

Abstract
Sulfadoxine/pyrimethamine is recommended for intermittent preventative treatment of malaria during pregnancy. Data from 98 women during pregnancy and 77 after delivery in four African countries were analyzed using nonlinear mixed‐effects modeling to characterize the effects of pregnancy, postpartum duration, and other covariates such as body weight and hematocrit on sulfadoxine/pyrimethamine pharmacokinetic properties. During pregnancy, clearance increased 3‐fold for sulfadoxine but decreased by 18% for pyrimethamine. Postpartum sulfadoxine clearance decreased gradually over 13 weeks. This finding, together with hematocrit‐based scaling of plasma to whole‐blood concentrations and allometric scaling of pharmacokinetics parameters with body weight, enabled site‐specific differences in the pharmacokinetic profiles to be reduced significantly but not eliminated. Further research is necessary to explain residual site‐specific differences and elucidate whether dose‐optimization, to address the 3‐fold increase in clearance of sulfadoxine in pregnant women, is necessary, viable, and safe with the current fixed dose combination of sulfadoxine/pyrimethamine.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Inconsistent findings are reported in previous studies on the pharmacokinetics of sulfadoxine/pyrimethamine in pregnancy.
WHAT QUESTION DOES THIS STUDY ADDRESS?
☑ How do pregnancy, time after delivery, and other clinically important covariates affect the pharmacokinetic properties of sulfadoxine/pyrimethamine across different countries?
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ Clearance was 17.5% lower for pyrimethamine and 3‐fold higher for sulfadoxine during pregnancy. This change for sulfadoxine is similar to other studies and for pyrimethamine it is similar to the study in Kenya. In addition, changes in sulfadoxine clearance after delivery appear to follow a gradual time‐course of 13 weeks.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ We present methods to investigate some of the site differences observed in the effect of pregnancy on sulfadoxine‐pyrimethamine pharmacokinetics, to scale whole‐blood to plasma concentrations for more efficient use of DBS data and to systematically identify outlier‐samples for exclusion. Further research (using paired plasma – DBS samples) is necessary to explain residual site‐specific differences and elucidate whether dose‐optimization (possibly region‐specific), up to 13 weeks after delivery is necessary.
In 2015, there were an estimated 438,000 malaria deaths (236,000–635,000) worldwide with most (90%) occurring in the African Region.1 Pregnant women are at a particularly high risk of contracting malaria, and their disease outcomes are generally worse than the general population due to a modulated immune response and accumulation of parasites in the placenta. In sub‐Saharan Africa alone, ∼25 million pregnant women are exposed to malaria infection, and 10,000 maternal deaths are attributed to malaria annually.2 Malaria during pregnancy also poses a risk to the fetus, as parasites in the placenta could lead to intrauterine growth restriction, resulting in low birth weight, congenital malaria, premature delivery, spontaneous abortion, and stillbirth.3 The fetus also has a higher risk of disease later in life because the abnormal intrauterine environment could affect mental, metabolic, and anthropometric development.4 Pregnant women therefore form the chief group of adults at risk from malaria infection.
Significant physiological changes occur during pregnancy, which may affect the pharmacokinetic properties of drugs, including those used in the treatment and prevention of malaria. Pregnant women have increased renal function,5 with increased glomerular filtration rate and renal tubular activity, usually resulting in increased clearance of renally eliminated drugs. The activity of drug‐metabolizing enzymes is altered during pregnancy, with some (e.g., CYP2D6 and CYP3A4) increased and others (e.g., CYP1A2) decreased, possibly affecting the exposure of drugs cleared by those metabolic pathways.5 Other factors could further complicate the picture, most notably lower plasma albumin concentrations, which can affect the plasma protein binding and thus volume of distribution and total drug clearance.5 Since all these physiological changes could lead to either drug under‐ or overexposure, it is important to investigate the pharmacokinetics of drugs widely used during pregnancy and determine if dosing regimens should be adjusted.
Sulfadoxine/pyrimethamine (SP) is recommended by the WHO as intermittent preventative treatment (IPTp) for the prevention of malaria in pregnancy in sub‐Saharan Africa.6 IPTp with SP is administered in at least two doses, starting in the second trimester of pregnancy after quickening, with an interval of at least 4 weeks.4 Each SP dose given as IPTp is intended to clear existing asymptomatic infection from the placenta and provide posttreatment prophylaxis of ∼4 weeks, thereby aiming for an effective reduction in maternal clinical malaria and low birth weight.7
Both sulfadoxine and pyrimethamine in healthy adults are absorbed well (bioavailability of >90%) and reach peak concentrations of 183 μg/mL for sulfadoxine and 0.55 ng/mL for pyrimethamine 2–8 h after oral administration. Both drugs are 85–90% protein bound in plasma. The mean terminal half‐lives determined in healthy adults are 8.33 for sulfadoxine and 4.16 days and pyrimethamine.4, 8
The literature suggests that SP concentrations in plasma are higher than red blood cells.9, 10 Only unbound drug in plasma is immediately available for elimination and distribution.11
Sulfadoxine is primarily cleared renally through glomerular filtration; however, 70% of filtrated sulfadoxine undergoes tubular reabsorption, which contributes to the long‐elimination half‐life. Minor changes in urine pH significantly affect the amount of ionized and unionized species, which in turn influence reabsorption and total clearance.12 Sulfadoxine undergoes limited phase II metabolism into the more water‐soluble glucuronide (2–3%) and acetylate (5%) metabolites, and its acetylated form, metabolized by N‐acetyltransferase 2 (NAT2), is excreted primarily in urine, with a small fraction excreted via the stool.13
Pyrimethamine is primarily metabolized in the liver into numerous unknown metabolic products, and about 15–30% is excreted unchanged in urine.4
Even though SP is the only recommended regimen for IPTp in pregnancy,3 and is used extensively throughout Africa, there is limited understanding of its pharmacokinetic properties in pregnancy. Previous reports generally show that sulfadoxine concentrations are lower in pregnant women when compared to nonpregnant adults and are inconsistent for pyrimethamine.5, 14, 15
In this work, we present a population pharmacokinetic analysis of previously reported data, pooled from a multicenter trial in four African countries.8 The aim of this study was to characterize the pharmacokinetic properties of SP during pregnancy and after delivery by using nonlinear mixed‐effects modeling.
METHODS
Pharmacokinetic data was available for pregnant women from the African countries of Mali, Zambia, Mozambique, and Sudan, as previously presented by Nyunt et al.8 During pregnancy, women received a single, oral fixed dose combination of 75 mg of pyrimethamine and 1,500 mg of sulfadoxine as part of IPTp. Pharmacokinetic data were collected during either the second or third trimester of pregnancy. Several weeks after delivery and after stopping IPTp, the women were given another ad‐hoc dose and were sampled for a second time.
Capillary blood samples were collected by spotting 100 μL of whole‐blood on filter paper, air‐dried, and stored at 4°C. Samples were collected at the following timepoints: in Mali and Zambia, immediately before dosing and 3, 6, and 12 h, 1, 3, 7, 14, 21, and 28 days postdosing, both during pregnancy and after delivery, and in Mozambique and Sudan, immediately before dosing and 1, 2, 3, 7, 14, 21, and 28 days postdosing, during pregnancy and 7 days postdosing postpartum. Dried blood spot concentration measurements were performed using liquid chromatography and tandem mass spectrometry, as described previously.16 The entire blood spot was cut out of the filter paper, and drug molecules extracted using organic solvent. The lower limits of quantification (LLOQ) of the assays were 10 ng/mL and 10 μg/mL for pyrimethamine and sulfadoxine, respectively. The coefficient of variation was 9.8% at 65 μg/mL for sulfadoxine and 12.7% at 165 ng/mL for pyrimethamine. The study was approved by local communities and Institutional Ethics Committees at all study sites, and it was conducted in accordance with the Guidelines for Good Clinical Practice in the Conduct of Clinical Trials in Human Participants in South Africa and Good Clinical Practice Guideline E6: International Conference on Harmonisation (ICH)/WHO Good Clinical Practice standards for the conducts of clinical trials in drug research.8
Pharmacokinetic data were analyzed using nonlinear mixed‐effects modeling in the software NONMEM 7.3,17 and all parameters were estimated using the first‐order conditional with eta–epsilon interaction algorithm. The pharmacokinetics of sulfadoxine and pyrimethamine were first modeled independently to determine the structural model and covariate effects, and then combined into one model to investigate possible correlations between the pharmacokinetic parameters of the two drugs and to better account for the correlation in the uncertainty affecting the observed concentrations. One‐, two‐, and three‐compartment disposition models with first‐order absorption or transit‐compartment absorption were evaluated for the structural model. Between‐subject (BSV) and between‐occasion variability (BOV) was evaluated on the pharmacokinetic parameters assuming a log‐normal distribution. A combined error model with both additive and proportional components was used for the residual unexplained variability (RUV). The effect of body size was taken into account using allometric scaling with total body weight to adjust all volumes (allometric exponent fixed to 1) and clearance and flow rates to and from peripheral compartments (allometric exponent fixed to 0.75), as suggested by Anderson et al.18 Unfortunately, no height information was available for the patients, so testing the use of fat‐free mass or adjustments for body composition were not possible. The effect of study site, age, anemia, mg/kg dose, pregnancy status, estimated gestational age, and time after delivery were tested as predefined covariates, with a significance threshold of <0.01. The NONMEM objective function value (OFV), goodness of fit plots, and visual predictive checks (n = 1,000) guided the model development.
A nonparametric bootstrap with replacement (n = 500) was performed on the final model to assess the precision and robustness of the final parameter estimates.
For the data from the sites in Zambia and Mali, all concentrations lower than 7 ng/mL for pyrimethamine and 7 μg/mL for sulfadoxine were censored and thus the original values not available for the analysis, while all the measured values were available in the data from Mozambique and Sudan. All censored values were imputed to 3.5 ng/mL for pyrimethamine and 3.5 μg/mL for sulfadoxine, i.e., half of the censoring threshold. These imputed data were handled using an approach similar to the M6 suggested by Beal et al.19: if a series of censored values were present in a pharmacokinetic profile, only the first imputed value was retained in the analysis, while all trailing ones were excluded from the model fit, and included in the simulation‐based diagnostics only.
Capillary whole‐blood was the matrix used for drug concentration measurements. However, pyrimethamine and sulfadoxine do not partition equally between red blood cells (RBC) and plasma.9, 10 An attempt to scale capillary whole‐blood measurements to plasma concentrations was therefore evaluated in the model in order to describe individual and site differences as well as enable a comparison with published literature.
Baseline hemoglobin was recorded during pregnancy and after delivery, and it was converted to hematocrit by using the following formula:
| (1) |
HCT denotes the hematocrit as a fraction of 1 and HB denotes the hemoglobin in g/dL.20 The conversion described above has been proposed for individuals who are infected with Plasmodium falciparum malaria, but were similar (data not shown) to hematocrit and model parameter values obtained with formula proposed for healthy subjects21; therefore, Eq. 1 was selected to enable easier comparison of the pharmacokinetic results with infected individuals.
The model was parameterized in terms of pharmacokinetics of plasma concentrations, and the concentration values in the central compartment were rescaled to obtain the model prediction for whole‐blood capillary concentrations, which was fitted to the observed data. The formula used for the conversion is given next:
| (2) |
CWB denotes the whole‐blood drug concentration; CPL denotes the plasma drug concentration; θRBC/PL denotes the RBC‐plasma ratio, and HCT denotes hematocrit. The value of θRBC/PL for each drug was estimated in the model using a log‐normal Bayesian prior with typical values of 0.42 for pyrimethamine and 0.16 for sulfadoxine, as previously reported,9, 10 with 30% uncertainty included to account for potential population differences.
Pregnancy‐related changes in pharmacokinetics
The changes in pharmacokinetic parameters during pregnancy and after delivery were modeled taking into account estimated gestational age during pregnancy and the time from delivery for the postpartum pharmacokinetic visits. Linear, exponential, and sigmoidal models were tested to explore the time‐course of changes in pharmacokinetic parameters. Parameter values during pregnancy were considered as reference as more data points were available during pregnancy than after delivery (two sites only had day 7 concentrations postpartum). Moreover, the time after delivery (postpartum period) was different among the four sites.
RESULTS
Pharmacokinetic data were collected in 98 women during pregnancy, 77 of whom were sampled again after delivery, although at different frequencies and times after delivery in different sites. Data including 1,276 blood concentrations for both pyrimethamine and sulfadoxine were available for analysis, of which 210 (16.4%) of pyrimethamine and 107 (8.3%) of sulfadoxine concentrations were censored. There were 34 (19.1%) and 31 (17.7%) samples with detectable predose concentrations of sulfadoxine and pyrimethamine, respectively, and most of these values (100% and 74%, respectively) were below the nominal LLOQ of the assay. For individuals who did not have a predose sample collected postpartum, concentrations were assumed to be zero at the time of the dose. Sixteen samples (1.25%) were excluded from the dataset as outliers (explained in the Supplementary Material), including four samples below LLOQ.
As reported in the previous analysis,8 the observed drug concentrations were highly variable between study sites.
Baseline characteristics were recorded for all women during the pregnancy phase and 67 women after delivery; these are shown in Table 1 , stratified by study site. The baseline characteristics include age, weight, hemoglobin, alanine aminotransferase, and creatinine, with the addition of gestational age, gravidity, and parity in the pregnancy phase and time from delivery in the after delivery phase.8 Weight and hemoglobin data were missing postpartum for 10 individuals (13.5%) from the sites in Zambia and Sudan and was imputed as the median values recorded in that site and occasion.
Table 1.
Baseline characteristics expressed as median (interquartile range) stratified by study site
| Country | Parameter | During pregnancy | After delivery |
|---|---|---|---|
| Mali | Age (years) | 26 (22‐32) | |
| Number | 18 | 18 | |
| Weight (kg) | 60 (56‐65) | 59 (52‐64) | |
| Hemoglobin (g/dl) | 9.9 (9.4‐11.4) | 12.4 (11.6‐13.2) | |
| Gestational age/Time after delivery (weeks) | 28 (25‐29) | 8.3 (8.1‐8.6) | |
| Mozambique | Age (years) | 24 (22‐27) | |
| Number | 31 | 22 | |
| Weight (kg) | 61 (56‐65) | 55 (51‐63) | |
| Hemoglobin (g/dl) | 10.8 (9.6‐12) | 11.6 (11.2‐12.9) | |
| Gestational age/Time after delivery (weeks) | 27 (24‐29) | 46 (44‐51) | |
| Sudan | Age (years) | 28 (26‐32) | |
| Number | 24 | 9 | |
| Weight (kg) | 66 (57‐71) | 68 (67‐82) | |
| Hemoglobin (g/dl) | 9.2 (8.5‐10.9) | 10.5 (9.5‐10.8) | |
| Gestational age/Time after delivery (weeks) | 27 (21‐33) | 15 (14‐16) | |
| Zambia | Age (years) | 31 (25‐36) | |
| Number | 25 | 18 | |
| Weight (kg) | 60 (56‐66) | 61 (55‐66) | |
| Hemoglobin (g/dl) | 11.3 (10.5‐12.1) | 13.1 (11.6‐14.9) | |
| Gestational age/Time after delivery (weeks) | 27 (24‐28) | 6.4 (6.0‐6.9) | |
A combined model with first‐order absorption, first‐order elimination, and three disposition compartments for pyrimethamine and two for sulfadoxine provided the best fit to the observed concentration–time data. The final model parameter values are shown in Table 2 , and visual predictive checks of drug concentration vs. time are shown in Figure 1. Additionally, observed day 7 drug concentrations during pregnancy and the postpartum are shown for the Mozambique and Sudan sites (samples were only collected on day 7 postpartum) in Figure 2 to illustrate the pregnancy effect in the sites with the longest postpartum period (46 and 15 weeks).
Table 2.
Final pharmacokinetic parameter values for pyrimethamine and sulfadoxine during pregnancy and after delivery
| Parameter | Sulfadoxine | Pyrimethamine | ||
|---|---|---|---|---|
| Estimate | 95% CIa | Estimate | 95% CIa | |
| F | 1 FIXED | — | 1 FIXED | — |
| CL/F during pregnancy [L/h]b | 0.0303 | 0.0185, 0.0349 | 1.35 | 1.12,1.38 |
| Vc/F [L]b | 14.1 | 13.2, 14.4 | 163 | 151, 166 |
| ka [/h] | 0.531 | 0.464, 0.565 | 1.31 | 1.11, 2.70 |
| Qp1/F [L/h]b | 0.0252 | 0.0136, 0.0269 | 1.45 | 0.72, 1.61 |
| Vp1/F [L]b | 179 | 82, 212 | 29.8 | 23.9, 32.1 |
| Qp2/F [L/h]b | — | — | 0.122 | 0.064, 0.166 |
| Vp2/F [L]b | — | — | 251 | 142, 317 |
| θRBC/PL [fraction of one] | 0.155 | 0.023, 0.189 | 0.324 | 0.106, 0.525 |
| Change in CL when non‐pregnant [%] | −75.7 | −88.7, −66.6 | 21.2 | 12.3, 24.9 |
| T50 [weeks] | 6.35 | 5.47, 6.75 | — | — |
| γ – shape factor | 4.90 | 2.90, 7.41 | — | — |
| Difference in clearance in Mozambique [%] | — | — | −20.2 | −28.4, −17.4 |
| Site effect (scaling on observations) in Mozambique [%] | 21.2 | 8.2, 24.6 | 57.6 | 41.5, 60.6 |
| Site effect (scaling on observations) in Sudan [%] | 15.5 | 4.8, 20.0 | 33.2 | 19.6, 35.6 |
| Site effect (scaling on observations) in Zambia [%] | −24.8 | −30.7, −22.2 | −5.40 | −12.1, −3.9 |
| Between subject variability in CL [%]c | 31.3 | 21.8, 51.2 | 12.3 | 7.1, 16.9 |
| Between occasion variability in F [%]c | 20.7 | 16.7, 22.9 | 17.6 | 12.9, 21.5 |
| Between occasion variability in CL [%]c | — | — | 16.9 | 11.8, 22.3 |
| Between occasion variability in ka [%]c | 56.4 | 42.4, 70.1 | — | — |
| Correlation in bioavailability of the two drugs [%] | 67.9 | 55.9, 71.9 | * | * |
| Additive error [mg/mL pyra – and ug/mL for sulfa] | 2.13 | 2.00, 2.21 | 2.45 | 2.01, 2.68 |
| Proportional error [%] | 17.0 | 14.8, 17.5 | 18.0 | 15.1, 18.7 |
| Correlation in random unexplained error of the two drugs [%] | 61.3 | 54.2, 63.9 | * | * |
Pharmacokinetic parameter values are expressed referring to plasma. CI, confidence interval; F, relative bioavailability; CL/F, elimination clearance; VC/F, apparent volume of distribution of central compartment; ka, first order absorption rate constant; QP1/F, flow rate to and from shallow peripheral compartment; VP1/F, apparent volume of distribution of shallow peripheral compartment; QP2/F, flow rate to and from deep peripheral compartment; VP2/F, apparent volume of distribution of deep peripheral compartment; θRBC/PL, red blood cells to plasma ratio; T50, time at which of 50% post‐delivery effect; γ, post‐delivery effect shape parameter.
95% confidence interval denoted as 2.5‐97.5 percentiles of the estimates from 500 iterations of a nonparametric bootstrap.
All volumes and flow rates (clearance and flow rates to and from peripheral compartments) were allometrically scaled with total body weight centered on the median body weight (60kg).
BSV and BOV were assumed as log‐normally distributed and are reported here as approximate CV%
*This parameter has the same value for both S and P.
Figure 1.
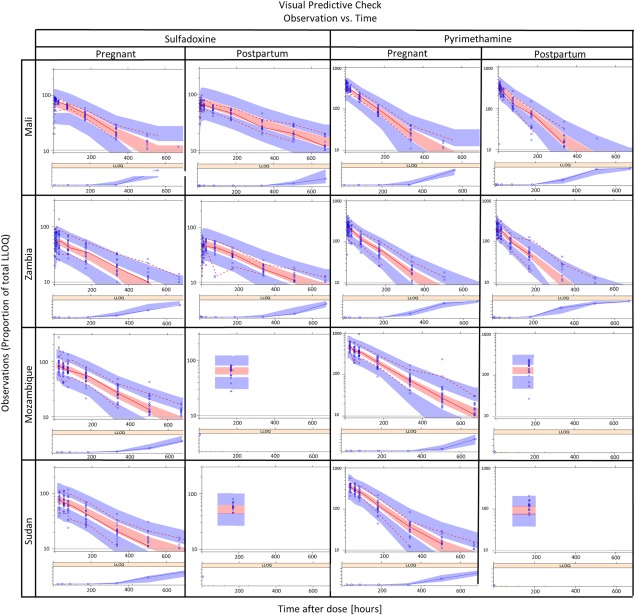
Visual predictive check for the combined final model, stratified by study site, drug, and pharmacokinetic visit. The observed data are plotted as blue circles while the lines represent the 5th, 50th, and 95th percentiles of the observed data. The shaded areas represent the 95% confidence intervals for the same percentiles, as predicted by the model.
Figure 2.
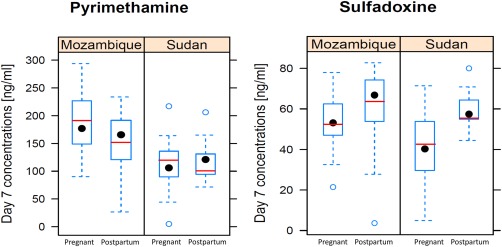
Observed day 7 concentrations in the Sudan and Mozambique sites for the pregnant and postpartum women. The boxplot summarizes the observed concentrations, while the model‐predicted median concentrations are shown as a red line.
Pregnancy proved to have a significant effect on the plasma clearance of both drugs. Compared to during pregnancy, pyrimethamine plasma clearance was found to be 21.2% higher after delivery. In contrast, sulfadoxine clearance progressively decreased after delivery, resulting in a total reduction of 75.8% around 3 months' postpartum. This time‐dependent change in clearance for sulfadoxine from pregnancy to after delivery was modeled using a sigmoid function and is shown in Figure 3.
Figure 3.
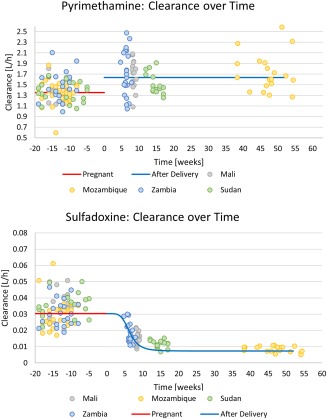
Pregnancy effect on plasma clearance vs. time after delivery for pyrimethamine (top panel) and sulfadoxine (bottom panel). The line represents the population typical value, while the dots represent the individual post‐hoc values from the final model after adjusting for the effect of weight, hematocrit, and the Mozambique site effect (for pyrimethamine only).
Additionally, pyrimethamine plasma clearance was found to be 20.9% lower in the subjects from Mozambique, both in the pregnancy and postdelivery visits.
Modeling sulfadoxine clearance as a function of time after delivery proved better than assuming a different pregnancy effect parameter for each site (ΔOFV = 12.4).
The use of hematocrit to scale whole‐blood concentrations to plasma resulted in a significantly improved model fit (ΔOFV = 14.2 and 14.4 for pyrimethamine and sulfadoxine, respectively) and decreased the variability between the four sites and pharmacokinetic visits (pregnancy and postpartum). The reestimated values of the RBC‐plasma ratio (θRBC/PL) were similar to the prior (−23% and −3% bias for pyrimethamine and sulfadoxine, respectively).
Allometric scaling with total body weight improved the model fit substantially (ΔOFV = 88.5) and decreased (albeit only slightly) the pregnancy effect, BSV in clearance, and the BOV in relative bioavailability for both drugs.
Even after adjusting for hematocrit, body size, and time after delivery, there were still significant site‐specific differences between the pharmacokinetic profiles. This was captured in the model by using the Mali site as a reference and introducing a scaling factor for all the concentrations from the other sites. This improved the model fit significantly for both pyrimethamine (ΔOFV = 227.7) and sulfadoxine (ΔOFV = 54.6). No other covariate effects (estimated gestational age, age, mg/kg dose) were found to improve the fit.
The model supported BSV in clearance and BOV in bioavailability for both drugs, with the addition of BOV in clearance for pyrimethamine and BOV in the absorption rate constant for sulfadoxine.
When the two drugs were analyzed in a joint model, their random BSV or BOV in bioavailability was found to be correlated (r = 0.677), and the inclusion of this improved the model (ΔOFV = 76.2). The RUV of the two drugs was found to be correlated (r = 0.613; ΔOFV = 315.7), reflecting that since the concentrations were measured in the same blood sample, some of the error was common between the two observations.
The post‐hoc estimates of the individual parameters from the final model were then used to calculate individual exposure values, shown in Table 3.
Table 3.
Summary of individual exposure parameters for pyrimethamine and sulfadoxine during pregnancy and after delivery, stratified by site
| Country | Parameter | Pyrimethamine | Sulfadoxine | ||
|---|---|---|---|---|---|
| Pregnancy | After delivery | Pregnancy | After delivery | ||
| Mali | Sample size (%) | 18 | 18 (100) | 18 | 18 (100) |
| Cmax (mg/L) | 361 (337, 399) | 308 (286, 362) | 78.8 (75.4, 82.0) | 69.4 (65.7, 73.2) | |
| Tmax (h) | 3.30 (3.22, 3.34) | 3.18 (3.16, 3.25) | 8.72 (6.78, 11.6) | 9.64 (8.95, 11.3) | |
| AUCinf (mg·h/L) | 41.2 (37.0, 45.9) | 31.6 (23.9, 34.5) | 29659 (26958, 39623) | 64128 (54276, 83819) | |
| Cday7 (mg/L) | 81.9 (74.8, 99.5) | 62.8 (41.7, 68.5) | 40.5 (37.8, 44.4) | 44.6 (42.2, 49.6) | |
| Mozambique | Sample size (%) | 31 | 22 (71) | 31 | 22 (71) |
| Cmax (mg/L) | 589 (556, 646) | 670 (599, 736) | 87.5 (77.5, 97.9) | 92.0 (82.3, 98.6) | |
| Tmax (h) | 3.37 (3.35, 3.42) | 3.27 (3.18, 3.32) | 9.43 (9.23, 9.57) | 10.4 (10.3, 10.5) | |
| AUCinf (mg·h/L) | 83.2 (74.2, 94.7) | 77.8 (62.1, 93.3) | 43048 (34749, 52655) | 161419 (142185, 194938) | |
| Cday7 (mg/L) | 186 (167, 211) | 149 (121, 182) | 49.5 (43.6, 55.1) | 66.0 (58.8, 69.4) | |
| Sudan | Sample size (%) | 23 | 15 (65) | 23 | 15 (65) |
| Cmax (mg/L) | 443 (426, 509) | 436 (411, 464) | 78.3 (72.4, 87.3) | 75.2 (73.4, 80.3) | |
| Tmax (h) | 3.32 (3.28, 3.38) | 3.29 (3.26, 3.34) | 9.15 (9.11, 9.33) | 10.3 (10.3, 10.4) | |
| AUCinf (mg·h/L) | 54.6 (50.9, 65.3) | 56.6 (44.7, 61.5) | 30664 (27050, 39969) | 107707 (97106, 133040) | |
| Cday7 (mg/L) | 124 (98.7, 143) | 107 (91.2, 118) | 40.6 (37.0, 48.8) | 53.4 (51.5, 54.9) | |
| Zambia | Sample size (%) | 25 | 22 (88) | 25 | 22 (88) |
| Cmax (mg/L) | 236 (216, 246) | 216 (194, 239) | 59.7 (46.4, 67.1) | 47.4 (42.0, 52.4) | |
| Tmax (h) | 3.26 (3.25, 3.35) | 3.23 (3.16,3.29) | 8.88 (7.37, 10.4) | 9.86 (8.50, 17.5) | |
| AUCinf (mg·h/L) | 28.5 (23.3, 35.1) | 21.4 (16.9, 26.6) | 29037 (23293, 35084) | 35467 (29468, 42818) | |
| Cday7 (mg/L) | 58.4 (45.2, 65.3) | 39.3 (29.9, 54.4) | 31.4 (26.5, 36.9) | 29.5 (26.8, 33.9) | |
| Total | Sample size (%) | 97 | 77 (79) | 97 | 77 (79) |
| Cmax (mg/L) | 426 (304, 565) | 378 (264, 570) | 77.8 (67.1, 87.0) | 72.2 (53.7, 81.9) | |
| Tmax (h) | 3.33 (3.25, 3.38) | 3.25 (3.16, 3.30) | 9.29 (8.77, 9.59) | 10.3 (9.71, 10.6) | |
| AUCinf (mg·h/L) | 50.8 (34.3, 76.9) | 39.3 (23.7, 60.6) | 33068 (27010, 43638) | 88380 (45962, 140733) | |
| Cday7 (mg/L) | 107 (72.4, 162) | 72.9 (44.9, 120) | 41.8 (35.5, 49.9) | 49.4 (34.6, 58.7) | |
The values were generated using post‐hoc individual parameter values from the final model. Post‐hoc parameter values are expressed in median (interquartile range). AUCinf, area under whole‐blood concentration‐time curve extrapolated to infinity; Cday7, whole‐blood concentration on day 7; Cmax, maximum whole‐blood concentration; Tmax, time of maximum whole‐blood concentration.
DISCUSSION
In this study, we report and compare the pharmacokinetic parameters of sulfadoxine and pyrimethamine in pregnant women and after delivery using nonlinear mixed‐effects modeling. We found that clearance during pregnancy was significantly higher for sulfadoxine and slightly lower for pyrimethamine when compared to after delivery. For sulfadoxine, the clearance was found to change progressively after delivery, resulting in a total reduction of 75.7% at ∼13 weeks after delivery compared to during pregnancy.
Sulfadoxine is primary renally cleared via glomerular filtration, with 70% tubular reabsorption seen in healthy adults. The increase in sulfadoxine clearance observed is consistent with the increase in glomerular filtration rate (GFR) and decreases in proximal tubular reabsorption that occur during pregnancy.22 Unfortunately, creatinine clearance was not available for most of the subjects to confirm this. Additionally, plasma albumin concentrations are expected to decrease during pregnancy, potentially increasing the free fraction and enhancing total clearance.23 It would be informative to measure and compare free drug concentrations for pregnant and nonpregnant women to determine to what extent the higher clearance during pregnancy is due to protein binding. If lower protein binding is the major factor, then dose adjustment may not be needed, as the unbound concentration, which elicits the drug effect, may remain unchanged. The progressive decline in clearance in the first weeks after delivery suggests that these changes experienced during pregnancy take some time to normalize after delivery. Our model estimated the pregnancy effects to disappear after around 13 weeks, which is consistent with literature suggesting that the GFR and renal blood flow reach the prepregnant state 6–12 weeks after delivery.24
For pyrimethamine, clearance was found to be lower during pregnancy than after delivery (21.2%). Literature suggests pyrimethamine is metabolized along multiple pathways by currently unknown metabolites. Pregnancy is known to increase the activity of CYP2A6, CYP2C9, CYP3A4, and CYP2D6 and decrease the activity of CYP1A2 and CYP2C19, although this seems to be dependent on the concentration of female hormones.5 The change in clearance during pregnancy could therefore be due to pregnancy‐related changes in metabolizing enzyme activity as well as polymorphisms within individual CYP subfamilies. Confirmation of this theory is difficult since pyrimethamine metabolism is not well characterized.
The previous analysis of these data15 also found pyrimethamine clearance to be lower during pregnancy than after delivery and sulfadoxine clearance to be slightly higher during pregnancy than after delivery. However, due to the limited sampling schedule, the pregnancy effect could only be investigated in the sites in Zambia and Mali, which incidentally are those where the postpartum sampling was at the shortest time after delivery (4.7–11.8 weeks). The original study also identified the large variability in the disposition of both drugs between the study sites during pregnancy and after delivery. The model‐based analysis we propose here was also able to quantify the change in clearance for the Mozambique and Sudan sites, which only had day 7 concentrations and could therefore show the time‐course change in clearance for sulfadoxine after delivery. The sulfadoxine pregnancy effect described in our model reaches a plateau only 13 weeks after delivery, and this could not be captured in the previous analysis, where only the Zambia and Mali sites were investigated.
We used hematocrit to scale whole‐blood concentrations to plasma to explain some of the variability between study sites. Hematocrit measurements were only available at baseline for each pharmacokinetic period, but longitudinal changes are expected during pregnancy, possibly affecting pharmacokinetics. This lack of information may have limited our capacity to accurately scale between whole‐blood and plasma concentrations in each sample. Scaling whole‐blood concentrations to plasma concentrations using hematocrit is beneficial in drugs that have a concentration in RBCs that is different from the concentration in plasma, especially in patient groups where hematocrit values are expected to change significantly over time or differ significantly between individuals, as is the case with pregnancy. This method could be used in other patient groups where pharmacokinetic samples are collected as dried blood spots, such as children who often have anemia due to comorbidities such a malnutrition, symptomatic malaria, or concurrent helminth infections.25 A concern with the use of dried blood spots is that hematocrit affects the viscosity of blood and thus the thickness and density of the drop on the filter paper. If the concentration is determined from a fixed size punch of the blood spot, this may affect the concentration reading, as the volume of blood will differ.26 In the current study, the entire blood spot was cut out and analyzed, thus preventing this having an effect.
Allometric scaling was also able to explain some of the variability between study sites. The absence of height information about the participants was a limitation, which meant that total body weight was the only available size descriptor for allometric scaling. Body weight is not an accurate predictor of the size of drug‐metabolizing organs in a population with varying degrees of body fat, and fat‐free mass is arguably a better predictor.18 However, without height information, no assessment of the effect of body composition was possible. The situation is further complicated by changes related to pregnancy.
As previously mentioned, the time after delivery at which the women were sampled to assess changes in pharmacokinetic parameters postpartum depended on the study site, with the shortest of 4.7 to 7.9 weeks in Zambia and the longest of 38.3 to 63.4 weeks in Mozambique. Thus, the timing of the postpartum pharmacokinetic visit and the site effect may be confounded. Moreover, the sites with later pharmacokinetic visits only collected day 7 samples, further hindering the description of the pregnancy effect in these sites. That said, these differences enabled explaining the time‐course of changes in clearance, which when introduced in the model, improved the model fit significantly (ΔOFV = 17.7), and this modeling strategy was more parsimonious than including different pregnancy effects in different study sites.
Despite these limitations, we were able to reduce the large between‐site variability observed in pharmacokinetic parameters by using weight, hematocrit, and postpartum period. When comparing the variability of individual exposures (AUC) after delivery, the “site effect” quantified in the model decreased from 146% to 58%, 79% to 33%, and 32% to 6% for pyrimethamine and from 151% to 21%, 67% to 16%, and 45% to 25% for sulfadoxine for Mozambique, Sudan, and Zambia, respectively, using Mali as the reference site. The variability that our model could only attribute to study site may be due to heterogeneity in the study population in terms of pharmacogenetics, diet, and widespread concurrent use of undocumented traditional medicines.27, 28 However, the fact that this remaining site‐specific variability could be described reasonably well using a simple scaling factor possibly points to differences in bioavailability, different drug production batches from the same drug manufacturer or systematic differences in the sample collection, preparation, and handling of the dried blood spots at the various study sites. The latter could have been excluded by the collection of matched plasma samples in a subset of patients from each site. Quantifying and understanding site‐specific differences in sulfadoxine and pyrimethamine pharmacokinetic parameters is valuable for informing studies to optimize dosing during pregnancy.
Another benefit is that the model is able to describe the pharmacokinetics of the two drugs concomitantly, and thereby account for the correlation in the pharmacokinetic parameters and in the observed drug measurements. The fact that concentrations for both drugs were measured in the same sample also strengthened our model‐based approach to exclude outliers, as we discarded values with extreme normalized prediction distribution errors for both drugs. This approach is meant to detect issues with sampling labeling/handling that would affect the concentrations of both drugs. This method is particularly useful in studies using dried blood spots, where potential errors involved in sample collection such as paper contamination and ineffective sample drying may affect drug concentration values.26
To our knowledge, there have been three other published studies evaluating the pharmacokinetics of SP in pregnant women. A study from Papua New Guinea12 compared 30 pregnant women to 30 age‐matched nonpregnant women. They found the clearance of sulfadoxine to be 67% and pyrimethamine to be 47% higher during pregnancy. Another study13 comparing the pharmacokinetics of HIV‐positive and HIV‐negative pregnant women to the same women used as their own controls postpartum (only 11 out of 33 return postpartum) in Kenya found the median exposure to sulfadoxine to be 40% lower during pregnancy and no significant difference in the exposure to pyrimethamine. They did not find a significant difference between the HIV+ and HIV– women in either the pregnancy or postpartum phase. A study from Uganda29 compared 199 pregnant women to 34 nonpregnant women. They found clearance of sulfadoxine and pyrimethamine to be almost 5‐fold and 58% higher during pregnancy, respectively. The higher clearance of sulfadoxine during pregnancy found in our study is similar to the trends found in the other three studies. Our findings of pyrimethamine clearance during pregnancy is similar to the finding of the study in Kenya. The differences between the results of their studies and ours could possibly be due to the fact that some of the women included in these studies were parasitemic (43% in Papua New Guinea and 33% in Kenya), and chloroquine was coadministered in the study from Papua New Guinea. Other possible explanations could be differences in pharmacogenetics between the study population, estimated gestational age, hemoglobin, postpartum duration, study design, and methods of drug assay and analysis. As the studies in Papua New Guinea and Uganda did not use the same women as controls, between‐subject variability could have affected their results.
CONCLUSION
This study reports changes in the pharmacokinetics of SP during pregnancy and after delivery as well as presenting a model accounting for the effect of body‐size and hematocrit. We found that the clearance of sulfadoxine during pregnancy is significantly increased, although it is unclear if this is due to increased renal function or difference in plasma protein binding. Further investigation into free concentrations may help to inform the model to determine whether dose‐optimization is necessary. Pregnancy‐related changes in pyrimethamine are small and not expected to be clinically relevant.
Supporting information
Supporting Information S1
Supporting Information S2
Acknowledgments
We thank Dr Kassoum Kayentao (Malaria Research and Training Center, University of Bamako, Mali), Dr Yasmin Cassam (Mozambique Ministry of Health), and Dr Philip Thuma (Malaria Institute at Macha, Zambia) as local principal investigators who contributed substantially to the conception, study design, execution, and data analysis in the primary publication. We acknowledge the World Wide Antimalarial Resistance Network (WWARN) for collating, curating, and making the data available for this analysis. WWARN is funded by a Bill & Melinda Gates Foundation grant. The studies in Mali and Zambia were funded by the Johns Hopkins Malaria Research Institute and the Bloomberg Foundation. The study in Mozambique by a Global Fund for Fighting AIDS, Tuberculosis, and Malaria grant to the Lebombo Spatial Development Initiative. The Development Partnerships in Higher Education program of the UK Department for International Developments funded collaboration among researchers conducting studies on antimalarial pharmacokinetics in pregnancy. The Division of Clinical Pharmacology at the University of Cape Town gratefully acknowledges Novartis Pharma for their support of the development of pharmacometric skills in Africa. These funders did not participate in the study protocol development or the writing of the article.
Conflict of Interest
K.I.B. is a member of the WHO Technical Expert Group (TEG) on Malaria Chemotherapy and of the WHO TEG on Drug Resistance and Containment. The remaining authors declare that no competing interests exist.
Author Contributions
P.D., M.C.dK., J.T., K.B., wrote the article; P.D., M.C.dK., K.B., designed the research; M.N., I.A., performed the research; P.D., M.C.dK., L.W., analyzed the data.
References
- 1. World Health Organization . World Malaria Report 2015 (WHO, Geneva, Switzerland: ). [Google Scholar]
- 2. Dellicour S. et al Quantifying the number of pregnancies at risk of malaria in 2007: a demographic study. PLoS Med. 7, 1–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Desai M. et al Epidemiology and burden of malaria in pregnancy. Lancet Infect Dis. 7, 93–104 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Agomo C.O. et al Parasitologic assessment of two‐dose and monthly intermittent preventive treatment of malaria during pregnancy with sulphadoxine‐pyrimethamine. Malar. Res. Treat. 2011, 932895 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jeong H. Altered drug metabolism during pregnancy: hormonal regulation of drug‐metabolizing enzymes. Expert Opin. Drug Metab. Toxicol. 6, 689–699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braun, V. et al Lack of effect of intermittent preventive treatment for malaria in pregnancy and intense drug resistance in western Uganda. Malar. J. 14, 372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kayentao, K. et al Intermittent preventive therapy for malaria during pregnancy using 2 vs 3 or more doses of sulfadoxine‐pyrimethamine and risk of low birth weight in Africa. Clin. Corner. 309, 594–604 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nyunt, M.M. et al Pharmacokinetics of sulfadoxine and pyrimethamine in intermittent preventive treatment of malaria in pregnancy. Clin. Pharmacol. Ther. 87, 226–234 (2010). [DOI] [PubMed] [Google Scholar]
- 9. Dua, V.K. et al Sulphadoxine concentrations in plasma, red blood cells and whole blood in healthy and Plasmodium falciparum malaria cases after treatment with Fansidar using high‐performance liquid chromatography. J. Pharm. Biomed. Anal. 12, 1317–1323 (1994). [DOI] [PubMed] [Google Scholar]
- 10. Rudy, A.C. et al Binding of pyrimethamine to human plasma proteins and erythrocytes. Pharm. Res. 7, 1055–1060 (1990). [DOI] [PubMed] [Google Scholar]
- 11. Schmidt, S. et al Significance of protein binding in pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 99, 1107–1022 (2010). [DOI] [PubMed] [Google Scholar]
- 12. Charpiat, B. et al for the Eurotoxo Group (panel 2). Systematic search and analysis of published pharmacokinetic data related to sulfadoxine [Unpublished report]. Bordeaux (France): The Eurotoxo Group; 2004.
- 13. Hickman, M.R. et al Pharmacokinetics and Pharmacodynamics of Antimalarial Drugs Used in Combination Therapy, 1st edn. Bentham Science Publishers, Sharjah, UAE, 2015. [Google Scholar]
- 14. Karunajeewa, H.A. et al Pharmacokinetic properties of sulfadoxine‐pyrimethamine in pregnant women. Antimicrob. Agents Chemother. 53, 1403–1408 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Green, M.D. et al Pharmacokinetics of sulfadoxine‐pyrimethamine in HIV‐infected and uninfected pregnant women in Western Kenya. J. Infect. Dis. 196, 1468–1476 (2007). [DOI] [PubMed] [Google Scholar]
- 16. Barnes, K.I. et al Sulfadoxine‐pyrimethamine pharmacokinetics in malaria: pediatric dosing implications. Clin. Pharmacol. Ther. 80, 582–596 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Beal, S. et al NONMEM Users Guides (1989‐2009) ICON Development Solutions, Ellicott City, MD, 2009. [Google Scholar]
- 18. Anderson, B.J. et al Mechanism‐based concepts of size and maturity in pharmacokinetics. Annu. Rev. Pharmacol. Toxicol. 48, 303–332 (2008). [DOI] [PubMed] [Google Scholar]
- 19. Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28, 481–504 (2001). [DOI] [PubMed] [Google Scholar]
- 20. Lee, S.J. et al The relationship between the haemoglobin concentration and the haematocrit in Plasmodium falciparum malaria. Malar. J. 7, 149 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grgac, K. et al Hematocrit and oxygenation dependence of blood 1H2O T1 at 7 Tesla. Magn. Reson. Med. 70, 1153–1159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheung, K.L. et al Renal physiology of pregnancy. Adv. Chronic Kidney Dis. 20, 209–214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Isoherranen, N. et al Drug metabolism and transport during pregnancy: How does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab. Dispos. 41, 256–262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davison, J.M. et al 24‐Hour creatinine clearance during the third trimester of normal pregnancy. J. Obstet. Gynaecol. 87, 106–109 (1980). [DOI] [PubMed] [Google Scholar]
- 25. Osazuwa, F. et al A significant association between intestinal helminth infection and anemia burden in children in rural communities of Edo state, Nigeria. N. Am. J. Med. Sci. 3, 30–34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Edelbroek, P.M. et al Dried blood spot methods in therapeutic drug monitoring: methods, assays, and pitfalls. Ther. Drug Monit. 31, 327–336 (2009). [DOI] [PubMed] [Google Scholar]
- 27. Leon‐Cachon, R.B. et al Application of genomic technologies in clinical pharmacology research. Rev. Invest. Clin. 67, 212–218 (2015). [PubMed] [Google Scholar]
- 28. Thomford, N. et al Pharmacogenomics implications of using herbal medicinal plants on African populations in health transition. Pharmaceuticals 8, 637–663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Odongo, C.O. et al Trimester‐specific population pharmacokinetics and other correlates of variability in sulphadoxine‐pyrimethamine disposition among Ugandan pregnant women. Drugs Res. Dev. 15, 351–362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1
Supporting Information S2