

Abstract
Within replisomes for DNA replication, the primosome is responsible for unwinding double-stranded DNA and synthesizing RNA primers. Assembly of the bacteriophage T4 primosome on individual molecules of ssDNA or forked DNA (fDNA) has been studied by using FRET microscopy. On either DNA substrate, an ordered process of assembly begins with tight 1:1 binding of ssDNA-binding protein (gp32) and helicase-loading protein (gp59) to the DNA. Magnesium adenosine 5′-O-(3-thiotriphosphate) (MgATPγS) mediates the weak binding of helicase (gp41) to DNA coated with gp32 and gp59, whereas MgATP induces gp32 and gp59 to dissociate, leaving gp41 bound to the DNA. Finally, primase (gp61) binds to the gp41·DNA complex. Ensemble studies were used to determine protein stoichiometries and binding constants. These single-molecule studies provide an unambiguous description of the pathway for assembly of the primosome on the lagging strand of DNA at a replication fork.
Keywords: DNA replication, fluorescence microscopy, fluorescence-resonance energy transfer
Primosomes are essential units within replisomes for DNA replication. Primosomes function to advance the replication fork through DNA duplex unwinding and formation of the oligoribonucleotides required for Okazaki-fragment synthesis on the lagging strand. In general, the constitutive proteins of primosomes are functionally conserved and include a DNA helicase, a specialized RNA polymerase or primase, an accessory assembly protein, and an ssDNA-binding protein (1). The replicative helicases, minichromosome maintenance proteins (eukaryotes/archaea), DnaB (Escherichia coli), simian virus 40 tumor antigen, gp4 protein (bacteriophage T7), and gp41 (T4), have common hexameric architecture and similar biochemical properties (2). The primases, however, do not yield equivalent RNA products and have been divided into two major families: bacterial/bacteriophage DnaG-type and eukaryotic/archeal Pritype enzymes. The former have been thought to act primarily as monomers synthesizing short 4- to 12-nt oligoribonucleotides; the latter are multiprotein complexes that include a DNA polymerase (polα-primase), consequently producing chimeric primers with 5′ RNA and 3′ DNA ends (3). Ultimately, both serve to prime the lagging-strand polymerase for processive elongation.
Studies on the T4 bacteriophage have been especially informative in providing insights into replisome mechanisms and functions (4). The primosome of the T4 bacteriophage is assembled from the product of phage genes 41 (DNA helicase), 61 (primase), 59 (helicase-loading protein), and 32 (ssDNA-binding protein). The gp41 protein is a dimer that assembles into a proposed ring-shaped hexamer of asymmetric dimers upon nucleoside triphosphate binding (5, 6). The gp59 protein, which has a high affinity for forked DNA (fDNA) (7, 8), mediates the assembly of gp41 onto ssDNA coated with gp32 protein and binds to both gp41 and gp32 proteins on or off DNA (9–11). With information from the crystal structures of the gp32 protein (12) and the gp59 protein (7), the sites of their interaction have been determined by site-specific crosslinking (13). Similarly, mapping by crosslinking has established the site on the gp59 protein that contacts the gp41 protein (14). Both gp32 and gp41 proteins have been found to induce higher oligomeric complexes of gp59 whose subunits are arranged in a head-to-head orientation.
Kinetic measurements of the ATP-dependent DNA unwinding by gp41 showed maximal activity when gp59 and gp41 were present in a 1:1 mole ratio (hexamer of gp41:hexamer of gp59) (15), reminiscent of the complex of hexameric DnaC (analogous to gp59) and hexameric DnaB helicase in E. coli (16). The gp61 protein, in turn, reaches a maximal priming rate in a 1:1 mole ratio with gp41 and forms higher oligomeric complexes in the presence of DNA, implying that the active gp61 protein is also a hexamer, possibly with a ring-like structure (17). The analogous T7 gene-4 protein combines helicase and primase activity in two functional domains within a single polypeptide that forms a hexamer (18) and a heptamer (19).
From this background, we have defined the pathway of primosome assembly on an ssDNA or fDNA substrate by using single-molecule total internal-reflection fluorescence microscopy and FRET. Interpretations with ensemble experiments are equivocal because of confounding protein–protein interactions both on and off the DNA. The order in which the various proteins assemble to build up the primosome substructure was monitored by introducing donor/acceptor fluorophore pairs selected for FRET at specific loci within the proteins. Because FRET signals fall off as the reciprocal of the sixth power of the distance between the donor and acceptor fluorophores, a separation of <100 Å between the fluorophores is necessary to produce a FRET signal. Thus, detection of FRET implies intimate protein–protein contacts.
Experimental Methods
DNA Substrates. For ensemble studies, the ssDNA substrate was 45 nt long, containing a GTT-priming site (5′-GGGTGGGAGGGAGGTTTGCAACTGATCGATGATAGTACGTCTGTG-3′). For single-molecule studies, the ssDNA substrate was 52 nt long, containing a GTT priming site, and biotinylated at the 5′ end (5′-biotin-AGGGTGGGTGGGAGGGTGGGTTGGAGGGAGTGGGATGATAGTACGTCTGTGT-3′). fDNA substrates were constructed, as described in ref. 20, from a 34-mer primer (5′-ACTCCTTCCGCACGTAATTTTTGACGCACGTTGT-3′) annealed to a 62-mer leading-strand template (5′-ACACAGACGTACTATCATGACGCATCAGACAACGTCGTGCAAAAATTACGTGCGGAAGGAGT-3′) and a partially complementary 50-mer lagging-strand template containing a GT T priming site (5′-GGTGGGTGGGAGGGTGGGTTGGAGGGAGTGGGATGATAGTACGTCTGTGT-3′). For single-molecule studies, the fDNA substrate was biotinylated at the 3′ end of the lagging strand.
Labeling Proteins with Fluorophores. Proteins were purified by chitin-based affinity chromatography as described in ref. 17. The fluorescent labels, 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM), Oregon green 488 maleimide, Alexa Fluor 488 C5 maleimide (A488), and Alexa Fluor 555 C2 maleimide (A555) were attached to accessible cysteine residues on gp32, gp59, gp41, and gp61, as required. Proteins were exchanged into labeling buffer [25 mM Hepes (pH 7.3)/150 mM NaOAc/10% glycerol] and labeled with three-molar excess of the appropriate fluorescent dye for 14–16 h at 4°C. Attachment of the fluorophore on gp32 occurs selectively at C166, the only solvent-accessible cysteine (13). Two cysteine residues, C42 and C215, on gp59 are able to react with the maleimide fluorescent dyes (13). Attachment of a fluorophore to gp41 is predicted to occur at C316, based on crosslinking data reported in ref. 14. Fluorophore labeling of gp61 was shown previously to occur at only two of the six possible cysteines (C124 and C144); C144 is preferentially labeled in the absence of DNA (17). Free dye was removed by chromatography on a cation-exchange resin for gp59 and gp61 and on hydroxyapatite for gp32 and gp41. In each case, the free dye flowed through the column, whereas the protein was retained and eluted with either a salt or phosphate gradient. The labeled proteins stored with 10 mM 2-mercaptoethanol were frozen in aliquots at -70°C.
Characterization of Fluorophore-Labeled Proteins. The extent of labeling was calculated by using the absorbance of the fluorescent dye, and the protein concentrations were determined by the Bradford method. The extent of labeling ranged from 0.5 to 1.2 labels per protein for gp32 and gp41 with one accessible cysteine and from 1.2 to 2.0 labels per protein for gp59 and gp61 with two accessible cysteines.
The activity of the labeled proteins was confirmed by using conventional activity assays. An ATPase assay was used to test the activity of labeled gp32 and labeled gp59 in combination with unlabeled gp41 (10); no significant difference was found in the ATPase activity of gp41 combined with labeled or unlabeled gp32 and gp59. Many labeling reactions of gp41 were carried out; the labeled gp41 used in this work had 10% of the activity of unlabeled gp41 in an unwinding assay (21) and similar activity in the ATPase assay. Studies were carried out with both partially active labeled gp41 and fully active unlabeled gp41, as indicated in the text and figure legends. The priming activity of labeled gp61 was tested by using the priming assay (17); labeling of gp61 resulted in a 20–50% loss in priming activity compared with unlabeled gp61.
Ensemble FRET. Ensemble FRET experiments were performed by using a Fluoromax-2 spectrofluorometer (ISA, Edison, NJ). The complex buffer used in all fluorescence experiments consisted of 25 mM TrisOAc (pH 7.5), 150 mM KOAc, and 10 mM Mg(OAc)2. FRET experiments involving CPM as the donor were excited at 390 nm and monitored at 475 nm for CPM quenching due to energy transfer to Oregon green 488; acceptor sensitization was simultaneously monitored at 519 nm. Likewise, experiments using A488 and A555 as the FRET pair were excited at 488 nm and monitored at 515 nm for donor quenching and at 566 nm for acceptor sensitization. A series of solutions, ranging from 0.1 to 1 μM for the primosome proteins and from 0.05 to 0.1 μM for ssDNA substrate, were combined and allowed to equilibrate at 25°C for ≥10 min to generate the various assemblies. When gp41 was included in the experiment, the effect of adding either 1 mM MgATP or magnesium adenosine 5′-O-(3-thiotriphosphate) (MgATPγS) was examined. Emission spectra were recorded for each protein mixture and corrected for background fluorescence due to the direct excitation of the acceptor at the donor-excitation wavelength. To quantify the background fluorescence of the acceptor, emission spectra were recorded exactly as above, except that unlabeled protein was substituted for the donor-labeled protein at identical concentrations. Binding constants and stoichiometries were determined from plots of the donor quenching and acceptor sensitization vs. concentration of acceptor-labeled protein by using the following equation, where D is the concentration of donor-labeled protein and A is the concentration of acceptor-labeled protein (22).
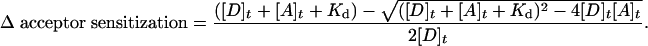 |
Single-Molecule FRET. Single molecules were observed with a home-built f luorescence microscope using total internal-reflection optics. A Zeiss microscope with a ×100, 1.45 numerical aperture, oil-immersion lens and a Pentamax intensified charge-coupled device camera (Roper Instruments, Trenton, NJ) were used to detect single molecules. The microscope filters were set to observe fluorescence from three different sources: F1, emission from donor A488 (excitation at 488 nm/emission at 510–540 nm); F2, emission from energy transfer between A488 and A555 (excitation at 488 nm/emission at 595–645 nm); and F3, emission from acceptor A555 (excitation at 514 nm/emission at 535–585 nm). Experiments were performed at ambient temperature (≈25°C). All single-molecule experiments were performed in triplicate, and, in all cases, well resolved fluorescent spots were observed.
The DNA substrates were attached through a biotin–avidin interaction to the surface of a glass slide prepared with avidin on the surface (23). A solution containing 100 nM ssDNA or fDNA, 15 μM BSA, and 0.1 M sodium phosphate (pH 7.0) was passed three times through the space between the coverslip and the avidin-coated slide. The slide was allowed to incubate for 30 min and then was washed three times with phosphate buffer (without BSA). The concentration of avidin on the slide was adjusted to observe well separated fluorescent spots in the microscope field judged to be due to individual strands of DNA coated with fluorescently labeled proteins. Previously, the identical conditions for coating the slides with avidin resulted in the presence of single molecules established by catastrophic photobleaching (23). This confirmation was not possible for these experiments because multiple protein molecules may be bound to a single strand of DNA. However, slides prepared with 5′-biotinylated 33-mer ssDNA labeled with fluorescein at the 3′ end bleached as single molecules with a half-life of ≈8 s. The mean-fluorescence intensity of individual fluorescein-labeled ssDNA was the same, ±10%.
The first protein to be added in a given experiment was passed three times over a slide with bound DNA substrate and allowed to stand for 10 min. Depending on the experiment, 100–400 nM fluorophore-labeled or unlabeled protein in phosphate buffer was used. The protein not bound to the DNA was then washed away with three aliquots of buffer. Subsequent protein additions were carried out on the microscope stage without moving the slide in three similar applications. This procedure served to wash away the previous unbound protein, and only an excess of the final protein was left on the slide. Fluorescent spots were observed only in the presence of biotinylated-DNA substrate. No fluorescence is seen if DNA without biotin is used, demonstrating that biotin is required and that nonspecific binding of proteins to the slide does not occur.
Primosome Activity on Immobilized ssDNA Substrate. The ssDNA substrate was a 5′-biotinylated 109-mer containing two GTT priming sites (5′-biotin-AGAGGATGATATGGAGGAGAGGTATAGGAGAGAGAAGTATGTGGAGGTTATGGTGGAGTGGTATAGTGGAGTGAAGTGAGGTGAAGGGTTGATGGTGAATATTGGGGAG-3′). ssDNA (≈1.5 pmol) was immobilized per well of a Reacti-Bind NeutrAvidin-coated clear strip plate (Pierce) and washed with coupling buffer [50 mM TrisOAc (pH 7.5)/150 mM NaCl]. A mixture of primosome proteins (1 μM each gp59, gp41, and gp61) in assay buffer [25 mM TrisOAc (pH 7.5)/150 mM KOAc/10 mM Mg(OAc)2/1 mM DTT] with 100 μM guanosine 5′-O-(3-thiotriphosphate) was preincubated in the ssDNA-coated wells for 10 min at room temperature. The protein solution was then removed, and the plate was washed with buffer. The priming reaction was initiated by adding a reaction mixture of 1 mM ATP and 200 μM each CTP, GTP, and UTP, including [α-32P]CTP (≈3,000 Ci/mmol) in assay buffer (1 Ci = 37 GBq). The reaction was incubated at room temperature for 1 min, and 5-μl aliquots were removed and quenched with an equal volume of 0.5 M EDTA. The quenched reaction mixture was separated by denaturing 20% gel electrophoresis followed by analysis using phosphorimaging techniques. Immobilization of ssDNA before incubation with the primosome proteins was required for primer synthesis. The presence of additional primosome proteins in the reaction mixture introduced after the wash step resulted in the synthesis of even more RNA primers.
Results and Discussion
Stoichiometric Complex Between gp32 and gp59. The initial experiments examined the interaction between gp32 and gp59 on ssDNA or fDNA; the results were the same with both types of DNA substrates. When A488-labeled gp32 was added to the slide with bound fDNA, single spots were observed with filter set 1, designed for detection of A488, but not with the other filter sets (Fig. 1A a–c). When A555-labeled gp59 is added, no fluorescence is seen with filter set 1, but fluorescence is seen with both filter sets 2 and 3, designed for detection of FRET between A488 and A555 and detection of A555, respectively (Fig. 1 A d–f). There is a one-to-one correspondence between the position and number of spots in all the frames, indicating that all the gp59 bound to the fDNA associates closely with gp32. The efficiency of energy transfer is ≥50% because the signal-to-noise ratio is ≥2. If A555-labeled gp59 is added to the fDNA first, fluorescence is seen only with filter set 3, revealing that gp59 also binds to fDNA in the absence of gp32 (Fig. 1 A g–i). When A488-labeled gp32 is then added, fluorescence is seen with both filter sets 2 and 3 (data not shown).
Fig. 1.

Protein–protein interactions between gp32 and gp59 on fDNA. (A) The fluorescence from individual molecules of fDNA with the proteins bound in the order as indicated at the side of each row. The gp32 protein is labeled with A488 (gp32D) and the gp59 protein is labeled with A555 (gp59A). The filter sets are described in Experimental Methods: F1 is for A488 emission, F2 for FRET between A488 and A555, and F3 for A555 emission. (B) Ensemble FRET studies of Oregon-green-488-maleimide-labeled gp59 titrated into a solution of 400 nM CPM-labeled gp32 and 100 nM fDNA. The fluorescence spectra of 400 nM CPM-gp32 alone (black line), the endpoint of the titration at 1 μM Oregon-green-488-maleimide-gp59 (dark gray line), and several intermediate spectra (light gray lines) are shown. (C) Analysis of the donor quenching and acceptor sensitization plotted against the gp59 concentration determines the stoichiometry among gp32, gp59, and fDNA to be 1:1:1 with a calculated binding constant of ≈40 nM.
If unlabeled gp59 (500 nM) is added to the fluorescent A488-labeled gp32·A555-labeled gp59·ssDNA complex, most of the fluorescence is displaced from the ssDNA, although not quantitatively. This decrease in signal is consistent with strong protein–protein interactions between gp32 and gp59, both on and off ssDNA. A similar effect is seen if ssDNA (52-mer, 500 nM) without biotin is added to the fluorescent complex, indicating that the two proteins can be transferred to a competing strand of ssDNA (data not shown).
Although the absolute stoichiometry of proteins bound to the ssDNA or fDNA was not determined from single-molecule measurements, the relative intensity of the fluorescence (on a scale of 1–10 ± 25%) can be determined by using National Institutes of Health image software. For a given experiment, the distribution of relative intensities was similar for A488-labeled gp32, A555-labeled gp59, and A488-lableled gp32·A555-labeled gp59 bound to ssDNA with ≈80% of the relative intensities lying in the 2–3 range (data not shown). These results indicate that a heterogeneous population of protein complexes containing multiple gp32 and gp59 molecules is found on individual ssDNA molecules.
In ensemble studies, the interaction between gp32 and gp59 in the absence of DNA did not produce an observable FRET signal. Shown in Fig. 1B is the appearance of a FRET signal when a bulk solution of labeled gp32 bound to ssDNA or fDNA was titrated with labeled gp59. The binding stoichiometry among gp32, gp59, and ssDNA or fDNA is 1:1:1 (Fig. 1C). Normally, gp32 is considered to bind ssDNA with very tight cooperativity and an 8- to 10-nt footprint size. However, because of the short DNA substrates used in our studies, we observed a very low degree of cooperativity and predominantly only a 1:1 stoichiometry between gp32 and DNA substrate. The calculated binding constants are ≈30 nM on ssDNA substrate and ≈40 nM on fDNA substrate. However, the protein and DNA concentrations in these titrations are well above this Kd value; therefore, these binding constants are only approximations. This is in accord with previous data (24).
ATP-Dependent Binding of gp41. The next set of experiments examined the introduction of gp41 by gp59 onto ssDNA or fDNA; again, similar results were obtained, with both DNA substrates coated with gp32. The addition of active unlabeled gp41 to the f luorescent A488-labeled gp32·A555-labeled gp59·fDNA complex (Fig. 2A a–c) causes no major changes in complex fluorescence in the absence of nucleotide and only a small decrease in fluorescence in the presence of 500 μM MgATPγS to promote hexameric gp41 formation while preventing movement of the helicase (Fig. 2 A d–f). However, addition of gp41 to the complex in the presence of 500 μM MgATP results in the disappearance of the A488-labeled gp32 and A555-labeled gp59 proteins (Fig. 2 A g–i). These results suggest that hexameric gp41 in the presence of a nonhydrolyzable nucleotide analog binds only weakly to DNA with minimal disruption of the gp32·gp59 interaction. However, loading of gp41 protein onto DNA is more efficient in the presence of a hydrolyzable nucleotide displacing both gp32 and gp59. The displacement of gp32 and gp59 by gp41 may be the result of their dissociation from the primosome, actively being displaced by translocation of gp41 requiring ATP hydrolysis, or may simply be due to the lack of space to bind on short DNA substrates. In these single-molecule studies, rebinding of gp32 and gp59 to the DNA is not observed because of their extremely low concentrations on the slide.
Fig. 2.

The ATP-dependent loading of gp41 by gp59 onto fDNA coated with gp32. The fluorescence from individual molecules of fDNA with the proteins bound in the order as indicated at the side of each row. (A) The gp32 protein is labeled with A488 (gp32D), the gp59 protein is labeled with A555 (gp59A), and the gp41 protein is unlabeled. MgATPγS (500 μM) is present for the sample in row 2, and 500 μM MgATP is present for the sample in row 3. (B) The gp32 protein is unlabeled, the gp59 protein is labeled with A555 (gp59A), and the gp41 is labeled with A488 (gp41D). MgATPγS (500 μM) is present for the sample in row 2, and 500 μM MgATP is present for the sample in row 3 (5 min after addition of gp41 and nucleotide) and in row 4 (30 min after addition of gp41 and nucleotide). The filter sets are described in the legend to Fig. 1.
In ensemble studies with ssDNA, the gp59·gp32 FRET signal was partially reduced by the addition of gp41 only in the presence of MgATPγS (data not shown). There was no reduction in FRET signal with either gp41 alone or with MgATP, as in the single-molecule experiments. These disparate results can be explained by the experimental design. In the presence of MgATPγS, the partial reduction in the ensemble FRET signal can be attributed to the displacement of gp32 and gp59 from the DNA by gp41 for a fraction of the complex population. In the presence of MgATP, from the single-molecule experiments, we expect that gp41 displaced all of the gp32 and gp59 from the ssDNA. However, the high concentration of gp32 and gp59 in ensemble studies allowed them to rebind the DNA, whereas the very low concentration of gp32 and gp59 on the slides in single-molecule studies did not permit them to rebind to the DNA. Additionally, after gp41 hydrolyzed all of the ATP in the ensemble studies, the lack of nucleotide would prevent the gp41 from oligomerizing and disrupting any of the gp32·gp59 FRET signal.
The binding of gp41 was tracked directly by repeating the above experiments with A488-labeled gp41, which was added to unlabeled gp32·A555-labeled gp59·fDNA complex. The initial gp32·A555-labeled gp59·fDNA complex displays fluorescence with filter set 3 (Fig. 2B j–l). No change in fluorescence is observed when A488-labeled gp41 is added in the absence of nucleotide (data not shown). When A488-labeled gp41 is added in the presence of 500 μM MgATPγS, no change in fluorescence is observed with filter set 3, and a small amount of fluorescence is observed with filter set 2 (Fig. 2B m–o). Likewise, when A488-labeled gp41 is added in the presence of 500 μM MgATP, fluorescence is observed initially with both filter sets 2 and 3 (Fig. 2B p–r). But after 30 min, fluorescence is observed with filter set 1, indicating that only A488-labeled gp41 is bound to the DNA (Fig. 2B s–u). These data are in accord with the expected assembly of the dimeric gp41 into a hexameric form in the presence of nucleotide, followed by its loading onto DNA by the gp59 protein. Only a relatively small portion of the fDNA has bound A488-labeled gp41 after the addition of MgATPγS. If the slide is washed with buffer three times or if the buffer contains 15 μM BSA, no A488-labeled gp41 is seen bound to the DNA. We estimate that Kd > 200 nM. However, the amount of A488-labeled gp41 bound to fDNA after addition of MgATP is significantly greater than that found with MgATPγS. In addition, a transient FRET signal is observed between gp59 and gp41 followed by the loss of gp32 and A555-labeled gp59 based on the earlier experiments. The gp41 cannot be displaced by washing with buffer and/or BSA after the 30-min treatment with MgATP. We conclude that nucleotide binding to gp41 induces a conformational change that promotes close association of gp41 with gp59 (as judged by the FRET) but that ATP hydrolysis is required for more efficient binding to the DNA and the eventual loss of gp32 and gp59 from the complex.
Under the conditions in Fig. 2 a–o, the images were unchanged from 5 min to >30 min. The time required for complete loss of gp32 and gp59 is approximately the same for unlabeled and labeled gp41.
Ensemble FRET signals were observed between gp59 and gp41; however, significant FRET signals were also observed for this complex in the absence of DNA, thus preventing protein complexes on the DNA from being distinguished from those in solution. This ambiguity, coupled with the transient nature of the FRET signal on DNA, prevented the determination of stoichiometry or a binding constant for this complex.
In separate experiments, we asked whether both gp32 and gp59 must be present on the DNA to load gp41. Binding of A488-labeled gp41 to either a complex of gp59·DNA or gp32·DNA was not observed even in the presence of 100 μM MgATP (data not shown). This result suggests that gp41 loading is promoted by the presence of both gp32 and gp59.
Binding of gp61 Completes Primosome Assembly. To complete the primosome assembly, A488-labeled gp41 and A555-labeled gp61 were added to the nonfluorescent gp32·gp59·fDNA complex in the presence of MgATP. The final complex fluoresces with filter sets 2 and 3 but not with filter set 1, requiring that both A488-labeled gp41 and A555-labeled gp61 are bound to the fDNA (Fig. 3A a–f). A repeat of the above experiment with A555-labeled gp59, unlabeled gp41, and A488-labeled gp61 in the presence of 500 μM MgATP resulted in the loss of A555-labeled gp59 fluorescence and no addition of A488-labeled-gp61 fluorescence (data not shown). The absence of any signal suggests that the gp41 displaced A555-labeled gp59 and that the gp41·A488-labeled gp61 complex may have translocated off the DNA because of helicase movement. Additionally, A555-labeled gp61 was not able to bind gp32 and gp59-coated DNA in the presence or absence of MgATP (Fig. 3A g–i). We conclude that only gp41 can promote gp61 binding and that the gp41·gp61·DNA complex is readily captured only when gp41 has marginal unwinding activity (A488-labeled gp41 has ≈10% the activity of unlabeled gp41). Unfortunately, the presence of significant FRET signals between gp41 and gp61 in the absence of ssDNA hampered our ability to determine the stoichiometry or a binding constant for the gp41·gp61 complex in ensemble studies.
Fig. 3.

Completed primosome formation by the addition of gp61 or an alternative, direct formation of the gp41/gp61 primosome. The fluorescence from individual molecules of fDNA with the proteins bound in the order as indicated at the side of each row. (A) The gp32 and gp59 proteins are unlabeled, the gp41 protein is labeled with A488 (gp41D), and the gp61 protein is labeled with A555 (gp61A) in the presence of 500 μM MgATP. (B) The gp41 protein is labeled with A488 (gp41D), and the gp61 protein is labeled with A555 (gp61A) in the absence of MgATP. The filter sets are described in the legend to Fig. 1.
Finally, we asked whether the primosome now defined as the complex of gp41·gp61 bound to fDNA could be reached directly. In the absence of gp32 and gp59, both A488-labeled gp41 and A555-labeled gp61 bind separately to fDNA, even in the absence of nucleotide (Fig. 3B j–o). A FRET signal is observed when both A488-labeled gp41 and A555-labeled gp61 are present (Fig. 3B p–r), suggesting close association of gp41 and gp61.
Primosome complexes formed on an immobilized ssDNA substrate were able to synthesize pentaribonucleotide primers in a 96-well plate-assay format (data not shown). Thus assembly of the primosome in the single-molecule system generates a primosome that can carry out its normal biological functions. However, the possibility that the low activity of the labeled gp41 may be influencing the results merits discussion. Both the partially active, labeled gp41 and the fully active, unlabeled gp41 displace gp32 and gp59 from DNA only in the presence of MgATP. Tight binding of unlabeled gp41 requires MgATP, indicating that ATPase activity is essential for binding and subsequent events. The labeled gp41 has a lower, but not negligible, activity as a helicase/ATPase that is sufficient for primosome formation. Consequently, observations with either unlabeled or labeled gp41 are self-consistent, so the deduced assembly process is physiologically relevant.
Conclusions
The satisfactory agreement among data compiled from crosslinking, isothermal calorimetry, and ensemble FRET titrations now coupled to the results from the single-molecule FRET studies provides the mechanism for primosome assembly shown in Fig. 4. Thus the primosome-assembly sequence, as visualized on an individual molecule of fDNA, is as follows. The gp32 protein binds to fDNA with either subsequent or concurrent binding of gp59. These two proteins form a stoichiometric, closely associated complex on the fDNA in accord with data presented in refs. 11 and 25. Subsequently, gp41 binds to gp59 and is loaded onto the fDNA in the presence of nucleotide. ATP hydrolysis and unwinding activity are required for gp41 to displace gp32 and gp59, by either dissociation or translocation. The gp61 protein then binds and interacts closely with gp41 on fDNA. Other pathways for primosome assembly in the absence of gp32 may also exist, for example, the direct binding of gp41 and gp61 to fDNA or, alternatively, by gp59. However, these pathways are less relevant to the action of the replisome in the cellular environment.
Fig. 4.

Assembly mechanism of the T4 lagging-strand primosome on fDNA. The gp32 protein binds to fDNA with either subsequent or concurrent binding of gp59. Subsequently, gp41 binds to gp59 and is loaded onto the fDNA in the presence of nucleotide. ATP hydrolysis is required for gp41 to displace gp32 and gp59, either directly or by translocation. The gp61 protein then binds and interacts closely with gp41 on fDNA. In the absence of gp32 and gp59, both gp41 and gp61 bind to fDNA.
One can ask whether the same assembly pathway applies to primosome function in the context of a replication fork, the difference being the presence of the polymerase and clamp proteins. Electron microscopy studies have shown that gp32-covered ssDNA on the lagging-strand template is organized into bobbin-like structures that may also contain the gp59 protein (26). Our data suggest the primosome that drives replication-fork unwinding and priming of lagging-strand synthesis consists of simply the gp41 and gp61 proteins that are formed at a gp32·gp59 interface in an ATP-driven process. Because gp61 alone does not bind at this interface, gp59 imbedded at other loci in the bobbin may reflect an abortive complex or may act to coordinate the action of the leading- and lagging-strand holoenzymes (27).
Acknowledgments
This work was supported by National Institutes of Health Grants GM65128 (to G.G.H.), GM13306 (to S.J.B.), and GM071130 (to M.M.S.).
Abbreviations: fDNA, forked DNA; CPM, 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin; A488, Alexa Fluor 488 C5 maleimide; A555, Alexa Fluor 555 C2 maleimide; MgATPγS, magnesium adenosine 5′-O-(3-thiotriphosphate).
References
- 1.Benkovic, S. J., Valentine, A. M. & Salinas, F. (2001) Annu. Rev. Biochem. 70, 181-208. [DOI] [PubMed] [Google Scholar]
- 2.Yu, X., Jexewska, M. J., Bujalowski, W. & Egelman, E. H. (1996) J. Mol. Biol. 259, 7-14. [DOI] [PubMed] [Google Scholar]
- 3.Keck, J. L. & Berger, J. M. (2001) Nat. Struct. Biol. 8, 2-4. [DOI] [PubMed] [Google Scholar]
- 4.Baker, T. A. & Bell, S. P. (1998) Cell 92, 295-305. [DOI] [PubMed] [Google Scholar]
- 5.Liu, C. C. & Alberts, B. M. (1981) J. Biol. Chem. 256, 2813-2820. [PubMed] [Google Scholar]
- 6.Dong, F., Gogol, E. P. & von Hippel, P. H. (1995) J. Biol. Chem. 270, 7462-7473. [DOI] [PubMed] [Google Scholar]
- 7.Mueser, T. C., Jones, C. E., Nossal, N. G. & Hyde, C. C. (2000) J. Mol. Biol. 296, 597-612. [DOI] [PubMed] [Google Scholar]
- 8.Jones, C. E., Mueser, T. C. & Nossal, N. G. (2000) J. Biol. Chem. 275, 27145-27154. [DOI] [PubMed] [Google Scholar]
- 9.Yonesaki, T. (1994) J. Biol. Chem. 269, 1284-1289. [PubMed] [Google Scholar]
- 10.Morrical, S. W., Hempstead, K. & Morrical, M. D. (1994) J. Biol. Chem. 269, 33069-33081. [PubMed] [Google Scholar]
- 11.Lefebvre, S. D., Wong, M. L. & Morrical, S. W. (1999) J. Biol. Chem. 274, 22830-22838. [DOI] [PubMed] [Google Scholar]
- 12.Shamoo, Y., Friedman, A. M., Parsons, M. R., Konigsberg, W. H. & Steitz, T. A. (1995) Nature 376, 362-366. [DOI] [PubMed] [Google Scholar]
- 13.Ishmael, F. T., Alley, S. C. & Benkovic, S. J. (2001) J. Biol. Chem. 276, 25236-25242. [DOI] [PubMed] [Google Scholar]
- 14.Ishmael, F. T., Alley, S. C. & Benkovic, S. J. (2002) J. Biol. Chem. 277, 20555-20562. [DOI] [PubMed] [Google Scholar]
- 15.Raney, K. D., Carver, T. E. & Benkovic, S. J. (1996) J. Biol. Chem. 271, 14074-14081. [DOI] [PubMed] [Google Scholar]
- 16.San Martin, C., Radermacher, M., Wolpensinger, B., Engel, A., Miles, C. S., Dixon, N. E. & Carazo, J. M. (1998) Structure 6, 501-509. [DOI] [PubMed] [Google Scholar]
- 17.Valentine, A. M., Ishmael, F. T., Shier, V. K. & Benkovic, S. J. (2001) Biochemistry 40, 15074-15085. [DOI] [PubMed] [Google Scholar]
- 18.Kato, M., Frick, D. N., Lee, J., Tabor, S., Richardson, C. C. & Ellenberger, T. (2001) J. Biol. Chem. 276, 21809-21820. [DOI] [PubMed] [Google Scholar]
- 19.Toth, E. A., Li, Y., Sawaya, M. R., Cheng, Y. & Ellenberger, T. (2003) Mol. Cell 12, 1113-1123. [DOI] [PubMed] [Google Scholar]
- 20.Kaboord, B. F. & Benkovic, S. J. (1993) Proc. Natl. Acad. Sci. USA 90, 10881-10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raney, K. D. & Benkovic, S. J. (1995) J. Biol. Chem. 270, 22236-22242. [DOI] [PubMed] [Google Scholar]
- 22.Segel, H. I. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems (Wiley, New York), pp. 72-77.
- 23.Rajagopalan, P. T., Zhang, Z., McCourt, L., Dwyer, M., Benkovic, S. J. & Hammes, G. G. (2002) Proc. Natl. Acad. Sci. USA 99, 13481-13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu, H., Wang, Y., Bleuit, J. S. & Morrical, S. W. (2001) Biochemistry 40, 7651-7661. [DOI] [PubMed] [Google Scholar]
- 25.Ma, Y., Wang, T., Villemain, J. L., Giedroc, D. P. & Morrical, S. W. (2004) J. Biol. Chem. 279, 19035-19045. [DOI] [PubMed] [Google Scholar]
- 26.Chastain, P. D., Jr., Makhov, A. M., Nossal, N. G. & Griffith, J. (2003) J. Biol. Chem. 278, 21276-21285. [DOI] [PubMed] [Google Scholar]
- 27.Jones, C. E., Mueser, T. C. & Nossal, N. G. (2004) J. Biol. Chem. 279, 12067-12075. [DOI] [PubMed] [Google Scholar]