

Abstract
In animals, sporadic injections of the mitochondrial toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) selectively damage dopaminergic neurons but do not fully reproduce the features of human Parkinson's disease. We have now developed a mouse Parkinson's disease model that is based on continuous MPTP administration with an osmotic minipump and mimics many features of the human disease. Although both sporadic and continuous MPTP administration led to severe striatal dopamine depletion and nigral cell loss, we find that only continuous administration of MPTP produced progressive behavioral changes and triggered formation of nigral inclusions immunoreactive for ubiquitin and α-synuclein. Moreover, only continuous MPTP infusions caused long-lasting activation of glucose uptake and inhibition of the ubiquitin-proteasome system. In mice lacking α-synuclein, continuous MPTP delivery still induced metabolic activation, but induction of behavioral symptoms and neuronal cell death were almost completely alleviated. Furthermore, the inhibition of the ubiquitinproteasome system and the production of inclusion bodies were reduced. These data suggest that continuous low-level exposure of mice to MPTP causes a Parkinson-like syndrome in an α-synuclein-dependent manner.
Keywords: neurodegeneration, mitochondria, neuronal inclusions, Lewy bodies
To understand the pathogenesis of Parkinson's disease (PD), it is necessary to reproduce its biochemical, physiological, and morphological features in animal models. One hallmark of PD is neuronal inclusions called Lewy bodies (for reviews, see refs. 1–4), which are difficult to recreate in animal models. In mice, PD is classically modeled by application of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (5). MPTP is transported into brain where it is metabolized to MPP+ (6). MPP+ is selectively taken up into dopaminergic neurons (7) and acts as a potent inhibitor of mitochondrial complex I, thus poisoning dopaminergic neurons (8, 9). When injected into mice, MPTP causes a PD-like syndrome with massive loss of nigral dopaminergic neurons and striatal dopamine. Surprisingly, MPTP injections do not induce neuronal inclusions characteristic of PD (refs. 5 and 10; reviewed in ref. 1) even after repeated injections (11, 12). Besides MPTP, at least three other toxins, rotenone (13–15), ubiquitin-proteasome inhibitors (16, 17), and methamphetamine (18), produce a PD-like syndrome in mice, but unlike MPTP, these toxins also cause formation of α-synuclein-containing inclusions (13–18).
The current data suggest that oxidative damage (via inhibition of mitochondrial complex I) and impairments of the ubiquitin–proteasome system represent a final common pathway in the pathogenesis of PD-like syndromes (refs. 16 and 19; reviewed in refs. 2, 3,20, and 21). In support of this conclusion, the complex I inhibitor rotenone also impairs the ubiquitin proteasome system (19, 22), and α-synuclein and its fragments may inhibit the proteasome (23, 24). Furthermore, Lewy bodies in PD contain both ubiquitin and α-synuclein, consistent with a general failure of the ubiquitin proteasome system, and mutations in enzymes of the ubiquitin proteasome system are found in familial forms of PD (reviewed in refs. 1–4).
In view of these observations, it is puzzling that MPTP-induced damage to nigrostriatal neurons does not induce formation of Lewy bodies. In facing this discrepancy, one should recall that only continuous administration of rotenone triggers formation of neuronal inclusions (13), whereas discontinuous administration of rotenone even at high doses damages the basal ganglia but produces no inclusions (25, 26). This crucial observation indicates that inclusions may form only when mild and prolonged inhibition of the mitochondrial respiratory chain causes a chronic decrease of the ubiquitin-proteasome activity. If this hypothesis was correct, then other inhibitors of the mitochondrial respiratory chain, such as MPTP, should induce similar effects when delivered continuously at low doses.
To test this hypothesis, we examined whether continuous application of MPTP in mice produces formation of neuronal inclusions resembling Lewy bodies, and leads to a dose-dependent impairment of the ubiquitin proteasome system. We implanted mice chronically with osmotic minipumps and measured the pathogenic effects of continuously delivered MPTP in comparison with sporadic injections. We find that mice exposed to continuously administered MPTP exhibit a dose-dependent PD-like syndrome with formation of α-synuclein containing inclusions, and that mice lacking α-synuclein are largely protected against the chronic toxicity of continuously administered MPTP, but not against the acute toxicity of MPTP.
Experimental Procedures
Animal Experiments. Male C57BL/6J mice (9 weeks old; Charles River Breeding Laboratories) were treated with MPTP (Fluka) with two procedures:
Continuous minipump infusions. Mice were implanted with osmotic minipumps (Alzet, Cupertino CA, purchased from Charles River Breeding Laboratories) that release saline solution (control; n = 15 mice) or MPTP-HCl at 1 mg (n = 10), 5 mg (n = 10), or 30 mg (n = 20) per kg bodyweight daily (see Supporting Text, which is published as supporting information on the PNAS web site, for a detailed description), and analyzed 30 days after the start of MPTP infusions unless stated otherwise. When mice receiving 30 mg/kg MPTP started to exhibit behavioral impairments (after ≈1 week), a subgroup was treated with L-DOPA (100 mg/kg ip; Sigma; n = 5; ref. 27) or apomorphine (5 mg/kg daily delivered s.c. with osmotic Alzet minipumps; n = 5; ref. 28). In additional sets of mice, we measured MPTP and MPP+ concentrations as described (29), surgically removed pumps and striatum after 1–28 days of continuous MPTP infusions, and examined proteasome activities (16) (see Supporting Text for a detailed description). For 2-deoxyglucose (2-DG) uptake experiments, mice receiving 30 mg/kg daily MPTP were killed at 7 (n = 10), 14 (n = 8), and 28 (n = 8) days after pump implantation.
Single or multiple bolus injections of MPTP. Mice were injected i.p. with a single (30 mg/kg; n = 20) or four separate MPTP doses (4 × 20 mg/kg, 2 h apart; n = 20; refs. 5 and 30), killed 7 and 30 days after injections, and analyzed morphologically and neurochemically. Additional mice treated with bolus injections of MPTP (30 mg/kg) were killed at 1 h, 7 days, or 28 days after injections to assay 2-DG uptake (n = 8 for each time point), or killed at 30 min, 1 h, 2 h, 4 h, 6 h, and 12 h after injections to measure MPTP and MPP+ in the striatum (n = 5 for each time point). Proteasome activities were determined before treatment and at 2, 4, 12, 24, and 48 h after injections (n = 5 for each time point; see Supporting Text).
α-Synuclein-deficient mice. We analyzed the effects of continuous MPTP infusion (30 mg/kg) on α-synuclein-deficient and littermate control mice by using two lines of α-synuclein-deficient mice: α-synuclein knockout (KO) mice with a deletion of the first α-synuclein coding exon (ref. 31; n = 10 mice for measurements of monoamine levels and for light microscopy; n = 5 for electronmicroscopy, proteasome assays, and 2-DG uptake; n = 15 for locomotor activity measurements), and a spontaneous α-synuclein deletion that arose in Bl6 mice from a commercial vendor (Harlan–Winkelmann; see refs. 31 and 32; n = 5 for each assay).
2-DG uptake experiments. 2-DG uptake experiments were carried out essentially as described (33) 1 h or 7 and 28 days after sporadic MPTP administration (a single dose of 30 mg/kg MPTP; n = 8 for each group) or 7, 14, and 28 days after the beginning of the continuous MPTP administration (n = 5–10; see Supporting Text). Autoradiograms were analyzed by quantitative densitometry (at least eight sections per condition) by using corpus callosum as a control area.
Open field tests. Mice were housed in separate cages and adapted to the open-field test daily 1 week before MPTP infusions. Mice were examined daily between 9:00 and 12:00 a.m. from 3 days before until up to 21 days after starting the MPTP minipump infusions (see Supporting Text for details).
Biochemical Assays. Transmitter measurements. Transmitter measurements were performed by reverse-phase ion-pairing HPLC coupled with two electrochemical detectors (ref. 16; see Supporting Text).
Assay of proteasome activity. Proteasome activity was measured in substantia nigra homogenates by using the 20S Proteasome Activity Assay kit (Chemicon) for chymotrypsin-like activity, Cbz-Leu-Leu-Glu-AMC (Sigma) for postglutamyl peptidase activity (or peptidyl-glutamyl-peptide hydrolyzing, PGPH activity), and Boc-Leu-Ser-Thr-Arg-AMC (Sigma) for trypsin activity. Activities were monitored by detection of fluorescent 7-amido-4-methylcoumarin (AMC) after cleavage from the various synthetic fluorogenic peptides (see Supporting Text for details).
Morphological Experiments. Light and electron microscopy of native and immunostained samples were performed essentially as described (refs. 16, 31, and 34; see Supporting Text for a detailed description).
Statistics. Comparisons were analyzed by using the ANOVA test with Sheffè's post hoc analysis.
Results
Continuous MPTP Delivery via an Osmotic Minipump. To test whether continuous administration of MPTP via an implanted minipump is feasible, we first monitored the stability of MPTP in implanted minipumps in mice. In the minipumps, MPTP levels declined slowly in the first 21 days, decreased more rapidly thereafter, and became undetectable at 28 days (Fig. 7a, which is published as supporting information on the PNAS web site). Part of the loss of MPTP was due to spontaneous oxidation of MPP+, which gradually increased in the pump.
We then measured the concentration of MPP+ in the striatum of the same mice in which we monitored the pump concentrations of MPTP and MPP+, and additionally determined the striatal MPP+ concentrations of mice that had been injected sporadically with MPTP (once with 30 mg or four times with 20 mg of MPTP per kg of body weight). We found that continuous MPTP administration produced stable MPP+ levels in the striatum for up to 21 days. In contrast, sporadic MPTP injections resulted in up to 5-fold higher peak MPP+ concentrations that then rapidly declined (Fig. 7 b–d).
Stimulation of 2-DG Uptake by MPTP. We next examined the relative efficacy of continuous vs. sporadic MPTP administration in stimulating 2-DG uptake in brain. A bolus injection of MPTP (30 mg/kg bodyweight) dramatically enhanced 2-DG uptake, measured 1 h after the injection by using 14C-labeled 2-DG as a radioactive tracer, but had little effect on 2-DG uptake 7 days later (Fig. 1 a–c). In contrast, continuous MPTP pump infusions exerted a long-lasting activation of 2-DG uptake (Fig. 1d) that was evident in the neostriatum (Fig. 1e), globus pallidus (Fig. 1f), and thalamus (Fig. 1g). No such activation was observed over the frontal cortex or substantia nigra (Fig. 1 h–j).
Fig. 1.

MPTP-induced activation of [14C]2-DG uptake. (a–d) False-color autoradiographs of brain sections from mice 1 h after injection with [14C]2-DG. 2-DG uptake was monitored in control mice (a), mice that were analyzed 1 h (b) or 7 days (c) after injections with a single dose of MPTP (30 mg/kg), and mice that were analyzed after 28 days of continuous MPTP infusion (30 mg/kg daily). (e–j) 2-DG uptake was quantified by film densitometry (n = 8–10 mice per group) in the neostriatum (e), globus pallidus (f), thalamus (g), frontal cortex (h), substantia nigra pars compacta (i), and substantia nigra pars reticulata (j). Asterisks indicate that the treated sample is significantly different (P < 0.05; ANOVA) from the controls (single asterisk) or from both the control and the MPTP-bolus injected sample 7 days after the injection (double asterisk). All data shown in this in all subsequent figures represent means ± SEMs.
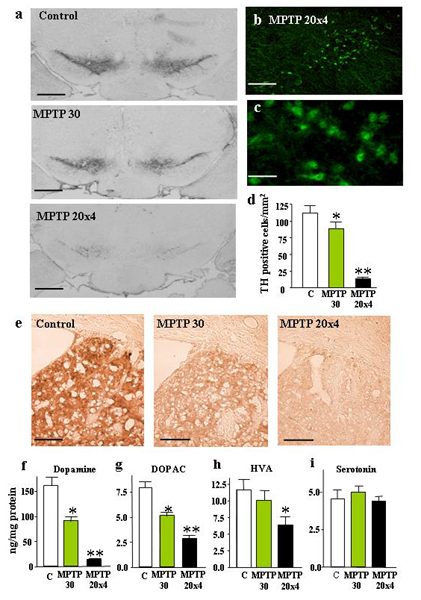
Neurotoxicity After Sporadic MPTP Administrations. Consistent with earlier studies (5, 29, 30), we found that sporadic MPTP injections (one dose of 30 mg/kg, or four doses of 20 mg/kg) caused nigral cell loss in mice, as documented by a large decrease in tyrosine hydroxylase (TH)-positive neurons in the substantia nigra and TH-positive fibers in the striatum (Fig. 8, which is published as supporting information on the PNAS web site). The nigral cell loss was caused by neurodegeneration as visualized by Fluoro-Jade B staining (34), and was associated with a decline in the levels of dopamine and DOPAC (a dopamine metabolite) by up to 85%, whereas the levels of serotonin were unchanged (Fig. 8). Furthermore, bolus injections of MPTP decreased the nigral ubiquitin proteasome system activity for a short time period (<24 h). The ubiquitin–proteasome activity rapidly recovered and increased above starting levels, probably because of a reactive increase in proteasome activity in glia cells (Fig. 2a). However, even in the presence of massive neuronal cell death, we never observed neuronal inclusion bodies in sporadically MPTP-treated mice.
Fig. 2.

Striatal proteasome activity after MPTP treatments. (a) Relative chymotrypsin-like, trypsin-like, and peptidyl-glutamyl-peptide hydrolyzing (PGPH) proteasome activities in mice exposed to a single (30 mg/kg) or four separate doses (20 mg/kg each) of MPTP (n = 5 mice at each time interval). Asterisks indicate statistically significant differences (P < 0.05) from baseline proteasome activity (single asterisk) or from both baseline proteasome activity and activity after single MPTP dose (double asterisk). (b) Delayed and prolonged inhibition of proteasome activity after continuous MPTP administration (1, 5, or 30 mg/kg MPTP daily) for the indicated time periods. Asterisks indicate statistically significant differences (P < 0.05) from baseline proteasome activity (single asterisk) or from both baseline proteasome activity and activity after lower MPTP doses (1 and 5 mg/kg, daily, double asterisk; n = 5 mice).
Neurotoxicity After Continuous MPTP Infusions. Similar to sporadic MPTP injections, continuous MPTP pump infusions (5 or 30 mg/kg daily) severely decreased the number of TH-positive cells in the substantia nigra and the density of TH-positive fibers in the striatum (Fig. 3 a–c), but had no effect on GABAergic neurons (data not shown). In parallel with the decrease in TH-positive cells, we observed an astrocytic tissue reaction (Fig. 9, which is published as supporting information on the PNAS web site), and detected FluoroJade B-positive degenerating neurons (Fig. 10, which is published as supporting information on the PNAS web site). The decrease in TH-positive neurons was accompanied by a marked decline in the levels of dopamine, DOPAC, and HVA, whereas the levels of serotonin were not altered (Fig. 3d). The dopamine decline was evident after continuous infusions of only 1 mg/kg MPTP, which caused no significant loss of TH-positive cells.
Fig. 3.

Neurotoxicity induced by continuous MPTP administration. (a) Representative tyrosine hydroxylase (TH)-stained sections of the substantia nigra from mice that were continuously treated for 28 days with control pump infusions or with infusions of 1, 5, or 30 mg MPTP/kg daily. (Scale bar, 600 μm.) (b and c) TH-positive cell counts in the substantia nigra (b) and semiquantitative densitometric measurements of the TH signal in striatum (c)(n = 10 mice per group). (d) Striatal monoamine levels in MPTP-treated mice (n = 10 mice per group). Asterisks indicate statistically significant differences (P < 0.05) of a sample compared to control (single asterisks) or to both the control and the lower MPTP dose (double asterisks).
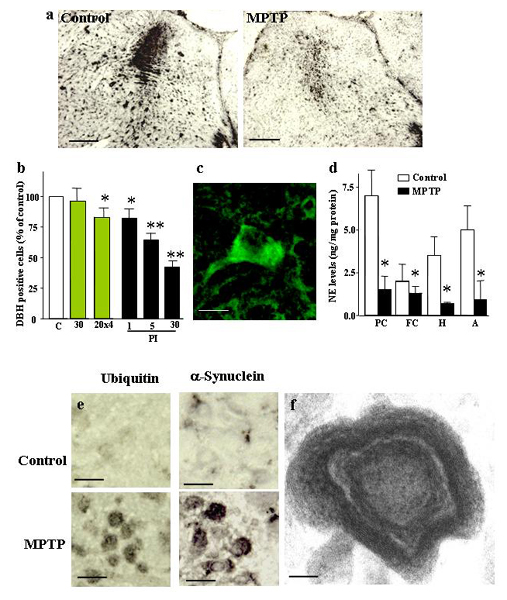
We next measured the activity of the ubiquitin proteasome system in the substantia nigra from mice continuously treated with MPTP infusions (Fig. 2b). Again, we observed a long-lasting impairment that was strongest for the maximal dose, but significant even after MPTP infusions of only 1 mg/kg. We then asked whether the decline in the ubiquitin–proteasome system correlated with other features of PD-like syndromes. Immunostaining of the substantia nigra with antibodies to ubiquitin (Fig. 4a) and α-synuclein (Fig. 4b) revealed clusters of immunoreactive material in neurons. When examined by electron microscopy at early time intervals (72 h), these clusters corresponded to “whorls” (Fig. 4c). Whorls are neuronal inclusions that consist of concentric membranes and contain α-synuclein immunoreactivity, as previously observed after administration of proteasome inhibitors (16). After 30 days of MPTP infusion, inclusions no longer contained membranes but were electron-dense and fibrillar and still stained for α-synuclein (Fig. 4d and Fig. 11, which is published as supporting information on the PNAS web site).
Fig. 4.

Continuous MPTP administration induces neuronal inclusions and a dopamine-dependent behavioral phenotype. (a and b) Inclusion bodies in the substantia nigra from mice treated for 28 days with continuous MPTP infusions (30 mg/kg daily). Inclusions were stained for ubiquitin (a) and α-synuclein (b). (Scale bars, 120 μm at Left and 35 μm at Right.) (c and d) Electron micrographs of inclusions observed 72 h (c) or 30 days (d) of MPTP infusion. (Scale bars, 0.1 μm.) Note that similar inclusions were observed in the locus coeruleus (Fig. 12). (e and f) Open field behaviors of mice infused continuously with control solution or intermediate (5 mg/kg daily) and high doses of MPTP (30 mg/kg daily). Suppression of open field activity (e) and rearing (f) by continuous MPTP administration was reversed by the mixed dopamine agonist apomorphine (APO). Asterisks indicate statistically significant differences from control mice (see Movies 1–3, which are published as supporting information on the PNAS web site).
The effects of continuous MPTP administration were not limited to dopaminergic neurons. As in human PD, we also observed neurodegeneration of noradrenergic neurons in the locus coeruleus, with a pathology similar to that of the substantia nigra: a marked loss of noradrenergic cells as visualized by dopamine β-hydroxylase staining, and a large decrease (up to 80%) in norepinephrine levels throughout the forebrain (Fig. 12, which is published as supporting information on the PNAS web site). In addition, we detected ubiquitin- and α-synuclein-positive inclusion bodies in noradrenergic neurons of the locus coeruleus.
Finally, we examined the motor activity of mice that were continuously exposed to MPTP by monitoring their horizontal and vertical movements in an open field (Fig. 4 e and f). MPTP infusions of both 5 and 30 mg/kg severely depressed motor activity in mice, although a subset of treated mice appeared to tolerate MPTP much better than the majority. This depression was fully reversed by addition of apomorphine, a dopamine agonist, suggesting that the decrease in activity is due to a loss of dopamine (see Movie 1).
Effects of Continuous MPTP Administration in α-Synuclein-Deficient Mice. When we infused α-synuclein KO and control littermate mice continuously with MPTP, we found that α-synuclein-deficient mice appeared to experience few behavioral impairments even at high MPTP doses (30 mg/kg daily; Fig. 5a). This was observed despite the fact that continuous MPTP infusion produced the same long-lasting metabolic activation, as measured by 2-DG uptake, in α-synuclein KO and wild-type control mice (Fig. 5b). However, the ubiquitin–proteasome system was significantly less inhibited in α-synuclein KO mice than in littermate controls (Fig. 5c). Furthermore, in α-synuclein KO mice, continuous MPTP infusion failed to induce a significant loss of TH-positive neurons in the substantia nigra and of TH-positive fibers in the striatum (Fig. 6 a–c). The decrease in dopamine and DOPAC levels was also occluded in α-synuclein KO mice (Fig. 6e). Finally, neuronal inclusions that are produced by continuous MPTP administration (both whorls and authentic nonmembrane limited inclusions) were decreased but not completely suppressed in α-synuclein-deficient mice (Fig. 6d). These studies were performed in two independent sets of α-synuclein KO mice, suggesting that they are not caused by genetic background effects.
Fig. 5.

Effect of an α-synuclein deletion on MPTP toxicity. (a) Spontaneous horizontal activity and rearing of control and MPTP-treated wildt-ype and α-synuclein KO mice monitored in the open field test (30 mg of MPTP per kg daily for 28 days; n = 8). (b) Uptake of [14C]2-DG in littermate wild-type and α-synuclein KO mice that were continuously infused for 7 days with control or MPTP (30 mg/kg daily) solution. Pictures display false-color autoradiograms. (c) Proteasome activity in control and α-synuclein KO mice continuously infused with MPTP (30 mg per kg of body weight daily). Proteasome activities in the substantia nigra are depicted as percent of control (means ± SEMs) as a function of time after beginning of the infusions (five mice per group). In a and c, asterisks indicate statistically significantly different values (P < 0.05) from controls.
Fig. 6.

Deletion of α-synuclein alleviates neurodegeneration caused by continuous MPTP infusion. (a) Representative TH-labeled sections of the striatum from wild-type and α-synuclein KO mice treated with control infusions or 30 mg MPTP/kg (scale bars = 2 mm). (b) Semiquantitative measurements of the TH immunoreactivity in the striatum by densitometry of light microscopy pictures in control and MPTP-treated wild-type and α-synuclein KO mice (n = 10 mice). (c) Quantitation of TH-positive cells in the substantia nigra from littermate wild-type and α-synuclein KO mice that were treated with either control or MPTP infusions (30 mg/kg daily; n = 5). (d) Electron microscopic quantitation of cells with “whorl” inclusion bodies (see Fig. 4c)in the substantia nigra of littermate wild-type and α-synuclein KO mice that were treated with either control or MPTP infusions (30 mg/kg daily; n = 5 mice). (e) Quantitation of striatal levels of dopamine, its metabolite DOPAC, and serotonin in wild-type and α-synuclein KO mice that were treated with control infusions or 30 mg of MPTP per kg of body weight daily (n = 10). Asterisks indicate that the respective value for the wild-type mice is statistically significantly different (P < 0.05) from that obtained for the other three conditions (control-treated wild-type and α-synuclein KO and MPTP-treated α-synuclein KO mice). MPTP-treated α-synuclein KO mice exhibit a statistically significant difference with control treated mice only for whorls (d), but even here the number of whorls in the KO mice is significantly less than in wild-type mice (indicated by a double asterisk).
Discussion
Our data, consistent with previous studies (e.g., refs. 5–8), show that discontinuous injections of MPTP in mice severely damage the dopaminergic system as evidenced by: (i) Brief activation of 2-DG uptake (Fig. 1 b, c, and e–j); (ii) brief inhibition of the ubiquitin proteasome system (Fig. 2a); (iii) loss of dopamine and its metabolites (Fig. 8); and (iv) essentially irreversible neuronal degeneration in the substantia nigra (Fig. 8). However, despite their toxicity, bolus injections of MPTP did not induce formation of inclusion bodies (data not shown). In contrast, continuous MPTP infusions caused less acute toxicity because the peak concentrations of MPP+ were lower (Fig. 7 b–d; see also expanded discussion in Supporting Text), but produced chronic impairments in dopaminergic neurons of the substantia nigra and noradrenergic neurons of the locus coeruleus. These impairments consisted of (i) degeneration of dopaminergic and noradrenergic neurons (Figs. 3, 9, 10, and 12 a–c), (ii) loss of dopamine and norepinephrine, but not of serotonin, from the target areas of the respective neurons (Figs. 3d and 12d), (iii) formation of inclusion bodies in the substantia nigra and locus coeruleus (Figs. 4 a–d and 12 e and f); (iv) behavioral changes that were reversed by dopamine receptor agonists (Fig. 4 e and f); and (v) long-lasting inhibition of the ubiquitin-proteasome system (Fig. 2). Continuous MPTP infusions thus recreate a disease state that mimics human PD better than acute MPTP bolus injections.
Previous studies (11, 12, 35) showed that deletion of α-synuclein may protect mice against the toxicity of repeated MPTP injections, but did not clarify whether α-synuclein is required for the acute and/or chronic toxicity of MPTP. Our current and earlier data (31) suggest that the acute toxicity of MPTP is largely independent of α-synuclein [e.g., metabolic activation of 2-DG uptake (Fig. 5c) was still observed in α-synuclein KO mice], whereas chronic MPTP toxicity requires α-synuclein. The latter conclusion is based on the following findings in α-synuclein-deficient mice: (i) Inhibition of the ubiquitin proteasome system was alleviated by deletion of α-synuclein (Fig. 5d); (ii) MPTP-induced behavioral abnormalities were abolished by deletion of α-synuclein (Fig. 5 a and b); (iii) loss of dopaminergic neurons in the substantia nigra was prevented by deletion of α-synuclein (Fig. 6 b and c); (iv) the generation of neuronal whorl inclusion bodies was decreased in α-synuclein-deficient mice (Fig. 6d); and (v) the decline in dopamine induced by MPTP administration was prevented in α-synuclein deficient mice (Fig. 6e). A plausible interpretation of our results is that continuous low-level mitochondrial damage produced by chronic MPTP application causes an α-synuclein-dependent neurotoxicity by a downstream mechanism. This toxicity leads to generation of neuronal inclusion bodies and degeneration of catecholaminergic neurons, similar to the effects of chronic administration of rotenone which is also a mitochondrial complex I inhibitor (3, 13, 19). Thus, continuous mitochondrial inhibition may impair the ubiquitin proteasome system, which in turn generates a “Parkinson-genic” cascade that involves α-synuclein and could represent a potential target for therapeutic approaches.
Several key questions arise. First, does the involvement of α-synuclein in chronic MPTP toxicity reflect a physiological function for α-synuclein that has been activated in the wrong context, or does α-synuclein produce an accidental pathogenicity that contributes to MPTP toxicity but is unrelated to the normal function of α-synuclein? In the former case, all synucleins should be similarly involved, and deletion of β-synuclein (which is coexpressed with α-synuclein in most neurons; ref. 31) should have similar effects as deletion of α-synuclein. In the latter case, only deletion of α-synuclein should have an effect. With the availability of β-synuclein KO mice (36), this question can now be tested. Second, what is the connection between α-synuclein, inhibition of mitochondrial complex I, and impairment of the ubiquitin proteasome system, all of which appear to be components of the Parkinson-genic cascade (see also expanded discussion in Supporting Text)? Third, why is the dopaminergic system particularly sensitive to inhibition of the ubiquitin proteasome system? Presumably, oxidative stress produced by dopamine and its metabolites plays a role here (4, 16), but it is not known whether this is a sufficient explanation for the vulnerability of dopaminergic neurons.
Supplementary Material
Acknowledgments
We thank Dr. L. Schmued (National Center for Toxicological Research, Jefferson, AR) for Fluoro Jade-B, and M. Dini and C. Policicchio for skillful technical assistance. This work was supported by Italian Ministry of Research Grant RBNE017555_001 (to F.F.) and by Public Health Service Grant R01 NS40057 (to T.C.S.).
Author contributions: F.F. and T.C.S. designed research; F.F., O.M.S., P.L., M.G., R.R., M.F., G.L., C.L.B., F.P., G.B., A. Pellegrini, F.N., S.R., and A. Paparelli performed research; O.M.S. and T.C.S. contributed new reagents/analytic tools; F.F. and T.C.S. analyzed data; and F.F. and T.C.S. wrote the paper.
Abbreviations: PD, Parkinson's disease; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; 2-DG, 2-deoxyglucose; KO, knockout; TH, tyrosine hydroxylase.
References
- 1.Dauer, W. & Przedborski, S. (2003) Neuron 39, 889-909. [DOI] [PubMed] [Google Scholar]
- 2.Giasson, B. I. & Lee, V. M. (2003) Cell 114, 1-8. [DOI] [PubMed] [Google Scholar]
- 3.Greenamyre, J. T. & Hastings, T. G. (2004) Science 304, 1120-1122. [DOI] [PubMed] [Google Scholar]
- 4.Lansbury, P. T. & Brice, A. (2002) Curr. Opin. Cell Biol. 14, 653-660. [DOI] [PubMed] [Google Scholar]
- 5.Heikkila, R. E., Hess, A. & Duvoisin, R. C. (1984) Science 224, 1451-1453. [DOI] [PubMed] [Google Scholar]
- 6.Heikkila, R. E., Manzino, L., Cabbat, F. S. & Duvoisin, R. C. (1984) Nature 311, 467-469. [DOI] [PubMed] [Google Scholar]
- 7.Javitch, J. A., D'Amato, R. J., Strittmatter, S. M. & Snyder, S. H. (1985) Proc. Natl. Acad. Sci. USA 82, 2173-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicklas, W. J., Vyas, I. & Heikkila, R. E. (1985) Life Sci. 36, 2503-2508. [DOI] [PubMed] [Google Scholar]
- 9.Ramsay, R. R., Salach, J. I. & Singer, T. P. (1986) Biochem. Biophys. Res. Commun. 134, 743-748. [DOI] [PubMed] [Google Scholar]
- 10.Forno, L. S., DeLanney, L. E., Irwin, I. & Langston, J. W. (1993) Adv. Neurol. 60, 600-608. [PubMed] [Google Scholar]
- 11.Dauer, W., Khplodilov, N., Vila, M., Trillat, A. C., Goodchild, R., Larsen, K. E., Staal, R., Tieu, K., Schmitz, Y., Yuan, C. A., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 14524-14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drolet, R. E., Behrouz, B., Lookingland, K. J. & Goudreau, J. L. (2004) Neurotoxicology 25, 761-769. [DOI] [PubMed] [Google Scholar]
- 13.Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V. & Greenamyre, J. T. (2000) Nat. Neurosci. 3, 1301-1306. [DOI] [PubMed] [Google Scholar]
- 14.Hoglinger, G. U., Feger, J., Prigent, A., Michel, P. P., Parain, K., Champy, P., Ruberg, M., Oertel, W. H & Hirsch, E. C. (2003b) J. Neurochem. 84, 491-502. [DOI] [PubMed] [Google Scholar]
- 15.Lee, H. J., Shin, S. Y., Choi, C., Lee, Y. H. & Lee, S. J. (2002) J. Biol. Chem. 277, 5411-5417. [DOI] [PubMed] [Google Scholar]
- 16.Fornai, F., Lenzi, P., Gesi, M., Ferrucci, M., Lazzeri, G., Busceti, C. L., Ruffoli, R., Soldani, P., Ruggieri, S., Alessandri, M. G. & Paparelli, A. (2003) J. Neurosci. 23, 8955-8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McNaught, K. S., Perl, D. P., Brownell, A. L. & Olanow, C. W. (2004) Ann. Neurol. 56, 149-162. [DOI] [PubMed] [Google Scholar]
- 18.Fornai, F., Lenzi, P., Gesi, M., Soldani, P., Ferrucci, M., Lazzeri, G., Capobianco, L., Battaglia, G., De Blasi, A., Nicoletti, F. & Paparelli, A. (2004) J. Neurochem. 88, 114-123. [DOI] [PubMed] [Google Scholar]
- 19.Hoglinger, G. U., Carrard, G., Michel, P. P., Medja, F., Lombes, A., Ruberg, M., Friguet, B. & Hirsch, E. C. (2003) J. Neurochem. 86, 1297-1307. [DOI] [PubMed] [Google Scholar]
- 20.Chung, K. K. K., Dawson, V. L. & Dawson, T. M. (2001) Trends Neurosci. 24, S7-S14. [DOI] [PubMed] [Google Scholar]
- 21.Ciechanover, A. & Brundin, P. (2003) Neuron 40, 427-446. [DOI] [PubMed] [Google Scholar]
- 22.Shamoto-Nagai, M., Maruyama, W., Kato, Y., Isobe, K., Tanaka, M., Naoi, M. & Osawa, T. (2003) J. Neurosci. Res. 74, 589-597. [DOI] [PubMed] [Google Scholar]
- 23.Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B. & Jensen, P. H. (2004) J. Biol. Chem. 279, 12924-12934. [DOI] [PubMed] [Google Scholar]
- 24.Snyder, H., Mensah, K., Theisler, C., Lee, J., Matouschek, A. & Wolozin, B. (2003) J. Biol. Chem. 278, 11753-11759. [DOI] [PubMed] [Google Scholar]
- 25.Heikkila, R. E., Nicklas, W. J., Vyas, I. & Duvoisin, R. C. (1985) Neurosci. Lett. 62, 389-394. [DOI] [PubMed] [Google Scholar]
- 26.Ferrante, R. J., Schulz, J. B., Kowall, N. W. & Beal, M. F. (1997) Brain Res. 753, 157-162. [DOI] [PubMed] [Google Scholar]
- 27.Fornai, F., Chen, K., Giorgi, F. S., Gesi, M., Alessandri, M. G. & Shih, J. C. (1999) J. Neurochem. 73, 2434-2440. [DOI] [PubMed] [Google Scholar]
- 28.Battaglia, G., Busceti, C. L., Cuomo, L., Giorgi, F. S., Orzi, F., De Blasi, A., Nicoletti, F., Ruggieri, S. & Fornai, F. (2002) Neuropharmacology 42, 367-373. [DOI] [PubMed] [Google Scholar]
- 29.Fornai, F., Alessandrì, M. G., Torracca, M. T., Bassi, L. & Corsini, G. U. (1997) J. Pharmacol. Exp. Ther. 283, 100-107. [PubMed] [Google Scholar]
- 30.Battaglia, G., Busceti, C. L., Molinaro, G., Biagioni, F., Storto, M., Fornai, F., Nicoletti, F. & Bruno, V. (2004) J. Neurosci. 24, 828-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlüter, O. M., Fornai, F., Alessandri, M. G., Takamori, S., Gaper, M., Jahn, R. & Südhof, T. C. (2003) Neuroscience 118, 985-1002. [DOI] [PubMed] [Google Scholar]
- 32.Specht, C. G. & Schoepfer, R. (2001) BMC Neurosci. 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sokoloff, L., Reivich, M., Kennedy, C., Des Rosiers, M. H., Patlak, C. S., Pettigrew, K. D., Sakurada, O. & Shinohara, M. (1977) J. Neurochem. 28, 897-916. [DOI] [PubMed] [Google Scholar]
- 34.Schmued, L. C. & Hopkins, K. J. (2000) Brain Res. 874, 123-130. [DOI] [PubMed] [Google Scholar]
- 35.Robertson, D. C., Schmidt, O., Ninkina, N., Jones, P. A., Sharkey, J. & Buchman, V. L. (2004) J. Neurochem. 89, 1126-1136. [DOI] [PubMed] [Google Scholar]
- 36.Chandra, S., Fornai, F., Kwon, H. B., Yazdani, U., Atasoy, D., Liu, X., Hammer, R. E., Battaglia, G., German, D. C., Castillo, P. E. & Südhof, T. C. (2004) Proc. Natl. Acad. Sci. USA 101, 14966-14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}