

Abstract
Poly (ADP-ribose) polymerase-1 (PARP1) plays a vital role in DNA repair and is expected to be an effective target in various malignancies. The aim of the present study was to investigate the clinical and biological significance of PARP1 expression in esophageal squamous cell carcinoma (ESCC). Immunohistochemical (IHC) staining was used to examine the association between PARP1 expression and the clinicopathological features of 86 patients with ESCC. The antitumor effect of small interfering RNA against PARP1 (siPARP1) was examined in a proliferation assay, and the mechanisms of this effect were investigated using western blot analysis and cell cycle assays. Cox multivariate analysis revealed that high expression of PARP1 in IHC staining was a statistically significant independent prognostic factor of poor overall survival (OS). The adjusted hazard ratio for OS in the group with high expression of PARP1 was 2.39 (95% confidence interval, 1.29–4.44; P=0.0051). In vitro assays showed that siPARP1 significantly decreased proliferation through G2/M arrest. Furthermore, western blot analysis showed that PARP1 was associated with the ataxia telangiectasia mutated-checkpoint kinase 2-cell division control 25c pathway. The present study suggests that PARP1 expression has a critical role in ESCC progression, and may be a clinical therapeutic target.
Keywords: poly (ADP-ribose) polymerase-1, cell cycle arrest, G2/M, molecular target drug, esophageal squamous cell carcinoma
Introduction
Esophageal cancer is the eighth most common malignancy and the sixth leading cause of cancer-associated mortality worldwide (1–3). Even subsequent to combined multimodality treatment, clinical outcomes remain extremely poor (4,5). More effective treatments based on novel mechanisms are required. DNA repair pathways play a vitally important role for maintaining genomic integrity. Failure of these pathways may lead to unrepaired DNA lesions, and the accumulation of such lesions is associated with genomic instability (6). In recent years, therefore, the strategy of inhibiting proteins associated with DNA repair has shown promise for new treatments of various malignancies.
Poly (ADP-ribose) polymerase-1 (PARP1) is a 113-kDa nuclear polymerase that modifies substrates by poly ADP-ribosylation, and can conjugate ADP from NAD+ to target proteins, such as histones (7,8). At present, it has been shown that PARP1 plays a role in the repair of DNA damage and is activated by DNA strand breaks, particularly those of single-stranded DNA (9–11). Numerous studies have reported that PARP1 inhibitors are effective in patients with breast and ovarian cancer, since the BRCA gene, which is another DNA repair gene, is frequently mutated (12–14); PARP1 expression is upregulated to compensate for the impaired DNA repair (12,15). Previously, PARP1 inhibitors have received attention in patients with malignancies other than breast and ovarian cancer (7,16). However, there are few studies investigating PARP1 in esophageal cancer, particularly esophageal squamous cell carcinoma (ESCC) (17).
The present study aimed to investigate the association between PARP1 expression and prognosis in patients with ESCC, as well as the effect of inhibiting PARP1 expression on the proliferation of ESCC cells.
Materials and methods
Clinical tissue samples
Between January 1998 and December 2011, 86 tissue samples were collected from patients who had undergone radical esophagectomy, without preoperative therapies such as chemotherapy or chemoradiotherapy, for primary ESCC at the Department of Gastroenterological Surgery, Osaka University Hospital (Osaka, Japan). Pathological tumor stage was evaluated using the seventh edition of the TNM classification established by the Union for International Cancer Control (18). The present study was approved by the Ethics Committee of Osaka University Hospital (Suita, Japan) and written consent was obtained from all the patients in the present study.
Antibodies
The primary antibodies used for western blot analysis were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA) and were as follows: Rabbit anti-checkpoint kinase 2 (Chk2) (catalog no. 2662S; dilution, 1:1,000); rabbit anti-phospho-Chk2 (Thr68) (catalog no. 2661S; dilution, 1:1,000); rabbit anti-cell division control (cdc) 25c (catalog no. 4688S; dilution, 1:1,000); rabbit anti-phospho-cdc25c (Thr48) (catalog no. 9527S; dilution, 1:1,000); mouse anti-cdc2 (catalog no. 9116S; dilution, 1:1,000); rabbit anti-phospho-cdc2 (Tyr15) (catalog no. 9111S; dilution, 1:1,000); and rabbit anti-cyclin B1 (catalog no. 4138S; dilution, 1:1,000). Mouse anti-PARP1 (catalog no. sc-8007; immunohistochemistry dilution, 1:50; western blotting dilution, 1:1,000) was obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Secondary antibodies used for western blot analysis were obtained from GE Healthcare Life Sciences (Little Chalfont, UK) and were as follows: Horseradish peroxidase (HRP)-conjugated sheep anti-mouse IgG (catalog no. NA931; dilution, 1:100,000) and HRP-conjugated donkey anti-rabbit IgG (catalog no. NA934; dilution, 1:100,000).
Immunohistochemical (IHC) staining
A mouse anti-PARP1 antibody (catalog no. sc-8007; dilution, 1:50; Santa Cruz Biotechnology, Inc.) was used. In brief, 4-µm-thick sections of 10% formalin-fixed and paraffin-embedded blocks were used for immunohistochemistry. These sections were deparaffinized in xylene, dehydrated in graded ethanol, and heated in 10 mM citrate buffer (pH 6.0) for 40 min at 95°C for antigen retrieval by autoclave. Endogenous peroxidase activity was blocked with 0.3% H2O2 in methanol. Nonspecific binding was blocked with horse serum for 20 min using the VECTASTAIN Elite ABC kit (catalog no. PK-6102, Vector Laboratories, Burlingame, CA, USA). The sections were incubated overnight with mouse anti-PARP1 antibody (catalog no. sc-8007; dilution, 1:50, Santa Cruz Biotechnology, Inc.) at 4°C in a moist chamber. Antibody staining was visualized with the VECTASTAIN Elite ABC kit, followed by 3,3′-diaminobenzidine tetrahydrochloride plus H2O2 for 2 min 30 sec. All sections were counterstained with Mayer's hematoxylin (catalog no. 131-09665; Wako, Osaka, Japan).
PARP1 expression was evaluated by the intensity of stained cancer samples, particularly nuclei, as previously reported (6). Tonsil tissues collected from patients who had undergone tonsillectomy between January and December 2011 at the Department of Otorhinolaryngology-Head and Neck Surgery, Osaka University Hospital (Osaka, Japan), were used as a positive control. The intensity was scored from 0 to 3 (0, none; 1, weak; 2, moderate; 3, strong). Expression was considered to be low when scores were 0 or 1 and high when scores were 2 or 3. Evaluation was performed by two double-blinded independent observers, who were unaware of the clinicopathological data and outcome. When a discrepant evaluation between the two independent observers was found, the evaluation was rechecked and discussed.
Cell lines and culture conditions
Human ESCC TE1, TE4, TE5, TE6, TE8, TE9, TE10 and TE11 cell lines were obtained from the Riken Bioresource Center Cell Bank (Tsukuba, Japan). All cell lines were cultured in RPMI-1640 (Nacalai Tesque, Inc., Kyoto, Japan), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Inc.). These cell lines were incubated in 5% CO2 at 37°C.
Small interfering RNA (siRNA) design
siRNA against PARP1 (siPARP1;catalog nos., sc-29437A-C) and nontargeting siRNA (negative control siRNA; catalog no., sc-37007) were purchased from Santa Cruz Biotechnology, Inc. The sequence of siPARP1 was designed as follows: sc-29437A sense, 5′-GAGUCAAGAGUGAAGGAAATT-3′ and antisense, 5′-UUUCCUUCACUCUUGACUCTT-3′; sc-29437B sense, 5′-GGUAUCAACAAAUCUGAAATT-3′ and antisense, 5′-UUUCAGAUUUGUUGAUACCTT-3′; sc-29437C sense, 5′-GCAACAAACUGGAACAGAUTT-3′ and antisense, 5′-AUCUGUUCCAGUUUGUUGCTT-3′. Cells were cultured to 50–60% confluency. The siRNA oligonucleotides (20 nM) in Opti-MEM (Thermo Fisher Scientific, Inc.) were transfected into cells using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, Inc.) and incubated in Opti-MEM, according to the manufacturer's protocol. Cells were incubated for 48 h (RT-qPCR and cell proliferation assay) and 72 h (for western blotting and cell cycle assay) subsequent to transfection. The transfection efficiency was confirmed by RT-qPCR and western blot analysis.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total cellular RNA was extracted from cell pellets with TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Relevant complementary DNA was amplified by PCR with the Reverse Transcription System A3500 (Promega Corporation, Madison, WI, USA). Reverse transcription was performed at 42°C for 60 min followed by heating at 95°C for 5 min.
The primer sequences were customized by Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) as follows: PARP1 (accession no. NM_001618) forward, 5′-GACGAGCTAAAGAAAGTGTGTTCAA-3′ and reverse, 5′-GGTCCAAGATCGCCGACTC-3′; GAPDH (accession nos. NM_002046, NM_0012,56799, NM_001289745 and NM_001289746) forward, 5′-CAACTACATGGTTTACATGTTC-3′ and reverse, 5′-AAATGAGCCCCAGCCTTC-3′, which was used as an internal control.
RT-qPCR was performed using the FastStart DNA Master SYBR Green I (Roche Diagnostics, Basel, Switzerland) and LightCycler System (Roche Diagnostics). The cycling conditions were as follows: 1 cycle at 95°C for 10 min followed by 45 cycles at 95°C for 10 sec, 54°C for 10 sec, and 72°C for 10 sec.
Western blot analysis
Cells were resuspended in ice-cold radioimmunoprecipitation assay buffer (Thermo Fisher Scientific, Inc.) supplemented with a cocktail of protease/phosphatase inhibitors (Thermo Fisher Scientific, Inc.). Cells were further incubated on ice for 10 min and centrifuged at 14,000 × g at 4°C for 20 min. Subsequent to determination of the protein concentration using Bio-Rad Protein Assay (catalog no., 5000006JA; Bio-Rad Laboratories, Inc., Hercules, CA, USA), the proteins in each sample were resolved on SDS-PAGE (10% gel; Bio-Rad Laboratories, Inc.) and transferred onto polyvinylidene difluoride membranes (Merck KGaA). The membranes were washed with Tris-buffered saline containing 0.1% Tween-20 (TBST) and blocked with Blocking One-P (Nacalai Tesque, Inc.) at room temperature. The membranes were incubated with respective primary antibodies against different targets at 4°C overnight. Subsequent to incubation with primary antibodies, secondary antibodies were added and incubated for 1 h at room temperature. Subsequent to incubation with secondary antibodies, signals were detected with ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences, Little Chalfont, UK).
Cell proliferation assay
Cells were seeded into 96-well plates at a density of 5×103 cells/100 µl/well (Costar; Corning Inc., Corning, NY, USA) for 24 h, and then transfected with siPARP (catalog nos. sc-29437A-C) or negative control siRNA (catalog no. sc-37007). Cell viability was quantified subsequent to 0, 24, 48 and 72 h of transfection by the WST-8 assay using the Cell Counting Kit-8 (catalog no. 343-07623; Dojindo Laboratories, Kumamoto, Japan), according to the manufacturer's protocol. Absorbance was measured at 450 nm with the Model 680XR Microplate Reader (Bio-Rad Laboratories, Inc.).
Cell cycle assay
The cell cycle was assessed by flow cytometry. First, 96 h prior to analysis, cells were seeded into 6-well plates at a density of 5×105 cells/2 ml/well. A total of 72 h prior to analysis, cells were transfected with siPARP1 or negative control. Subsequent to treatment with siRNA, cells were harvested, washed twice with PBS, and suspended with 70% cold ethanol (500 µl) for 30 min on ice. Cells were washed with PBS, then resuspended with 100 µg/ml RNase A (Qiagen, Hilden, Germany) for 20 min at 37°C and 25 µg/ml propidium iodide (PI; Dojindo Laboratories) for 20 min on ice. Subsequent to treatment, PI fluorescence was analyzed using FACS Canto II (BD Biosciences, Franklin Lakes, NJ, USA). Data from at least 10,000 cells were analyzed using BD FACSDIVA 7.0 (BD Biosciences). Cell cycle distribution was calculated with FlowJo software version 8.8.7 (Tree Star, Inc., San Carlos, CA, USA).
Statistical analysis
Associations between PARP1 expression and clinicopathological factors were analyzed using the χ2 test for categorical variables and the Mann-Whitney U test for continuous variables. Overall survival (OS) was defined as the period from the date of surgery to the date of mortality from any cause. Survival was estimated using the Kaplan-Meier method and compared using the log-rank test. The hazard ratio (HR) for recurrence or mortality in the group with high expression of PARP1 was estimated using a Cox proportional hazards model. Multivariate Cox regression analysis was performed to adjust for potential confounding factors. A statistically significant difference was indicated by P<0.05. All reported P-values were two-tailed. All statistical analyses were performed with JMP Pro 11.2.1 software (SAS Institute, Cary, NC, USA).
Results
Patient characteristics and expression of PARP1 in ESCC tissues
The present study examined the expression of PARP1 in ESCC tissues by IHC staining. The expression PARP1 in cancer cells was observed in the cytoplasm and nucleus, but predominantly in the nucleus (Fig. 1A-D). Among the 86 patients with ESCC, low expression of PARP1 was observed in 54 patients (62.8%) and high expression was observed in 32 patients (37.2%). No significant difference in clinicopathological features was observed between the patients with low and the patients with high expression of PARP1 (Table I).
Figure 1.
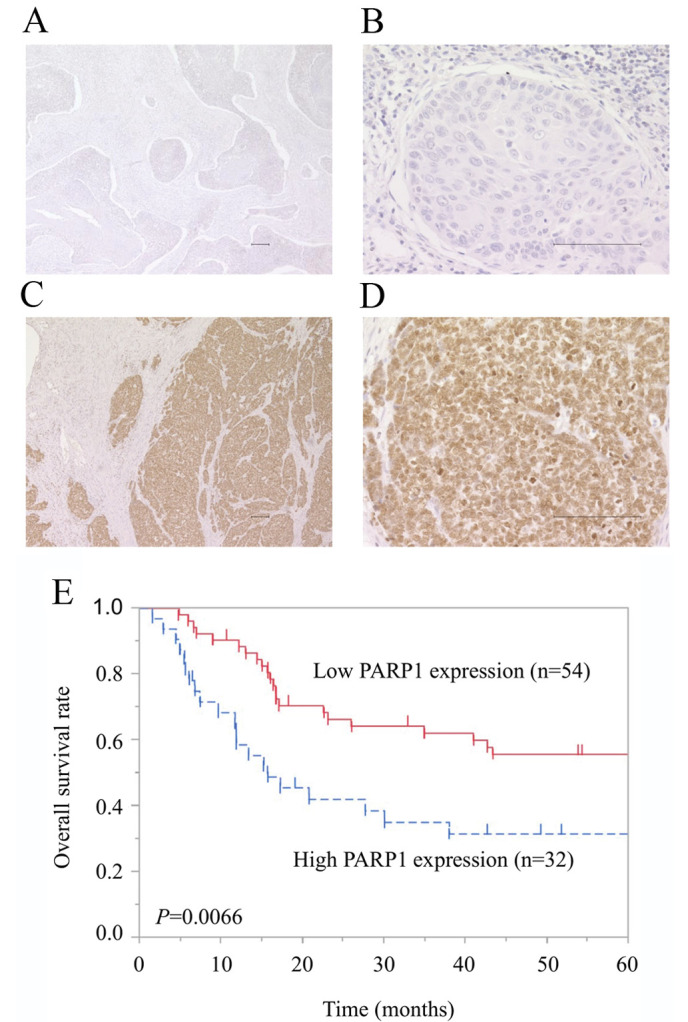
Effect of PARP1 expression on prognosis of patients with esophageal squamous cell carcinoma. (A and B) Low PARP1 expression. (C and D) High PARP1 expression. Immunohistochemical staining of PARP1 in esophageal squamous cell carcinoma is shown at magnifications of (A and C) ×40 and (B and D) ×200. Scale bar, 100 µm. (E) The Kaplan-Meier survival curve indicates that the overall survival of patients with high expression of PARP1 was significantly worse than that of patients with low expression of PARP1 (P=0.0066). PARP, poly (ADP-ribose) polymerase-1.
Table I.
Association between PARP1 expression and clinicopathological features in 86 patients with ESCC.
| PARP1 expression, n | ||||
|---|---|---|---|---|
| Characteristic | Low | High | Total, n | P-value |
| Total | 54 | 32 | 86 | |
| Age, years | 0.7295 | |||
| <65 | 24 | 13 | 37 | |
| ≥65 | 30 | 19 | 49 | |
| Sex | 0.4653 | |||
| Male | 46 | 29 | 75 | |
| Female | 8 | 3 | 11 | |
| Esophageal location | 0.2792 | |||
| Upper | 7 | 7 | 14 | |
| Middle/lower | 47 | 25 | 72 | |
| Histology of SCC | 0.6725 | |||
| Well/moderately differentiated | 40 | 25 | 65 | |
| Poorly differentiated | 14 | 7 | 21 | |
| Venous invasion | 0.8681 | |||
| No | 26 | 16 | 42 | |
| Yes | 28 | 16 | 44 | |
| Lymphatic invasion | 0.6351 | |||
| No | 5 | 4 | 9 | |
| Yes | 49 | 28 | 77 | |
| Depth of tumor invasion | 0.9500 | |||
| pT1-2 | 24 | 14 | 38 | |
| pT3-4 | 30 | 18 | 48 | |
| Lymph node metastasis | 0.3370 | |||
| pN0 | 17 | 7 | 24 | |
| pN1-3 | 37 | 25 | 62 | |
PARP1, poly (ADP-ribose) polymerase-1; ESCC, esophageal squamous cell carcinoma.
Association between prognosis and expression of PARP1
The mean follow-up time for all patients in the present study was 45.3±41.8 months. The group with high PARP1 expression had a significantly worse OS compared with patients with low expression [HR 2.25; 95% confidence interval (CI), 1.23–4.11; P=0.0092; Fig. 1E]. The 5-year OS rate was 31.6% in the high-expression group and 55.7% in the low-expression group. Multivariate Cox regression analysis revealed that high expression of PARP1 was a statistically significant independent prognostic factor of poor OS, along with pT3 and 4 and pN1-3 disease (Table II). The adjusted HR for OS in the group with high PARP1 expression was 2.39 (95% CI, 1.29–4.44; P=0.0051).
Table II.
Univariate and multivariate Cox regression analyses for overall survival in 86 patients with ESCC.
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| Characteristic | HR | 95% CI | P-value | HR | 95% CI | P-value |
| Age, ≥65 vs. <65 years | 1.24 | 0.68–2.36 | 0.4846 | |||
| Sex, male vs. female | 4.02 | 1.23–24.67 | 0.0169 | 3.29 | 0.97–20.55 | 0.0578 |
| Location, middle/lower vs. upper | 1.31 | 0.69–2.41 | 0.3949 | |||
| Histology of SCC, poorly vs. well/moderately differentiated | 1.48 | 0.73–2.82 | 0.2631 | |||
| Venous invasion, positive vs. negative | 1.47 | 0.80–2.71 | 0.2112 | |||
| Lymphatic invasion, positive vs. negative | 2.57 | 0.79–15.75 | 0.1315 | |||
| pT, 3–4 vs. 1–2 | 2.68 | 1.42–5.35 | 0.0021 | 2.10 | 1.06–4.42 | 0.0335 |
| pN, 1–3 vs. 0 | 3.36 | 1.52–8.88 | 0.0018 | 2.66 | 1.14–7.29 | 0.0219 |
| PARP1 expression, high vs. low | 2.25 | 1.23–4.11 | 0.0092 | 2.39 | 1.29–4.44 | 0.0061 |
PARP1, poly (ADP-ribose) polymerase-1; ESCC, esophageal squamous cell carcinoma; HR, hazard ratio; CI, confidence interval.
PARP1 mRNA expression in ESCC cell lines
RT-qPCR analysis was performed to compare the mRNA expression of PARP1 in the ESCC TE1, TE4, TE5, TE6, TE8, TE9, TE10 and TE11 cell lines. The expression of PARP1 mRNA was the highest in TE9 cells and the second highest in TE6 cells (Fig. 2A). Accordingly, TE6 and TE9 cells were selected for subsequent analysis in a PARP1 inhibition assay. RT-qPCR and western blot analysis showed that siPARP1 significantly reduced the expression of PARP1 mRNA and protein compared with negative control cells (Fig. 2B).
Figure 2.

Suppression of PARP1 inhibited ESCC cell growth. (A) RT-qPCR analysis of PAPR1 mRNA expression in the ESCC TE1, TE4, TE5, TE6, TE8, TE9, TE10 and TE11 cell lines. The expression of PARP1 mRNA was the highest in TE9 cells and the second highest in TE6 cells. (B) RT-qPCR and western blot analysis showed that siPARP1 significantly reduced the expression of PARP1 mRNA and protein compared with the NC in TE6 and TE9 cells. (C) Cell proliferation assay of ESCC cells (TE6 and TE9) comparing NC and KD of PARP1. ESCC cells transfected with siPARP1 showed significantly decreased proliferation compared with NC (at 24, 48 and 72 h). For all experiments, n=5, and KD was compared with NC at each time-point (0, 24, 48, and 72 h). *P<0.05. PARP, poly (ADP-ribose) polymerase-1; ESCC, esophageal squamous cell carcinoma; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; siPARP1, small interfering RNA against PARP1; NC, negative control; KD, knock-down.
Inhibition of PARP1 reduces the proliferative activity of ESCC cell lines
The effects of PARP1 inhibition on the proliferative activity of ESCC TE6 and TE9 cell lines were examined. siPARP1 significantly inhibited cell growth compared with the negative control siRNA at 24, 48 and 72 h after transfection (Fig. 2C).
Inhibition of PARP1 induces cell cycle arrest in ESCC cell lines
Subsequently, the effect of siPARP1 on the cell cycle in ESCC TE6 and TE9 cells was examined with flow cytometry. siPARP1 significantly increased the ratio of cells in the G2/M phase compared with negative control siRNA (TE6, P<0.01; TE9, P<0.001) and significantly decreased the ratio of cells in the G0/G1 phase (TE6, P<0.001; TE9, P<0.01) (Fig. 3A). These results indicate that siPARP1 affected the G2/M checkpoint. Fig. 3B shows the schema of the G2/M checkpoint. siPARP1-induced G2/M arrest by was investigated by western blotting with monoclonal antibodies specific for several key regulators, consisting of Chk2, phosphorylated (p-)Chk2 (Thr68), cdc25c, p-cdc25c (Thr48), cdc2, p-cdc2 (Tyr15) and cyclin B1. Cells treated with siPARP1 showed a notable decrease in p-Chk2 (Thr68) and p-cdc25c (Thr48) expression, and a notable increase in p-cdc2 (Tyr15) expression compared with parental and negative control cells (Fig. 3C).
Figure 3.

Inhibition of PARP1-induced cell cycle arrest at the G2/M checkpoint. (A) Cell cycle analysis of TE6 and TE9 cells transfected with siPARP1 (n=3, cells transfected with siPARP1 were compared with the NC for each cell cycle). Compared with NC, siPARP1 significantly increased the ratio of cells in the G2/M phase and significantly decreased the ratio of cells in the G0/G1 phase. (B) Schema of ATM-Chk2-cdc25c pathways and cdc2/cyclin B1. (C) Western blot analysis showed that inhibition of PARP1 induced cell cycle arrest at the G2/M checkpoint through the ATM-Chk2-cdc25c pathway. PARP, poly (ADP-ribose) polymerase-1; siPARP1, small interfering RNA against PARP1; NC, negative control; KD, knock-down; cdc, cell division control; ATM, ataxia telangiectasia mutated; Chk2, checkpoint kinase 2.
Discussion
In the present study, IHC staining was used to demonstrate that the expression level of PARP1 was useful in predicting clinical outcomes in patients with ESCC. Furthermore, it was demonstrated that inhibition of PARP1 with siPARP1 reduced the proliferative activity of ESCC cells, and the effect of PARP1 inhibition induced cell cycle arrest at the G2/M phase. At present, although various anticancer drugs have been widely used in patients with ESCC, molecular targeted drugs have not been established as the treatment for patients with ESCC. The present study also suggests that new molecular targeted treatment for ESCC using PARP inhibitors may have potential in a clinical setting.
Previous studies have reported that PARP1 plays a critical role in responding to DNA damage by activating DNA repair pathways responsible for cellular survival (11,19). In breast and ovarian cancers in particular, PARP inhibitors are the most recent treatment and are highly regarded (12). Additionally, previous studies have indicated an association between PARP1 and the cell cycle. Jelinic and Levine (16) reported that PARP inhibitors did not affect homology-directed DNA damage, but did affect cell cycle arrest at the G2 phase in a human osteosarcoma cell line. Park et al (7) reported that PARP1 inhibition significantly attenuated growth and colony formation, and induced G2/M arrest in gastric cancer cells. Overall, it was hypothesized that PARP1 inhibition suppressed proliferation and regulated the cell cycle at the G2/M checkpoint in ESCC. The present study supported this hypothesis by analyzing experimental data from proliferation and cell cycle assays. Flow cytometry showed that PARP1 inhibition induced cell cycle arrest at the G2/M phase. By contrast, no significant difference in apoptosis was observed between the negative control group and the siPARP1-treated group (data not shown). These results were supported by a previous study (7).
In addition, western blotting was used to examine the detailed mechanisms of G2/M arrest induced by PARP1 inhibition. This analysis showed that PARP1 inhibition inhibited the phosphorylation of Chk2 and cdc25c, the latter of which is responsible for removal of phosphates at Thr14 and Tyr15 and the subsequent activation of cdc2 (20,21). Therefore, these results revealed that siPARP1 induced cell cycle arrest at the G2/M phase through the ataxia telangiectasia mutated (ATM)-Chk2-cdc25c pathway, suggesting that PARP1 may interact with the ATM-Chk2 pathway. PARP1 inhibition has potential in ESCC therapy by acting via the induction of cell cycle arrest at the G2/M phase, through the ATM-Chk2-cdc25c pathway.
There are several limitations to the present study. One is the relatively small number of tissue samples from patients with ESCC, thus restricting the IHC analysis. Additional multicenter studies involving more patients are required. Another limitation is that the present study was conducted strictly in vitro. Additional studies focusing on the effect of PARP inhibition in vivo are required to investigate the potential clinical application of the present findings in patients with ESCC.
In conclusion, the present IHC analysis showed that PARP1 may be an independent prognostic marker in ESCC, and experiments using ESCC cells demonstrated that PARP1 inhibition could induce cell cycle arrest at the G2/M phase through the ATM-Chk2-cdc25c pathway. With respect to personalized treatments, PARP inhibitors may be of use in patients with ESCC that show high PARP1 expression in the future.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura M, Iwahashi M, Nakamori M, Ojima T, Katsuda M, Iida T, Hayata K, Kato T, Yamaue H. New prognostic score for the survival of patients with esophageal squamous cell carcinoma. Surg Today. 2014;44:875–883. doi: 10.1007/s00595-013-0628-z. [DOI] [PubMed] [Google Scholar]
- 3.Okumura H, Uchikado Y, Setoyama T, Matsumoto M, Owaki T, Ishigami S, Natsugoe S. Biomarkers for predicting the response of esophageal squamous cell carcinoma to neoadjuvant chemoradiation therapy. Surg Today. 2014;44:421–428. doi: 10.1007/s00595-013-0580-y. [DOI] [PubMed] [Google Scholar]
- 4.Borghesi S, Hawkins MA, Tait D. Oesophagectomy after definitive chemoradiation in patients with locally advanced oesophageal cancer. Clin Oncol (R Coll radiol) 2008;20:221–226. doi: 10.1016/j.clon.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Makino T, Yamasaki M, Miyata H, Tanaka K, Takahashi T, Kurokawa Y, Nakajima K, Takiguchi S, Mori M, Doki Y. Solitary lymph node recurrence of esophageal squamous cell carcinoma: Surgical failure or systemic disease? Ann Surg Oncol. 2016;23:2087–2093. doi: 10.1245/s10434-015-5086-y. [DOI] [PubMed] [Google Scholar]
- 6.Alexander BM, Wang XZ, Niemierko A, Weaver DT, Mak RH, Roof KS, Fidias P, Wain J, Choi NC. DNA repair biomarkers predict response to neoadjuvant chemoradiotherapy in esophageal cancer. Int J tadiat Oncol Biol Phys. 2012;83:164–171. doi: 10.1016/j.ijrobp.2011.05.033. [DOI] [PubMed] [Google Scholar]
- 7.Park SH, Jang KY, Kim MJ, Yoon S, Jo Y, Kwon SM, Kim KM, Kwon KS, Kim CY, Woo HG. Tumor suppressive effect of PARP1 and FOXO3A in gastric cancers and its clinical implications. Oncotarget. 2015;6:44819–44831. doi: 10.18632/oncotarget.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 9.Juarez-Salinas H, Sims JL, Jacobson MK. Poly(ADP-ribose) levels in carcinogen-treated cells. Nature. 1979;282:740–741. doi: 10.1038/282740a0. [DOI] [PubMed] [Google Scholar]
- 10.Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283:593–596. doi: 10.1038/283593a0. [DOI] [PubMed] [Google Scholar]
- 11.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoeijmakers JH. DNA damage, aging and cancer. N Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 13.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 14.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs; Proc Natl Acad Sci USA; 2008; pp. 17079–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 16.Jelinic P, Levine DA. New insights into PARP inhibitors' effect on cell cycle and homology-directed DNA damage repair. Mol Cancer Ther. 2014;13:1645–1654. doi: 10.1158/1535-7163.MCT-13-0906-T. [DOI] [PubMed] [Google Scholar]
- 17.Sakogawa K, Aoki Y, Misumi K, Hamai Y, Emi M, Hihara J, Shi L, Kono K, Horikoshi Y, Sun J, et al. Involvement of homologous recombination in the synergism between cisplatin and poly (ADP-ribose) polymerase inhibition. Cancer Sci. 2013;104:1593–1599. doi: 10.1111/cas.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobin LH, Compton CC. TNM seventh edition: what's new, what's changed: Communication from the international union against cancer and the American Joint committee on cancer. Cancer. 2010;116:5336–5339. doi: 10.1002/cncr.25537. [DOI] [PubMed] [Google Scholar]
- 19.Gradwohl G, Ménissier de Murcia JM, Molinete M, Simonin F, Koken M, Hoeijmakers JH, de Murcia G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA; Proc Natl Acad Sci USA; 1990; pp. 2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atherton-Fessler S, Liu F, Gabrielli B, Lee MS, Peng CY, Piwnica-Worms H. Cell cycle regulation of the p34cdc2 inhibitory kinases. Mol Biol Cell. 1994;5:989–1001. doi: 10.1091/mbc.5.9.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter T. Protein kinases and phosphatases: The yin and yang of protein phosphorylationand signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]