

Abstract
Thermophilic environments represent an interesting niche. Among thermophiles, the genus Thermus is among the most studied genera. In this study, we have sequenced the genome of Thermus parvatiensis strain RL, a thermophile isolated from Himalayan hot water springs (temperature >96°C) using PacBio RSII SMRT technique. The small genome (2.01 Mbp) comprises a chromosome (1.87 Mbp) and a plasmid (143 Kbp), designated in this study as pTP143. Annotation revealed a high number of repair genes, a squeezed genome but containing highly plastic plasmid with transposases, integrases, mobile elements and hypothetical proteins (44%). We performed a comparative genomic study of the group Thermus with an aim of analysing the phylogenetic relatedness as well as niche specific attributes prevalent among the group. We compared the reference genome RL with 16 Thermus genomes to assess their phylogenetic relationships based on 16S rRNA gene sequences, average nucleotide identity (ANI), conserved marker genes (31 and 400), pan genome and tetranucleotide frequency. The core genome of the analyzed genomes contained 1,177 core genes and many singleton genes were detected in individual genomes, reflecting a conserved core but adaptive pan repertoire. We demonstrated the presence of metagenomic islands (chromosome:5, plasmid:5) by recruiting raw metagenomic data (from the same niche) against the genomic replicons of T. parvatiensis. We also dissected the CRISPR loci wide all genomes and found widespread presence of this system across Thermus genomes. Additionally, we performed a comparative analysis of competence loci wide Thermus genomes and found evidence for recent horizontal acquisition of the locus and continued dispersal among members reflecting that natural competence is a beneficial survival trait among Thermus members and its acquisition depicts unending evolution in order to accomplish optimal fitness.
Keywords: thermophiles, Thermus parvatiensis, CRISPR, Pilus genes, natural transformation, phage resistance
Introduction
The genus Thermus belongs to the vast group of extreme thermophiles that have held biochemical and industrial attention. The discovery of Thermus aquaticus in 1969 (Brock and Freeze, 1969), and subsequently the multi-billion-dollar industry of Taq DNA polymerase has revolutionized the field of extremophile research. Not only do the extremophiles provide understanding of life at extreme habitats, but they also serve as model organisms to study protein structure and functions. Members of this genus have been isolated from hot water springs all over the world (Chung et al., 2000; Ming et al., 2014). The proteins encoded by Thermus spp. have high stability and have been used in various industries, DNA polymerases (Carballeira et al., 1990; Engelke et al., 1990; Rao and Saunders, 1992) being chief among them; along with xylanases (Blank et al., 2014), amylases (Shaw et al., 1995), lipases (Kretza et al., 2012) and many other enzymes. The genus is well known for bioremediation of heavy metals thus lowering the toxicity at heavy metal contaminated sites. T. scotoductus for instance, has been shown to reduce Cr(VI) aerobically (Opperman and van Heerden, 2006).
The phylogenetic relationships amongst the members of the genus have been as dynamic as the genetic constitution of its members (Kumwenda et al., 2014). Members of the genus Meiothermus, which were earlier classified to the genus Thermus, were later moved to constitute a new genus to accommodate these moderately thermophilic and strictly aerobic group (Nobre et al., 1996). The members of the genus Thermus are a part of the phylum Deinococcus-Thermus, along with Deinococcus, Deinobacter, Deinobacterium, Truepera, Marinithermus, Meiothermus, Oceanithermus, Rhabdothermus, and Vulcanithermus. Members of the genus Thermus form yellow to orange-yellow colonies and generally have small genome sizes of less than 2.5 Mb with extrachromosomal elements of common occurrence (Henne et al., 2004; Bruggemann and Chen, 2006). Previous genome analyses of the group have revealed highly plastic nature of the genome of Thermus species with plasmids and megaplasmids being the center for such plasticity (Bruggemann and Chen, 2006). Genome wide rearrangements have been instrumental in shaping the genomes of these thermophiles (Kumwenda et al., 2014). Elements that are considered to belong to the mobilome, i.e., insertion sequence (IS) elements, transposons and prophages, occur widely in the genomes of Thermus species (Kumwenda et al., 2014). Along with this, thermophilic organisms are known to thrive under viral selection pressure. Selective forces continually acting at extreme environments bring about a strong evolutionary streamlining of these genomes (Sabath et al., 2013).
Among the drivers of evolution; genetic recombination, rearrangement, horizontal gene transfer, conjugation, transformation and mutations are key players for this genus (Kumwenda et al., 2014). One of the most likely explanations for this is the highly evolved competence system of Thermus which serves as an efficient arrangement for the uptake of alien genetic material from the environment (Lorenz and Wackernagel, 1994). In most organisms, competence is not a constitutive phenomenon, but tightly controlled by factors related to the cell cycle (induced/artificial competence). In contrast to artificial competence, some organisms (including Thermus species) are constitutively competent (Friedrich et al., 2002); reviewed by Averhoff (2009). The exact mechanism of uptake of free DNA from the environment varies from species to species. In case of Thermus species, the type IV pilus (T4P) system has been implicated in natural transformation, although the link between piliation and natural transformation seems unclear. Amongst all naturally competent species, Thermus thermophilus HB27 has the most efficient (40 kb/s per cell) natural competence system and a robust, non-selective competence machinery (Averhoff, 2009). The development of competence machinery in Thermus is of great evolutionary significance and explains the dynamism in Thermus genomes. We provide a brief introduction of the genes involved in imparting natural competence, followed by a genus-wide analysis of competence loci in Thermus.
The genus Thermus comprises 17 validly published species, the genome sequences of which are available in public databases. T. parvatiensis strain RLT (Dwivedi et al., 2015) was isolated from a hot water spring located atop (altitude ~1,700 m) the Himalayan ranges at Manikaran, India. The hot spring water has high temperature (90–98°C) (Dwivedi et al., 2012) and circum-neutral pH. Low O2 potential (4.8 ± 0.2 cm3 STP/L), low dissolved CO2 (14.7 ± 0.1 cm3 STP/L) and high concentration of arsenic (140 ppb) (Sangwan et al., 2015) prevalent at the niche further provide strong selection pressures. T. parvatiensis forms yellow colonies on polypeptone yeast extract agar at 60-80°C and demonstrates protease activity (Dwivedi et al., 2015). Previously, strain RL was sequenced using Roche 454 GS (FLX Titanium) system and Sanger shotgun sequencing. The raw data generated was assembled into 17 contigs (Dwivedi et al., 2012). In order to fill the gaps and generate a complete genome record, we determined the entire genome sequence using single molecule real time (SMRT) sequencing method. Here, we present the complete genome of T. parvatiensis and perform a comparative genomic analysis of available Thermus genomes. Our study is designed to uncover the phylogenetic relatedness among members based on phylogenomic methods, the core-pan genome structure as well as conserved genome features with the help of metagenomic recruitments. Further, we have analyzed genus specific evolutionary dynamics which are facilitated by the highly efficient natural competence system of this genus and a possible link with predominance of viral signatures found in these genomes.
Materials and methods
Genome sequencing, assembly, and annotation of T. parvatiensis replicons
SMRT genome sequencing was performed using PacBio RSII system at McGill University and Genome Quebec Innovation Centre, Canada. Genomic DNA was extracted using CTAB method (Doyle and Doyle, 1990) followed by quality assessment on gel and quantification by ND1000 Nanodrop spectrophotometer. Sheared large insert library preparation was followed by generation of raw reads with an average read length of 9,878 nt. A total of 2,488 GB raw data was generated with 224,211 reads encompassing 857,926,800 bases with an average sequencing depth of 428 × (Supplementary Table 1). De novo assembly was performed at the Genome Quebec, Canada using the HGAP assembler (Chin et al., 2013) (coverage cut-off 30 ×). Assembly validation was performed by aligning raw reads onto finished contigs using the Burrows-Wheeler Aligner version 0.7.9a (Li and Durbin, 2009). Visual inspection of the assembly was performed using Tablet version 1.14.04.10 (Milne et al., 2010). Ends of contigs were searched for overlaps using the formatdb and BLAST functions of Ugene (Okonechnikov et al., 2012). Circularized replicons were uploaded on RAST server (Aziz et al., 2008) for general genome annotations. RNAmmer version 1.2 (Lagesan et al., 2007) was used to detect rRNA operons. Phages were scanned using online tools PHAST (Zhou et al., 2011) and PHASTER (Arndt et al., 2016). For detailed annotations of the phage regions, analysis was extended to include probable phages and associated regions using Phage Search Tool against viral and prophage databases (http://www.phantome.org/Downloads/). Further, PHAST tool was also used to decipher the completeness of the phage genome followed by annotation using BLASTx against the ORFs predicted from prophage databases. Aragorn (Laslett and Canback, 2004) online tRNA database was used to detect tRNAs in the genome. The WebMGA (Wu et al., 2011) server was used for general COG category assignment. An approach integrating the Z-curve analysis, dnaA box location and genes surrounding the OriC was used to identify the origin of replication on the chromosome using the Ori-Finder server (Gao and Zhang, 2008). T. parvatiensis genome was searched against DNA box database to locate the origin of replication and for this, DNA box repeat sequence (TGTGGATAA) of T. thermophilus (closest relative of T. parvatiensis) was used as reference to guide the BLAST search.
Phylogenomic assessments
For comparative analyses, sequenced genomes of the genus Thermus (17 genomes) were downloaded from the NCBI GenBank database. The genomes included in this study are, T. thermophilus HB27 (Henne et al., 2004), T. thermophilus HB8, T. parvatiensis RL (Dwivedi et al., 2012, 2015), T. scotoductus SA-01 (Gounder et al., 2011), T. oshimai JL-2 (Murugapiran et al., 2013), Thermus species CCB_US3_UF1 (Teh et al., 2012), T. aquaticus Y51MC23, T. antranikianii HN3-7, T. filiformis Wai33 A1, T. thermophilus JL-18 (Murugapiran et al., 2013), T. thermophilus SG0.5JP17-16, T. islandicus PRI3838, T. caliditerrae YIM 77777, T. igniterrae RF-4, T. amyloliquefaciens YIM 77409 (Yu et al., 2015; Zhou et al., 2016), T. tengchongensis YIM 77401 (Mefferd et al., 2016), and T. brockianus GE-1. Accession numbers of genomes included in this study and other general genome features are included in Table 1. Among the genomes selected for comparisons, 10 were complete genomes and seven were draft genomes. All complete genomes selected were found to harbor 1–4 plasmid(s) which were downloaded as separate sequences.
Table 1.
General genome features of organisms belonging to the genus Thermus.
| Strain | NCBI Accession No. | Source of Isolation | Genome Size | Plasmid (s) | G+C (%) | Predicted CDS | tRNA | rRNA operons | |
|---|---|---|---|---|---|---|---|---|---|
| T. parvatiensis | RL* | CP014141 | Hot spring, India | 2,016,098 | pTP143 (143,277) | 68.5 | 2,383 | 54 | 2 |
| T. thermophilus | HB27* | AE017221 | Hot spring, Japan | 2,127,482 | pTT27 (232,605) | 69.4 | 2,244 | 47 | 6 |
| T. thermophilus | HB8* | AP008226 | Hot spring, Japan | 2,197,207 | pTT27 (256,992), pTT8 (9,322), pVV8 (81,151) | 69.4 | 2,268 | 48 | 6 |
| T. thermophilus | JL-18* | CP003252 | Great Boiling Spring, USA | 2,311,212 | pTTJL1801 (265,886), pTTJL1802 (142,731) | 69.0 | 2,424 | 52 | 6 |
| T. thermophilus | SG0.5JP17-16* | CP002777 | Hot Spring | 2,303,227 | pTHTHE1601 (440,026) | 68.6 | 2,405 | 53 | 6 |
| T. scotoductus | SA-01* | CP001962 | Fissure water, South Africa | 2,355,186 | pTSC8 (8,383) | 64.9 | 2,514 | 47 | 6 |
| T. oshimai | JL-2* | CP003249 | Great Boiling Spring, USA | 2,401,329 | pTHEOS01 (271,713), pTHEOS02 (57,223) | 68.6 | 2,521 | 59 | 6 |
| T. sp. | CCB_US3_UF1* | CP003126 | Hot spring, Malaysia | 2,263,488 | pTCCB09 (19,716) | 68.6 | 2,228 | 48 | 6 |
| T. aquaticus | Y51MC23* | CP010822 | Hot spring, USA | 2,338,641 | pTA14 (14,448), pTA16 (16,597), pTA69 (69,906), pTA78 (78,727) | 68.0 | 2,436 | 55 | 3 |
| T. brockianus | GE-1* | CP016312 | Kamchatka, Russia | 2,388,273 | pTB1 (342,792), pTB2 (10,299) | 66.9 | 2,789 | 47 | 2 |
| T. antranikianii | DSM 12462 (HN3-7) | AUIW01000000 | Hot spring, Iceland | 2,163,625 | ND | 64.8 | 2,321 | 47 | 4 |
| T. filiformis | ATCC 43280 (Wai33 A1) | JPSL02000000 | Hot spring, New Zealand | 2,386,081 | ND | 69.0 | 2,338 | 47 | 6 |
| T. islandicus | DSM 21543 (PRI 3838) | ATXJ01000000 | Hot spring, Iceland | 2,263,010 | ND | 68.4 | 2,470 | 47 | 6 |
| T. igniterrae | ATCC 700962 (RF-4) | AQWU01000000 | Hot spring, Iceland | 2,225,983 | ND | 68.8 | 2,379 | 43 | 6 |
| T. caliditerrae | YIM 77777 | JQNC01000000 | Hot spring, China | 2,218,114 | ND | 67.2 | 2,327 | 50 | 3 |
| T. amyloliquefaciens | YIM 77409 | JQMV00000000 | Hot spring, China | 2,160,855 | ND | 67.4 | 2,313 | 48 | 6 |
| T. tengchongensis | YIM 77401 | JQLK01000000 | Geothermally heated soil, China | 2,562,314 | ND | 66.4 | 2,750 | 47 | 2 |
Smallest genome/plasmid sequences and GC content are underlined whereas the largest are in bold.
Complete genome; ND-Not determined.
Phylogenetic analysis based on traditional 16S rRNA gene sequences was performed, for which 16S rRNA genes were fetched from the respective genomes using RNAmmer version 1.2 server (Lagesan et al., 2007). Multiple sequence alignment was performed using Muscle (Edgar, 2004). Unaligned sequences were trimmed from the edges. Phylogenetic tree was constructed using Maximum-likelihood (ML) algorithm (Felsenstein, 1993) employed in Mega version 6 (Tamura et al., 2013). Although 16S rRNA gene is a well-established marker for tracing phylogeny, dependence on just one gene may lead to biased phylogenetic projections. Hence, evolutionary relationships were reconstructed using multiple conserved marker genes extracted from the genomes. For this, 31 conserved bacterial single copy genes were extracted from each genome using AmphoraNet server (Kerepesi et al., 2014). Individual marker gene sequences for individual genomes were concatenated. Alignment was performed using Muscle (Edgar, 2004). Further, 400 conserved bacterial marker genes were retrieved from each genome using PhyloPhlAn (Segata et al., 2013). For the above three sequence based analyses, ML trees were rendered with 1,000 bootstrap revaluations. To trace phylogeny using whole genome data, well established phylogenomic approaches were employed. Average nucleotide identity (ANI) values were calculated using the BLASTALL algorithm (ANIb) of JSpecies v 1.2.1 (Richter and Rosello-Mora, 2009). A two-way matrix containing pairwise ANI scores was used to perform hierarchical clustering using Pearson correlation (average linkage). Similar dendrogram was generated using a two-way matrix of tetranucleotide frequencies calculated using regression analysis by JSpecies v 1.2.1. To evaluate phylogeny on the basis of variable component of the genome, pan genome phylogeny was reconstructed by hierarchical clustering using information from a binary gene presence-absence (1/0) matrix generated by BPGA (Chaudhari et al., 2016). Gene presence-absence matrix constituted the information about presence or absence of the total gene complement (pan genome) for all Thermus species. In order to resolve the precariousness of sub-species level relationships, pairwise digital DNA-DNA hybridization (dDDH) values were calculated using the genome to genome distance calculator (ggdc.dsmz.de) (Auch et al., 2010).
Analysis of genome flexibility
Genome sequences were uploaded on RAST server (Aziz et al., 2008) and coding sequences were extracted from RAST predictions. Coding sequences (amino acids) were compared using formatdb and BLASTALL programs available in the package BLAST version 2.2.26 (Altschul et al., 1990). Genomic islands were predicted using IslandViewer 3 (Dhillon et al., 2015). Dot-plots and synteny maps were constructed to uncover the extent of rearrangements (duplications, deletions, insertions) occurring as a function of genome distance. Dot-plots were generated using BLASTN (Wheeler and Bhagwat, 2007) with T. parvatiensis as the reference. Synteny maps were constructed by identifying conserved locally collinear blocks (LCBs) among genomes, followed by whole genome alignments using progressiveMauve version 20150226 (Darling et al., 2010) at three spaced seed patterns and a high seed weight (seed weight = 15) for sensitive alignment of closely related genomes. Horizontally acquired regions on the megaplasmid pTP143 were detected by BLAST based comparison with all sequenced Thermus plasmids. These regions were confirmed using Alien Hunter (Vernikos and Parkhill, 2006) at default thresholds. Further, a mapping of syntenic regions on all Thermus plasmids was performed using progressiveMauve (Darling et al., 2010) for visual demonstration.
Core and pan genome analysis was performed using BPGA algorithm (Chaudhari et al., 2016). Usearch (Edgar, 2010), which is the default clustering algorithm of BPGA, was employed for orthologous gene identification and clustering. Core genome plot was rendered by plotting the total number of shared genes with each subsequent addition of a genome against the number of genomes. Pan genome plot was rendered by plotting the total number of distinct gene families identified with the addition of each genome vs. number of genomes. To avoid biasedness, median values of 20 random permutations were used for rendering these plots. Representative (seed) sequences of both core and pan genome were used for function based analysis.
Metagenomic sequence data recruitment was performed in order to gain insights into strain specific flexible repertoire harbored by T. parvatiensis, and to investigate whether this flexibility is strain specific or extended to other members of the genus as well. For this purpose, water samples were collected from the hot water spring at Manikaran, India, at two locations, namely MNW1 and MNW2 (intra-site distance: 100 m; 32°01′34.8″N, 077°20′50.3″E). DNA extraction from the water samples (10 L each, filtered through 0.45 μ filters) was carried out by PowerMax (R) Water DNA isolation kit (MoBio Laboratories Inc., Carlsbad, CA, USA) as per manufacturer's instructions. Metagenomic data was generated using Illumina GAII technology with an insert size of 170 bp (DDBJ/EMBL/GenBank accession number PRJEB19501). In order to delineate metagenomic islands (hereafter referred to as MGIs), metagenomic raw reads from hot spring water (MNW1 and MNW2) were recruited onto the chromosome and plasmid of T. parvatiensis using nucmer available with MUMmer (Kurtz et al., 2004) package. Coverage plots were generated using mummerplot available in MUMmer package. Regions with no or little mapping after metagenomic reads tilling at coverage cut-off of 80% were identified as MGIs (Steffen et al., 2012). Mapping coverage was determined using coords file generated by nucmer (identity cut-off: 80%). BLASTp algorithm was implemented to identify the presence of these regions on other Thermus genomes.
Detection of genus specific survival strategies
CRISPR analysis
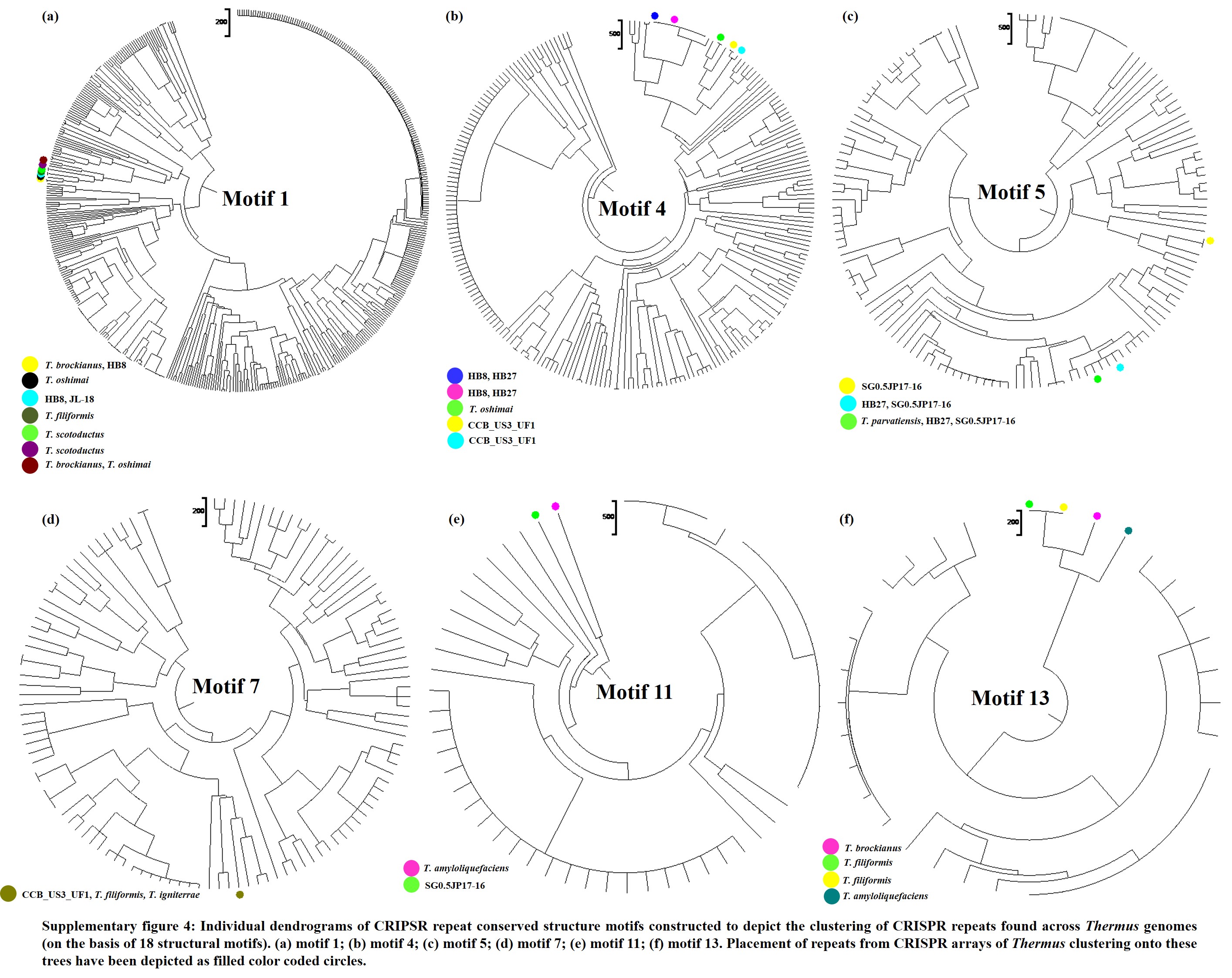
CRISPR arrays were extracted from genomes using CRISPRFinder (Grissa et al., 2007) online server which performs BLAST against dbCRISPR (CRISPR database; last updated on 2017-01-02). CRISPRFinder further classifies the identified CRISPR arrays as true or false based on whether or not they are associated with CRISPR associated genes (Cas) respectively. Cas genes were annotated using CRISPRone (Zhang and Ye, 2017). CRISPRs lacking Cas genes in the vicinity were designated as false/questionable CRISPRs. Only true CRISPRs were selected for analyses. CRISPR arrays have two components: repeats and spacers; both of which were analyzed to study the evolution and probable viral diversity respectively. Classification and clustering of CRISPR repeats and repeat-based Cas gene predictions were undertaken using CRISPRmap, a comprehensive cluster analysis method (based on Markov clustering) which clusters conserved sequence families and potential structure motifs (Lange et al., 2013). Repeats were classified based on 40 conserved sequence families and 33 probable structural motifs. Further, 24 families and 18 structural motifs were considered for the construction of repeat cluster maps. For prediction of potential viruses most frequently associated with Thermus genomes, spacer sequences from all genomes were extracted and BLAST against viral GenBank database of NCBI (Deng et al., 2007) with a threshold e-value of 1. For better stringency, among all matches, only those having 100% identity of more than 20 nucleotides were considered as valid hits.
Comparison of competence imparting genes
Genes involved in imparting competence (16 genes) to T. thermophilus HB27 were used as reference for extracting competence associated genes from individual genomes using BLAST. PilA1-A4 genes were aligned using Hirschberg (KAlign) algorithm (Lassmann and Sonnhammer, 2005). Visual alignment consensus was built at 70% threshold. Relationships among PilA1-A4 genes were inferred by PhyML (Guindon et al., 2009) maximum-likelihood method using HKY85 substitution model (Hasegawa et al., 1985). Median size estimations were made using boxplot function in R (https://cran.r-project.org/mirrors.html).
Results and discussion
Genome sequencing, assembly, and annotation of T. parvatiensis replicons
Genome assembly, finishing, and annotation
The genome of T. parvatiensis strain RL was initially assembled into three contigs (totally 2,066,435 bp, 1,886,121 bp, 159 Kbp and 21 Kbp) with G+C content of 68.5%. The 21 Kbp contig, mapped onto the chromosome (BLASTn and mapping with previously generated assembly; Dwivedi et al., 2012). The entire 21 Kbp region, seems to represent an integrated plasmid or a large genomic island incorporated into the genome, based on the annotation of mostly hypothetical genes and transposable elements among genes identified. This was supplemented by differences in mean G+C% of this region (67.3%) as compared to the rest of the genome (68.5%). In an attempt to circularize the largest contig, its ends were aligned against each other and an overlapping region of 13,300 nt was removed. Similarly, in order to circularize the 159 Kbp contig, an overlapping region of 15,853 bp region was removed from the ends of the contig. Finally, two replicons were reconstructed: a chromosome (1,872,821 bp) and a megaplasmid (143,277 bp) (Figure 1) (total size: 2,016,098 bp) (Table 2). The chromosomal origin of replication was located at 158,478–158,778. A total of 12 DNA boxes with consensus sequence of TGTGGATAA were identified spanning the 301 nt OriC region. The total number of predicted coding sequences were 2,383. The genome was found to harbor two rRNA operons and 54 tRNAs and tmRNAs. COG functional category assignment placed a large number of genes to amino acid transport and metabolism (11.04%), general function prediction (12.95%), energy production and conservation (7.54%) and translation, ribosomal structure and biogenesis (7.40%). A number of genes were classified into the unknown function category (7.78%). A large proportion of the genome is strictly attributed to genes needed by the organism for essential cellular processes. T. parvatiensis thrives at a high arsenic concentration (140 ppb). We investigated the presence of arsenic resistance mechanisms in this thermophilic organism. Arsenate reductase gene arsC (1 copy), arsenic efflux pump protein arsB (2 copies) and arsR transcriptional regulator (4 copies) were identified. The above genes belong to the ars (arsenic resistance) operon responsible for the efflux of As(III) out of the cells (Yang and Rosen, 2016). Genes for the oxidation of arsenic (aox operon) were not identified. The mechanism of arsenic detoxification in T. parvatiensis thus involves extrusion of arsenic out of the cells (rather than oxidation).
Figure 1.

Replicon maps of Thermus parvatiensis strain RL (A) ORFs on the chromosome have been mapped on both strands, the origin of replication is marked with a red arrow. From outside to inside: genes on the negative strand, genes on the positive strand, GC percentage and GC skew. (B) Detailed map of T. parvatiensis megaplasmid pTP143 marked with prominent categories of genes in different colors. Genes representing DNA repair genes (green), mobile element genes (red), transcriptional regulators (magenta) and gene clusters (purple; denitrification gene cluster, cobalamin biosynthetic gene cluster, carotenoid synthesis gene cluster) have been specifically highlighted.
Table 2.
Genomic features of the chromosome and plasmid of T. parvatiensis strain RL.
T. parvatiensis strain RL uniquely harbored three integrated phages in its genome (Supplementary Figure 1)—two on the chromosome and one on the plasmid. This was in contrast to all its close phylogenetic neighbors taken into account in this study namely, T. thermophilus HB8, T. thermophilus HB27, T. thermophilus JL18, and T. thermophilus SG0.5JP17-16 in which no phages could be identified. The first phage (20.3 Kbp) on the chromosome revealed the presence of phage structural proteins such as tail assembly protein and coat protein along with three heat shock proteins which can be directly implicated to the environment, i.e., hot spring water (surface water temperature >95°C). The phage region was associated with two hybrid histidine kinases, which are implicated in two-component regulatory system (Khorchid and Ikura, 2006). The second chromosomal phage (23.2 Kbp) was annotated and revealed a probable integron with attL and attR sites along with integrase encoding gene and flanking tRNA. The gene cassette of this integron had 23 hypothetical proteins and 16 viral proteins. These results also indicate that this integron might denote a super-integron with 37 ORFs captured. Phage regions have been known to play a role in horizontal gene transfer by specialized as well as generalized transduction (Touchon et al., 2017). Genes harbored on the integrated phage regions corresponding to two-component system and heat shock proteins reflect the dispersal of these genes might be an active phenomenon among the population.
Plasmid pTP143
Plasmids, including small plasmids, as well as large megaplasmids of up to 440 Kbp are known to be present in Thermus genomes (Table 1). The plasmids of Thermus are known to be the center of plasticity, harboring genes for mobile elements, transposons and a number of biosynthetic clusters (Henne et al., 2004; Bruggemann and Chen, 2006). The megaplasmid pTP143 of T. parvatiensis, contained 181 coding sequences. A large number of the genes harbored on the plasmid were, however, genes belonging to integrases, transposases, mobile elements and hypothetical protein coding genes (81 genes, constituting 44% of the total plasmid genes) (Figure 1) which denote the plastic nature of the plasmid. A low coding density (87.65%) was observed for pTP143, as compared to the chromosome (94.12%). Genetic analysis of T. parvatiensis megaplasmid pTP143 revealed a cobalamin biosynthetic cluster, a denitrification cluster and a carotenoid biosynthesis cluster (responsible for imparting the yellow pigment to the organism). A thermophilic lifestyle demands a robust DNA repair system. Genes required for thermophilic existence most suitably correspond to an elevated number of DNA repair genes (Bruggemann and Chen, 2006). Consequently, a recQ helicase, reverse gyrases, photolyase phrB, sbcC, and sbcD nucleases (implicated in deleting hairpin structures) were found on the megaplasmid. Genes related to stress response, surE (involved in nucleic acid pool maintenance; Proudfoot et al., 2004) and cytochrome P450 (Kelly and Kelly, 2013) were also identified. A number of transcriptional factors known to modulate stress conditions were found on the plasmid. These included a transcriptional regulator IcIR involved in regulation of responses to quorum sensing and toxic stress (Molina-Henares et al., 2005). A transcriptional regulator Crp/Fnr known to be responsive to environmental changes (Körner et al., 2003; Zhou et al., 2012), such as oxidative stress, carbon dioxide concentrations and heavy metal impositions was annotated. Crp/Fnr regulators act by regulating the expression of genes involved in alleviating the respective stress conditions. MerR, a heavy metal modulating transcriptional regulator (Brown et al., 2003), which activates promotors of genes in response to heavy metal influx was annotated. T. parvatiensis harbors a plastic plasmid with mobile and hypothetical gene components. Constituting genes for DNA repair, stress response and transcriptional regulators, we believe that the megaplasmid has an indispensable role to play for the thermophilic survival of T. parvatiensis. Not only essential for survival, the megaplasmid demonstrates a potentially crucial role in communicating and modulating temperature stress via an appropriate response carried out by the transcriptional regulators it harbors.
Phylogenomic assessments
The novel phylogeny of strain RL has already been discussed based on multi locus gene analysis (Dwivedi et al., 2015). We describe here, the microbial phylogeny within the genus Thermus using the traditional 16S rRNA gene, 31 bacterial single copy genes and 400 conserved bacterial marker genes. To strengthen the analyses, we have used whole genome patterns established by ANI scores, tetra-nucleotide scores, pan genome and dDDH values. The phylogenetic tree based on 16S rRNA gene sequences placed strain RL along with members of the T. thermophilus clade into a single monophyletic clade, closely bifurcating with SG0.5JP17-16 (Figure 2), and residing with strains of T. thermophilus, i.e., HB27, HB8, JL-18, and SG0.5JP17-16. This was expected from over 99% identity of 16S rRNA gene sequence of T. parvatiensis with members of T. thermophilus. A single gene, however, is not able to provide the required phylogenetic resolution, hence, phylogenetic relationships were further investigated on the basis of conserved marker genes. For this, 31 bacterial single copy genes and 400 bacterial conserved marker genes were used. The phylogenetic tree constructed using 31 essential single copy genes placed T. parvatiensis in the same clade as T. thermophilus (Figure 2). However, the phylogenetic tree constructed using 400 conserved bacterial marker genes placed T. parvatiensis at an outlier position with respect to T. thermophilus group with a strong bootstrap support (100%) (Figure 2). Correlations based on gene distances scored on the basis of ANI scores, placed T. parvatiensis along with SG0.5JP17-16. However, these two strains did not fall into the T. thermophilus clade, but clustered with strain CCB_US3_UF1 and T. igniterrae instead (Figure 2). The novel species status of strain RL was also reflected in its ANI scores with T. thermophilus members (95.03–95.57%), which fall on the borderline for species delineation based on ANI cut-off (95–96%) (Konstantinidis and Tiedje, 2005) (Supplementary Table 2). This is in contrast to the high ANI scores among members of the T. thermophilus group (>96%). Tetra-nucleotide based correlations also placed T. parvatiensis as an outlier, lying just outside the tight T. thermophilus group (Figure 2). The same was reflected in the dendrogram based on pan genes presence-absence (1/0) matrix (Figure 2). Digital DDH values (Supplementary Table 3) were able to separate T. parvatiensis from T. thermophilus clearly. T. parvatiensis vs. T. thermophilus dDDH values were in the range 61.0–64.6%, which were below the 70% DDH cut-off for species delineation. This was in contrast to the high intra-species scores among the T. thermophilus group (68.9–89%).
Figure 2.

Inference of evolutionary relationships among Thermus spp. based on phylogenetic and phylogenomic methods. Phylogenetic analysis based on (A) 16S rRNA gene sequences; (B) 31 single copy genes; and (C) 400 conserved bacterial marker genes, of the species under study using maximum likelihood method. Bars represent the number of substitutions per nucleotide position. Percentage bootstrap values (≥70%) are shown next to the nodes. Phylogenomic dendrograms showing hierarchical clustering of species under study constructed using (D) whole genome distance matrix based on ANI scores; (E) tetranucleotide frequencies; and (F) pan genes presence-absence matrix. Gradation of colors from black to yellow in (D) depicts increasing genome distance on the basis two-way ANI matrix. Black denotes minimum distance and yellow denotes maximum distance. Organisms have been grouped together into clades on the basis of minimum distance (black). Blue shade (in A,B) depicts clustering of T. parvatiensis within T. thermophilus group. Light brown shade (in C–F) depicts the position of T. parvatiensis separately from other closely clustered Thermus members (shaded gray in C–F).
Above analyses suggest that species diversification for the genus Thermus has taken place by acquisition/deletion/rearrangement of regions which cannot be reflected in the 16S rRNA gene. Conserved genes (31 and 400) are better able to reflect the phylogenetic relationships among the members. However, a drawback of the above methods is that they fall short of taking in account the intragenomic heterogeneity that is quite high among Thermus members due to extensive gene shuffling. For this reason, whole genome based methods such as ANI, tetranucleotide frequency and DDH values should be regarded as more accurate phylogenomic methods for estimating phylogenetic relationships within this genus. We have been able to resolve the phylogeny of T. parvatiensis by whole genome methods. In spite of having a high percentage similarity with the T. thermophilus group, based on 16S rRNA gene sequences, T. parvatiensis represents a different species based on whole genome methods, which are more reliable as compared to gene based methods. Thus, T. parvatiensis, in course of evolution, has accumulated genome wide differences that have led to its bifurcation with the T. thermophilus group, and represents a genetically unique species.
Analysis of genome flexibility
Genome organization
All species belonging to the genus Thermus, as described in this study, have been isolated from thermophilic environments (mostly hot spring waters) from all over the world, i.e., from USA (Murugapiran et al., 2013), Japan (Henne et al., 2004), India (Dwivedi et al., 2012), South Africa (Gounder et al., 2011), China (Yu et al., 2015; Zhou et al., 2016), Malaysia (Teh et al., 2012), New Zealand (Hudson et al., 1987; Mefferd et al., 2016), and Iceland (Chung et al., 2000). This illustrates that the genus is spread across the globe, thriving at the most extreme environments. Being confined to stressed niches, these G+C rich (64.8–69.4%) organisms possess small genomes, ranging from 2.01 Mb of T. parvatiensis to 2.5 Mb of T. tengchongensis YIM 77401 (mean genome size: 2.25 Mb). Considering the powerful evolutionary forces that have been constantly shaping their genomes, in geographically diverse niches, the genome size of Thermus shows low variability (Table 1). Already known to shed and rearrange genes that are not uniquely essential for survival, the members of the genus have been successful in maintaining their genome sizes close to the average genome size of the genus. A large part of the genomes is comprised of genomic islands, varying from 0 to 5.85% (Supplementary Table 4).
To demonstrate the extent of genomic shuffling, synteny maps of the 10 complete genomes were generated (Figure 3). T. parvatiensis and SG0.5JP17-16 revealed a conserved organizational synteny with each other and lack of inversions and rearrangements. Synteny was conserved among members of T. thermophilus group, however, large blocks of inversions were observed in relation to strains CCB_US3_UF1, T. scotoductus, T. aquaticus, T. oshimai, and T. brockianus. These demonstrate the huge genome wide rearrangements occurring at the genus level (Figure 3). The same observation was reflected in dot-plot comparisons of T. parvatiensis with other members (Supplementary Figure 2).
Figure 3.

Organizational (synteny) comparisons of T. parvatiensis with nine representatives of Thermus. Only members with complete genomes have been considered for this analysis. Synteny maps showing (A) comparison of the chromosome of T. parvatiensis against the chromosomes of other genomes; and (B) comparison of plasmid pTP143 against plasmid sequences (concatenated wherever number of plasmids >1) of other Thermus members. Boxes of different colors represent locally collinear blocks (LCBs) (or locally conserved regions) connected via lines of the same color to their corresponding positions on other genomes. For each genome, the LCBs above and below the reference line (indicated by black triangle) denote the orientation of the LCBs with respect to the reference sequence (LCBs below the black reference line denote inversions). Black lines (in B) below LCBs represent the position of coding sequences. Red arrows on plasmid pTP143 (in B) mark the regions that could not be mapped on other Thermus plasmids.
The genes on Thermus plasmids are known to undergo vast rearrangement events. In some cases, they have been observed to shift from the plasmid to the chromosome and get stabilized there. In other cases, large plasmids with a majority of non-essential but potentially benefit imparting clusters have been discerned. The former case has been observed in T. scotoductus and CCB_US3_UF1, both of which have small plasmids as most of the genes got incorporated on the chromosomes, leaving plasmids with diminished configurations. pTSC8 (8,383 bp), for example, has discarded many non-essential genes like cobalamine biosynthesis pathway genes, plasmid stability genes and chromosome partitioning genes, but retained genes for aerobic and anaerobic respiration to attain a much more compact conformation (Gounder et al., 2011). The latter situation has been observed for plasmid pVV8 (81,151 bp) which was found to be enriched in phosphonate metabolism genes, which are not of common occurrence in Thermus genomes (Ohtani et al., 2012). To evaluate this trend in pTP143 and other plasmids, we surveyed genetic clusters commonly observed on Thermus plasmids. We observed the presence of advantageous traits such as cobalamin biosynthesis on pTP143 and pTHEOS01 (T. oshimai plasmid) and nitrate reduction on pTP143, pTHEOS01 and T. thermophilus JL-18 plasmid. To assess possibly laterally acquired regions on pTP143, we performed a comparative mapping of plasmids based on BLASTn comparisons (identity cut-off: 80, coverage cut-off: 80, e-value: 1e-30) across all the Thermus plasmids. The plasmid pTP143 of T. parvatiensis showed highest identity to the plasmids pTHTHE1601 (identity: 99.3%, query cover: 79%), pTTJL1801 (identity: 97.5%; query cover: 78%), pTT27 of HB27 (identity: 99.0%; query cover: 47%) and pTT27 of HB8 (identity: 98.9%; query cover: 49%). A region on pTP143 that failed to show any homologous regions with any of the plasmids was analyzed as a putatively horizontally transferred region (Supplementary Table 5). This region harbored 3 genes for mobile element proteins, 9 hypothetical protein coding genes, sbcC and sbcD (hairpin structure resolving nucleases) (Figure 4). The genes present on this locus were identified using BLASTp (identity cut-off: 70%, e-value cut-off: 1.00E-15) on the chromosomes of T. aquaticus, T. brockianus, HB8, SG0.5JP17-16, JL-18, CCB_US3_UF1 and even on the chromosome of T. parvatiensis.
Figure 4.

(A) Detailed map of plasmid pTP143 with genes assigned to COG categories and depicted in a color-coded manner. Rings from outside to inside represent positions of genomic islands (GI) (red); coding sequences (brown); GC content (black); GC skew (purple) and COG category assignment (multiple colors according to the color key) (B) GIs found on the megaplasmid pTP143 (linear view) have been highlighted. These regions contain hypothetical protein coding genes, some plasmid specific genes, a toxin-antitoxin system and repair genes. Most of the genes on the GIs were placed into COG category “L” (replication and repair).
The genes on the plasmids of Thermus are under movement and involved in shifting from the plasmid to the chromosome, in order to get stabilized. In case of Thermus species, plasmids perhaps contribute more to the flexibility of genome by either acquiring or shedding of the genes. Incoming genes/pathways may first be incorporated on the plasmid and later be stabilized either on the plasmid itself or through integration into the chromosome. The presence of a genomic island on plasmid pTP143 with multiple mobile elements suggests that it might get mobilized soon. In the process, some other genes may also get shifted from the plasmid to the chromosome. Across Thermus genomes, this process has led to either stabilization of the megaplasmid with megaplasmids acquiring a vast majority of genes (SG0.5JP17-16), or it has led to streamlining of the plasmid (T. scotoductus).
Conserved and variable gene repertoire of Thermus group
The core genome denotes the conserved functions, whereas, the pan genome denotes the entire genetic potential of a group (Tettelin et al., 2005). The total number of genes constituting the core genome for the genus Thermus were 1177. Functional annotation of genes constituting the core genome placed a high number of genes into categories coding for amino acid metabolism and transport (12.15%), translation (10.07%), energy production and conservation (7.75%) and coenzyme metabolism (6.07%), designating these as the conserved functions specific for the genus. The pan genome was estimated at 5188 and constituted accessory genes and unique genes (singletons). Accessory genes are the ones whose orthologs are present in two or more genomes, but not all the genomes. Singletons are genes that are unique to just one genome out of all those compared. The variability in accessory genome depicts the flexibility of the genome structure. Accessory gene number varied from 661 to 1,166 (mean: 946). High number of accessory genes were observed in T. tengchongensis (1,166), T. brockianus (1,094), JL-18 (1093), T. oshimai (1,053), T. scotoductus (1,032), SG0.5JP17-16 (1027), and T. aquaticus (1003) (Figure 5B). Variability in the number of unique genes (singletons) was observed from 30 to 229 genes (mean 98). A large number of singletons were identified in T. filiformis (229), T. islandicus (212), T. tengchongensis (175), and T. aquaticus (154) (Figure 5B). Out of the genomes having high number of accessory genes (higher than mean), 55.5% of the genomes had high number of singletons too. These genomes were T. aquaticus, T. brockianus, T. oshimai, T. scotoductus, and T. tengchongensis. The accessory and unique components of the genus were enriched in genes belonging to carbohydrate metabolism and transport (8.17 and 8.76% respectively), replication and repair (8.17 and 8.76% respectively), inorganic ion transport and metabolism (5.85 and 5.69% respectively) and signal transduction (5.71 and 5.40% respectively) which reflect the diverse functional counterparts harbored by these organisms. The pan genome of the genus was estimated as an “open” pan genome because a plateau was not observed (Figure 5A) after addition of all genomes to the pan genome plot. Addition of more genomes to the group will lead to expansion of the pan repertoire (Rouli et al., 2015). The genus has thus maintained a conserved core genome, but an expansive and sundry pan genome. The flexibility of the genomes is explained by the high influx of genes into these organisms via horizontal gene transfer. Although, a number of features might get recruited and incorporated in the genome, only those that have a survival benefit for the organism will be retained and the rest will be discarded by the highly active rearrangement events that are continually taking place in these genomes. In due course of time, extensive rearrangement events have led to the establishment of those genomic features that have benefitted the organism for better survival; other dispensable elements were either shed off or transferred to the plasmid. Overall, genome-wide differences and anticipation in accordance with specific gene repertoire required at a niche (niche specialization) can be considered to be driving forces in the evolution of the genus Thermus.
Figure 5.

Core and pan genome analysis of Thermus genus (A) Plot of pan and core genome. The plot represents a stabilized core structure but an open pan-genome; (B) Graph showing the number of core genes, accessory genes, unique genes and exclusively absent genes in all genomes under study.
In order to designate strain specific, potentially utilizable attributes of T. parvatiensis, we identified MGIs of T. parvatiensis. The term MGI encompasses those regions of the genome that are identified by mapping metagenomic data from an environment against the genome of an organism isolated from the same niche (Pašić et al., 2009). These regions stand out as “gaps” with little or no reads corresponding to these regions, thus highlighting the strain specific potential that these organisms have accumulated in contrast to the environmental counterparts. Such an analysis was performed for T. parvatiensis by aligning raw metagenomic data from hot spring water (Manikaran, India) onto the replicons (Figure 6). T. parvatiensis chromosome showed a relatively high coverage of reads (4 ×) with five regions with no coverage of reads (MGIs) (Figure 6). T. parvatiensis plasmid pTP143, however had thin coverage of reads (1.9 ×) and five MGIs. Thus, plasmid pTP143 seems to have accumulated more strain specific variations which denote high flexibility of the plasmid. This data further states that the chromosome in case of Thermus is more or less stable in terms of the genes it harbors, however, much influx and rearrangements occur via plasmid. The chromosomal MGIs harbored genes for arginine biosynthesis, iron-sulfur cluster assembly proteins, transcriptional regulators, two component system genes and hypothetical genes (Supplementary Table 6). A MGI (4,811 bp) on the plasmid was found to harbor genes specifically involved in environmental response to stress in general and oxidative stress in particular. These genes included radical SAM (S-adenosylmethionine) domain heme biosynthesis protein (heme is a co-factor for hemoproteins that functions in electron transport chain), cytochrome c552 which is particularly involved in electron transport at low aeration, peptide methionine sulfoxide reductase (MsrA) known to protect against reactive oxygen and nitrogen species (Weissbach et al., 2002). Cobalamin biosynthesis genes were largely detected on the plasmid MGIs, including uroporphyrinogen-III methyltransferase, BluB, adenosylcobalamine-phosphate synthase and CbiG. Other prominently represented genes included plasmid stability genes (ParB, Soj, and StbB) and DNA repair genes (reverse gyrase, sbcC and sbcD). These genes implicate the conservation of low aeration oxidation response, plasmid stability and DNA repair as strain specific features. In order to get insights about the prevalence of these specialized regions in other genomes, we performed BLASTn (identity > 95%, query coverage > 95%, e-value <1.00E-30) of MGI regions with other Thermus genomes. Whereas, genes prevalent on MGI1 of the chromosome did not show significant identity with other members of the genus, MGI2-5 of the chromosome were found to have homologous counterparts in JL-18, SG0.5JP17-16, HB27, and T. brockianus. Plasmid MGIs 1, 3, 4, and 5, similarly could be identified on JL-18, SG0.5JP17-16, HB8, HB27, however, plasmid MGI2 of pTP143 was unique in this respect and significant similarity could not be observed with other members. The genes thus annotated on MGIs, were conserved within the T. thermophilus group indicating close adaptive relatedness. Chromosomal MGI1 and plasmid MGI2 were identified as strain specific MGIs for T. parvatiensis. These attributes suggest the conservation of features that are not directly implicated with the niche, but are retained in the genome as anticipated survival benefits.
Figure 6.

Depiction of metagenomic islands recovered by recruitment of raw reads obtained from the metagenomic sequencing of hot spring water (Manikaran, India) onto T. parvatiensis plasmid (A) and chromosome (B). MGIs on the plasmid pTP143 (5 MGIs) are highlighted in gray; MGIs on the chromosome of T. parvatiensis (5 MGIs) are marked with arrows. Reads mapped to the reference (identity ≥ 80%) are represented as blue (MNW1) and red (MNW2) dots.
Genus specific survival strategies
Abundance of CRISPR arrays
Analysis of CRISPRs among Thermus species was performed to get insights into prevalence of viral defense system within the genus. All genomes harbored CRISPR loci, except T. antranikianii. Nine CRISPR arrays were identified in T. filiformis, HB8, HB27 and T. igniterrae, followed by T. aquaticus which had 7 CRISPR arrays (Table 3). T. parvatiensis, on the other hand, was found to harbor only 1 true CRISPR array. One questionable array was detected in T. islandicus. The CRISPR cassette/array number varied from 0-9 with DR length variation from 25 to 37 wide all genomes analyzed (mean CRISPR array count per genome: 5.529). The number of spacers harbored within CRISPR arrays denote the frequency of viral invasions. T. oshimai was found to carry the highest number of spacers (134 spacers). A high frequency of viral attacks in these thermophiles was indicated by a high mean number of spacers harbored by each genome (mean: 72.7 spacers per genome). Five Thermus genomes (29.4%) harbored >100 CRISPR spacers. Within T. oshimai itself, two CRISPR arrays harboring large number of spacers were uncovered. One of the CRISPR arrays harbored 88 copies of a single DR consensus (CGGTCCATCCCCACGGGCGTGGGGACTAC; DR length: 29 bp), with an equally high number of spacers. The other array (DR consensus = CTTTGAACCGTACCTATAAGGGTTTGAAAC; DR length: 30 bp) had acquired 67 spacers. On the contrary, within the same genus, an array containing only 3 spacers was also observed. This indicates high variation among CRISPR elements within the species. Cas genes were extracted from true CRISPRs and annotated (Table 3). The Cas system in Thermus was found to be composed of genes belonging to types I, III, and IV (Supplementary Figure 3). Universal type genes cas1 and cas2 were identified on CRISPR loci. Apart from this, the class I effector cas3 which is responsible for the helicase and DNase activity was also annotated (Table 3). Thus, cleavage of foreign entities entering Thermus genomes is brought about by type I, III, and IV mediated action. Analysis of repeats based on sequence and structure (Figure 7) was performed. Sequence families 1 and 18 were predominant (spotted in 7 and 6 genomes respectively) (Supplementary Table 8). Among structural motifs (based on the classification of motifs into 33 groups), motifs 25 and 5 were most represented (8 and 6 genomes respectively). Motif 6, motif 20, motif 23, and motif 31 were least represented (1 genome each) (Supplementary Tables 7, 8). Additionally, on the basis of repeat-cas binding projections, probable cas genes harbored by Thermus genomes were predicted and found to belong to types I, III, and IV. Separate cluster trees were constructed for structure motifs (based on the classification of structural motifs into 18 groups) to denote the placement of Thermus repeats among all consensus repeats present in the database. A majority of repeats (10 repeats) mapped onto motif 1 and occupied a close phylogenetic position within the cluster tree (Supplementary Figure 4). In order to discern the phages most frequently attacking Thermus genomes, we analyzed spacer matches with viral sequence database (Supplementary Table 9). Positive inferences were based only on the results that satisfied the stringency criteria. A number of phages of different families were detected to infect Thermus species (Figure 7B). Among these, phages of families Sphaerolipoviridae, Siphoviridae, Myoviridae, and Herpesviridae were identified as the most prominent bacteriophages. Earlier Siphoviridae, Myoviridae, Inoviridae, and Tectiviridae have been reported from Thermus species (Yu et al., 2006). However, our analysis revealed the dominance of Sphaerolipoviridae (28.1%), with known thermophilic phages P23-77 and IN93 being prominently detected. A detailed list of viruses, the invasion memory of which is incorporated within Thermus CRISPRs is mentioned in Supplementary Table 9. Our analysis thus reveals the abundant presence of defense mechanism and frequent viral encounters in this genus. Even though a large number of spacers (1,223) were analyzed, only 62 (5.07%) could be assigned significant matches to the viral database.
Table 3.
Summary of CRISPR elements found across all Thermus genomes under this study.
| Species | Confirmed CRISPRs | DR length | Spacers | Cas types | Cas genes |
|---|---|---|---|---|---|
| T. parvatiensis | 1 | 37 | 9 | Other | cas6, cas2 |
| T. thermophilus HB27 | 9 | 32-37 | 6+3+15+7+7+6+6+13+9 = 72 | I, III, IV | csa3, cas2, cas10, csm2gr11, csm3gr7, csx1, cmr3gr5, cmr4gr7, cmr5gr11, DinG, cas5, cas8c, cas7b, cas1, cas4, WYL, cas8b5, cas7, cas3, cas6 |
| T. thermophilus HB8 | 9 | 29-37 | 14+4+3+12+9+12+23+20+12 = 109 | I, III | csx1, cas1, cas2, cas10, csm2gr11, csm3gr7, csm3gr5, cmr4gr7, cmr5gr11, WYL, cas3HD, cas8e, cse2gr11, cas7, cas5, cas6e, cas1, cas8b5, cas3, cas6 |
| T. thermophilus JL-18 | 6 | 29-37 | 21+23+7+25+19+5 = 100 | I | WYL, cas3HD, cas8e, cse2gr11, cas7, cas5, cas6e, cas1, cas2, cas4, cas8b5, cas3, cas6, csa3, cas8U1, csb2gr5, casR |
| T. thermophilus SG0.5JP17-16 | 6 | 28-37 | 3+13+6+10+6+18 = 56 | I | casR, cas7, csb2gr5, cas3, cas8U1, cas4, csa3, cas2, cas1, WYL, cas8b5, cas5, cas6, cas8c, cas7b |
| T. scotoductus | 3 | 29-30 | 9+36+42 = 87 | I | cas2, cas1, cas6e, cas5, cas7, cse2gr11, cas8e, cas3HD, WYL |
| T. oshimai | 5 | 29-36 | 14+6+88+30+4 = 134 | I, III, IV | cas6, csx1, csm3gr7, csm4gr5, csm2gr11, cas10, cas2, csx1, WYL, cas3HD, cas8e, cse2gr11, cas7, cas6e, cas1, csa3, DinG, cas3 |
| T. sp. CCB_US3_UF1 | 7 | 28-36 | 3+17+14+23+18+9+12 = 96 | I, III | csm3gr7, csm5gr11, cmr4gr7, cmr3gr5, cas10, csx1, cas4, cas3HD, cas6, cas8b1, cas7, csm2gr11, csm4gr5, cas1 |
| T. aquaticus | 7 | 26-36 | 7+5+22+3+6+12+3 = 58 | I, III | cas2, cas1, csx1, csa3, cas5, cas7, cas8b1, cas3HD, cas4, cas10, csm2gr11, csm3gr7, csm4gr5, cas6, csm5gr11, csm4gr7 |
| T. brockianus | 8 | 29-37 | 9+14+7+10+12+8+25+27 = 112 | I, III | cas6, cas10, csm2gr11, csm3gr7, csm4gr5, csx1, cas2, cas1, cmr3gr5, cmr4gr7, cmr5gr11, cas1, cas4, cas3, cas3HD, cas8b1, cas7b, cas5, cas8c, cas8e, cse2gr11, cas7, cas5, cas6E |
| T. antranikianii | 0 | 0 | 0 | — | — |
| T. filiformis | 9 | 25-37 | 3+24+4+5+6+22+5+12+7 = 88 | I, III, IV | cas4, WYL, DinG, cas3HD, cas8e, cse2gr11, cas7, cas5, cas6e, csx1, cas10, cmr3gr5, csm3gr7, cmr4gr7, cmr4gr11, cas2, cas6, cmr2gr11, csm4gr5, casR, cas2, cas1, cas3D, cas8b1, cas7b |
| T. islandicus | 0 | 0 | 0 | I, III | cas1, csx1, cas6, cas3, cas7, cas8b5, WYL, cas8b1, cas7b, cas5, cas2, csa3, csm3gr7, csm4gr5, csm2gr11, cas10 |
| T. igniterrae | 9 | 28-36 | 5+23+3+11+3+4+15+17+28 = 109 | I, III | csm3gr7, csm4gr5, csm2gr11, cas10, csx1, WYL, cas3HD, cas8e, cse2gr11, cas7, cas5, cas6e, cas1 |
| T. caliditerrae | 4 | 35-36 | 21+18+9+10 = 58 | I, III | csm3gr7, cmr5gr11, cmr3gr5, cas10, cas6, csx1, csm2gr11, csm4gr5, cas2, cas1, cas3 |
| T. amyloliquefaciens | 5 | 29-36 | 15+5+9+6+18 = 53 | I, III | csx1, csm4gr5, csm3gr7, csm2gr11, cas10, cas6, csx1, cas2, csa3, cas1, cas6e, cas5, cas7, cse2gr11, cas8e, cas3HD, WYL |
| T. tengchongensis | 4 | 30-37 | 67+3+12+9 = 91 | I, III | csa3, csm2gr11, csa3, csa3, casR, cas7, cas8b1, cas3HD, cas4, cas3, cas5, cas8b5, WYL |
Figure 7.

CRISPR repeat (A) and spacer (B) analyses. (A) Dendrogram depicting clustering of CRISPR array repeats from all genomes of Thermus spp. against CRISPR repeats database (4,719 consensus repeats). The classification of repeats is based upon the division of all repeats into 24 conserved sequence families and 18 conserved structural motifs. The position of branches representing repeats from Thermus spp. is marked by red arrows and depicted by a number which corresponds to a genome, as follows: 1. T. aquaticus, 2. T. amyloliquefaciens, 3. T. brockianus, 4. T. caliditerrae, 5. Thermus sp. CCB_US3_UF1, 6. T. filiformis, 7. T. thermophilus HB8, 8. T. thermophilus HB27, 9. T. igniterrae, 10. T. thermophilus JL-18, 11. T. oshimai, 12. T. scotoductus, 13. T. tengchongensis, 14. T. parvatiensis, 15. T. thermophilus SG0.5JP17-16. Concentric circles are color coded and represent the following from inside out respectively: Structure motifs, sequence families, CAS subtypes, taxonomic groups, superclasses. Black and blue color branches represent bacterial and archaeal sequences respectively. (B) Pie diagram representing the relative abundance of viral families infecting Thermus genomes, predicted on the basis of CRISPR spacer analysis.
CRISPRs constitute the characteristic prokaryotic and archaeal adaptive as well as inheritable immune system composed of short repeat sequences (direct repeats/DRs) interspaced with short segments of nucleotides known as spacers. Spacers represent the memory of past invasions by foreign genetic elements like viruses (Barrangou et al., 2007) or plasmids (Marraffini and Sontheimer, 2008). Spacers are incorporated into CRISPR loci whenever a bacteriophage infects the organism. This way, a CRISPR array can be considered as a library of past viral invasions faced by an organism. CRISPR arrays are associated with CRISPR associated genes (Cas genes), which are present in the vicinity of CRISPR arrays. Together with virus specific spacers, Cas genes encode an arsenal of proteins and RNAs, which in conjugation, destroy the foreign element, the next time it invades (Mojica et al., 2005; Barrangou et al., 2007). The CRISPR system is thus, a defense mechanism against bacteriophage invasions on the bacterial genome. CRISPR arrays that lack the requisite Cas genes in their vicinity are known as questionable or false CRISPRs. Predominance of viruses in thermophilic niches and consequently prevalence of CRISPRs in thermophilic genomes is known (Anderson et al., 2011; Weinberger et al., 2012b). We analyzed the CRISPR loci from all Thermus genomes in this study in order to investigate the probable viruses infecting the genus and to uncover the organization of cas genes. Our examination divulged the ubiquity of CRISPR arrays within the genus Thermus, reflecting a resilient viral defense system. The predominance of CRISPRs among the Thermus group suggests presence and activity of phages in thermophilic environments. A wide scenario of phage invasion in T. oshimai was particularly denoted by high number of spacers present in this species. The uneven distribution of CRISPR arrays within a group can be explained by the hypothesis that probably the cost of harboring CRISPR elements in particular bacteria outweighs the benefits harnessed (Weinberger et al., 2012a). Cas genes, which are momentous to the functioning of CRISPRs, are divided into two classes (class I and II), depending on their mechanism of action. Class I CRISPR-Cas systems act by employing a number of Cas proteins which bring about the required action in a cascade of events. Class II systems, on the other hand, rely on single effector proteins for binding and cleavage of the target site. Based on the specific proteins involved, class I is further divided into types I, III and IV and class II is divided into types II and V. Widespread presence of type I (15 genomes) and type III (12 genomes) was observed in Thermus. Type IV systems were detected only in 3 genomes. Type IV systems have been known to be rare (<2%) in both bacteria and archaea (Makarova et al., 2015). Evolutionary relationships were uncovered on the basis of repeat sequences by clustering repeat sequences into conserved sequence families and structural motifs (Lange et al., 2013). CRISPR DRs transcribe repeat RNA sequences which serve as Cas protein binding templates. Repeat sequences show significant conservation in their sequence as well as hairpin structure forming motifs (Lange et al., 2013). Sequence conservation has been used as a criterion for clustering of DRs into families. Similarly, structural motif grouping is based on RNA loop structures. Using this analysis, we were able to identify the distribution of sequence families and structural families within this genus. Interestingly, motifs 20 and 31, which were identified in T. filiformis and HB8, have been known to constitute a mixture of both bacterial and archaeal domains. Viral diversity analysis delineated most probable viral predators for the Thermus group. A high proportion of spacers, however, were left un-assigned, implying the huge viral genosphere that is yet unexplored.
Genes imparting competence
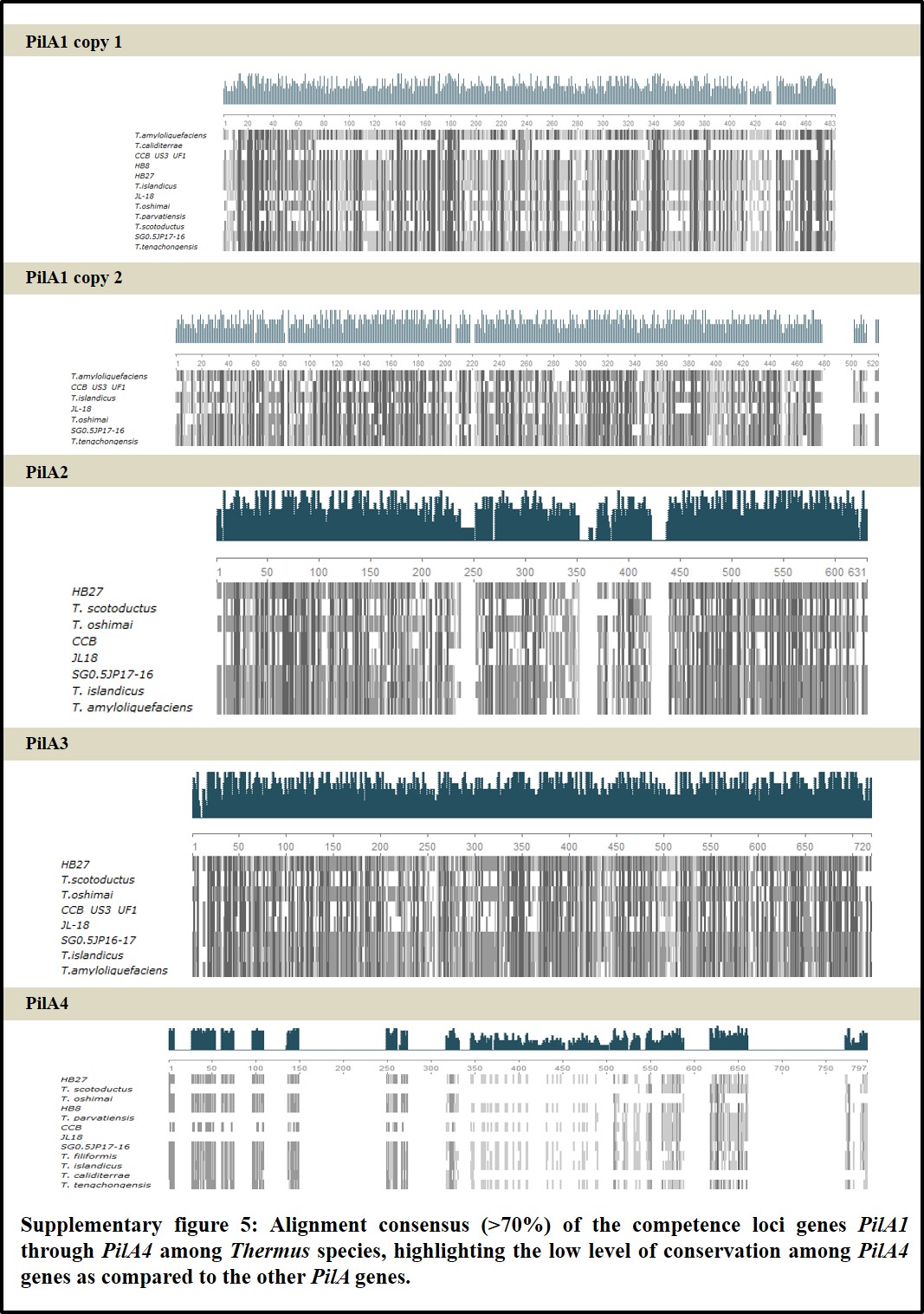
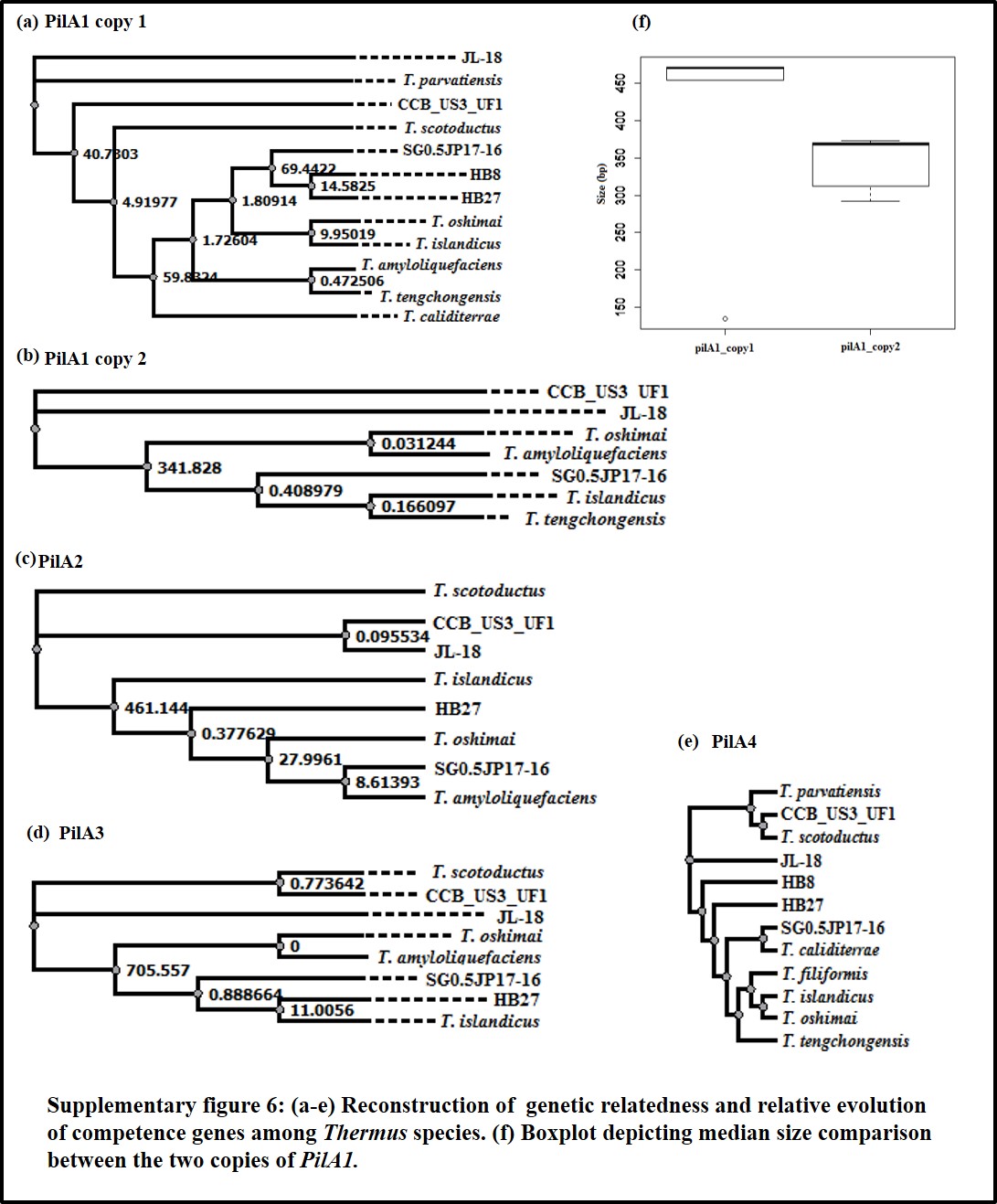
Genes implicated in natural transformability of HB27 include PilA1, PilA2, PilA3, PilA4, ComZ, CinA, DprA, ComEA, ComEC, PilF, PilC, PilM-N, PilN-O-W, ComF, PilQ, and PilD. Apart from HB27, all of the above genes were harbored by T. oshimai, CCB_US3_UF1, T. islandicus, JL-18, and SG0.5JP17-16. In this study, all Thermus genomes were found to show a homogeneous profile with respect to genes except PilA1-PilA4 and ComZ. Among competence genes, PilA gene plays a decisive role in efficient translocation (Schwarzenlander et al., 2009). PilA1-A4 genes and ComZ are present as a cluster in all genomes, however the arrangement and organization of genes show a difference across genotypes. The PilA-ComZ locus was found to be harbored in all Thermus genomes except T. igniterrae and T. antranikianii. Either these strains have not yet acquired the cluster or the cluster has been missed out during sequencing (draft genomes). The PilA-ComZ operon contains PilA1, A2, A3, A4, and ComZ as principal genes. The operon, however, also contains other genes with pilin/putative pilin/pseudopilin domains and genes coding for hypothetical proteins. The PilA-ComZ locus in Thermus was found to be comprised of 9–12 genes out of which 4–7 genes were pseudopilins with significant similarity to the PilA genes of strain HB27 (Figure 8). Apart from pseudopilin genes, the locus is comprised of genes coding for hypothetical proteins, chromosome segregation protein, apolipoprotein and DUF820 superfamily nucleases. PilA1 gene (present in 14 genomes) was found to be duplicated in 7 genomes. The two copies of PilA1 were, however, not identical and showed identity ranging from 67 to 81%. By convention, we have named the PilA1 of HB27 (471 bp) as copy 1. Copy 2 of PilA1 (present in 7 strains) is a smaller gene ranging from 272 to 373 bp (mean = 329 bp). In T. caliditerrae, only a fragmented copy (135 bp) of PilA1 was recovered. PilA2 (582–614 bp) was recovered in 7 strains as a complete copy. In case of T. scotoductus, identity at the C- and N- terminals and non-identity at the middle of the gene sequence was observed. A truncated version of PilA2 was identified in T. tengchongensis. PilA3 was recovered in a complete state (685–746 bp) in nine strains. In T. scotoductus, a truncated PilA3 was detected. The most deviant forms among PilA genes were observed for PilA4. Patterns of alignment among all PilA genes best denote this observation (Supplementary Figure 5). The alignment features are most consistent (>70%) in case of PilA1 through PilA3 genes. In case of PilA4, similarity among genes is observed only at the C- and N- termini of the genes. PilA4 of HB27 is 396 bp in size, however, in the other 13 strains harboring PilA4, only a small part (87–121 bp) showed significant similarity (>80%, e-value: 1E-15) to PilA4 of HB27. Additionally, truncated versions of PilA4 were observed in T. aquaticus, T. caliditerrae, and T. amyloliquefaciens.
Figure 8.

Organization of putatively horizontally transferred PilA-ComZ locus genes from all Thermus genomes in this study. Blue, Pilus family proteins; Gray, Nucleases; Tan, Hypothetical/Conserved proteins; Maroon, Transposase/Conjugative element; Rhomboid, unformed CDS; Navy blue, Toxin-antitoxin elements; Black triangles, tRNAs.
A complete PilA-ComZ locus was identified in HB27, T. oshimai, CCB_US3_UF1, JL-18, SG0.5JP17-16, and T. islandicus. However, in other genomes, we could identify the presence of “genetic scars.” Genetic scars are truncated genes or pseudogenes (without start/stop codons), but show significant identity to functional PilA gene regions of HB27 (taken as a reference for all comparisons here). Two genetic scars were identified in T. scotoductus (A3 and A2), two in T. caliditerrae (A1 and A4), one in T. amyloliquefaciens (A4), one in T. aquaticus (A4) and one in T. tengchongensis (A2). Interestingly, an IS4 family transposase, which is known to be found widely across the Thermus genomes was found incorporated in the PilA-ComZ locus of T. aquaticus. This can be an evidence for recent transposition activity at the locus. Another evidence is the presence of a conjugative protein in the PilA-ComZ locus of HB27. Interestingly, a toxin-antitoxin (TA) gene system was found to be incorporated into the competence locus of T. caliditerrae and showed similarity to a TA pair in Rhodothermus marinus. Horizontally transferred regions are generally found to be associated with tRNA genes (Darmon and Leach, 2014). As expected, tRNA genes were located in close proximity of the PilA-ComZ loci of 10 genomes in this study. Out of the ten genomes in which tRNA genes were associated with the competence locus, seven are still not fully formed, thus giving a strong boost to the hypothesis of recent horizontal acquisition of the locus. Genetic relatedness of Thermus strains on the basis of PilA1-A4 genes was reconstructed to infer the evolutionary history of PilA genes on the basis of their sequence development (Supplementary Figure 6). PilA1 copy1 and copy2 gene trees highlighted strain CCB_US3_UF1 and JL-18 as an outlier for both trees and T. parvatiensis as an additional outlier for PilA1 copy1 gene. CCB_US3_UF1 and JL-18 retained their outlier positions in all other trees. In case of PilA2 and PilA3 sequences, T. scotoductus emerged as an outlier. For PilA4, T. parvatiensis, CCB, T. scotoductus and JL-18 lie on the out branches of the dendrogram.
Genes responsible for imparting competence in the genus Thermus can be divided into two groups: the first group of genes are responsible for uptake of DNA and the second group is responsible for transport of DNA into the cell. Some of these genes belong to the T4P family of proteins which form a complex on the membrane of the cell, spanning the S-layer, Outer Membrane (OM), Secondary Cell Wall Polymers (SCWP) and Peptidoglycans (PG) which comprise the periplasm and the inner membrane (IM). Sixteen genes have been known to play a role in natural transformation in HB27, the transformation machinery of which is the most extensively studied. PilQ is a secretin which forms a macromolecular homopolymeric complex on the outer membrane (Burkhardt et al., 2011) and binds to the incoming DNA. Once DNA reaches the periplasmic space, the pseudopilus proteins (pilA1 through pilA4) come into play, forming subunits of the DNA translocator complex present in the periplasm. PilA1-PilA4 pilins form a shaft like structure in the periplasm which is attached to the outer membrane through PilQ (Burkhardt et al., 2011) and to the inner membrane through a motor ATPase PilF. PilF uses the energy of ATP hydrolysis to draw DNA toward the inner membrane (Rose et al., 2011; Collins et al., 2013). Double stranded DNA further binds to the inner membrane protein ComEA. An inner membrane channel protein, ComEC (co-transcribed along with ComEA) has recently been shown to modulate the expression of PilA4 and PilN in relation to environmental cues like nutrient limitation and low temperature (Salzer et al., 2016). The DNA passing through the IM is single stranded DNA. Hence, a nuclease has to be acting on double stranded DNA to make it single stranded. This nuclease has not been identified yet.
A stable arrangement of the PilA-ComZ locus could be observed in T. oshimai, CCB_US3_UF1, JL-18, SG0.5JP17-16, and T. islandicus. The PilA-ComZ locus represents a small horizontally acquired region (genomic islet) on the chromosome. It can be distinguished by high number of hypothetical protein coding genes, gene duplications and their association with tRNA genes (Darmon and Leach, 2014). Gene duplication events observed in case of PilA1 genes demonstrate species radiation forces and amenability of the genomes to evolutionary forces (Roth et al., 2007). The presence of a transposase in T. aquaticus, a conjugative element in HB27, TA element in T. caliditerrae and truncated PilA genes (genetic scars) on the locus indicate recent horizontal origins. Truncated PilA4 genes observed across this genus denote that it has either not developed or has undergone degradation, determined by the presence of genetic scars which show similarity to N-/C-terminals of PilA4 of HB27, but not to the complete gene. The close association of a TA system with PilA-ComZ cluster of T. caliditerrae reflects recent acquisition of this cluster. TA systems have been found to be associated with genomic islands and other mobile elements. Association with a TA system, promotes the maintenance of a horizontally acquired island and stabilization into the host genome (Rowe-Magnus et al., 2003; Iqbal et al., 2015). Generally, horizontally transferred loci are marked with pseudogenes as there is a strong selection pressure against these regions (Hao and Golding, 2010). Pseudogenes are non-functional versions of a previously functional gene, which are in the process of getting lost from the genome (Hao and Golding, 2010). Pseudogenes are known to be associated with recently laterally acquired regions or failed HGT events (Hao and Golding, 2010). The predominance of pseudogenes can be due to the high rates of gene turnover in laterally acquired regions. Some of the truncated genes may get stabilized and some eliminated in due course of time. The evidence thus provided leads us to believe that the competence machinery in Thermus is of horizontal origin and in course of evolution, may get stabilized or eliminated. The presence of a highly efficient transformation system however does not ensure the incorporation of incoming DNA into the host genetic material. Natural transformation in native conditions is activated during environmental challenges such as starvation, wherein DNA is taken up from the environment (Seitz and Blokesch, 2013). Most of the nucleic acid taken up during this process is used to fulfill nutritional requirements. In this process, some amount of DNA may get incorporated into the host genome, thus diversifying the host pan repertoire and expanding the already diverse arsenal of Thermus group.
Choice between natural competence and viral resistance
The pilus structure in Thermus imparted by the T4P genes plays a role not only in twitching motility and natural competence, but also in bacteriophage infection. PilA mutants have been shown to lose not only twitching motility and natural competence, but are also resistant to phage infection in HB27 and HB8 strains (Tamakoshi et al., 2011). On the basis of our comparative analysis for the Thermus group, we propose a link between pilus gene diversification and CRISPR abundance. Our data suggests continued acquisition and evolution of pilus gene structure among the analyzed Thermus genomes. Along with this, a CRISPR system with high number of spacers suggests a robust immune machinery against bacteriophages. In case of T. islandicus, only one questionable CRISPR array was observed, and no CRISPR array was observed in the case of T. antranikianii. Interestingly, a PilA-ComZ locus was also absent in T. antranikianii. The two observations, when coupled together suggest that T. antranikianii might be resistant to a huge proportion of viruses due to lack of pilus system genes which are implicated in phage entry, thus avoiding the need for harboring the CRISPR system. Other Thermus members have, on the other hand, chosen pilus mediated natural transformation as an important evolutionary trait, even though it makes them more susceptible to phage attacks, leading them to harbor more frequent CRISPR defense systems. Thus, natural transformation may be regarded as an overall benefit imparting trait in the small thermophilic genomes of Thermus. Natural transformation has played a role in survival of these organisms since long. Therefore, the dispersal of this system is a rather favorable phenomenon wide this genus even though it imposes an additional cost of harboring CRISPR machinery on them.
Conclusion
Organisms belonging to the genus Thermus have occupied a significant position and have diversified present knowledge about thermophilic survival. T. parvatiensis, in accordance with its affiliation to the genus has maintained a small genome and a plastic plasmid. Plasmids of Thermus are hotspots for genome dynamism, acting as centers of influx as well as efflux of genes and pathways in this genus. A dynamic pan genome along with strain specific gene reservoirs signify acquisition and conservation of favorable attributes. One of the factors contributing towards this dynamism is an active natural transformation system of this genus. The natural competence machinery in Thermus has proved to be an overall advantageous trait for the dual reason of nutrition limitation and genetic variability. It has, however, made these organisms susceptible to viral grazing, leading to the development of viral defense arsenal, known as CRISPRs. The efficacy of choices made has led to proficient sustenance of this genus in the face of adversity and beyond.
Ethics statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Author contributions
RL conceived the study and supervised manuscript preparation. CT performed the analysis except CRIPSR analysis. HM performed CRISPR analysis. HK prepared all tables and figures. VD, RN and KK helped in data interpretation and drafting of the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CT and HK gratefully acknowledge Council of Scientific and Industrial Research and Indian Council of Medical Research respectively for providing research fellowships.
Footnotes
Funding. This work was supported by grants from the Department of Biotechnology (Grant no. BT/PR15118/BCE/8/1141/2015), Government of India and National Bureau of Agriculturally Important Microorganisms (Grant no. NBAIM/AMAAS/2014-17/PF/9).
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01410/full#supplementary-material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Anderson R. E., Brazelton W. J., Baross J. A. (2011). Using CRISPRs as a metagenomic tool to identify microbial hosts of a diffuse flow hydrothermal vent. FEMS Microbiol. Ecol. 77, 120–133. 10.1111/j.1574-6941.2011.01090.x [DOI] [PubMed] [Google Scholar]
- Arndt D., Grant J. R., Marcu A., Sajed T., Pon A., Liang Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. 10.1093/nar/gkw387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auch A. F., Von Jan M., Klenk H. P., Göker M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. 10.4056/sigs.531120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averhoff B. (2009). Shuffling genes around in hot environments: the unique DNA transporter of Thermus thermophilus. FEMS Microbiol. Rev. 33, 611–626. 10.1111/j.1574-6976.2008.00160.x [DOI] [PubMed] [Google Scholar]
- Aziz R. K., Bartels D., Best A. A., DeJongh M., Disz T., Edwards R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- Blank S., Schröder C., Schirrmacher G., Reisinger C., Antranikian G. (2014). Biochemical characterization of a recombinant xylanase from Thermus brockianus, suitable for biofuel production. JSM Biotechnol. Biomed. Eng. 2:1027. [Google Scholar]
- Brock T. D., Freeze H. (1969). Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J. Bacteriol. 98, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N. L., Stoyanov J. V., Kidd S. P., Hobman J. L. (2003). The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 27, 145–163. 10.1016/S0168-6445(03)00051-2 [DOI] [PubMed] [Google Scholar]
- Bruggemann H., Chen C. (2006). Comparative genomics of Thermus thermophilus: plasticity of the megaplasmid and its contribution to a thermophilic lifestyle. J. Biotechnol. 124, 654–661. 10.1016/j.jbiotec.2006.03.043 [DOI] [PubMed] [Google Scholar]
- Burkhardt J., Vonck J., Averhoff B. (2011). Structure and function of PilQ, a secretin of the DNA transporter from the thermophilic bacterium Thermus thermophilus HB27. J. Biol. Chem. 286, 9977–9988. 10.1074/jbc.M110.212688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballeira N., Nazabal M., Brito J., Garcia O. (1990). Purification of a thermostable DNA polymerase from Thermus thermophilus HB8, useful in the polymerase chain reaction. Biotechniques 9, 276–281. [PubMed] [Google Scholar]
- Chaudhari N. M., Gupta V. K., Dutta C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep. 6:24373. 10.1038/srep24373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563–569. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- Chung A. P., Rainey F. A., Valente M., Nobre M. F., daCosta M. S. (2000). Thermus igniterrae sp. nov. and Thermus antranikianii sp. nov., two new species from Iceland. Int. J. Syst. Evol. Microbiol. 50, 209–217. 10.1099/00207713-50-1-209 [DOI] [PubMed] [Google Scholar]
- Collins R. F., Hassan D., Karuppiah V., Thistlethwaite A., Derrick J. P. (2013). Structure and mechanism of the PilF DNA transformation ATPase from Thermus thermophilus. Biochem. J. 450, 417–425. 10.1042/BJ20121599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. C. E., Mau B., Perna N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon E., Leach D. R. F. (2014). Bacterial genome instability. Microbiol. Mol. Biol. Rev. 78, 1–39. 10.1128/MMBR.00035-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W., Nickle D. C., Learn G. H., Maust B., Mullins J. I. (2007). ViroBLAST: a stand-alone BLAST web server for flexible queries of multiple databases and user's datasets. Bioinformatics 23, 2334–2336. 10.1093/bioinformatics/btm331 [DOI] [PubMed] [Google Scholar]
- Dhillon B. K., Laird M. R., Shay J. A., Winsor G. L., Lo R., Nizam F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, W104–W108. 10.1093/nar/gkv401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle J. J., Doyle J. L. (1990). Isolation of plant DNA from fresh tissue. Focus 12, 13–15. [Google Scholar]
- Dwivedi V., Kumari K., Gupta S. K., Kumari R., Tripathi C., Lata P., et al. (2015). Thermus parvatiensis RLT sp. nov., isolated from a hot water spring, located atop the Himalayan ranges at Manikaran, India. Indian J. Microbiol. 55:357 10.1007/s12088-015-0538-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi V., Sangwan N., Nigam A., Garg N., Niharika N., Khurana P., et al. (2012). Draft genome sequence of Thermus sp. RL isolated from hot water spring located atop the Himalayan ranges at Manikaran. Indian J. Bacteriol. 194:3534 10.1128/JB.00604-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and throughput. Nucleic Acids Res. 32, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Engelke D. R., Krikos A., Bruck M. E., Ginsburg D. (1990). Purification of Thermus aquaticus DNA polymerase expressed in Escherichia coli. Anal. Biochemi. 191, 396–400. 10.1016/0003-2697(90)90238-5 [DOI] [PubMed] [Google Scholar]
- Felsenstein J. (1993). PHYLIP (Phylogeny Inference Package), Version 3.5c. Distributed by the author. Seattle, WD: Department of Genome Sciences, University of Washington. [Google Scholar]
- Friedrich A., Prust C., Hartsch T., Henne A., Averhoff B. (2002). Molecular analyses of the natural transformation machinery and identification of pilus structures in the extremely thermophilic bacterium Thermus thermophilus strain HB27. Appl. Environ. Microbiol. 68, 745–755. 10.1128/AEM.68.2.745-755.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., Zhang C. T. (2008). Ori-Finder: a web based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics. 9:79. 10.1186/1471-2105-9-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounder K., Brzuszkiewicz E., Liesegang H., Wollherr A., Daniel R., Gottschalk G., et al. (2011). Sequence of the hyperplastic genome of the naturally competent Thermus scotoductus SA-01. BMC Genomics 12:577. 10.1186/1471-2164-12-577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I., Vergnaud G., Pourcel C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Delsuc F., Dufayard J. F., Gascuel O. (2009). Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 537, 113–137. 10.1007/978-1-59745-251-9_6 [DOI] [PubMed] [Google Scholar]
- Hao W., Golding G. B. (2010). Inferring bacterial genome flux while considering truncated genes. Genetics 186, 411–426. 10.1534/genetics.110.118448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Kishino H., Yano T. (1985). Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174. 10.1007/BF02101694 [DOI] [PubMed] [Google Scholar]
- Henne A., Bruggemann H., Raasch C., Wiezer A., Hartsch T., Liesegang H., et al. (2004). The genome sequence of the extreme thermophile Thermus thermophilus. Nat. Biotechnol. 22, 547–553. 10.1038/nbt956 [DOI] [PubMed] [Google Scholar]
- Hudson J. A., Morgan H. W., Daniel R. M. (1987). Thermus filiformis sp. nov., a filamentous caldoactive bacterium. Int. J. Syst. Evol. Microbiol. 37, 431–436. 10.1099/00207713-37-4-431 [DOI] [Google Scholar]
- Iqbal N., Guérout A. M., Krin E., Le Roux F., Mazel D. (2015). Comprehensive functional analysis of the 18 Vibro cholerae N16961 Toxin-Antitoxin systems substantiates their role in stabilizing the superintegron. J. Bacteriol. 197, 2150–2159. 10.1128/JB.00108-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S. L., Kelly D. E. (2013). Microbial cytochromes P450: biodiversity and biotechnology. Where do cytochromes P450 come from, what do they do and what can they do for us? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 368:20120476. 10.1098/rstb.2012.0476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerepesi C., Bánky D., Grolmusz V. (2014). AmphoraNet: the webserver implementation of the AMPHORA2 metagenomic workflow suite. Gene 533, 538–540. 10.1016/j.gene.2013.10.015 [DOI] [PubMed] [Google Scholar]
- Khorchid A., Ikura M. (2006). Bacterial histidine kinase as a signal sensor and transducer. Int. J. Biochem. Cell. Biol. 38, 307–312. 10.1016/j.biocel.2005.08.018 [DOI] [PubMed] [Google Scholar]
- Konstantinidis K. T., Tiedje J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. 10.1128/JB.187.18.6258-6264.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körner H., Sofia H. J., Zumft W. G. (2003). Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol. Rev. 27, 559–592. 10.1016/S0168-6445(03)00066-4 [DOI] [PubMed] [Google Scholar]
- Kretza E., Papaneophytou C. P., Papi R. M., Karidi K., Kiparissides C., Kyriakidis D. A. (2012). Lipase activity in Thermus thermophilus HB8: purification and characterization of the extracellular enzyme. Biotechnol. Bioprocess Eng. 17, 512–525. 10.1007/s12257-011-0481-0 [DOI] [Google Scholar]
- Kumwenda B., Litthauer D., Reva O. (2014). Analysis of genomic rearrangements, horizontal gene transfer and role of plasmids in the evolution of industrial important Thermus species. BMC Genomics 15:813. 10.1186/1471-2164-15-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesan K., Hallin P., Rødland E. A., Staerfeldt H. H., Rognes T., Ussery D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. 10.1093/nar/gkm160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S. J., Alkhnbashi O. S., Rose D., Will S., Backofen R. (2013). CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 41, 8034–8044. 10.1093/nar/gkt606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D., Canback B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. 10.1093/nar/gkh152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T., Sonnhammer E. L. L. (2005). KAlign - an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 6:298. 10.1186/1471-2105-6-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M. G., Wackernagel W. (1994). Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 58, 563–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K. S., Wolf Y. I., Alkhnbashi O. S., Costa F., Shah S. A., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nature Rev. Microbiol. 13, 722–736. 10.1038/nrmicro3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini L. A., Sontheimer E. J. (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. 10.1126/science.1165771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefferd C. C., Zhou E. M., Yu T. T., Ming H., Murugapiran S. K., Huntemann M., et al. (2016). High-quality draft genomes from Thermus caliditerrae YIM 77777 and T. tengchongensis YIM 77401, isolates from Tengchong, China. Genome Announc. 4:e00312-16. 10.1128/genomeA.00312-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I., Bayer M., Cardle L., Shaw P., Stephen G., Wright F., et al. (2010). Tablet-next generation sequence assembly visualization. Bioinformatics 26, 401–402. 10.1093/bioinformatics/btp666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming H., Yin Y. R., Li S., Nie G. X., Yu T. T., Zhou E. M., et al. (2014). Thermus caliditerrae sp. nov., a novel thermophilic species isolated from a geothermal area. Int. J. Syst. Evol. Microbiol. 64, 650–656. 10.1099/ijs.0.056838-0 [DOI] [PubMed] [Google Scholar]
- Mojica F. J., Díez-Villaseñor C., García-Martínez J., Soria E. (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60, 174–182. 10.1007/s00239-004-0046-3 [DOI] [PubMed] [Google Scholar]
- Molina-Henares A. J., Krell T., Guazzaroni M. E., Segura A., Ramos J. L. (2005). Members of the IcIR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30, 157–186. 10.1111/j.1574-6976.2005.00008.x [DOI] [PubMed] [Google Scholar]
- Murugapiran S. K., Huntemann M., Wei C.-L., Han J., Detter J. C., Han C., et al. (2013). Thermus oshimai JL-2 and T. thermophilus JL-18 genome analysis illuminates pathways for carbon, nitrogen, and sulfur cycling. Stand. Genomic Sci. 7, 449–468. 10.4056/sigs.3667269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobre M. F., Trüper H. G., da Costa M. S. (1996). Transfer of Thermus ruber (Loginova et al. 1984), Thermus silvanus (Tenreiro et al. 1995), and Thermus chliarophilus (Tenreiro et al. 1995) to Meiothermus gen. nov. as Meiothermus ruber comb. nov., Meiothermus silvanus comb. nov., and Meiothermus chliarophilus comb. nov., respectively, and emendation of the genus Thermus. Int. J. Syst. Bacteriol. 46, 604–606. 10.1099/00207713-46-2-604 [DOI] [Google Scholar]
- Ohtani N., Tomita M., Itaya M. (2012). The third plasmid pVV8 from Thermus thermophilus HB8: isolation, characterization, and sequence determination. Extremophiles 16, 237–244. 10.1007/s00792-011-0424-x [DOI] [PubMed] [Google Scholar]
- Okonechnikov K., Golosova O., Fursov M. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. 10.1093/bioinformatics/bts091 [DOI] [PubMed] [Google Scholar]
- Opperman D. J., van Heerden E. (2006). Aerobic Cr(VI) reduction by Thermus scotoductus strain SA-01. J. Appl. Microbiol. 103, 1907–1913. 10.1111/j.1365-2672.2007.03429.x [DOI] [PubMed] [Google Scholar]
- Pašić L., Rodriguez-Mueller B., Martin-Cuadrado A. B., Mira A., Rohwer F., Rodriguez-Valera F. (2009). Metagenomic islands of hyperhalophiles: the case of Salinibacter ruber. BMC Genomics 10:570. 10.1186/1471-2164-10-570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot M., Kuznetsova E., Brown G., Rao N. N., Kitagawa M., Mori H., et al. (2004). General enzymatic screens identify three new nucleotidases in Escherichia coli. Biochemical characterization of SurE, YfbR, and YjjG. J. Biol. Chem. 279, 54687–54694. 10.1074/jbc.M411023200 [DOI] [PubMed] [Google Scholar]
- Rao V. B., Saunders N. B. (1992). A rapid polymerase-chain-reaction-directed sequencing strategy using a thermostable DNA polymerase from Thermus flavus. Gene 113, 17–23. 10.1016/0378-1119(92)90665-C [DOI] [PubMed] [Google Scholar]
- Richter M., Rosello-Mora R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose I., Biuković G., Aderhold P., Müller V., Grüber G., Averhoff B. (2011). Identification and characterization of a unique, zinc-containing transport ATPase essential for natural transformation in Thermus thermophilus HB27. Extremophiles 15, 191–202. 10.1007/s00792-010-0343-2 [DOI] [PubMed] [Google Scholar]
- Roth C., Rastogi S., Arvestad L., Dittmar K., Light S., Ekman D., et al. (2007). Evolution after gene duplication: models, mechanisms, sequences, systems, and organisms. J. Exp. Zool. 306B, 58–73. 10.1002/jez.b.21124 [DOI] [PubMed] [Google Scholar]
- Rouli L., Merhej V., Fournier P. E., Raoult D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infec. 7, 72–85. 10.1016/j.nmni.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe-Magnus D. A., Guerout A. M., Biskri L., Bouige P., Mazel D. (2003). Comparative analysis of superintegrons: engineering extensive genetic diversity in the Vibrionaceae. Genome Res. 13, 428–442. 10.1101/gr.617103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath N., Ferrada E., Barve A., Wagner A. (2013). Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol. Evol. 5, 966–977. 10.1093/gbe/evt050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer R., Kern T., Joos F., Averhoff B. (2016). The Thermus thermophilus comEA/comEC operon is associated with DNA binding and regulation of the DNA translocator and type IV pili. Environ. Microbiol. 18, 65–74. 10.1111/1462-2920.12820 [DOI] [PubMed] [Google Scholar]
- Sangwan N., Lambert C., Sharma A., Gupta V., Khurana P., Khurana J. P., et al. (2015). Arsenic rich Himalayan hot spring metagenomics reveal genetically novel predator-prey genotypes. Environ. Microbiol. Rep. 7, 812–823. 10.1111/1758-2229.12297 [DOI] [PubMed] [Google Scholar]
- Schwarzenlander C., Haase W., Averhoff B. (2009). The role of single subunits of the DNA transport machinery of Thermus thermophilus HB27 in DNA binding and transport. Environ. Microbiol. 11, 801–808. 10.1111/j.1462-2920.2008.01801.x [DOI] [PubMed] [Google Scholar]
- Segata N., Börnigen D., Morgan X. C., Huttenhower C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Comm. 4:2304. 10.1038/ncomms3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz P., Blokesch M. (2013). Cues and regulatory pathways involved in natural competence and transformation in pathogenic and environmental Gram-negative bacteria. FEMS Microbiol. Rev. 37, 336–363. 10.1111/j.1574-6976.2012.00353.x [DOI] [PubMed] [Google Scholar]
- Shaw J. F., Lin F. P., Chen S. C., Chen H. C. (1995). Purification and properties of an extracellular α-amylase from Thermus sp. Bot. Bull. Acad. Sci. 36, 195–200. [Google Scholar]
- Steffen M. M., Li Z., Effler T. C., Hauser L. J., Boyer G. L., Wilhelm S. W. (2012). Comparative metagenomics of toxic freshwater cyanobacteria bloom communities on two continents. PLoS ONE 7:e44002. 10.1371/journal.pone.0044002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamakoshi M., Murakami A., Sugisawa M., Tsuneizumi K., Takeda S., Saheki T., et al. (2011). Genomic and proteomic characterization of the large Myoviridae bacteriophage ΦTMA of the extreme thermophile Thermus thermophilus. Bacteriophage 1, 152–164. 10.4161/bact.1.3.16712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh B. S., Rahman A. Y. A., Saito J. A., Hou S., Alam M. (2012). Complete genome sequence of the thermophilic bacterium Thermus sp. strain CCB_US3_UF1. J. Bacteriol. 194, 1240. 10.1128/JB.06589-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H., Masignani V., Cieslewicz M. J., Donati C., Medini D., Ward N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. 10.1073/pnas.0506758102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M., de Sousa J. A. M., Rocha E. P. C. (2017). Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr. Opin. Microbiol. 38, 66–73. 10.1016/j.mib.2017.04.010 [DOI] [PubMed] [Google Scholar]
- Vernikos G. S., Parkhill J. (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22, 2196–2203. 10.1093/bioinformatics/btl369 [DOI] [PubMed] [Google Scholar]
- Weinberger A. D., Sun C. L., Plucinski M. M., Denef V. J., Thomas B. C., Horvath P., et al. (2012a). Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput. Biol. 8:e1002475. 10.1371/journal.pcbi.1002475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger A. D., Wolf Y. I., Lobkovsky A. E., Gilmore M. S., Koonin E. V. (2012b). Viral diversity threshold for adaptive immunity in prokaryotes. mBio 3:e00456-12. 10.1128/mBio.00456-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbach H., Etienne F., Hoshi T., Heinemann S. H., Lowther W. T., Matthews B., et al. (2002). Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Arch. Biochem. Biophysic. 397, 172–178. 10.1006/abbi.2001.2664 [DOI] [PubMed] [Google Scholar]
- Wheeler D., Bhagwat M. (2007). BLAST QuickStart, in Comparative Genomics, Vol. 1, 2 Edn., ed Bergman N. H. (Totowa, NJ: Humana Press; ). [PubMed] [Google Scholar]
- Wu S., Zhu Z., Fu L., Niu B., Li W. (2011). WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics. 12:444. 10.1186/1471-2164-12-444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. C., Rosen B. P. (2016). New mechanisms of bacterial arsenic resistance. Biomed. J. 39, 5–13. 10.1016/j.bj.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M. X., Slater M. R., Ackermann H. W. (2006). Isolation and characterization of Thermus bacteriophages. Arch. Virol. 151, 663. 10.1007/s00705-005-0667-x [DOI] [PubMed] [Google Scholar]
- Yu T. T., Ming H., Yao J. C., Zhou E. M., Park D. J., Hozzein W. N., et al. (2015). Thermus amyloliquefaciens sp. nov., isolated from a hot spring sediment sample. Int. J. Syst. Evol. Microbiol. 65, 2491–2495. 10.1099/ijs.0.000289 [DOI] [PubMed] [Google Scholar]
- Zhang Q., Ye Y. (2017). Not all predicted CRISPR-Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinformatics. 18:92. 10.1186/s12859-017-1512-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A., Chen Y. I., Zane G. M., He Z., Hemme C. L., Joachimiak M. P., et al. (2012). Functional characterization of Crp/Fnr-type global transcriptional regulators in Desulfovibrio vulgaris hildenborough. Appl. Environ. Microbiol. 78, 1168–1177. 10.1128/AEM.05666-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou E. M., Murugapiran S. K., Mefferd C. C., Liu L., Xian W. D., Yin Y. R., et al. (2016). High-quality draft genome sequence of Thermus amyloliquefaciens type strain YIM 77409T with an incomplete denitrification pathway. Stand. Genomic Sci. 11:20. 10.1186/s40793-016-0140-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liang Y., Lynch K., Dennis J. J., Wishart D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.