

Abstract
Eukaryotic cells, including some human cancers, that lack telomerase can sometimes maintain telomeres by using recombination. It was recently proposed that recombinational telomere elongation (RTE) in a telomerase-deletion mutant of the yeast Kluyveromyces lactis occurs through a roll-and-spread mechanism as described in our previous work. According to this model, a tiny circle of telomeric DNA is copied by a rolling-circle mechanism to generate one long telomere, the sequence of which is then spread to all other telomeres by gene-conversion events. In support of this model, we demonstrate here that RTE in K. lactis occurs by amplification of a sequence originating from a single telomere. When a mutationally tagged telomere is of normal length, its sequence is spread to all other telomeres at a frequency (≈10%) consistent with random selection among the 12 telomeres in the cell. However, when the mutationally tagged telomere is considerably longer than other telomeres, cellular senescence is partially suppressed, and the sequence of the tagged telomere is spread to all other telomeres in >90% of cells. Strikingly, the transition between a state resistant to recombination and a state capable of initiating recombination is abrupt, typically occurring when telomeres are ≈3–4 repeats long. Last, we show that mutant repeats that are defective at regulating telomerase are also defective at regulating telomere length during RTE.
Keywords: alternative lengthening of telomeres, rolling circle
Telomeres are the specialized DNA–protein complexes at the ends of linear DNA molecules (1, 2). They are composed of tandem arrays of short, direct repeats and the specialized proteins that cap the telomeres and prevent them from eliciting the repair responses that are normally activated by broken DNA ends. Telomeres shorten with each cell division because of the end-replication problem of DNA polymerase (3, 4). This shortening is normally prevented by the ribonucleoprotein enzyme telomerase, which adds repeats onto telomeric ends by copying an RNA template.
The absence of a telomere-maintenance pathway leads to replicative senescence in most human cells (5). In contrast, almost all human cancers maintain telomeres, primarily through the presence of telomerase (6), but sometimes through activation of a recombinational mechanism (7). Yeasts, most notably Saccharomyces cerevisiae and Kluyveromyces lactis, have been useful models for understanding recombinational telomere elongation (RTE). Yeast mutants lacking telomerase undergo growth senescence, with most cells ultimately dying (8–10). Postsenescence survivors that emerge have elongated telomeres. Various mechanisms, including inter- and intratelomeric events, have been proposed for how recombination elongates telomeric-repeat arrays (8, 9, 11). We have previously proposed the roll-and-spread model to account for RTE in K. lactis (12). In this model, a tiny telomeric circle is used as a template for a rolling-circle event that elongates one telomere in a senescing cell. The elongated telomere's sequence is then spread to all other telomeres by intertelomeric gene conversions. Consistent with this model, DNA circles containing telomeric repeats can promote RTE in K. lactis (12, 13). In this report, we use K. lactis telomerase-deletion mutants that have a single telomere with mutationally tagged repeats to test key predictions of the roll-and-spread model.
Materials and Methods
Strains and Construction of Mutant Telomeres. All K. lactis strains used are derivatives of 7B520 (14). The ter1-deletion mutant was described in ref. 15. The plasmid containing TER1 (pJR31) is a derivative of pKL316 (15) that carries the HIS3 gene. Construction of cloned K. lactis telomeres containing 11.5 Bcl repeats and 11.5 Kpn repeats are described in refs. 16 and 17. Loss of the TER1 plasmid produced telomere shortening that eventually triggered RTE.
To construct an elongated telomere containing Bcl repeats, we took advantage of the observation that use of the QuikChange mutagenesis kit (Stratagene) for altering the sequence of a cloned K. lactis telomere often generated cloned telomeres with altered numbers of repeats (D. H. Underwood, W. McRae and M.J.M., unpublished observations). Sequences of the K. lactis telomeric repeat and base changes in the Bcl- and Kpn-mutant repeats are given in Fig. 1A. After two cycles of “mutagenesis” using oligonucleotides matching the Bcl repeat, a telomere clone with ≈11.5 telomeric repeats was converted into a clone with ≈40 repeats (≈1,000 bp).
Fig. 1.

Spreading of a telomeric sequence from a single telomere source during RTE. (A) Sequence of the K. lactis telomeric repeat and base changes present in the Bcl- and Kpn-mutant repeats. (B) Experimental outline. K. lactis ter1Δ cells carrying a HIS3 TER1 plasmid (15) were transformed with URA3-tagged telomeric fragments (18) containing ≈11 and ≈40 Bcl telomeric repeats. Bcl and WT repeats are shown as gray and white boxes, respectively. Loss of the TER1 plasmid produced telomere shortening and RTE. After RTE, spreading of the Bcl repeats to all other telomeres was observed in a minority of cases in cells originally containing the normal-length Bcl telomere and, in the great majority of cases, in cells originally containing the extra-long Bcl telomere. (C) Growth scores of ter1Δ strains containing one tagged telomere, either normal-length Bcl (♦), long Bcl (⋄), or long Kpn (•), after loss of TER1. Growth scores are based on average size and appearance of colonies on rich-media plates during serial restreaking done every 2 days (9). A score of 4 indicates growth indistinguishable from WT cells and a score of 1 indicates very small, rough colonies and typically high cell mortality. (D) BsrBI-digested genomic DNA of a strain with a normal-length Bcl telomere (lane 1) and a strain with an extra-long Bcl telomere (lane 2) before loss of telomerase. The normal-length Bcl telomere is among other telomeres in the major band, which is <500 bp. The extra-long Bcl telomere is indicated by the arrow. (E) Southern blot hybridized to a telomeric probe of seven postsenescence survivors from the normal-length Bcl strain. Genomic DNAs are shown digested with EcoRI (-) and EcoRI/BclI(+). (F) Nine postRTE isolates from the extra-long Bcl strain. Digests are as in E.
Transformation of K. lactis Cells with Mutant Telomeric Repeats. K. lactis ter1Δ cells carrying a HIS3 TER1 plasmid (15) were transformed with URA3-tagged telomeric fragments (18) containing ≈11 and ≈40 Bcl-telomeric repeats as outlined in Fig. 1B. Ura+ colonies were then examined by Southern blotting to confirm that the URA3-tagged telomere had replaced a single native telomere by means of subtelomeric recombination.
DNA and Hybridization Analysis. Hybridizations were carried out as described in ref. 19. The telomeric-hybridization probe was the Klac1–25 oligonucleotide (ACGGATTTGATTAGGTATGTGGTGT). BsrBI–BclI digests were separated on 4% NuSieve 3:1 (Cambrex Bio Science, Rockland, ME) agarose.
Results
The roll-and-spread model (12) predicts that K. lactis telomerase-deletion mutants will produce postsenescence survivors with elongated telomeres that are derived from a single source. We therefore tested whether a sequence from a single telomere can spread to all other telomeres during survivor formation. The strategy for doing this is shown in Fig. 1B. K. lactis ter1Δ cells containing TER1 on a plasmid were first constructed to have a single telomere composed of mutationally tagged but phenotypically normal “Bcl” telomeric repeats (15, 16, 20) (Fig. 1 A). We predicted that, if a sequence from a single, randomly selected telomere became amplified and spread to all telomeres in the cell, approximately 1 in 12 postsenescence survivors would have Bcl repeats at all 12 telomeres. When the TER1 plasmid was lost, the cells containing the Bcl telomere underwent gradual telomere shortening and growth senescence indistinguishable from typical ter1Δ mutants (Fig. 1C and data not shown). Similarly, the production of well growing postsenescence survivors that appeared after 75–100 cell divisions also appeared to be unaltered. Examination of telomere structure in survivors derived from the strain containing the Bcl telomere revealed that 175 of 195 (90%) had restriction patterns that were resistant to digestion with BclI (Fig. 1E). This finding indicates that Bcl repeats were absent or rare in the elongated telomeres. The majority of these survivors retained the single URA3-tagged telomere but a minority had lost it completely, presumably through gene conversion (data not shown) (18). In sharp contrast, 20 of 195 (10%) of the survivors had all of their telomeric fragments shortened by cleavage with BclI (Fig. 1E and Table 1), indicating that each of the telomeres in these survivors had acquired Bcl-telomeric repeats. As is typical with K. lactis ter1Δ postsenescence survivors, the extent of telomere elongation was highly variable between different telomeres and different survivors. Where spreading of Bcl repeats had occurred, BclI digestion was often seen to remove up to several hundred base pairs of DNA from telomeric fragments. Acquisition of Bcl repeats by a telomere originally lacking them occurred through both inter- and subtelomeric events. The former were detectable as telomeric fragments retaining WT repeats after BclI cleavage (Fig. 1E), whereas the latter involved acquisition of the URA3 gene along with the Bcl repeats (data not shown). These results demonstrate that a sequence from one telomere can be spread to all other telomeres and supports the argument that this routinely occurs during the formation of a postsenescence survivor.
Table 1. Frequency of spreading of sequence from a single tagged telomere to all other telomeres in K. lactis cells lacking telomerase.
| Tagged telomere | No. of clones | No. of clones with spreading of tag (%) |
|---|---|---|
| Bcl | 195 | 20 (10) |
| Long Bcl | 78 | 74 (94) |
| Long Kpn | 40 | 38 (95) |
K. lactis ter1Δ cells initially containing a single telomere tagged with mutant repeats were grown by serial streaking through 75–100 cell divisions on rich-media plates and examined for spreading of the mutant repeats to all other telomeres.
Another important prediction of the roll-and-spread model is that a key, and possibly rate-limiting, intermediate in the formation of a postsenescence survivor is the generation of a single elongated telomere by rolling-circle gene conversion. We therefore examined the effect of a single elongated telomere on a telomerase-deletion mutant. As described in Materials and Methods, it was possible to adapt a common mutagenesis procedure to randomly alter the number of telomeric repeats present in a cloned K. lactis telomere and generate Bcl telomeres of various sizes. A Bcl telomere engineered to be ≈1,000-bp long (≈2–3 times normal length) was transformed into K. lactis cells, where it replaced a single native telomere (Fig. 1D). Subclones from 10 independent transformants were then screened for loss of the plasmid containing TER1. Serial passaging of subclones of these transformants on solid media demonstrated that the subclones exhibited early-senescence phenotypes similar to other ter1Δ cells. However, the subclones typically did not exhibit the severe growth defects characteristic of late senescence of ter1Δ cells (Fig. 1C). This result indicated that the single long telomere partially suppressed senescence. Also, unlike the case with a normal-length Bcl telomere, cells starting with the long Bcl telomere appeared to be more resistant to loss of the subtelomeric URA3 tag (data not shown). We next examined the telomere structure of the subclones ≈75–100 cell divisions after loss of the TER1 gene, a point equivalent to that at which better growing postsenescence survivors emerge in typical ter1Δ cells. Our results showed that 94% (73 of 78) of the subclones had telomeres that were all cleaved with BclI. No spreading of Bcl repeats occurred if cells with the long telomere retained TER1 function (data not shown). These results demonstrated that a sequence from a single elongated telomere is highly preferentially spread to all other telomeres in ter1Δ cells. The suppression of senescence that is caused by the single long telomere is presumably accounted for by the apparent ease with which the sequence from one long telomere can spread to other telomeres.
The presence of some WT repeats at the basal regions of many telomeres, even after spreading of Bcl repeats had occurred (visible as telomeric hybridization after BclI digestion in clones 2, 5, and 7 in Fig. 1E and clones 1–9 in Fig. 1F), indicated that shortening WT telomeres had often initiated recombination events before complete loss of telomeric repeats. We determined the number of residual WT repeats that were present by examining DNA digested with BsrBI and BclI. The latter removes the acquired Bcl repeats and the former cuts 10 of 12 telomeres in K. lactis at a subtelomeric position just 3-bp internal to the start of the telomeric-repeat array (Fig. 2A). Results from this analysis (Fig. 2 B and C) showed that there was a clearly favored size at which telomeres recombined. In most clones examined, both from Bcl and long-Bcl backgrounds, most recombinant telomeres contained 3.5 WT repeats (detectable as a 93-bp band). Whereas clones from the long-Bcl background often had one or more telomere with 4.5 or 5.5 basal WT repeats, no clones derived from the normal-length-Bcl background had any telomeres with >3.5 WT repeats. These results are consistent with observations that short telomeres in yeast are prone to recombination (18, 21). These findings suggest further that the capping function that normally protects a K. lactis telomere from acquiring a sequence by means of recombination is lost quite abruptly as a telomere shortens to below ≈4 repeats.
Fig. 2.

Determination of the number of basal WT repeats remaining after spread of Bcl repeats from RTE. (A) Schematic of the fragments produced in B and C. Bcl and WT repeats are shown as gray and white boxes, respectively. Sequences shown in capital letters demarcate the BsrBI/BclI fragments, with the Bcl mutation underlined. A fragment containing one full WT repeat is 43 bp long, with additional repeats increasing in size by multiples of 25 bp. (B) Southern blot using a telomeric probe of BsrBI/BclI-digested genomic DNA from eight postsenescence survivors from the normal-length Bcl strain. Clone 3 did not spread Bcl repeats and has telomeres resistant to BclI cleavage. All other clones were among those in which all telomeres had acquired Bcl repeats. (C) Southern blot of digests, equivalent to B, of genomic DNA from post-RTE isolates from the extra-long Bcl strain, all of which had spread Bcl repeats.
We next examined the consequences to ter1Δ cells of a single long telomere composed of Kpn-mutant repeats (Fig. 1 A). The ter1-Kpn mutation is a member of a group of ter1-template mutations that alter a domain of the telomeric repeat that is adjacent to, but not within, the telomeric binding site of the Rap1 protein (17, 22). These mutations produce extreme telomere elongation but only after a period of growth, often very extended, in which telomeres are somewhat shorter than normal. Unlike the long Bcl telomere, which is only transiently long, a Kpn telomere in a cell with WT telomerase is stably long, with a WT telomeric-repeat array of nearly normal length being added onto the dysfunctional array of mutant repeats (17). As is seen with the long Bcl telomere, when the plasmid carrying TER1 was lost from cells carrying the long Kpn telomere, growth senescence was partially suppressed (Fig. 1C), and spreading of the tagged sequence to all other telomeres occurred at a 95% frequency (Table 1 and Fig. 3). Furthermore, telomeres that had acquired Kpn repeats through spreading retained similar numbers of basal WT repeats as did those with the long-Bcl-telomere experiment (data not shown). Strikingly, however, the telomeres in ter1Δ cells where Kpn repeats had spread to all telomeres were very long, extending to limit mobility in gels. This result is very different from cells of ter1Δ postsenescence survivors with all telomeres composed of Bcl or WT repeats, where such extreme elongation was never observed. These results indicate that Kpn repeats are defective, not only in the ability to regulate telomerase, but also in the ability to regulate RTE.
Fig. 3.
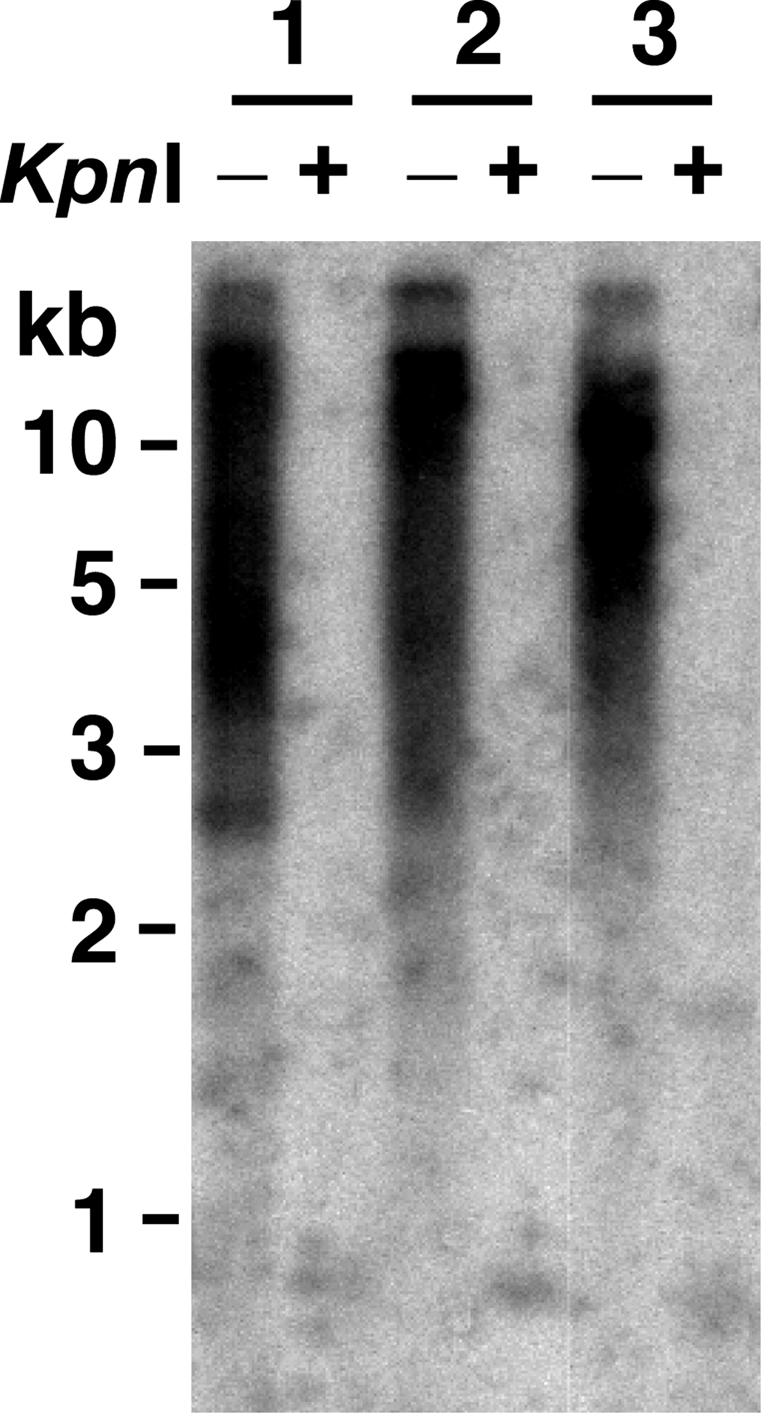
Greatly elongated telomeres produced by RTE in cells that have Kpn repeats. Shown is a Southern blot, hybridized to a telomeric probe, of three post-RTE clones derived from a strain originally having one Kpn telomere. Genomic DNAs are shown digested with EcoRI (-) and EcoRI/KpnI (+).
Discussion
Our results here support the argument that the telomere elongation that generates postsenescence survivors in K. lactis telomerase-deletion mutants usually occurs through a single telomere source. When all telomeres are the same size, a sequence from one particular telomere spreads to all others at a frequency (10%) proportional to its abundance (1 of 12 telomeres). In contrast, when a telomere is substantially longer than other telomeres, its sequence is spread to all other telomeres in 90–95% of cell lineages. These results are fully consistent with the roll-and-spread model, which proposes that a single, tiny telomeric circle is copied by a rolling-circle event to generate the first long telomere and that other telomeres become elongated by copying the sequence from that telomere. Our results are, however, inconsistent with other models of RTE that have been proposed (9, 11, 23). RTE based solely on intratelomeric-recombination events cannot account for the spread of Bcl repeats to other telomeres. And models based on intertelomeric recombination in the absence of a rolling-circle event cannot easily explain how a cell with only extremely short telomeres could specifically amplify and spread a sequence from only one of those telomeres to all others.
Whereas our data here do not directly address the possible involvement of telomeric circles in RTE, substantial other circumstantial evidence suggests this possibility. Survivors generated in ter1Δ cells with two kinds of telomeric repeats were found to have telomeres with repeating patterns common to most, or all, elongated telomeres. The 100-bp periodicity of some of these patterns suggested the possibility that they might have been generated by copying telomeric circles as small as 100 bp (12). Telomeric circles of this size that were built in vitro and transformed into K. lactis cells promoted RTE to lengths (hundreds of base pairs) similar to that seen in ter1Δ cells (13). Other experiments (12) showed that single molecules of 1.6-kb circles containing telomeric repeats and a selectable marker gene promoted very long (>10 kb) extensions of telomeres. Although small telomeric circles have yet to be observed in ter1Δ cells, both double-stranded and single-stranded telomeric circles as small as ≈100 bp (or nt) have been shown to be very abundant in a K. lactis mutant with long, dysfunctional telomeres (C. Groff-Vindman, A. Cesare, S. Natarajan, J. Griffith, and M.J.M., unpublished data). How such small telomeric circles form is not clear. However, their dependence on RAD52 indicates that they are a product of homologous recombination. One possible mechanism for the formation of these circles is an intratelomeric loop-out initiated by strand invasion of the 3′ end of a telomere into its own, more internal, telomeric repeats. Such a mechanism has been proposed to underlie telomere rapid deletion (24).
Our results suggest that spreading of a sequence from a long telomere to a short telomere is initiated by the transition of the short telomere to a recombination-prone state. This transition occurs within a relatively narrow telomeric size range, at ≈4 repeats in length. This size is substantially shorter than the minimum observed size of telomeres in TER1 cells, indicating that this transition is different from the transition of a shortening telomere to a telomerase-accessible state (21). A telomere in a recombination-prone state presumably permits the resectioning of the 5′ strand that occurs at DNA double-strand breaks (25). The exposed 3′ tail of the short telomere could then bind recombination proteins, including Rad51 and Rad52, and strand-invade another chromosome end. Extension of the invaded 3′ end by a DNA polymerase would then copy the sequence of the longer telomere onto the shorter telomere.
The ease with which the sequence of a long telomere can be spread to other telomeres appears to affect the nature of the recombination events involved. Among ter1Δ clones in which spreading of Bcl repeats has occurred, fewer instances of co-spreading of the subtelomeric URA3 marker gene occur in cells with a long Bcl telomere than in cells with a short Bcl telomere (data not shown). Also, the other telomeres in a long-Bcl-telomere strain tended to retain a slightly higher average number of WT repeats (Fig. 2C). These data likely indicate that a senescing ter1Δ strain without a preexisting long telomere often suffers appreciable telomeric-sequence loss between the point when the transition to a recombination-prone state occurs and when recombination actually succeeds at lengthening the telomere. We predict that the presence of one long telomere in senescing ter1Δ cells bypasses the need for a rolling-circle event and often permits spreading to occur at an earlier point than would otherwise be the case. This would explain why the presence of a single long telomere reduces the severity of senescence in ter1Δ cells.
How telomeres are capped to prevent them from eliciting a DNA-repair response is not well understood. Proteins known to be involved in capping in yeast include the single-strand telomere-binding complex Cdc13–Stn1–Ten1 (26–29) and the double-strand telomere-binding protein Rap1 (30). Mutations in CDC13 and STN1 have recently been shown to cause or contribute to the ability of telomeres to undergo RTE even in the presence of telomerase (ref. 31 and S. Iyer and M.J.M., unpublished data). Capping at some telomeres, including those of mammalian cells, appears to also involve formation of a t-loop, a displacement loop formed by strand-invasion of the 3′ overhang into a more internal double-stranded telomeric sequence (32). Why reducing telomere length in K. lactis to ≈100 bp causes a transition to a recombination-prone state is not known. We suggest that the loss of a higher-order structure such as a foldback or t-loop (32–34) may underlie the abrupt switch to the recombination-prone state. A related question is whether the transition to a recombination-prone state represents a complete or partial loss of the protective telomere-capping function. A 100-bp-long telomere should still be able to bind telomere-binding proteins that might partially protect it from initiating a recombination event and perhaps completely block it from participating in nonhomologous-end joining. Retention of some capping function would help explain why covalent fusions between telomeres appear to be rare in ter1Δ cells, although they are common in mutants with terminal repeats defective at binding Rap1 (30). The transition to a fully uncapped state is therefore likely to require even greater loss of telomeric sequence than is required for the transition to a recombination-prone state.
The lengthened telomeres present in K. lactis ter1Δ survivors are seldom longer than 1 kb (9). This finding indicates that RTE is regulated in these cells in a way that prevents runaway telomere elongation. Two things expected to contribute to this regulation are the length-dependence of the recombination-prone state and the limited lengthening capacity of the key events governing survivor formation. Because a recombination-prone state is produced in a telomere composed of WT repeats only when that telomere is reduced less than ≈4 repeats in size, any telomere that is appreciably lengthened to above this size should be resistant to any further recombination. This finding suggests that telomere length in ter1Δ survivors is largely a consequence of how much a telomere can be elongated by a single recombination event. According to the roll-and-spread model, the critical event affecting telomere length in survivors is the length of sequence that could be generated by one instance of rolling-circle replication of a telomeric circle. Data with transformed telomeric circles suggest that very small telomeric circles may be much poorer templates for extensive DNA synthesis than are larger circles. Whereas K. lactis telomeres are routinely extended by >10 kb in cells transformed with a 1.6-kb telomere/URA3 circle (12), they are extended by only hundreds of base pairs when acquiring a sequence from a 100-nt telomeric circle (13).
The highly elongated telomeres present in ter1Δ cells that have spread mutant-Kpn-telomeric repeats demonstrate that the Kpn mutation disrupts a mechanism that limits telomere length in normal ter1Δ survivors. The Kpn mutant is a member of the delayed-elongation class of mutations within the K. lactis telomeric repeat that cluster to one side of the Rap1-binding site and disrupt the ability of the internal region of the telomere (away from the terminus) to regulate the addition of sequences by telomerase at the end (17, 22). The nature of the defect in Kpn repeats is unknown, but it does not involve a disruption of Rap1 binding to the telomeric repeat (35). It is likely that the Kpn mutation leads to a disruption in telomere structure that permits increased access by both telomerase and proteins that bring about recombination. We propose that telomeres composed of Kpn repeats are disrupted in their ability to form the higher-order structures needed to protect them from initiating recombination, and that this structure is necessary but not sufficient for generating the telomere state (formed by relatively long telomeres) that is resistant to being elongated by telomerase. With such a defect, even long telomeres composed of Kpn repeats would have a capping defect and be prone to continued recombination events that could lengthen them uncontrollably. Interestingly, the long telomeres of ter1Δ cells with Kpn repeats resemble those of human alternative-lengthening-of-telomeres (ALT) cells (36). This finding may suggest that telomere-capping defects such as defects in certain telomere-binding proteins may be required for triggering the ALT state.
In summary, K. lactis telomeres are likely to undergo two distinct transitions that occur at different lengths and that regulate different aspects of telomere function. At ≈4 repeats, telomeres can form a structure that is resistant to initiating recombination but is still accessible to telomerase. Only at a much larger size (likely ≥20 repeats), would they be able to assume a structure that is also inaccessible to telomerase. With these two transitions, a telomere can retain a protective cap that distinguishes it from a broken end over a wide range of sizes that could still allow access by telomerase.
Whether the copying of telomeric circles or the spreading of a sequence from a single telomere source will be found to occur in other instances of RTE is not known. Copying telomeric circles has been proposed to be responsible for the sudden emergence of type-II survivors (which have elongated tracts of telomeric repeats) in S. cerevisiae (37) and could also account for the telomere-Y′-element amplifications of type-I survivors (8). The spreading of a marker gene inserted into a telomeric sequence to some other telomeres has been observed in human ALT cells (38). The production of telomeric circles of a wide range of sizes has recently been found to be a characteristic of human ALT cells (39, 40). This finding clearly suggests that circles could contribute to RTE in some human cancers. Much further work will be needed to fully understand the similarities and differences of RTE in its various guises.
Acknowledgments
We thank Will McRae for technical support and C. Groff-Vindman for comments on the manuscript. This work was supported by National Institutes of Health Grant GM61645-01.
Author contributions: Z.T. and M.J.M. designed research; Z.T., K.N., C.D., and M.J.M. performed research; Z.T. and M.J.M. analyzed data; and Z.T. and M.J.M. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: RTE, recombinational telomere elongation; ALT, alternative lengthening of telomeres.
References
- 1.McEachern, M. J., Krauskopf, A. & Blackburn, E. H. (2000) Annu. Rev. Genet. 34, 331-358. [DOI] [PubMed] [Google Scholar]
- 2.Chan, S. R. & Blackburn, E. H. (2004) Philos. Trans. R. Soc. London B 359, 109-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harley, C. B., Futcher, A. B. & Greider, C. W. (1990) Nature 345, 458-460. [DOI] [PubMed] [Google Scholar]
- 4.Hastie, N. D., Dempster, M., Dunlop, M. G., Thompson, A. M., Green, D. K. & Allshire, R. C. (1990) Nature 346, 866-868. [DOI] [PubMed] [Google Scholar]
- 5.Shay, J. W. & Wright, W. E. (2001) Science 291, 839-840. [DOI] [PubMed] [Google Scholar]
- 6.Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, M. D., Ho, P. L., Coviello, G. M., Wright, W. E., Weinrich, S. L. & Shay, J. W. (1994) Science 266, 2011-2015. [DOI] [PubMed] [Google Scholar]
- 7.Henson, J. D., Neumann, A. A., Yeager, T. R. & Reddel, R. R. (2002) Oncogene 21, 598-610. [DOI] [PubMed] [Google Scholar]
- 8.Lundblad, V. & Blackburn, E. H. (1993) Cell 73, 347-360. [DOI] [PubMed] [Google Scholar]
- 9.McEachern, M. J. & Blackburn, E. H. (1996) Genes Dev. 10, 1822-1834. [DOI] [PubMed] [Google Scholar]
- 10.Teng, S. C. & Zakian, V. A. (1999) Mol. Cell. Biol. 19, 8083-8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, Q., Ijpma, A. & Greider, C. W. (2001) Mol. Cell. Biol. 21, 1819-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Natarajan, S. & McEachern, M. J. (2002) Mol. Cell. Biol. 22, 4512-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Natarajan, S., Groff-Vindman, C. & McEachern, M. J. (2003) Eukaryot. Cell 2, 1115-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wray, L. V., Witte, M. M., Dickson, R. C. & Riley, M. I. (1987) Mol. Cell. Biol. 7, 1111-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roy, J., Fulton, T. B. & Blackburn, E. H. (1998) Genes Dev. 12, 3286-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Underwood, D. H. & McEachern, M. J. (2001) BioTechniques 5, 934-935, 938. [DOI] [PubMed] [Google Scholar]
- 17.Underwood, D. H., Carroll, C. & McEachern, M. J. (2004) Eukaryot. Cell 3, 369-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McEachern, M. J. & Iyer, S. (2001) Mol. Cell 7, 695-704. [DOI] [PubMed] [Google Scholar]
- 19.Church, G. M. & Gilbert, W. (1984) Proc. Natl. Acad. Sci. USA 81, 1991-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McEachern, M. J., Underwood, D. H. & Blackburn, E. H. (2002) Genetics 160, 63-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teixeira, M. T., Arneric, M., Sperisen, P. & Lingner, J. (2004) Cell 117, 323-335. [DOI] [PubMed] [Google Scholar]
- 22.McEachern, M. J. & Blackburn, E. H. (1995) Nature 376, 403-409. [DOI] [PubMed] [Google Scholar]
- 23.Wang, S. S. & Zakian, V. A. (1990) Nature 345, 456-458. [DOI] [PubMed] [Google Scholar]
- 24.Bucholc, M., Park, Y. & Lustig, A. J. (2001) Mol. Cell. Biol. 21, 6559-6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White, C. I. & Haber, J. E. (1990) EMBO J. 9, 663-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garvik, B., Carson, M. & Hartwell, L. (1995) Mol. Cell. Biol. 15, 6128-6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nugent, C. I., Hughes, T. R., Lue, N. F. & Lundblad, V. (1996) Science 274, 249-252. [DOI] [PubMed] [Google Scholar]
- 28.Grandin, N., Reed, S. I. & Charbonneau, M. (1997) Genes Dev. 11, 512-527. [DOI] [PubMed] [Google Scholar]
- 29.Grandin, N., Damon, C. & Charbonneau, M. (2001) EMBO J. 20, 1173-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McEachern, M. J., Iyer, S., Fulton, T. B. & Blackburn, E. H. (2000) Proc. Natl. Acad. Sci. USA 97, 11409-11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grandin, N. & Charbonneau, M. (2003) Mol. Cell. Biol. 23, 3721-3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., Moss, H. & de Lange, T. (1999) Cell 97, 503-514. [DOI] [PubMed] [Google Scholar]
- 33.Grunstein, M. (1997) Curr. Opin. Cell Biol. 9, 383-387. [DOI] [PubMed] [Google Scholar]
- 34.de Bruin, D., Kantrow, S. M., Liberatore, R. A. & Zakian, V. A. (2000) Mol. Cell. Biol. 20, 7991-8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krauskopf, A. & Blackburn, E. H. (1996) Nature 383, 354-357. [DOI] [PubMed] [Google Scholar]
- 36.Reddel, R. R. (2003) Cancer Lett. 194, 155-162. [DOI] [PubMed] [Google Scholar]
- 37.Teng, S., Chang, J., McCowan, B. & Zakian, V. A. (2000) Mol. Cell 6, 947-952. [DOI] [PubMed] [Google Scholar]
- 38.Dunham, M. A., Neumann, A. A., Fasching, C. L. & Reddel, R. R. (2000) Nat. Genet. 26, 447-450. [DOI] [PubMed] [Google Scholar]
- 39.Wang, R. C., Smogorzewska, A. & de Lange, T. (2004) Cell 119, 355-368. [DOI] [PubMed] [Google Scholar]
- 40.Cesare, A. J. & Griffith, J. D. (2004) Mol. Cell. Biol. 24, 9948-9957. [DOI] [PMC free article] [PubMed] [Google Scholar]