

Abstract
Ovarian cancer is one of the major threats to female health. Identifying cancer cases at an early stage and selecting effective therapeutic drugs for patients is challenging. The number of studies concerning long non-coding RNAs (lncRNAs) is increasing rapidly; there is a large body of evidence indicating that lncRNAs are crucial in oncogenic and tumor-suppression mechanisms. Therefore, in the present study, lncRNA expression in ovarian cancer was considered. All of the existing ovarian cancer microarray datasets in the Gene Expression Omnibus database were assessed and two met the criteria for the present study; these were designated the training and validation sets. A re-annotation pipeline method was established to annotate lncRNAs from existing probe sets. When comparing ovarian cancer with normal ovarian tissues, seven lncRNAs from the RefSeq database, based on their combined ability to classify tissue in the training set, were identified and validated with the validation set. Research into the molecular functions of the seven identified lncRNAs may contribute to the understanding of ovarian cancer oncogenesis; they may also be candidates for novel ovarian cancer biomarkers.
Keywords: long non-coding RNA, ovarian cancer, microarray
Introduction
Ovarian cancer (OC) is one of the most common types of malignant carcinoma and is associated with a high mortality rate (1). Due to of the lack of an effective early-stage biomarker for cancer screening and detection, >70% of ovarian cancer cases are diagnosed at an advanced stage with tumor metastasis to other organs; the 5-year survival rate for patients with metastatic OC is <30% (2). Although multiple biomarkers have been developed to categorize the molecular fingerprint of OC by identifying recurring genetic defects, using the existing biomarkers to achieve an accurate diagnosis of ovarian cancer stage and assign a specific, optimized treatment regimen to each patient remains an unsolved challenge (3).
Up to 74.7% of the human genome does not code for proteins and instead gives rise to non-protein-coding RNAs (ncRNAs) (4). Long non-coding RNAs (lncRNAs) are ncRNAs longer than 200 nucleotides; they are RNA polymerase II transcripts that lack an open reading frame (5). LncRNAs are the largest class of ncRNAs; ~15,931 lncRNA genes have been annotated in humans (6). Unlike the smaller non-coding microRNAs, functions for the majority of lncRNAs have yet to be elucidated. However, with improvements in transcriptome profile technology and research, there is increasing evidence demonstrating that certain lncRNAs serve important functions in cancer development (7). These lncRNAs may regulate gene expression at the transcriptional, post-transcriptional and epigenetic levels by interacting with DNA, RNA or protein molecules, or by causing transcriptional interference (8). They serve functions in oncogenic and tumor-suppressive pathways (9).
In the present study, the lncRNA expression signatures in OC were profiled via the analysis of a cohort of previously published OC gene expression profiles from the Gene Expression Omnibus (GEO). The results may provide novel information on lncRNA expression profiles.
Materials and methods
GEO OC gene expression data
OC expression data used in the present study were obtained from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/gds). Datasets used in this study were selected using the following criteria: i) They were datasets from patients with OC; ii) they used OC tumor tissue and superficial scrapings from normal ovary tissue samples for comparison; iii) they used the same platform (Human Genome U133 Plus 2.0 microarray, Affymetrix, Inc., Santa Clara, CA, USA); and iv) they contained >3 samples. Based on these criteria, two datasets were selected: GSE36668 (10) and GSE18520 (11), as described in Table I. In the present study, the training-validation approach was adapted for screening potential biomarkers, a standard strategy for microarray-based classification analysis (12). GSE36668 was used as the training set and contained four ovarian borderline carcinoma (SBOC) samples, four serous ovarian carcinoma (SC) samples and four superficial scraping from normal ovary (SNO) samples. GSE18520 contained 53 advanced-stage, high-grade primary ovarian carcinoma specimens (OCS) and 10 normal ovarian surface epithelium brushings (Normal). Dataset GSE18520 served as a validation set for the data derived from GSE36668. The whole study design is presented in Fig. 1.
Table I.
Details of the two microarray datasets used in the present study.
| Dataset | Control samples | Tumor samples | Author, year | Platform | (Refs.) |
|---|---|---|---|---|---|
| GSE36668 | 4 | 8 | Elgaaen et al, 2012 | GPL570 | (10) |
| GSE18520 | 10 | 53 | Mok et al, 2009 | GPL570 | (11) |
GPL570, Affymetrix Human Genome U133 Plus 2.0 array.
Figure 1.
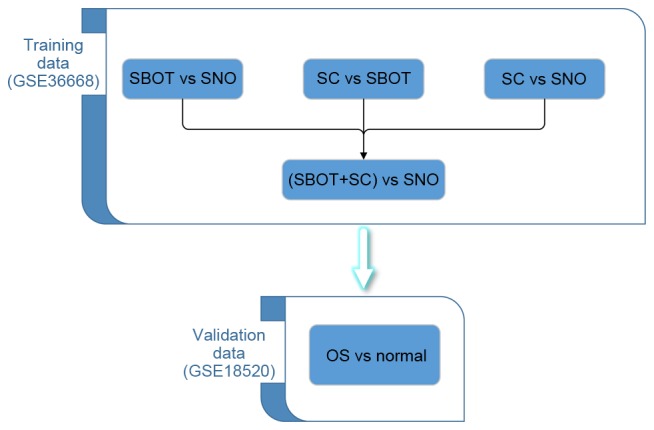
Organization of the training and validation datasets. Based on specific criteria, two datasets of OCS microarray data were selected from the Gene Expression Omnibus. Each dataset included data from OCS and normal ovarian tissue. OC, ovarian cancer; SBOC, ovarian borderline carcinoma specimens; SNO, normal ovary specimens; SC, ovarian serous carcinoma specimens; OCS, ovarian carcinoma specimens.
Individual data processing
The raw CEL files of the datasets were normalized and background adjusted using the robust multichip average method, as this was previously demonstrated to be an effective method for normalizing lncRNA profiling data (13). The normalized data were then analyzed with linear model for microarray data (LIMMA), a modified t-test incorporating the Benjamini-Hochberg multiple hypotheses correction technique (14). These steps were performed with R version 3.1.4 (15). In the analysis of the training dataset GSE36668, SBOC data and SC data were combined and compared with SNO data (Fig. 1). When analyzing data from the GSE18520 validation set, OCS sample data were compared with normal sample data. Adjusted P<0.01 combined with a ≥2-fold expression level difference was defined as a significant difference between probe sets.
Probe set re-annotation and the lncRNA classification pipeline
As novel lncRNAs and their functions are identified and studied every year, the annotation file downloaded from the Affymetrix website is not completely accurate and may be obsolete for screening target lncRNAs. Probe sets without annotation or annotated as coding RNAs are likely to be newly identified lncRNAs, and may potentially be crucial in various biological processes (16). In order to avoid annotation error and lower the false positive rate, an lncRNA classification pipeline was developed to identify the lncRNAs represented on the Affymetrix array. Initially, the sequences of all probe sets were downloaded from the official Affymetrix website (http://www.affymetrix.com/Auth/analysis/downloads/data/HG-U133_Plus_2.probe_fasta.zip). NCBI BLAST-2.2.30+ (17) was used to perform an alignment search of all the probe sequences obtained from the Affymetrix probe sequence file against the RefSeq database. Then probe sets were filtered and re-annotated using the BLAST output, with the following criteria: i) The probe should perfectly hit a target gene (E-value, <2×10−6; query coverage, 100%; identity, 100%); ii) any probe that perfectly hit multiple targets was eliminated; and iii) the accession number of the target gene should start with ‘NR_’, as NR indicates non-coding RNA in the RefSeq database. This was then matched to the lncRNAs probe set datasets. The probe set re-annotation and lncRNA classification pipeline were performed using in-house Perl scripts in Perl 5.18 (https://www.perl.org/).
Data analysis
To visually inspect the result of a ‘leave one dataset out’ cross-validation, the differentially expressed lncRNAs of GSE36668 and GSE18520 were used for a hierarchical clustering analysis (HCA) in Cluster and TreeView (https://sourceforge.net/projects/jtreeview/files/) (18) and ‘gplots’ packages in R. Principal component analysis (PCA) was also performed using TM4 (19) and R 3.1.4 using the princomp function; the ‘plot3d’ package in R was used to draw a three-dimensional plot. The HCA grouped samples by their similarities in gene expression profiles, whereas the PCA summarized the most important variables in a dataset as principal components, and classified the samples using as few variables as possible (8). In HCA cluster analysis, the euclidean distance method was used to cluster arrays (http://CRAN.R-project.org/package=gplots).
Results
The present study included five cohorts of gene expression data from two datasets, GSE36668 and GSE18520, as described in Table I. Through a training-validation approach, differentially expressed probe sets from each dataset were obtained; the total number of these is included in Table II. Subsequent to re-annotating the probe sets and classifying lncRNAs, differentially expressed lncRNAs were identified from the two datasets, the totals of which are presented in Table III. HCA analysis of all samples in the datasets revealed clear distinctions between OC tumor tissue and normal tissue samples based on the expression of the identified lncRNAs (Fig. 2). After aggregating the groups of lncRNA signatures, seven lncRNAs were identified with PCA, as described in Table IV. The fold change of each of these seven lncRNAs is indicated in Fig. 3. Furthermore, the PCA results were suitable for distinguishing OC from normal tissue samples; the PCA results of all samples from the datasets indicated that the OC and normal tissue samples could be distinguished by the combined differential expression of the seven lncRNAs (Fig. 4). Thus, it was determined that the lncRNA signatures identified in the training set were representative.
Table II.
Numbers of all probe sets differentially expressed in each microarray dataset.
Table III.
Number of all lncRNAs differentially expressed in each dataset, following the re-annotation of existing probe sets.
| Dataset | Upregulated probe sets | Downregulated probe sets | Total differential probe sets |
|---|---|---|---|
| GSE36668 | 20 | 17 | 37 |
| GSE18520 | 94 | 25 | 119 |
lncRNA, long non-coding RNA.
Figure 2.

All detected lncRNA signatures in dataset (A) GSE36668 and (B) GSE18520. Samples are in columns and lncRNAs are in rows. Gene expression levels are indicated as follows: Red, upregulated; green, downregulated. lncRNA, long non-coding RNA; SBOC, ovarian borderline carcinoma specimens; SNO, normal ovary specimens; SC, ovarian serous carcinoma specimens.
Table IV.
lncRNAs of which the combined differential expression was sufficient to distinguish cancerous and normal tissue.
| lncRNAs | Chr | Description |
|---|---|---|
| LEMD1-AS1 | 1q32.1 | LEMD1 antisense RNA 1 |
| LOC100506834 | 7 | Uncharacterized LOC100506834 |
| NR2F1 | 5q14 | NR2F1 antisense RNA 1, transcript variant 5 |
| RNF157 | 17q25.1 | RNF157 antisense RNA 1 |
| IPW | 15q11.2 | Imprinted in Prader-Willi syndrome |
| LOC100507387 | 7 | Uncharacterized LOC100507387 |
| LOC100507564 | 1 | Uncharacterized LOC100507564 |
lncRNA, long non-coding RNA; Chr, chromosome.
Figure 3.

FC of the expression levels of the seven identified differentially expressed lncRNAs in (A) GSE36668 and (B) GSE18520. The combined differential expression of the identified lncRNAs was sufficient to distinguish cancer and normal tissues. lncRNA, long non-coding RNA; FC, fold change.
Figure 4.

Principle component analysis results of samples in (A) the training set, GSE36668 (green, serous carcinoma; red, borderline carcinoma; blue, normal ovary tissue) and (B) the validation set, GSE18520 (red, ovarian cancer; green, normal ovary tissue), based on the combined differential expression levels of seven lncRNAs. lncRNA, long non-coding RNA.
Discussion
Ovarian cancer is a great threat to female health worldwide. Early detection of ovarian cancer at its localized stage can increase the 5-year survival rate to ~90% (20). Hence, identifying novel targets that could serve as biomarkers for early diagnosis and treatment of ovarian cancer is urgently required. Previous studies investigating effective early ovarian cancer diagnostic markers have revealed that the development of ovarian cancer is associated with the aberrant expression of specific lncRNAs, including GAS5 (21), SPRY4-IT1 (22), C17orf91 (23) and CCAT2 (24).
In the present study, a training-validation approach and a probe set re-annotation and lncRNA classification pipeline were used to identify seven lncRNAs that were differentially expressed in OC tumor tissue from the two datasets. All the identified lncRNAs have annotations in the NCBI RefSeq database, meaning that the sequences of each lncRNA are completely known, improving the ease of examining the function and downstream target genes of each lncRNA. The identified lncRNAs may interact with various genes to affect the gene expression profile of cells and lead to oncogenesis. The identified lncRNAs may also be candidates for novel ovarian cancer detection biomarkers. Further in vivo and in vitro experiments are required to reveal the function of these lncRNAs and their association with ovarian cancer development.
As the Affymetrix Human Genome U133 Plus 2.0 array has a large number of probe sets, lncRNA expression data can be extracted with probe set re-annotation and the lncRNA classification pipeline, as performed in the present study. This is advantageous as the HG U133 Plus 2.0 array series is one of most commonly used commercial microarrays in human cancer profiling; prior to performing an lncRNA microarray, the information from existing published expression profile data should, therefore, be considered.
In conclusion, seven differentially expressed lncRNAs in ovarian carcinoma have been identified in the present study; their combined differential expression was sufficient to distinguish normal and cancerous tissue. A novel method for mining lncRNA data from pre-existing expression microarrays, instead of using a specialized lncRNA microarray, has been established. This method is comparatively low-cost and could potentially be applied in a wide range of other research areas.
Acknowledgements
The present study was supported by the Science and Technology Project of Shandong Province (grant no. 2014GGH218027).
References
- 1.Rooth C. Ovarian cancer: Risk factors, treatment and management. Br J Nurs. 2013;22(Suppl):23–30. doi: 10.12968/bjon.2013.22.Sup17.S23. [DOI] [PubMed] [Google Scholar]
- 2.Omura GA, Brady MF, Homesley HD, Yordan E, Major FJ, Buchsbaum HJ, Park RC. Long-term follow-up and prognostic factor analysis in advanced ovarian carcinoma: The gynecologic oncology group experience. J Clin Oncol. 1991;9:1138–1150. doi: 10.1200/JCO.1991.9.7.1138. [DOI] [PubMed] [Google Scholar]
- 3.Goetsch CM. Genetic tumor profiling and genetically targeted cancer therapy. Semin Oncol Nurs. 2011;27:34–44. doi: 10.1016/j.soncn.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59. doi: 10.1186/1741-7007-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang G, Lu X, Yuan L. LncRNA: A link between RNA and cancer. Biochim Biophys Acta. 2014;1839:1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Sun S, Pu JK, Tsang AC, Lee D, Man VO, Lui WM, Wong ST, Leung GK. Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiol Dis. 2012;48:1–8. doi: 10.1016/j.nbd.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, Ansell PJ, Zhao J, Weng C, Klibanski A. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282:24731–24742. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 10.Elgaaen BV, Olstad OK, Sandvik L, Odegaard E, Sauer T, Staff AC, Gautvik KM. ZNF385B and VEGFA are strongly differentially expressed in serous ovarian carcinomas and correlate with survival. PLoS One. 2012;7:e46317. doi: 10.1371/journal.pone.0046317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mok SC, Bonome T, Vathipadiekal V, Bell A, Johnson ME, Wong KK, Park DC, Hao K, Yip DK, Donninger H, et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: Microfibril-associated glycoprotein 2. Cancer Cell. 2009;16:521–532. doi: 10.1016/j.ccr.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet. 2005;365:488–492. doi: 10.1016/S0140-6736(05)17866-0. [DOI] [PubMed] [Google Scholar]
- 13.Yu F, Jiang Q, Zhou Y, Yang Z, Yu X, Wang H, Liu Z, Wang L, Fang W, Guo S. Abnormal expression of matrix metalloproteinase-9 (MMP9) correlates with clinical course in Chinese patients with endometrial cancer. Dis Markers. 2012;32:321–327. doi: 10.1155/2012/269279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen X, Yue M, Meng F, Zhu J, Zhu X, Jiang Y. Microarray analysis of differentially-expressed genes and linker genes associated with the molecular mechanism of colorectal cancer. Oncol Lett. 2016;12:3250–3258. doi: 10.3892/ol.2016.5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R Development Core Team, corp-author. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. [Google Scholar]
- 16.Zhi H, Ning S, Li X, Li Y, Wu W, Li X. A novel reannotation strategy for dissecting DNA methylation patterns of human long intergenic non-coding RNAs in cancers. Nucleic Acids Res. 2014;42:8258–8270. doi: 10.1093/nar/gku575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han L, Zhu J. Using matrix of thresholding partial correlation coefficients to infer regulatory network. Biosystems. 2008;91:158–165. doi: 10.1016/j.biosystems.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 20.Li K, Hüsing A, Fortner RT, Tjønneland A, Hansen L, Dossus L, Chang-Claude J, Bergmann M, Steffen A, Bamia C, et al. An epidemiologic risk prediction model for ovarian cancer in Europe: The EPIC study. Br J Cancer. 2015;112:1257–1265. doi: 10.1038/bjc.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Huang H, Li Y, Li L, Hou W, You Z. Decreased expression of long non-coding RNA GAS5 promotes cell proliferation, migration and invasion, and indicates a poor prognosis in ovarian cancer. Oncol Rep. 2016;36:3241–3250. doi: 10.3892/or.2016.5200. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Liu C, Lu Z, Chen L, Wang J, Li Y, Ma H. Upregulation of the long non-coding RNA SPRY4-IT1 indicates a poor prognosis and promotes tumorigenesis in ovarian cancer. Biomed Pharmacother. 2017;88:529–534. doi: 10.1016/j.biopha.2017.01.037. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Yu H, Xi M, Lu X. Long noncoding RNA C17orf91 is a potential prognostic marker and functions as an oncogene in ovarian cancer. J Ovarian Res. 2016;9:49. doi: 10.1186/s13048-016-0258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang S, Qing C, Huang Z, Zhu Y. The long non-coding RNA CCAT2 is up-regulated in ovarian cancer and associated with poor prognosis. Diagn Pathol. 2016;11:49. doi: 10.1186/s13000-016-0499-x. [DOI] [PMC free article] [PubMed] [Google Scholar]