation constants (KD) for CAV(1-104) of 155 nM or better, such as ligand 4. This phage-based, molecular evolution represented a 1000-fold improvement in KD relative to T20.20
We have undertaken further design refinement of this T20-derived CAV ligand, 4.20 Iterative cycles of synthesis and assay guided the design to yield an 80% reduction in length with 7.5-fold higher affinity (Figure 1). By synthesizing and screening carefully designed peptide library arrays, we also identified key residues, minimized ligand size, and optimized the sequence (Figures 2–4). We report that this ligand, 1, has high selectivity and affinity for its target (Figures 5–6), and can disrupt CAV oligomers (Figure 7).
Figure 1.
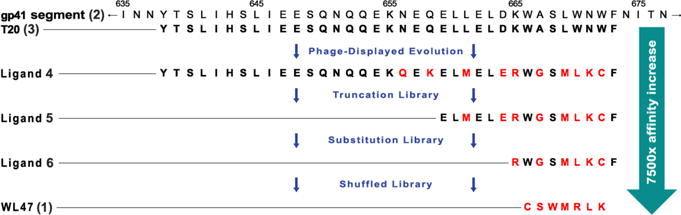
Evolution of CAV ligands. Sequence alignment showing the progression of peptide ligands from the native gp41 segment sequence (2) to T20 (3), to ligands 4, 5, and 6, and ultimately to ligand 1, which binds CAV(1-104) with 7500-fold higher affinity despite an 80% decrease in length compared to 3. Residues mutated from 3 are in red. Numbers above 2 indicate the residue numbers from the original protein.
Figure 2.
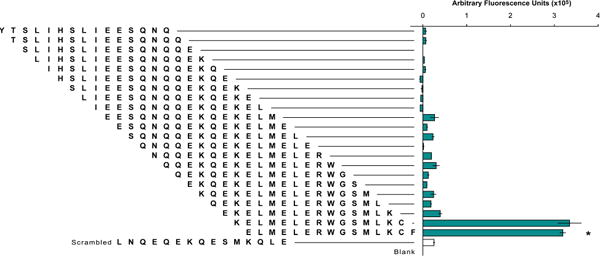
Initial identification of CAV interacting region. The sequence of ligand 4 was truncated to generate 22 unique peptide sequences of 15 amino acids each. Only two truncation sequences, consisting of residues 21–35 and residues 22–36, respectively, bound CAV(1-104) more than 2-fold above the level of the negative control. The negative control was a scrambled sequence of the randomly selected 11th truncation. An additional, blank negative control had a linker region but lacked a peptide. The sequence corresponding to residues 22–36 (indicated by asterisk) was chosen as the template for subsequent libraries. and designated ligand 5.
Figure 4.
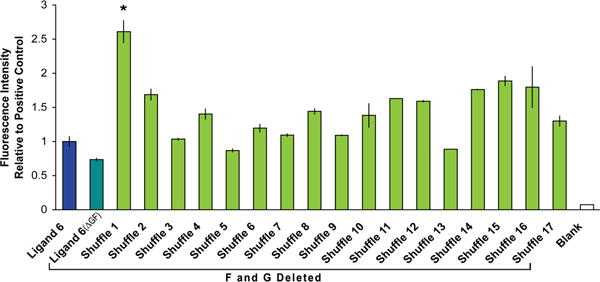
Unnecessary residues removed, and sequences shuffled. Direct removal of Gly9 and Phe15 from the ligand 6 sequence to create ligand 6(ΔGF) decreased binding. When the sequence lacking Gly and Phe is shuffled, however, many library members retain or increase binding ability. The 7-mer shuffled sequence (indicated by asterisk) with the highest binding was used for subsequent studies.
Figure 5.
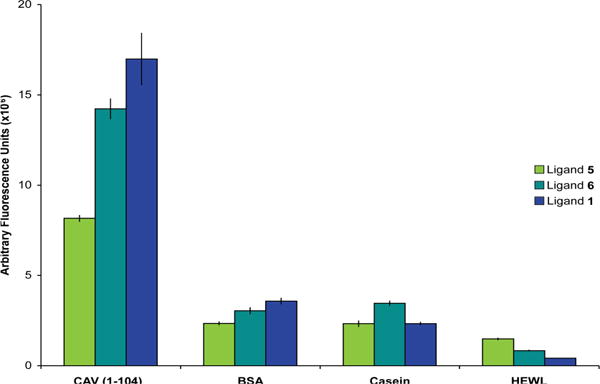
Demonstration of selectivity. CAV(1-104) and several control proteins were labeled with rhodamine and screened against duplicate SPOT sheets containing ligands 5, 6, and 1 to demonstrate selectivity of the ligand. Bovine serum albumin (BSA), casein, and hen egg white lysozyme (HEWL) all bound ligand 1 with reduced apparent affinity relative to CAV(1-104) as measured by fluorescence. Blank SPOTs containing only the double β-Ala linker were included as negative controls, and these baseline signals were subtracted from the corresponding signal for ligands 5, 6 and 1 SPOTs on each sheet.
Figure 6.
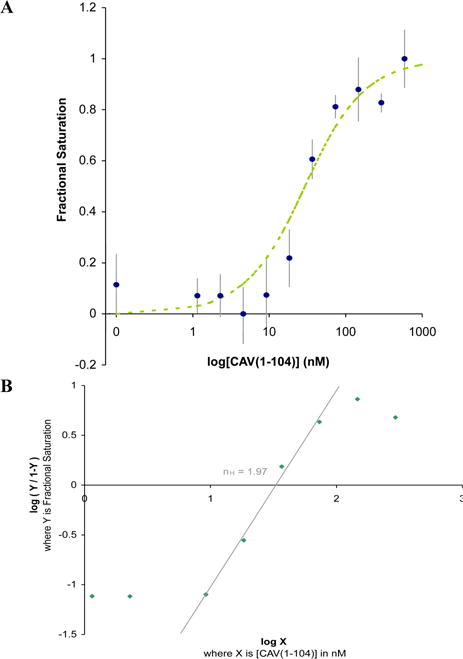
Binding affinity and cooperativity. a) Fluorescence anisotropy was measured for Mantyl-1 dimer incubated with the indicated concentrations of CAV(1-104). Using equation 1, a best fit binding curve was fit to the raw experimental data using a weighted method of least squares, and assuming each ligand 1 dimer functions as a single ligand. The KD for this binding interaction was calculated to 23 nM. b) A Hill plot yields a Hill coefficient (nH) of 1.97. This is the slope of the linear region of the plot as it crosses the x-axis (R2=0.992). Based on the two-site binding model, this coefficient indicates virtually complete positive cooperativity. Thus, upon binding one molecule of 1 (one half of the dimer), a second molecule (the other half of the dimer) binds essentially instantaneously the second site. This observation supports our decision to treat the 1 dimer as a single ligand for the purpose of KD calculations.
Figure 7.
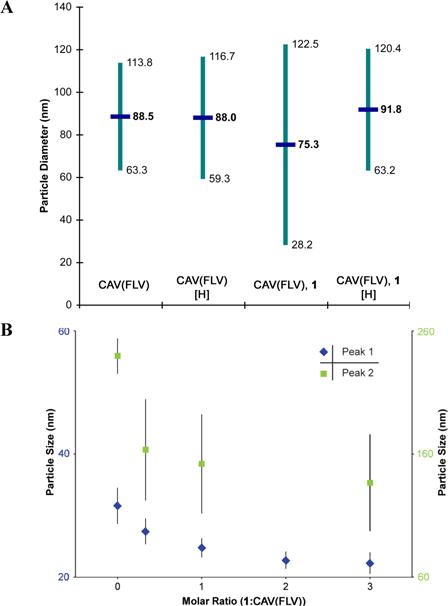
Disruption of oligomers. a) CAV(FLV) spontaneously forms oligomers which can be measured by dynamic light scattering (DLS). In this experiment, these oligomers had an apparent average diameter near 90 nm with polydispersity (indicated in teal) corresponding to approximately a 55 nm range. Incubating these oligomers with ligand 1 resulted in reduced average diameter and increased polydispersity, which are the expected outcomes of deoligomerization. With the disulfide dimerization of the ligand disrupted by reducing conditions, no deoligomerization effect was observed. b) In a separate DLS experiment, CAV(FLV) oligomeric peaks at 32 nm (blue, left axis) and 240 nm (green, right axis) each showed a dose-dependent reduction in diameter when incubated with varied concentrations of ligand 1. A maximum size reduction of 30% and 40% was observed for the respective peaks.
RESULTS AND DISCUSSION
Development of ligand 1 proceeded in three stages (Figure 1). First, regions of the starting ligand 4 contributing to the recognition of CAV were identified. Second, this functional region was trimmed though mutagenesis and screening to eliminate non-essential residues. Third, the remaining key residues were shuffled to identify the most promising arrangement.
First, truncation libraries identified key regions of the starting T20-derived ligand 4 (Figure 2). Such information could reduce the length of the ligand required for binding to CAV. All 22 possible 15-mer sequences within the 36-mer peptide were synthesized and screened as C-terminal adducts to cellulose. This technique, termed SPOT synthesis, allows rapid synthesis and screening of peptides on a positionally addressable array.23 Analogous to varying display levels in a phage-based library, the concentration of peptide in each SPOT can vary due to differences in amino acid coupling efficiency during synthesis. Nonetheless, our investigation of the level of variation in concentration between SPOTs confirmed that such variation is small compared to the differences in binding that we observed (Table S1). This method can thus provide reliable comparisons of binding affinity between peptides on the same array.24, 25
SPOT synthesis provided a dependable method to guide the development of ligand 1. CAV(1-104) bearing a fluorescent rhodamine tag was incubated with the peptide array and the degree of fluorescence measured for each library member revealed its relative apparent binding affinity. This experiment identified the C-terminal region of ligand 4 as contributing at least 98% of the observed binding to CAV(1-104). Furthermore, eliminating the non-essential region at the ligand N-terminus reduced ligand size from a 36- to a 15-mer peptide, ligand 5.
Additional trimming and mutagenesis next honed the CAV ligand. With the 15-mer peptide 5 as a template, a subsequent library featured similar and dissimilar substitutions at each position. This approach defines sidechain contributions, if any, to binding. Screening every member of a chemically synthesized library circumvents the problem of survivorship bias, which is inherent to molecular evolution approaches: only the most successful library members are analyzed following selections. By including every library member in the data set, the results from those members with poor apparent binding affinity can still contribute to a deeper understanding of the functionalities that control CAV ligand affinity.
This substitution library uncovered peptides with clear preferences for amino acid sidechains in specific positions. Library members with Lys or His substituting Arg7 retained apparent binding affinity to CAV(1-104), but substitution with the neutral sidechains of Gln or Ala at this site generated a peptide with reduced apparent affinity for CAV(1-104). Substitution of Lys13 had similar but more drastic changes, with Arg substitution retaining complete potency and His, Gln, or Ala substitution abolishing binding. Taken together, these data demonstrate the importance of the two positively charged Lys and Arg sidechains (Figure 3a).
Figure 3.
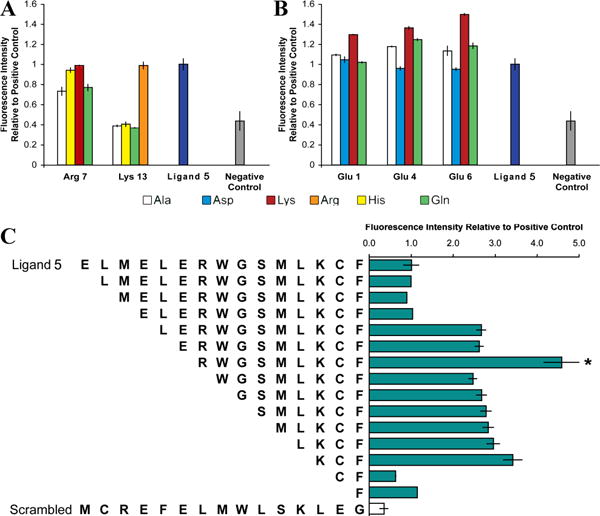
Identification of key residues, and removal of detrimental amino acids. a) In the library of similar and dissimilar substitutions of amino acids in ligand 5, at Arg7, ligand binding was retained for Lys and His substituents but was abolished by neutral Gln or Ala substituents. At Lys13, ligand binding was eliminated for all substituents except Arg. We conclude that both these residues contribute to binding primarily via positive charge. b) For Glu1, ligand binding was retained for negatively charged Asp and neutral Gln, and was slightly elevated for Ala. For Glu4 and Glu6, ligand binding increased moderately for neutral Gln and Ala. In short, positively charged residues are optimal, followed by neutral residues. Ligand binding was reduced by substitution with negatively charged Asp, and all Lys substitutions at these three sites increased ligand binding. These trends suggest that these Glu are not optimal for binding. c) Truncation of the six N-terminal residues, which included all three Glu, without removing any of the positively charged residues, yielded a peptide (indicated by asterisk) designated ligand 6 that became the template for subsequent library design. All libraries include a scrambled ligand 5 sequence as a negative control.
Furthermore, the three negatively charged Glu residues clustered at the N-terminus of ligand 5 adversely affected binding. Ala substitution at Glu1, Glu4, or Glu6 resulted in peptides with an unexpected increase in apparent binding affinity (Figure 3b). Removing six residues at the N-terminus, including these three negatively charged Glu, produced a 9-mer peptide with an approximately 3-fold increase in apparent binding affinity. Other truncations were less successful. For example, further truncation of the N-terminus eliminated the positively charged Arg7, and produced a sharp reduction in binding (Figure 3c). This result reemphasizes the importance of the positively charged sidechains. In summary, this SPOT-synthesized library truncated six residues to yield a 9-mer peptide, ligand 6, with increased apparent affinity for CAV(1-104).
Reduced ligand length made it feasible to synthesize a library with more substitutions. Each position of ligand 6 was systematically varied to further examine tolerance to substitution and sidechain requirements for binding. A library was synthesized by SPOT synthesis to include 19 amino acid substitutions for each of the nine individual sites remaining of ligand 6, yielding 171 unique sequences each bearing a single substitution, along with unsubstituted ligand 6 as a positive control. Sequences anchored to the cellulose solid support can benefit from their close proximity, and this may lead scaffold-bound monomeric peptides to bind the target with higher apparent affinity than monomeric peptides in solution.
The initial library data also identified residues not contributing significantly to ligand function (Figure S1). Such information guided trimming of the ligand to its minimum length. Up to this point, unnecessary amino acids were easily removed by simple truncation. For example, Phe15 could be clipped from the N-terminus and leave essential residues untouched, a desirable change since aromatic sidechains can contribute to nonspecific binding to other targets.26 However, direct removal of an internal residue such as Gly9 could disrupt the spacing of amino acids in the ligand.
Furthermore, the evolution and library screening to this point identified key sidechain functionalities. With this in mind, a library was synthesized wherein Gly and Phe were removed and the remaining residues shuffled using the GenScript Scrambled Library Peptide Library Design Tool.27 A total of 17 shuffled sequences were randomly chosen for synthesis and screening (sequences provided in Table S2). As expected, simple removal of Gly and Phe without otherwise altering the sequence (ligand 6(ΔGF)) resulted in a roughly 25% reduction in apparent binding affinity relative to ligand 6. However, a sequence seven residues in length and with 2.6-fold increased apparent binding affinity relative to ligand 6 was isolated from this library (Figure 4). This sequence, ligand 1, was chosen as the lead compound for further analysis.
A parallel SPOT screening assay next examined the specificity of ligand 1 for CAV. In this assay, rhodamine-labeled CAV(1-104) and three other rhodamine-labeled proteins chosen for their solubility, ubiquity, and known nonspecific binding properties were each incubated with identical SPOT arrays on which ligands 5, 6, and 1 had been synthesized along with blank SPOTs bearing only the dual β-Ala linker. These sheets were washed, blocked, and their fluorescence quantified as described above. ligand 1 bound CAV(1-104) with significantly stronger binding than the other proteins. Furthermore, ligand 6 shows an increase in CAV(1-104) binding relative to ligand 5, and ligand 1 has increased binding relative to ligand 6, confirming that the iterative process used to develop ligand 1 is effective at creating ligands with enhanced target binding. Conversely, little change in binding to control proteins is seen across the three ligand generations, affirming that specific rather than nonspecific binding has been enhanced during ligand evolution (Figure 5).
The KD of the binding interaction between CAV(1-104) and ligand 1 was determined using the measured increase in fluorescence anisotropy of Mantyl-1 upon binding to its target. Slightly smaller than Phe, the N-Methylanthranilyl “Mantyl” fluorophore offers minimal bulk to reduce potential binding disruption.28 This fluorophore was installed to create Mantyl-1. Varying concentrations of CAV(1-104) ranging from 0 to 586 nM were incubated with 12 nM Mantyl-1 dimers. Data were fit to the following equation described previously:29
| (1) |
Where [L] is the total concentration of Mantyl-1 dimer, [R] is the total concentration of CAV(1-104), and [LR] is the concentration of Mantyl-1 dimer bound to CAV(1-104). While considerably more complex than other common binding equations, equation 1 does not ignore binding site depletion, which is crucial for obtaining an accurate model when the ligand concentration is within an order of magnitude of the final KD value.29 By this method, a KD of roughly 23 nM was determined; with a binding affinity ranging from 44 to 3 nM with 95% confidence (Figure 6a). A Hill plot of the same data set yields a Hill coefficient (nH) of 1.97. This coefficient indicates virtually complete positive cooperativity in a two-site binding model; binding of one ligand 1 sequence (first half of dimer) to the first binding site induces the immediate binding of second ligand 1 sequence (second half of dimer) to the second site.30 In essence, both binding sites become occupied simultaneously. This observation supports our decision to perform KD calculations with dimerized ligand 1 considered as a single ligand (Figure 6b).
Demonstrating ligand 1 activity requires in vitro CAV oligomers and a method for measuring the degree of oligomerization. A recently developed full-length, soluble variant of CAV (CAV(FLV)) that spontaneously oligomerizes to form CAV nanoparticles with diameters consistent with those seen for caveolae in vivo (JNS and GAW, unpublished results) was used to examine deoligomerization by ligand 1. Ligand 1 effectively disrupts these nanoparticles. In the presence of a reducing agent, incubation of CAV(FLV) with ligand 1 does not disrupt oligomerization, which is consistent with our the hypothesis that ligand 1 function requires dimerization by disulfide bond (Figure 7a, Figure S1). This deoligomerization activity was also demonstrated to be dose-dependent (Figure 7b). Oligomerization and deoligomerization are a well-known aspect of CAV activity and caveolae formation.18,19 The overlapping nature of the oligomerization and scaffolding domains suggests the regulatory function of CAV relies upon its oligomeric state, as shown previously through oligomer complementation.31
Analysis of ligand 1 as a dimerized ligand has allowed for more critical analysis of previous work, and suggests that both sites of a two-site system must be filled in order for deoligomerization of CAV to occur. For example, isothermal calorimetry (ITC) data from Majumdar et. al. suggested a two-site binding model through negative cooperativity between the ligand and CAV.20 For dimerized ligand 1, one half of the dimer interacting with the first binding site could bring the second half of the ligand close enough to the second binding site to force binding via increased effective concentration, overcoming the previously observed negative coopertivity. Based on the large positive ΔS2, Majumdar et. al. also observed that binding to the second site must be entropically driven, likely by the “disruption of cavolin oligomers upon binding.”20 This matches our observation that deoligomerization only occurs when ligand 1 is dimerized such that the second binding site is filled. Thus data from the dimerized ligand 1 reinforces and clarifies the conclusions drawn from previous research on ligand 4.
As the first validated synthetic ligand for CAV, ligand 1 contributes to the ongoing debate over the existence and identity of a specific CAV binding motif that mediates the interaction between CAV and its many binding partners. Research in 1997 by Couet et. al. proposed that this binding motif consisted of a characteristic spacing of multiple aromatic amino acids as determined by examining the sequencing results of multiple phage display libraries that had been subject to selections against CAV residues (1-101).32 In subsequent research, many (but not all) CAV binding proteins were found to contain similar sequences.1 However, a 2012 paper by Byrne et. al. used statistical methods to investigate the incidence of such characteristic aromatic spacing in CAV binding proteins and in the proteome at large and found no statistical enrichment of these aromatic sequences in CAV binding proteins.33 The ligand 1 sequence does not have multiple aromatic residues and thus does not conform to the aromatic richness and spacing of the canonical CAV binding motif, suggesting that whether this motif mediates CAV binding for some proteins or not, it is clearly not the only way in which CAV may be targeted. Since CAV has many binding partners, the interaction between CAV binding motifs and the CAV scaffolding domain likely evolved as a panoply of low affinity interactions such that CAV could maintain its multiple partners.34 As a high affinity ligand, 1 would not be expected to conform to this pre-existing pattern. Notably, the initial phage display process produced a higher affinity binder with fewer aromatic residues, and subsequent optimization resulted in the removal of an additional aromatic residue.
CONCLUSION
The CAV ligand WL47 (1) is 80% smaller in length and has 7500-fold greater affinity than the original T20 parent sequence, while also demonstrating selectivity and deoligomerization activity. This ligand was developed through a generalizable peptide ligand optimization process involving iterative library design, synthesis, and screening. Ligand 1 provides a new tool for research that will allow investigators to probe the functions of CAV with unprecedented selectivity, and its application to live cell assays is currently underway.
Beyond providing a new tool, the development of ligand 1 demonstrates the ways in which small molecule medicinal chemistry principles can be successfully applied to the creation of peptide ligands. Recent novel peptide ligands have been derived exclusively from direct mimicry of existing sequences from known proteins.35,36 Conversely, the creation of novel small molecule drugs has involved greater experimentation and rational design. The application of these varied strategies ultimately allowed us to progress from our lead compound T20, which had an interaction with CAV too weak to be successfully measured by ITC, to a ligand sequence that binds CAV with a KD of 23 nM as measured by fluorescence anisotropy experiments.
Supplementary Material
Acknowledgments
We are grateful to the NIH, National Institute of General Medical Sciences (R01GM078528-01, 1R0RGM100700-01A1) and to Vertex Pharmaceuticals for financial support of Amanda J. H. Gilliam through a Vertex Scholarship. We thank Michael R. Lawson, Tivoli J. Olsen, Mark B. Richardson and Timothy R. Valentic for discussions and helpful advice.
ABBREVIATIONS USED
- CAV
caveolin-1
- CAV(1-104)
truncated caveolin-1
- CAV(FLV)
a soluble, full length variant of caveolin-1
- DLS
dynamic light scattering
- ITC
isothermal calorimetry
Footnotes
Supporting Information.
Experimental Materials and Methods.
Variation in peptide concentration between SPOTs.
Sequences for peptide library corresponding to Figure 4.
Rationale for removal of Gly9 and Phe15 and investigation of Cys14 supporting dimerization hypothesis, HPLC traces demonstrating 1 and Mantyl-1 purity.
Supplemental References
References
- 1.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of Caveolae and Caveolins in Health and Disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 2.Shvets E, Ludwig A, Nichols BJ. News from the Caves: Update on the Structure and Function of Caveolae. Curr Opin Cell Biol. 2014;29:99–106. doi: 10.1016/j.ceb.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Strålfors P. Caveolins and Caveolae, Roles in Insulin Signalling and Diabetes. Adv Exp Med Biol. 2012;729:111–126. doi: 10.1007/978-1-4614-1222-9_8. [DOI] [PubMed] [Google Scholar]
- 4.Zou H, Stoppani E, Volonte D, Galbiati F. Caveolin-1, Cellular Senescence and Age-Related Diseases. Mech Ageing Dev. 2011;132:533–542. doi: 10.1016/j.mad.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machado FS, Rodriguez NE, Adesse D, Garzoni LR, Esper L, Lisanti MP, Burk RD, Albanese C, Van Doorslaer K, Weiss LM, Nagajyothi F, Nosanchuk JD, Wilson ME, Tanowitz HB. Recent Developments in the Interactions between Caveolin and Pathogens. Adv Exp Med Biol. 2012;729:65–82. doi: 10.1007/978-1-4614-1222-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, Shirani J, Razani B, Tang B, Jelicks LA, Factor SM, Weiss LM, Tanowitz HB, Lisanti MP. Caveolin-1/3 Double-Knockout Mice Are Viable, but Lack Both Muscle and Non-Muscle Caveolae, and Develop a Severe Cardiomyopathic Phenotype. Am J Pathol. 2002;160:2207–2217. doi: 10.1016/S0002-9440(10)61168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP. Caveolin-1 Mutations (P132L and Null) and the Pathogenesis of Breast Cancer: Caveolin-1 (P132L) Behaves in a Dominant-Negative Manner and Caveolin-1 (−/−) Null Mice Show Mammary Epithelial Cell Hyperplasia. Am J Pathol. 2002;161:1357–1369. doi: 10.1016/S0002-9440(10)64412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlegel A, Lisanti MP. A Molecular Dissection of Caveolin-1 Membrane Attachment and Oligomerization - Two Separate Regions of the Caveolin-1 C-Terminal Domain Mediate Membrane Binding and Oligomer/oligomer Interactions in Vivo. J Biol Chem. 2000;275:21605–21617. doi: 10.1074/jbc.M002558200. [DOI] [PubMed] [Google Scholar]
- 9.Hailstones D, Sleer LS, Parton RG, Stanley KK. Regulation of Caveolin and Caveolae by Cholesterol in MDCK Cells. J Lipid Res. 1998;39:369–379. [PubMed] [Google Scholar]
- 10.Stuart ES, Webley WC, Norkin LC. Lipid rafts, Caveolae, Caveolin-1, and Entry by Chlamydiae into Host Cells. Exp Cell Res. 2003;287:67–78. doi: 10.1016/s0014-4827(03)00059-4. [DOI] [PubMed] [Google Scholar]
- 11.Lajoie P, Nabi IR. Lipid Rafts, Caveolae, and Their Endocytosis. Int Rev Cell Mol Biol. 2010;282:135–163. doi: 10.1016/S1937-6448(10)82003-9. [DOI] [PubMed] [Google Scholar]
- 12.Murata M, Peränen J, Schreiner R, Wieland F, Kurzchalia TV, Simons K. VIP21/caveolin Is a Cholesterol-Binding Protein. Proc Natl Acad Sci. 1995;92:10339–10343. doi: 10.1073/pnas.92.22.10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothberg KG, Heuser JE, Donzell WC, Ying Y-S, Glenney JR, Anderson RGW. Caveolin, a Protein Component of Caveolae Membrane Coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 14.Sargiacomo M, Scherer PE, Tang Z, Kübler E, Song KS, Sanders MC, Lisanti MP. Oligomeric Structure of Caveolin: Implications for Caveolae Membrane Organization. Proc Natl Acad Sci USA. 1995;92:9407–9411. doi: 10.1073/pnas.92.20.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harvey RD, Calaghan SC. Caveolae Create Local Signalling Domains through Their Distinct Protein Content, Lipid Profile and Morphology. J Mol Cell Cardiol. 2012;52:366–375. doi: 10.1016/j.yjmcc.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu P, Rudick M, Anderson RG. Multiple Functions of Caveolin-1. J Biol Chem. 2002;277:41295–41298. doi: 10.1074/jbc.R200020200. [DOI] [PubMed] [Google Scholar]
- 17.Hovanessian AG, Briand JP, Said EA, Svab J, Ferris S, Dali H, Muller S, Desgranges C, Krust B. The Caveolin-1 Binding Domain of HIV-1 Glycoprotein gp41 Is an Efficient B Cell Epitope Vaccine Candidate against Virus Infection. Immunity. 2004;21:617–627. doi: 10.1016/j.immuni.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Sinha B, Köster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler-Browne G, Vedie B, Johannes L, Morone N, Parton RG, Raposo G, Sens P, Lamaze C, Nassoy P. Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell. 2011;144:402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nassoy P, Lamaze C. Stressing Caveolae New Role in Cell Mechanics. Trends Cell Biol. 2012;22:381–389. doi: 10.1016/j.tcb.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Majumdar S, Hajduczki A, Vithayathil R, Olsen TJ, Spitler RM, Mendez AS, Thompson TD, Weiss GA. In Vitro Evolution of Ligands to the Membrane Protein Caveolin. J Am Chem Soc. 2011;133:9855–9862. doi: 10.1021/ja201792q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Champagne K, Shishido A, Root MJ. Interactions of HIV-1 Inhibitory Peptide T20 with the gp41 N-HR Coiled Coil. J Biol Chem. 2009;284:3619–3627. doi: 10.1074/jbc.M809269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashkenazi A, Wexler-Cohen Y, Shai Y. Multifaceted Action of Fuzeon as Virus–cell Membrane Fusion Inhibitor. Biochim Biophys Acta. 2011;1808:2352–2358. doi: 10.1016/j.bbamem.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 23.Frank R. Spot-Synthesis: An Easy Technique for the Positionally Addressable, Parallel Chemical Synthesis on a Membrane Support. Tetrahedron. 1992;48:9217–9232. [Google Scholar]
- 24.Weiser A, Or-Guil M, Tapia V, Leichsenring A, Schuchhardt J, Frömmel C, Volkmer-Engert R. SPOT Synthesis: Reliability of Array-based Measurement of Peptide Binding Affinity. Anal Biochem. 2005;342:300–300. doi: 10.1016/j.ab.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 25.Kramer A, Reineke U, Dong L, Hoffmann B, Hoffmüller U, Winkler D, Volkmer-Engert R, Schneider-Mergener J. Spot Synthesis: Observations and Optimizations. J Pept Res. 1999;54:319–327. doi: 10.1034/j.1399-3011.1999.00108.x. [DOI] [PubMed] [Google Scholar]
- 26.Ritchie TJ, Macdonald SJ. The Impact of Aromatic Ring Count on Compound Developability - Are Too Many Aromatic Rings a Liability in Drug Design? Drug Discovery Today. 2009;14:1011–1020. doi: 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 27.Peptide Screening Tools at www.genscript.com http://www.genscript.com/peptide_screening_tools.html (accessed Sep 12, 2012)
- 28.Anumula KR, Schulz RP, Back N. Fluorescent (Mantyl) Tag for Peptides: Its Application in Subpicomole Determination of Kinins. Peptides. 1992;13:663–669. doi: 10.1016/0196-9781(92)90170-8. [DOI] [PubMed] [Google Scholar]
- 29.Pollard TD. A Guide to Simple and Informative Binding Assays. Mol Biol Cell. 2010;21:4061–4067. doi: 10.1091/mbc.E10-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss JN. The Hill Equation Revisited: Uses and Misuses. FASEB J. 1997;11:835–841. [PubMed] [Google Scholar]
- 31.Levin AM, Coroneus JG, Cocco MJ, Weiss GA. Exploring the Interaction between the Protein Kinase A Catalytic Subunit and Caveolin-1 Scaffolding Domain with Shotgun Scanning, Oligomer Complementation, NMR, and Docking. Protein Sci. 2006;15:478–486. doi: 10.1110/ps.051911706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of Peptide and Protein Ligands for the Caveolin-Scaffolding Domain. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 33.Byrne DP, Dart C, Rigden DJ. Evaluating Caveolin Interactions: Do Proteins Interact with the Caveolin Scaffolding Domain through a Widespread Aromatic Residue-Rich Motif? PLoS One. 2012;7:e44879. doi: 10.1371/journal.pone.0044879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levin AM, Murase K, Jackson PJ, Flinspach ML, Poulos TL, Weiss GA. Double Barrel Shotgun Scanning of the Caveolin-1 Scaffolding Domain. ACS Chem Biol. 2007;2:493–500. doi: 10.1021/cb700055t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaltiel-Karyo R, Frenkel-Pinter M, Egoz-Matia N, Frydman-Marom A, Shalev DE, Segal D, Gazit E. Inhibiting α-Synuclein Oligomerization by Stable Cell-Penetrating β-Synuclein Fragments Recovers Phenotype of Parkinson’s Disease Model Flies. PLoS One. 2010;5:e13863. doi: 10.1371/journal.pone.0013863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saludes JP, Morton LA, Ghosh N, Beninson LA, Chapman ER, Fleshner M, Yin H. Detection of Highly Curved Membrane Surfaces Using a Cyclic Peptide Derived from Synaptotagmin-I. ACS Chem Biol. 2012;7:1629–1635. doi: 10.1021/cb3002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.