

Abstract
The stereospecifically labeled 6-monodeuterio methyl 2,6-diamino-2,6-dideoxy-α- and β-D-glucopyranosides were synthesized with a view to determining their side chain conformations. NMR studies in D2O at pH 5 and pH 11 reveal both anomers to adopt very predominantly the gt conformation consistent with the gauche conformation of 2-aminoethanol and its acetate salt. In contrast, as also revealed with the help of stereospecifically-labelled monodeuterio isotopomers, the methyl 2-amino-2-deoxy-α- and β-D-glucopyranosides are an approximately 1:1 mixture of gg and gt conformers as is found in glucopyranose itself.
Keywords: Side chain conformation, deuterium labelling, conformational analysis, aminosugars
1. Introduction
Ongoing projects in our laboratories necessitated knowledge of the side chain conformation of the 2-amino-2-deoxy- and especially the 2,6-diamino-2,6-dideoxyglucopyranosides. On the basis of numerous X-ray crystallographic and NMR spectroscopic studies the hydroxymethyl side chain of glucopyranose and its glycosides is considered to populate an approximately 60:40 equilibrium mixture of the staggered gg and gt conformations; the tg conformer being destabilized by dipolar repulsions between the C6-O6 and C4-O4 bonds (Figure 1).1–3
Figure 1.

Staggered Conformations of the Side Chain in the Hexopyranoses Illustrated for Methyl α-D-Glucopyranoside
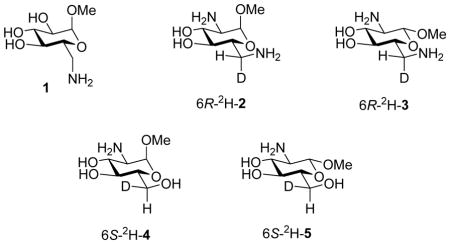
Analysis of the H5-H6R and H5-H6S coupling constants of ~9.2 and 3.1 Hz, respectively, led Bock and Duus to conclude that the side chain of methyl 6-amino-6-deoxy-β-D-glucopyranoside 1 exists as an 80:16:4 mixture of the gg, gt, and tg conformers in acidic solution (pH <1 and 4.75), and as a 70:30 mixture of gg, and gt conformers in aqueous base (pH >12).1 However, in the absence of the crucial rigorous assignment of the H6R and H6S resonances the coupling constants are also consistent with the tg conformer being the most populated state of 1.1 This ambiguity precludes the use of 1 as model for the side chain conformation of the 2,6-diamino-2,6-dideoxyglucopyranosides and prompted the synthesis and conformational analysis of both anomers, 2 and 3, of methyl 2,6-diamino-2,6-dideoxy-D-glucopyranoside stereospecifically deuterium labeled at the pro-6R site that we report here. Also reported are the syntheses and conformational analysis of both anomers, 4 and 5, of 6S-monodeuterio-2-amino-2-deoxy-D-glucopyranoside.
2. Results and discussion
Although shorter methods for the stereospecific introduction of deuterium into the hydroxymethyl group of the hexopyranosides based on asymmetric reduction of the corresponding aldehydes exist,4–6 we have preferred to adapt the earlier Meguro route based on the photobromination and deuteriodebromination of 1,6-anhydrohexose derivatives because of its unambiguous nature.7–10 Accordingly, white light stimulated bromination of 1,6-anhydro-D-glucopyranose triacetate 6 in α,α,α-trifluorotoluene11 gave 78% of the 6-exo-bromide 7, which was reduced with tributyltin deuteride in hot toluene to give the 6-exo-deuterio 1,6-anhydroglucose 8 in 82% yield (Scheme 1). Zemplen deacetylation then gave a triol which on reaction with toluenesulfonyl chloride in pyridine gave the 2,4-di-O-tosylate 9 in 82% yield for the two steps. Subsequent exposure to sodium methoxide in methanol afforded 77% of the galacto-configured epoxide 10 that on treatment with benzyl alcohol under Lewis acidic conditions gave the 4-O-benzyl derivative 11 in 78% yield. Treatment of 11 with sodium methoxide resulted in the manno-epoxide 12 in 88% yield, which on heating with sodium azide in hot aqueous DMF, followed by benzylation of the intermediate alcohol gave the 1,6-anhydroglucosamine derivative 13 in 83% yield. Opening of the 1,6-anhydro ring with trifluoroacetic acid in acetic anhydride then gave the stereoselectively mono-deuteriated glucosamine derivative 14 in 83% yield.
Scheme 1.

Synthesis of the 6S-deuterio-2-azido-2-deoxyglucose derivative 14.
Subsequent treatment of 14 with HCl in hot methanol gave the methyl α- and β-glycosides 15α and 15β in 42 and 25% yield, respectively, which, on hydrogenolysis over Pearlman’s catalyst gave the target compounds 6S-2H-4 and 6S-2H-5 in good yield (Scheme 2).
Scheme 2.

Synthesis of the 6S-deuteriated methyl α- and β-glucosaminides 4 and 5.
Sulfonylation of anomers 15α and 15β gave the 6-O-sulfonate esters 16 and 18 in excellent yield, which, on exposure to sodium azide in hot aqueous DMF gave the respective 6-azido-6-deoxy derivatives 17 and 19 in excellent yield with clean inversion of configuration (Scheme 3). Finally, hydrogenolysis of 17 and 19 afforded the methyl 2,6-dideoxy-2,6-diamino-α- and β-glucosides 6R-2H-2 and 6R-2H-3 in good yield (Scheme 3). Authentic samples of non-deuteriated 2, 4 and 5 were either commercial or were obtained by literature methods, while an authentic sample of 3 was prepared by routine methods as described in Scheme 4 from the known12 N-benzyloxycarbonyl derivative 20 of methyl β-D-glucosaminide.
Scheme 3.

Synthesis of the 6R-deuteriated methyl α- and β-glucosaminides 2 and 3.
Scheme 4.

Preparation of Unlabeled 3.
1H-NMR spectra of 2-5, of 6R-2H-2 and 6R-2H-3, and of 6S-2H-4 and 6S-2H-5 were obtained in D2O at pH 5 and pH 11 leading to the complete assignment of the H5, H6R and H6S resonances and their associated coupling constants laid out in Tables 1 and 2, respectively.
Table 1.
Diagnostic Chemical Shifts and Coupling Constants for Compounds 2-5 and their Selectively Monodeuteriated Analogs in D2O at pH 5.
| 2/6R-2H-2 | 3/6R-2H-3 | 4/6S-2H-4 | 5/6S-2H-5 | |
|---|---|---|---|---|
| δ H5 | 3.84/3.84 | 3.65/3.65 | 3.53/3.53 | 3.34/3.34 |
| δ H6R | 3.13/- | 3.12/- | 3.62/3.60 | 3.59/3.57 |
| δ H6S | 3.42/3.41 | 3.44/3.42 | 3.71/- | 3.77/- |
| J5,6R (Hz) | 8.8/- | 9.2/- | 4.9/5.1 | 5.5/5.5 |
| J5,6S (Hz) | 2.8/2.9 | 2.9/2.8 | 2.2/- | 1.8/- |
| J6S,6R (Hz) | 13.2/- | 13.3/- | 13.2/- | 12.1/- |
Table 2.
Diagnostic Chemical Shifts and Coupling Constants for Compounds 2-5 and their Selectively Monodeuteriated Analogs in D2O at pH 11.
| 2/6R-2H-2 | 3/6R-2H-3 | 4/6S-2H-4 | 5/6S-2H-5 | |
|---|---|---|---|---|
| δ H5 | 3.55/3.55 | 3.38/3.38 | 3.32/3.33 | 3.18/3.17 |
| δ H6R | 2.58/- | 2.60/- | 3.45/3.44 | 3.39/3.39 |
| δ H6S | 3.00/3.01 | 3.03/3.01 | 3.57/- | 3.60/- |
| J5,6R (Hz) | 9.1/- | 9.0/- | 5.3/5.3 | 5.8/5.7 |
| J5,6S (Hz) | 2.6/2.7 | 2.7/2.7 | 2.2/- | 2.1/- |
| J6S,6R (Hz) | 12.5/- | 12.7/- | 12.5/- | 12.4/- |
The H5-H6R coupling constants for 2 and 3 of 8.8 and 9.2 Hz and the corresponding and H5-H6S coupling constants of 2.8 and 2.9 Hz in aqueous acetic acid (Table 1), are comparable to those (~9.2 and 3.1 Hz) recorded for methyl 6-amino-6-deoxy-β-D-glucopyranoside 1 by Bock and Duus at pH 4.75.1 Accordingly, we conclude that i) the H6R and H6S resonances of 1 were correctly assigned and ii) that the side chains of 1, 2 and 3 populate very similar equilibrium mixtures of conformers, calculated on the basis of the Haasnoot-Altona equation13 to be a 16:80:4 gg:gt:tg ratio for 1 by Bock and Duus.1 On going from pH 5 (Table 1) to pH 11 (Table 2) there is only minimal change in the H5-H6R and H5-H6S coupling constants for 2 and 3, indicating that the predominance of the gt conformation for these glycosides is maintained in basic solution. Similarly, the H5-H6R and H5-H6S coupling constants for 4 and 5 in both aqueous acetic acid (Table 1) and basic D2O (Table 2) deviate little from those reported1 for the methyl α-D- and β-D-glucopyranosides in water indicating that similar side chain conformational preferences are adopted and approximate the 53:47:0 and 47:53:0 gg:gt:tg ratios computed by Bock and Duus for the α- and β-glucosides.1 We further conclude that replacement of the 2-hydroxy group in the α- and β-D-glucopyranosides and their 6-amino derivatives by a protonated amino group has only a minimal effect on the side chain conformation. The increased population of the gt conformer in the 6-amino series in acidic media compared to the corresponding 6-hydroxy compounds is consistent with the predominantly gauche conformation (~92%) adopted by ethanol amine hydrochloride in aqueous solution determined by Roberts and coworkers.14 The major gt conformation of 2 and 3 in aqueous base is also consistent with the predominant gauche conformation of neutral ethanol amine as determined by NMR spectroscopy,14 and computationally,15 and as determined by vibrational spectroscopy in the gas phase.16
Regarding the influence of anomeric configuration on side chain conformation, comparison of the coupling constant data for 2 and 3 in both acidic and basic media (Tables 1 and 2) reveals only minor differences suggesting that the side chain amine is the determining factor. For 3 and 4, which lack the 6-amino group, the changes, while still modest, are more pronounced in both acidic and basic media (Tables 1 and 2) and are consistent with a slightly higher population of the gt conformer for the equatorial anomer. This pattern follows that observed for the anomers of glucopyranosides by multiple groups.1,17,18
3. Conclusion
The side chains of both anomers of the methyl glycosides of 2,6-diamino-2,6-dideoxy-D-glucopyranose very predominantly adopt the gt conformation in aqueous solution at pH 5 and pH 11 as revealed by the stereospecific synthesis of 6-monodeuterio isotopomers. In contrast, anomers of the methyl glycosides of 2-amino-2-deoxy-D-glucopyranoside populate, like the methyl glycosides of glucopyranose itself, approximately 1:1 mixtures of the gg and gt conformations. The preference for the gt conformation in the 2,6-diamino series is therefore a consequence of the 6-amino group, and is consistent with the gauche conformation adopted by 2-aminoethanol under similar conditions.
4. Experimental section
4.1. General
All reactions were conducted under an atmosphere of nitrogen or argon. Extracts were dried over sodium sulfate and chromatography was carried out over silica gel. Sephadex chromatography was carried out over Sephadex C25. Specific rotations were measured on an automatic polarimeter with a path length of 10 cm in chloroform solution unless otherwise stated. High-resolution (HRMS) mass spectra were recorded in the electrospray mode using a time of flight mass analyzer (ESI-TOF).
4.2.1 1,6-Anhydro-2,3,4-tri-O-acetyl-6-(S)-bromo-β-D-glucopyranose (7)
1,6-Anhydro-2,3,4-tri-O-acetyl-β-D-glucopyranose 619 (10.0 g, 0.035 mol) was dissolved in α,α,α-trifluoromethylbenzene (600 mL) and N-bromosuccinimide (24.9 g, 0.14 mol) was added at ambient temperature. The reaction mixture was heated to reflux with irradiation by a 300 watt incandescent bulb for 6 h, monitoring by TLC (Hexane:ethyl acetate 1:1, Rf = 0.55). After completion, the reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate (200 mL) and was washed with saturated aqueous sodium bicarbonate. The aqueous layer was extracted with ethyl acetate twice and the combined organic layer was washed with brine, dried, and concentrated to dryness. The residual syrup was purified by silica gel column chromatography (hexane:ethyl acetate 5:1 to 2:1) to give the 6-(S)-bromo compound 720 as a colorless syrup (9.9 g, 0.027 mol, 78%). (c = 1.0), 1H NMR (600 MHz, CDCl3): δ = 6.41 (s, 1H, H6); 5.77 (br s, 1H, H1); 4.83 (br s, 1H, H3); 4.79 (br s, 1H, H5); 4.66 (br s, 1H, H4); 4.50 (br s, 1H, H2); 2.11 (s, 3H, CH3); 2.09 (s, 3H, CH3); 2.05 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ = 169.76, 169.50, 168.87 (-COCH3 x3); 102.08 (C1); 84.19 (C5); 79.36 (C6); 69.29 (C3); 67.93 (C2); 67.67 (C4); 20.77 (CH3 x2); 20.71 (CH3). ESI-HRMS: m/z calcd for C12H15O8BrNa [M+Na]+ 388.9848, found 388.9853.
4.2.2 1,6-Anhydro-2,3,4-tri-O-acetyl-6-(S)-deuterio-β-D-glucopyranose (8).7
Compound 7 (8.3 g, 0.023 mol), tributyltin deuteride prepared according to Neumann21 (21.0 g, 0.069 mol) and azobisisobutyronitrile (365 mg, 0.0023 mol) were dissolved in toluene (830 mL) and heated to reflux, with monitoring by TLC (hexane:ethyl acetate 1:1, Rf = 0.65). After 1 h, the reaction mixture was cooled to ambient temperature then was concentrated under reduced pressure. The residue was dissolved in a minimum of toluene and dichloromethane (1:1 mixture) and was purified by column chromatography (hexane:ethyl acetate 3:1 to 1:1) to give the white solid 6-(S)-deuterio 8 (5.5 g, 0.019 mol, 82%). (c = 0.15), 1H NMR (600 MHz, CDCl3): δ = 5.36 (br s, 1H, H1); 4.75 (m, 1H, H3); 4.55 (br s, 1H, H4); 4.52 (br s, 1H, H5); 4.49 (br s, 1H, H2): 3.99 (s, 1H, H6); 2.07 (s, 3H, CH3); 2.05 (s, 3H, CH3); 2.02 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ = 169.87, 169.51, 168.95 (-COCH3 x3); 99.04 (C1); 73.54 (C5); 70.27 (C4); 69.59 (C3); 69.03 (C2); 64.95 (t, J C-D = 23.6Hz, C6); 20.86 (-COCH3); 20.76 (-COCH3 x2). ESI-HRMS: m/z calcd for C12H15DO8Na [M+Na]+ 312.0806, found 312.0815.
4.2.3 1,6-Anhydro-2,4-di-O-p-toluenesulfonyl-6-(S)-deuterio-β-D-glucopyranose (9)
Compound 8 (6.0 g 0.021 mol) was dissolved in dry methanol (60 mL) and treated with sodium methoxide (200 mg, 0.37 mmol) at ambient temperature, with monitoring by TLC (chloroform:methanol 5:1, Rf = 0.4). After 5 h the reaction mixture was neutralized with Amberlite® IR120, then filtered, concentrated, and dried under reduced pressure. The residue was dissolved in pyridine (18 mL) and treated with a solution of p-toluenesulfonyl chloride (8.7 g, 0.046 mol) in 1:1 pyridine:chloroform (55 mL) in an ice bath. The reaction mixture was allowed to warm to ambient temperature, and stirred for 12 h, with monitoring by TLC (hexane:ethyl acetate 1:1, Rf = 0.6). The reaction mixture was quenched with methanol (5 mL), then was concentrated under reduced pressure. The residue was filtered through silica gel (hexane:ethyl acetate 2:1) and concentrated under vacuum to give 9 as a light yellow oil (8.1g, 0.017 mol, 82%), whose spectra were consistent with the literature22–24 for the non-deuteriated isotopomer and that was used in the next reaction without further purification. ESI-HRMS: m/z calcd for C20H21DO9S2Na [M+Na]+ 494.0666, found 494.0659.
4.2.4 1,6;3,4-Bisanhydro-2-O-p-toluenesulfonyl-6-(S)-deuterio-β-D-galactopyranose (10)
A solution of 9 (7.0 g, 0.015 mol) in dry methanol (140 mL) and was treated with 0.6 M methanolic sodium methoxide (94 mL) in an ice bath. The reaction mixture was allowed to warm to ambient temperature and was stirred for 10 h, with monitoring by TLC (hexane:ethyl acetate 1:1, Rf = 0.65). After completion the reaction mixture was concentrated and the residue was taken up in ethyl acetate (150 mL) and washed with water (100 mL). The aqueous layer was extracted with ethyl acetate and the combined organic layer was washed with brine, dried, and concentrated. The residue was purified by column chromatography (hexane:ethyl acetate 3:1 to 1:1) to give 10 as a white amorphous solid (3.6 g, 0.012 mol, 77%), whose spectra were consistent with the literature for the non-deuteriated isotopomer.23,24 (c = 1.2). 1H NMR (400MHz, CDCl3): δ = 7.84 (d, J = 8.3 Hz, 2H, arom.); 7.38 (d, J = 8.3 Hz, 2H, arom.); 5.16 (br s, 1H, H1); 4.83 (d, J5-4 = 4.8 Hz, 1H, H5); 4.39 (br s, 1H, H2); 3.92 (s, 1H, H6); 3.60 (t, J4-3 = 4.4 Hz, J4-5 = 4.4 Hz, 1H, H4); 3.13 (dd, J3-4 = 4.0 Hz, J3-N.D. = 1.5 Hz, 1H, H3); 2.46 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ = 146.63, 132.80, 138.19, 127.98 (arom.); 98.05 (C1); 71.78 (C2); 71.53 (C5); 64.44 (t, JC-D = 24.4Hz, C6); 52.82 (C4); 47.63 (C3); 21.71 (CH3). ESI-HRMS: m/z calcd for C13H13DO6SNa [M+Na]+ 322.0472, found 322.0481.
4.2.5 1,6-Anhydro-4-O-benzyl-2-O-p-toluenesulfonyl-6-(S)-deuterio-β-D-glucopyranose (11)
A solution of epoxide 10 (4.0 g, 0.013 mol) in benzene (80 mL), was treated with benzyl alcohol (7 mL, 0.04 mol) and 35 % boron trifluoride diethyl etherate (1 mL, 0.002 mol) at ambient temperature. The solution was heated to 50 °C with stirring for 12 h and monitored by TLC (hexane:ethyl acetate 1:1, Rf = 0.55). After completion the reaction mixture was poured into saturated aqueous sodium bicarbonate (100 mL), the aqueous layer was separated and extracted with ethyl acetate. The combined organic layer was washed with brine, dried, and concentrated under reduced pressure. The residue was purified by column chromatography (hexane:ethyl acetate 3:1 to 1:1) to give 11 (4.1 g, 0.010 mol, 78%) as a white solid, whose spectra were consistent with the literature for the non-deuteriated isotopomer.23,24 (c = 2.0). 1H NMR (600 MHz, CDCl3): δ = 7.81 (d, J = 8.4 Hz, 2H, arom.); 7.37-7.25 (m, 7H, arom.); 5.31 (br s, 1H, H1); 4.67 (d, JCH2a-CH2b = 12.1 Hz, 1H, CH2a); 4.59 (d, JCH2b-CH2a = 12.1 Hz, 1H, CH2b); 4.55 (s, 1H, H5); 4.21 (d, J2-3 = 3.4 Hz, 1H, H2); 3.96 (t, J3-2 = 3.6 Hz, J3-4 = 3.6 Hz, 1H, H3); 3.87 (s, 1H, H6); 3.31 (d, J4-3 = 3.6 Hz, 1H, H4); 2.43 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ = 145.29, 137.49, 133.06, 129.98, 128.52, 127.96, 127.84 (arom.); 99.88 (C1); 79.18 (C2); 78.40 (C4); 75.13 (C5); 71.73 (CH2); 70.12 (C3); 66.10 (t, JC-D = 23.5 Hz, C6); 21.69 (CH3). ESI-HRMS: m/z calcd for C20H21DO7SNa [M+Na]+ 430.1047, found 430.1039.
4.2.6 1,6;2,3-Bisanhydro-4-O-benzyl-6-(S)-deuterio-β-D-mannopyranose (12)
A solution of 11 (4.3 g, 0.011 mol) in chloroform (25 mL) was treated with 1.0 M methanolic sodium methoxide (25 mL) in an ice bath, then was allowed to warm to ambient temperature, and stirred for 5 h with monitoring by TLC (hexane:ethyl acetate 1:1, Rf = 0.60). The reaction mixture was poured into a mixture of water (150 mL) and chloroform (150 mL). The aqueous layer was separated and extracted with chloroform and the combined organic layer was washed with brine, dried and concentrated. The residue was purified by column chromatography (hexane:ethyl acetate 3:1 to 1:1) to give 12 (2.3 g, 8.14 mmol, 88%) as a colorless oil, whose spectra were consistent with the literature for the non-deuteriated isotopomer.23 (c = 1.3). 1H NMR (600 MHz, CDCl3): δ = 7.40-7.20 (m, 5H, arom.); 5.69 (d, J1-2 = 3.1 Hz, 1H, H1); 4.72 (s, 2H, CH2); 4.49 (br s, 1H, H5); 3.64 (br s, 1H, H6); 3.43 (t, J2-1 = 3.4 Hz, J2-3 = 3.3 Hz, 1H, H2); 3.18 (dd, J3-2 = 3.6 Hz, J3-N.D. = 1.5 Hz, 1H, H3). 13C NMR (150 MHz, CDCl3): δ = 137.27, 128.61, 128.12, 127.82 (arom.); 97.57 (C1); 73.31 (C4); 72.13 (CH2); 71.53 (C5); 65.46 (t, JC-D = 23.6 Hz, C6); 54.37 (C2); 47.80 (CH3). ESI-HRMS: m/z calcd for C13H13DO4Na [M+Na]+ 258.0853, found 258.0862.
4.2.7 1,6-Anhydro-2-azido-2-deoxy-3,4-di-O-benzyl-6-(S)-deuterio-β-D-glucopyranose (13)
A solution of 12 (2.0 g, 0.0085 mol) 9:1 water:DMF (40 mL), was treated with sodium azide (5.5g, 0.085 mol) and ammonium chloride (3.4g, 0.064 mol), and heated to reflux with stirring for 10 h, and monitoring by TLC (hexane:ethyl acetate 1:1, Rf = 0.50). The reaction mixture was concentrated under reduced pressure and the residue taken up in ethyl acetate (100 mL) and washed with water. The aqueous layer was extracted with ethyl acetate, and the combined organic layer was washed with brine, dried, and concentrated. The concentrate (2.0 g, 0.0072 mol, ESI-HRMS: m/z calcd for C13H14DN3O4Na [M+Na]+ 301.1023, found 301.1015) was dissolved in tetrahydrofuran (20 mL) and treated with sodium hydride (60% dispersion in oil, 430 mg, 0.011 mol) at ambient temperature. After stirring for 0.5 h benzyl bromide (1.8 mL, 0.014 mol) was added at ambient temperature and the reaction mixture stirred for 3 h with monitoring by TLC (hexane:ethyl acetate 2:1, Rf = 0.45). After completion methanol (1 mL) was added and the volatiles were removed under reduced pressure. The residue was dissolved in ethyl acetate and washed with water. The aqueous layer was extracted with ethyl acetate, and the combined organic layer washed, dried, and concentrated. The residue was purified by column chromatography (hexane:ethyl acetate 5:1 to 2:1) to give the colorless oil 13 (2.2 g, 0.0060 mol, 83%), whose spectra were consistent with the literature for the non-deuteriated isotopomer.25 (c = 0.5). 1H NMR (600 MHz, CDCl3): δ = 7.40-7.26 (m, 10H, arom.); 5.49 (s, 1H, H1); 4.62 (d, J5-4 = 2.2 Hz, 1H, H5); 4.61-4.48 (m, 4H, CH2 x2); 3.99 (s, 1H, H6); 3.65 (br s, 1H, H3); 3.38 (br s, 1H, H4); 3.28 (br s, 1H, H2). 13C NMR (150 MHz, CDCl3): δ = 137.39, 137.24, 128.57, 128.56, 128.07, 128.02, 127.91, 127.79 (arom.); 100.53 (C1); 76.18 (C3); 75.84 (C4); 74.31 (C5); 72.35 (CH2); 71.33 (CH2); 65.01 (t, JC-D = 23.6 Hz, C6); 59.88 (C2). ESI-HRMS: m/z calcd for C20H20DN3O4Na [M+Na]+ 391.1493, found 391.1488.
4.2.8 1,6-Di-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-6-(S)-deuterio-α-D-glucopyranose (14)
Compound 13 (2.1 g, 0.0057 mol) was dissolved in acetic anhydride (33 mL), treated with trifluoroacetic acid (3.3 mL) at ambient temperature and stirred for 1.5 h, with monitoring by TLC (hexane:ethyl acetate 2:1, Rf = 0.40). The reaction mixture was poured into saturated aqueous sodium carbonate (300 mL), and was extracted with ethyl acetate. The combined organic layer was washed with brine, dried, and concentrated. The residue was purified by column chromatography (hexane:ethyl acetate 4:1 to 2:1) to give the colorless oil 14 (2.2 g, 4.73 mmol, 83%), whose spectra were consistent with the literature for the non-deuteriated isotopomer.26 (c = 1.2). 1H NMR (600 MHz, CDCl3): δ = 7.45-7.26 (m, 10H, arom.); 6.23 (d, J1-2 = 3.7 Hz, 1H, H1); 4.94 (d, JCHa-CHb = 10.3 Hz, 1H, -O-CH2aPh); 4.91; (d, JCHb-CHa = 10.3 Hz, 1H, -O-CH2bPh); 4.88 (d, JCHa′-CHb′ = 10.6 Hz, 1H, -O-CH2a′Ph); 4.59 (d, JCHb′-CHa′ = 10.6 Hz, 1H, -O-CH2b′Ph); 4.25 (d, J6-5 = 4.0 Hz, 1H, H6); 3.98 (t, J3-2 = J3-4 = 9.2 Hz, 1H, H3); 3.93 (dd, J5-4 = 9.4 Hz, J5-6 = 4.1 Hz, 1H, H5); 3.64 (t, J4-3 = J4-5 = 9.2 Hz, 1H, H4); 3.60 (dd, J2-1 = 3.7 Hz, J2-3 = 9.6 Hz, 1H, H2); 2.15 (s, 3H, CH3); 2.03 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ = 170.53, 168.75 (2C, C=O x2); 137.44, 137.22 (2C, arom.); 128.62, 128.57, 128.22, 128.11, 128.08 (10C, arom.); 90.37 (C1); 80.55 (C3); 76.87 (C4); 75.68, 75.29 (2C, -CH2- x2); 71.22 (C5); 62.71 (C2); 62.04 (t, JC-D = 23.0 Hz, C6); 20.94, 20.78 (2C, -COCH3). ESI-HRMS: m/z calcd for C24H26DN3O7Na [M+Na]+ 493.1809, found 493.1798.
4.2.9 Methyl 2-azido-2-deoxy-3,4-di-O-benzyl-6-(S)-deuterio-α-and β-D-glucopyranose (15α) and (15β)
The 1,6-di-O-acatate 14 (520 mg, 0.318 mmol) was dissolved in 10% w/w methanolic hydrochloric acid (20 mL, prepared by addition of acetyl chloride (3.0 mL) to dry methanol (17 mL) in an ice bath) at ambient temperature and stirred for 8 h at 75 °C, with monitoring by TLC (hexane:ethyl acetate 2:1, Rf = 0.35 for 15αβ and Rf = 0.15 for the intermediate 1,6-diol) and ESI mass spectrometry. After conversion of the intermediate pyranose to the methyl glycosides, the reaction mixture was concentrated under reduced pressure, taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate. The aqueous layer was extracted with ethyl acetate, and the combined organic layer was washed with brine, dried, and concentrated. The residue was dissolved in the minimum volume of toluene for purification by column chromatography (hexane:ethyl acetate 4:1 to 2:1) which gave the colorless oils 15α (53.4 mg, 0.134 mmol, 42 %) and 15β (32.0 mg, 0.080 mmol, 25 %), whose spectra were consistent with the literature for the non-deuteriated isotopomers.27,28 15α: (c = 1.0). 1H NMR (600 MHz, CDCl3): δ = 7.43-7.27 (m, 10H, arom.); 4.93-4.85 (m, 3H, -CH2a,bPh, -CH2a′Ph); 4.78 (d, J1-2 = 3.7 Hz, 1H, H1); 4.68 (d, JCH2b′-CH2a′ = 11.0 Hz, 1H, -CH2b′Ph); 4.01 (dd, J3-2 = 9.9 Hz, J3-4 = 9.0 Hz, 1H, H3); 3.72 (d, J6-5 = 3.9 Hz, 1H, H6); 3.70 (dd, J5-4 = 9.5 Hz, J5-6 = 4.0 Hz, 1H, H5); 3.62 (t, J4-3 = J4-5 = 9.3 Hz, 1H, H4); 3.41 (s, 3H, -OCH3); 3.60 (dd, J2-1 = 3.7 Hz, J2-3 = 10.1 Hz, 1H, H2). 13C NMR (150 MHz, CDCl3) δ = 137.83, 137.79 (2C, arom.); 128.72-127.72 (10C, arom.); 98.69 (C1); 80.35 (C3); 77.86 (C4); 75.52, 75.04 (2C, -CH2- x2); 71.19 (C5); 63.67 (C2); 61.16 (t, JC-D = 22.4 Hz, C6); 55.24 (-OCH3). ESI-HRMS: m/z calcd for C21H24DN3O5Na [M+Na]+ 423.1755, found 423.1761. 15β: (c = 0.3). 1H NMR (600 MHz, CDCl3): δ = 7.39-7.27 (m, 10H, arom.); 4.89 (d, JCHa-CHb = 10.6 Hz, 1H, -CH2aPh); 4.85; (d, JCHa′-CHb′ = 11.0 Hz, 1H, -CH2a′Ph); 4.83 (d, JCHb-CHa = 10.6 Hz, 1H, -CHbPh); 4.66 (d, JCHb′-CHa′ = 11.0 Hz, 1H, -CH2b′Ph); 4.21 (d, J1-2 = 8.1 Hz, 1H, H1); 3.71 (d, J6-5 = 4.4 Hz, 1H, H6); 3.59 (t, J4-3 = J4-5 = 9.4 Hz, 1H, H4); 3.57 (s, 3H, -OCH3); 3.46 (t, J3-2 = J3-4 = 9.5 Hz, 1H, H3); 3.37 (dd, J2-1 = 8.1 Hz, J2-3 = 9.7 Hz, 1H, H2); 3.33 (dd, J5-4 = 9.6 Hz, J5-6 = 4.4 Hz, 1H, H5). 13C NMR (150 MHz, CDCl3): δ = 137.78, 137.69 (2C, arom.); 128.60-127.87 (10C, arom.); 103.01 (C1); 82.93 (C3); 77.35 (C4); 75.57(-CH2PH); 75.17 (C5); 75.12 (-CH2PH); 66.32 (C2); 61.33 (t, JC-D = 23.6 Hz, C6); 57.31 (-OCH3). ESI-HRMS: m/z calcd for C21H24DN3O5Na [M+Na]+ 423.1755, found 423.1747.
4.2.10 Methyl 6-(S)-deuterio-α-D-glucosaminide monoacetate (6S-2H-4)
To a solution of 15α (30 mg, 0.075 mmol) in 1,4-dioxane (1 mL) was added the same weight of 10% palladium hydroxide on activated charcoal. The reaction mixture was stirred 6 H under 1 atm of hydrogen atmosphere. Water (0.1 mL) was then added and the pressure adjusted to 3 atm of hydrogen. After stirring for 12 h the catalyst was filtered off and washed with water until the washings gave a negative ninhydrin test. The filtrate was concentrated and dried under reduced pressure. The residue was dissolved in methanol (0.5 mL) and slurried with silica gel. The methanol was removed in vacuo and the powder applied to a silica gel column that was eluted with chloroform:methanol:ammonium hydroxide 3:1:0.5, Rf = 0.25). The concentrated fractions were taken up in the minimum volume of 10% aqueous acetic acid and purified by Sephadex C-25 column (eluent: water then 2% ammonium hydroxide). Glacial acetic (50 μL) acid was added to the product-containing fractions, followed by drying under vacuum to give the title compound 6S-2H-4 as colorless oil (10.4 mg, 0.041 mmol, 55%), whose non-deuteriated isotopomer was previously reported as the free base.29 (c = 0.1, water). 1H NMR (600 MHz, D2O): δ = 4.85 (d, J1-2 = 3.7 Hz, 1H, H1); 3.86 (t, J3-2 = J3-4 = 9.7 Hz, 1H, H3); 3.60 (d, J6-5 = 5.1 Hz, 1H, H6); 3.53 (dd, J5-4 = 9.8 Hz, J5-6 = 5.1 Hz, 1H, H5); 3.31 (t, J4-3 = J4-5 = 9.7 Hz, 1H, H4); 3.28 (s, 3H, -OCH3); 3.16 (dd, J2-1 = 3.7 Hz, J2-3 = 9.9 Hz, 1H, H2). 13C NMR (150 MHz, D2O): δ = 176.63 (-C(O)OH); 96.03 (C1); 71.72 (C6); 69.82 (C3); 69.35 (C4); 59.84 (t, JC-D = 22.0 Hz, C6); 55.13 (-OCH3); 53.87 (C2); 20.35 (CH3C(O)OH). ESI-HRMS: m/z calcd for C7H15DNO5 [M+H]+ 195.1091, found 195.1098.
4.2.11 Methyl 6-(S)-deuterio-β-D-glucosaminide monoacetate (6S-2H-5)
Compound 15β (26.1 mg, 0.065 mmol) was subjected to hydrogenolysis and purification analogously to 15α to give the title compound 6S-2H-5 as colorless oil (6.8 mg, 0.027 mmol, 41%), whose non-deuteriated isotopomer was previously reported as the free base.30 (c = 0.1, water). 1H NMR (600 MHz, D2O): δ = 4.49 (d, J1-2 = 8.6 Hz, 1H, H1); 3.57 (d, J6-5 = 5.5 Hz, 1H, H5); 3.51 (dd, J3-2 = 10.6 Hz, J3-4 = 9.2 Hz, 1H, H3); 3.42 (s, 3H, -OCH3); 3.34 (dd, J5-4 = 9.4 Hz, J5-6 = 5.5 Hz, 1H, H5); 3.30 (t, J4-3 = J4-5 = 9.4 Hz, 1H, H4); 2.86 (dd, J2-1 = 8.6, J2-3 = 10.6 Hz, 1H, H2); 1.91 (s, 3H, CH3C(O)-). 13C NMR (150 MHz, D2O): δ = 176.90 (-C(O)OH); 99.75 (C1); 76.02 (C5); 71.90 (C3); 69.66 (C4); 59.99 (t, JC-D = 24.0 Hz, C6); 57.34 (-OCH3); 55.64 (C2); 20.52 (CH3C(O)OH). ESI-HRMS: m/z calcd for C7H15DNO5 [M+H]+ 195.1091, found 195.1083.
4.2.12 Methyl 2-azido-2-deoxy-3,4-di-O-benzy-6-O-p-toluenesulfonyl-6-(S)-deuterio-α-D-glucopyranoside (16)
A solution of 15α (55.0 mg, 0.137 mmol) and p-toluenesulfonyl chloride (39.2 mg, 0.206 mmol) in pyridine (0.5 mL) was stirred at ambient temperature for 2 h, with monitoring by TLC (hexane:ethyl acetate 2:1, Rf = 0.40). The reaction mixture was diluted with ethyl acetate and washed with 1 N hydrochloric acid. The aqueous layer was extracted with ethyl acetate and the combined extracts were washed with saturated aqueous sodium bicarbonate and brine, dried, concentrated and purified by silica gel column chromatography (hexane:ethyl acetate 4:1 to 2:1) to give the tosylate 16 as a colorless oil (58.4 mg, 0.105 mmol, 77%). (c = 0.5). 1H NMR (600 MHz, CDCl3): δ = 7.78 (d, J = 8.4 Hz, 2H, arom); 7.38-7.27 (m, 10 H, arom); 7.18 (dd, J = 2.2 Hz, 7.7 Hz, 2H, arom); 4.88 (d, JCH2a-CH2b = 10.6 Hz, 1H, -CH2aPh); 4.82 (d, JCH2b-CH2a = 10.6 Hz, 1H, -CH2bPh); 4.81 (d, JCH2a′-CH2b′ = 10.6 Hz, 1H, -CH2a′Ph); 4.47 (d, J1-2 = 3.7 Hz, 1H, H1); 4.47 (d, JCH2b′-CH2a′ = 10.6 Hz, 1H, -CH2b′Ph); 4.21 (d, J6-5 = 4.4 Hz, 1H, H6); 3.94 (dd, J3-2 = 9.9 Hz, J3-4 = 9.0 Hz, 1H, H3); 3.80 (dd, J5-4 = 10.1 Hz, J5-6 = 4.4 Hz, 1H, H5); 3.51 (dd, J4-3 = 9.1 Hz, J4-5 = 10.1, 1H, H4); 3.36 (s, 3H, -OCH3); 3.34 (dd, J2-1 = 3.7 Hz, J2-3 = 10.1 Hz, 1H, H2); 2.41 (s, 3H, -SO2PhCH3). 13C NMR (150 MHz, CDCl3) δ = 144.97, 137.60, 137.42, 132.81 (4C, arom); 130.00-127.75 (14C, arom); 98.53 (C1); 80.30 (C3); 77.43 (C4); 75.48 (CH2); 75.02 (CH2′); 68.90 (C5); 67.97 (t, JC-D = 22.4 Hz, C6); 63.40 (C2); 55.40 (-OCH3); 21.64 (PhCH3). ESI-HRMS: m/z calcd for C28H30DN3O7SNa [M+Na]+ 577.1843, found 577.1851.
4.2.13 Methyl 2,6-diazido-2,6-dideoxy-3,4-di-O-benzyl-6-(R)-deuterio-α-D-glucopyranoside (17)
A solution of 16 (85.0 mg, 0.153 mmol) in 9:1 N,N-dimethylformamide:water (1.2 mL) mixture was treated sodium azide (119 mg, 1.84 mol) and heated to 85 °C for 8 h with stirring, and monitoring by TLC (hexane:ethyl acetate 3:1, Rf = 0.45). On completion the reaction mixture was diluted with ethyl acetate and washed with water. The aqueous layer was extracted with ethyl acetate and the combined organic layer was washed with brine, dried, concentrated, and purified by column chromatography (hexane:ethyl acetate 6:1 to 3:1) to give 17 as white foam (52.2 mg, 0.123 mmol, 80%). (c = 0.5). 1H NMR (600 MHz, CDCl3): δ = 7.42-7.22 (m, 10H, arom); 4.89 (d, JCH2a-CH2b = 10.6 Hz, 1H, -CH2aPh); 4.88 (d, JCH2a′-CH2b′ = 11.0 Hz, 1H, -CH2a′Ph); 4.84 (d, JCH2b-CH2a = 10.6 Hz, 1H, -CH2bPh); 4.80 (d, J1-2 = 3.4 Hz, 1H, H1); 4.59 (d, JCH2b′-CH2a′ = 11.0 Hz, 1H, -CH2b′Ph); 3.97 (dd, J3-2 = 10.3 Hz, J3-4 = 9.2 Hz, 1H, H3); 3.81 (dd, J5-4 = 9.8 Hz, J5-6 = 2.2 Hz, 1H, H5); 3.52 (t, J4-3 = J4-5 = 9.5, 1H, H4); 3.46 (d, J6-5 = 2.2 Hz, 1H, H6); 3.44 (s, 3H, -OCH3); 3.42 (dd, J2-1 = 3.5 Hz, J2-3 = 10.3 Hz, 1H, H2). 13C NMR (150 MHz, CDCl3): δ = 135.05, 135.01 (2C, arom); 126.08-125.14 (10C, arom); 96.00 (C1); 77.74 (C3); 76.21 (C4); 72.97(CH2); 72.60 (CH2′); 67.82 (C5); 61.03 (C2); 52.84 (-OCH3); 48.19 (t, JC-D = 21.9 Hz, C6). ESI-HRMS: m/z calcd for C21H23DN6O4Na [M+Na]+ 448.1819, found 448.1811.
4.2.14 Methyl 2,6-diamino-2,6-dideoxy-6-(R)-deuterio-α-D-glucopyranoside diacetate (6R-2H-2)
The diazide 17 (25.0 mg, 0.059 mmol) was subjected to hydrogenolysis and purification as described for 15α to give the title compound, whose non-deuteriated isotopomer was previously reported as the HCl salt,31,32 as its diacetate salt in the form of a colorless oil (12.3 mg, 0.039 mmol, 67%). (c = 0.5, water). 1H NMR (600 MHz, D2O): δ = 5.01 (d, J1-2 = 3.0 Hz, 1H, H1); 3.84 (d, J5-4 = 9.5 Hz, 1H, H5); 3.81 (t, J3-2 = J3-4 = 10.0 Hz, 1H, H3); 3.41 (s, 1H, H6); 3.41 (s, 3H, -OCH3); 3.36 (t, J4-3 = J4-5 = 9.8 Hz, 1H, H4); 3.32 (dd, J2-1 = 3.0 Hz, J2-3 = 10.1 Hz, 1H, H2); 1.91 (s, 6H, CH3COOH). 13C NMR (150 MHz, CDCl3): δ = 179.91 (-C=O-); 96.16 (C1); 71.06 (C4); 69.36 (C3); 67.67 (C5); 55.49 (-OCH3); 53.59 (C2); 39.87 (t, JC-D = 23.6 Hz, C6); 22.32 (CH3COOH). ESI-HRMS: m/z calcd for C7H16DN2O4 [M+H]+ 194.1251, found 194.1248.
4.2.15 Methyl 2-azido-2-deoxy-3,4-di-O-benzyl-6-O-methanesulfonyl-6-(S)-deuterio-β-D-glucopyranoside (18)
A solution of 15β (50.0 mg, 0.125 mmol) in dichloromethane (1 mL) and was stirred with N,N-diisopropylethylamine (108 μL, 0.625 mmol) and methanesulfonyl chloride (20 μL, 0.250 mmol) at ambient temperature for 2 h, with monitoring by TLC (hexane:ethyl acetate 2:1, Rf = 0.40). Methanol (0.5 mL) was added and the mixture was concentrated under reduced pressure. The residue was diluted with ethyl acetate and washed with 1 N aqueous hydrochloric acid, saturated sodium bicarbonate, and brine. The organic layer was dried, concentrated, and purified by column chromatography (hexane:ethyl acetate 4:1 to 2:1) to give 18 (52.0 mg, 0.109 mmol, 87%) as a colorless oil. (c = 0.3). 1H NMR (600 MHz, CDCl3): δ = 7.40-7.26 (m, 10H, arom); 4.92 (d, JCH2a-CH2b = 11.0 Hz, 1H, -CH2aPh); 4.88 (d, JCH2a′-CH2b′ = 10.6 Hz, 1H, -CH2a′Ph); 4.82 (d, JCH2b-CH2a = 11.0 Hz, 1H, -CH2bPh); 4.64 (d, JCH2b′-CH2a′ = 10.6 Hz, 1H, -CH2b′Ph); 4.32 (d, J6-5 = 4.4 Hz, 1H, H6); 4.20 (d, J1-2 = 8.1 Hz, 1H, H1); 3.56 (s, 3H, -OCH3); 3.53 (dd, J4-3 = 8.2 Hz, J4-5 = 9.7 Hz, 1H, H4); 3.50 (dd, J5-4 = 9.7 Hz, J5-6 = 4.4 Hz, 1H, H5); 3.46 (dd, J3-2 = 9.7 Hz, J 3-4 = 8.3 Hz, 1H, H3); 3.38 (dd, J2-1 = 8.1 Hz, J2-3 = 9.7 Hz, 1H, H2); 3.02 (s, 3H, -SO2CH3). 13C NMR (150 MHz, CDCl3): δ = 137.56, 137.25 (2C, arom); 128.83-127.80 (10C, arom); 102.91 (C1); 82.84 (C3); 76.79 (C4); 75.64 (-CH2-); 76.23 (-CH2′-); 72.88 (C5); 67.27 (t, JC-D = 21.9 Hz, C6); 66.17 (C2); 57.34 (-OCH3); 37.70 (-SO2CH3). ESI-HRMS: m/z calcd for C22H26DN3O7SNa [M+Na]+ 501.1530, found 501.1526.
4.2.16 Methyl 2,6-diazido-2,6-dideoxy-3,4-di-O-benzyl-6-(R)-deuterio-β-D-glucopyranoside (19)
A stirred solution of 18 (50.0 mg, 0.105 mmol) in N,N-dimethylformamide:water (1.0 mL) was heated at 85 °C for 14 h with sodium azide (81.9 mg, 1.26 mmol), with monitoring by TLC (hexane:ethyl acetate 3:1, Rf = 0.45). The mixture was diluted with ethyl acetate and was washed with water. The aqueous layer was extracted with ethyl acetate and the combined organic layer was washed with brine, dried, concentrated, and purified by column chromatography (hexane:ethyl acetate 6:1 to 3:1) to give 19 (36.0 mg, 0.085 mmol, 81%) as a colorless oil (c = 0.2). 1H NMR (600 MHz, CDCl3): δ = 7.39-7.23 (m, 10H, arom); 4.91 (d, JCH2a-CH2b = 10.6 Hz, 1H, -CH2aPh); 4.87 (d, JCH2a′-CH2b′ = 11.0 Hz, 1H, -CH2a′Ph); 4.80 (d, JCH2b-CH2a = 10.6Hz, 1H, -CH2bPh); 4.58 (d, JCH2b′-CH2a′ = 11.0 Hz, 1H, -CH2b′Ph); 4.22 (d, J1-2 = 7.7 Hz, 1H, H1); 3.58 (s, 3H, -OCH3); 3.49-3.39 (m, 4H, H2, H3, H4 and H5); 3.39 (br s, 1H, H6). 13C NMR (150 MHz, CDCl3): δ = 137.65, 137.41 (2C, arom); 129.06-127.57 (10C, arom); 102.75 (C1); 82.96 (C3); 78.24 (C4); 75.61 (-CH2-); 75.19 (-CH2′-); 74.82 (C5); 66.29 (C2); 57.58 (-OCH3); 50.83 (t, JC-D = 21.3 Hz, C6). ESI-HRMS: m/z calcd for C21H23DN6O4Na [M+Na]+ 448.1819, found 448.1815.
4.2.17 Methyl 2,6-diamino-2,6-dideoxy-6-(R)-deuterio-β-D-glucopyranoside diacetate (6R-2H-3)
Hydrogenolysis of 19 (36.0 mg, 0.085 mmol) and purification as described for 15α gave the title compound as a colorless oil (19.1 mg, 0.061 mmol, 72 %). (c = 0.3, water). 1H NMR (600 MHz, D2O): δ = 4.62 (d, J1-2 = 8.8 Hz, 1H, H1); 3.65 (dd, J5-4 = 9.4 Hz, J5-6 = 2.7, 1H, H5); 3.63 (dd, J3-2 = 10.6 Hz, J3-4 = 9.2 Hz, 1H, H3); 3.52 (s, 3H, -OCH3); 3.42 (d, J6-5 = 2.8 Hz 1H, H6); 3.35 (t, J4-3 = J4-5 = 9.3 Hz, 1H, H4); 3.01 (dd, J2-1 = 8.7 Hz, J2-3 = 10.6 Hz, 1H, H2); 1.87 (s, 6H, CH3COOH). 13C NMR (150 MHz, D2O): δ = 180.31 (-C=O-); 99.76 (C1); 71.74 (C5); 71.48 (C3); 71.31 (C4); 57.40 (-OCH3); 55.34 (C2); 39.89 (t, JC-D = 21.3 Hz, C6); 22.32 (CH3COOH). ESI-HRMS: m/z calcd for C7H16DN2O4 [M+H]+ 194.1251, found 194.1259.
4.3.1 Methyl 6-azido-2,6-dideoxy-2-NH-benzyloxycarbonyl-β-D-glucopyranoside (21)
Methyl 2-NH-Cbz glucosaminide 2012 (40 mg, 0.12 mmol) was dissolved in pyridine (0.5 mL) and treated with 2,4,6-triisopropylbenzenesulfonyl chloride (145 mg, 0.49 mmol) at ambient temperature and with monitoring by TLC (chloroform:methanol 10:1, Rf = 0.4). After 9 h, the reaction was quenched with methanol (0.5 mL) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (chloroform : methanol 20:1 to 5:1) to give the 6-O-trisyl derivative (50 mg, 0.083 mmol, 68%), which was dissolved in N,N-dimethylformamide (1.5 mL), treated with sodium azide (65 mg, 0.99 mmol), and heated to 80 °C with stirring for 6 h and monitoring by TLC (chloroform:methanol 10:1, Rf = 0.7). The solvent was removed under reduced pressure, and the residue purified by silica gel column chromatography (chloroform : methanol 20:1 to 5:1) to give the title compound 21 (22.8 mg, 0.065 mmol, 78%) as a colorless oil. (c = 0.5 methanol). 1H NMR (600 MHz, CD3OD): δ = 7.48-7.28 (m, 5H, arom); 5.04 (s, 2H, -CH2Ph); 4.32 (d, J1-2 = 8.7 Hz, 1H, H1); 3.65 (dd, J6a-5 = 1.9 Hz, J6a-6b = 12.1 Hz, 1H, H6a); 3.53 (dd, J6b-5 = 5.6 Hz, J6b-6a = 11.9 Hz, 1H, H6b); 3.53 (s, 3H, -OCH3); 3.41 (br.t, J3-2 = J 3-4 = 8.9 Hz, 1H, H3); 3.32 (m, 1H, H2); 3.31 (m, 1H, H4); 3.30 (m, 1H, H5). 13C NMR (150 MHz, CD3OD): δ = 160.33 (-C=O-); 138.80, 129.82, 129.31 (6C, arom.); 104.47 (C1); 78.02 (C5); 76.53 (C3); 72.60 (C4); 69.48 (-CH2-); 60.71 (-OCH3); 57.59 (C2); 54.31 (C6). ESI-HRMS: m/z calcd for C15H20N4O6Na [M+Na]+ 375.1281, found 375.1292.
4.3.2 Methyl 2,6-diamino-2,6-dideoxy-β-D-glucopyranoside diacetate (3)
A solution of 21 (15 mg, 0.046 mmol) in a mixture of 1,4-dioxane (0.8 mL) and 10% v/v aqueous acetic acid (0.1 mL) was treated with 10% palladium hydroxide on activated charcoal (15 mg), then was stirred for 12 h with monitoring by TLC (chloroform:methanol:ammonium hydroxide 3:1:0.7, Rf = 0.40) under a hydrogen atmosphere (45 psi). The reaction mixture was filtered through Celite®, the filter pad washed with water, and the combined filtrate was concentrated and dried under reduced pressure to give 23 as the diacetate salt in the form of a colorless oil (12.6 mg, 0.040 mmol, 88%). (c = 0.3 water). 1H NMR (600 MHz, D2O): δ = 4.59 (d, J1-2 = 8.8 Hz, 1H, H1); 3.65 (dt, J5-4 = 9.5 Hz, J5-6a = 2.9 Hz, J5-6b = 9.2 Hz, 1H, H5); 3.60 (dd, J3-2 = 10.6 Hz, J3-4 = 9.2 Hz, 1H, H3); 3.52 (s, 3H, -OCH3); 3.44 (dd, J6a-5 = 2.9 Hz, J6a-6b = 13.1 Hz, 1H, H6a); 3.34 (t, J4-3 = J4-5 = 9.4 Hz, 1H, H4); 3.12 (dd, J6b-5 = 9.2 Hz, J6b-6a = 13.1 Hz, 1H, H6b); 2.97 (dd, J2-1 = 8.8 Hz, J2-3 = 10.6 Hz, 1H, H2); 1.84 (s, 3H, CH3OOH). 13C NMR (150 MHz, D2O): δ = 181.21 (CH3COOH); 100.10 (C1); 71.81 (C5); 71.81 (C3); 71.34 (C4); 57.39 (-OCH3); 55.41 (C2); 40.19 (C6); 23.11 (CH3COOH). ESI-HRMS: m/z calcd for C7H17N2O4 [M+Na]+ 193.1188, found 193.1197.
Supplementary Material
Acknowledgments
We thank the NIH (AI 123352) for support of this work, and acknowledge support from the NSF (MRI-084043) for the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
References
- 1.Bock K, Duus JO. J Carbohydr Chem. 1994;13:513. [Google Scholar]
- 2.Rao VSR, Qasba PK, Balaji PV, Chandrasekaran R. Conformation of Carbohydrates. Harwood Academic Publishers; Amsterdam: 1998. [Google Scholar]
- 3.Grindley TB. In: Glycoscience: Chemistry and Chemical Biology. Fraser-Reid B, Tatsuta K, Thiem J, editors. Vol. 1. Springer; Berlin: 2001. p. 3. [Google Scholar]
- 4.Midland MM, Asirwatham G, Cheng JC, Miller JA, Morel LA. J Org Chem. 1994;59:4438. [Google Scholar]
- 5.Xu L, Price NPJ. Carbohydr Res. 2004;339:1173. doi: 10.1016/j.carres.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Falcone-Hindley ML, Davis JT. J Org Chem. 1998;63:5555. [Google Scholar]
- 7.Ohrui H, Horiki H, Kishi H, Meguro H. Agric Biol Chem. 1983;47:1101. [Google Scholar]
- 8.Ohrui H, Nishida Y, Meguro H. Agric Biol Chem. 1984;48:1049. [Google Scholar]
- 9.Nishida Y, Ohrui H, Meguro H. Tetrahedron Lett. 1984;25:1575. [Google Scholar]
- 10.Ohrui H, Nishida Y, Watanabe M, Hori H, Meguro H. Tetrahedron Lett. 1985;26:3251. [Google Scholar]
- 11.Ogawa A, Curran DP. J Org Chem. 1997;62:450. doi: 10.1021/jo9620324. [DOI] [PubMed] [Google Scholar]
- 12.Miller DC, Carbain B, Beale GS, Alhasan SF, Reeves HL, Baisch U, Newell Dr, Golding BT, Griffin RJ. Org Biomol Chem. 2015;13:5279. doi: 10.1039/c5ob00211g. [DOI] [PubMed] [Google Scholar]
- 13.Haasnoot CAG, De Leeuw FAAM, Altona C. Tetrahedron. 1980;36:2783. [Google Scholar]
- 14.Smith TD, Gerken JB, Jog PV, Roberts JD. Org Lett. 2007;9:4555. doi: 10.1021/ol7020077. [DOI] [PubMed] [Google Scholar]
- 15.Radom L, Lathan WA, Hehre WJ, Pople JA. J Am Chem Soc. 1973;95:693. [Google Scholar]
- 16.Lane JR, Schrøder SD, Saunders GC, Kjaergaard HG. J Phys Chem A. 2016;120:6371. doi: 10.1021/acs.jpca.6b05898. [DOI] [PubMed] [Google Scholar]
- 17.Padrón JI, Vázquez JT. Chirality. 1997:626. [Google Scholar]
- 18.Thibaudeau C, Stenutz R, Hertz B, Klepach T, Zhao S, Wu Q, Carmichael I, Serianni AS. J Am Chem Soc. 2004;126:15668. doi: 10.1021/ja0306718. [DOI] [PubMed] [Google Scholar]
- 19.Bourke DG, Collins DJ, Hibberd AI, McLeod MD. Aust J Chem. 1996;49:425. [Google Scholar]
- 20.Ferrier RJ, Furneaux RH. Aust J Chem. 1980;33:1025. [Google Scholar]
- 21.Albert HJ, Neumann WP. Synthesis. 1980;942 [Google Scholar]
- 22.White JD, Bolton GL, Dantanarayana AP, Fox CMJ, Hiner RN, Jackson RW, Sakuma K, Warrier US. J Am Chem Soc. 1995;117:1908. [Google Scholar]
- 23.Grindley TB, Reimer GJ, Kralovec J, Brown RG, Anderson M. Can J Chem. 1987;65:1065. [Google Scholar]
- 24.Rasmussen TS, Jensen HH. Org Biomol Chem. 2010;8:433. doi: 10.1039/b918576c. [DOI] [PubMed] [Google Scholar]
- 25.Oikawa M, Shintaku T, Harald S, Fukase K, Kusumoto S. Bull Chem Soc Jpn. 1999;72:1857. [Google Scholar]
- 26.van der Klein PAM, Filemon W, Boons GJPH, Veeneman GH, van der Marel GA, van Boom JH. Tetrahedron. 1992;48:4649. [Google Scholar]
- 27.Shie CR, Tzeng ZH, Wang CC, Hung SC. J Chin Chem Soc. 2009;56:510. [Google Scholar]
- 28.Westerduin P, van Boom JH, van Boeckel CAA, Beetz T. Carbohydr Res. 1985;137:C4. [Google Scholar]
- 29.Jeffs PW, Chan G, Sitrin R, Holder N, Roberts GD, DeBrosse C. J Org Chem. 1985;50:1726. [Google Scholar]
- 30.Fujinaga M, Matsushita Y. Bull Chem Soc Jpn. 1966;39:185. [Google Scholar]
- 31.Iwata R, Sudo M, Nagafuji K, Wada T. J Org Chem. 2011;76:5895. doi: 10.1021/jo200951p. [DOI] [PubMed] [Google Scholar]
- 32.Gao F, Yan X, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, Wright GD, Auclair K. J Med Chem. 2006;49:5273. doi: 10.1021/jm060732n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.