

Abstract
Non-small cell lung cancer etiology and its treatment failure are due to epidermal growth factor receptor (EGFR) kinase domain mutations at amino acid position 790. The mutational change from threonine to methionine at position 790 (T790M) is responsible for tyrosine kinase inhibition failure. Using molecular dynamic simulation, the present study investigated the architectural changes occurring at the atomic scale. The 50-nsec runs using a GROMOS force field for wild-type and mutant EGFR's kinase domains were investigated for contrasting variations using Gromacs inbuilt tools. The adenosine triphosphate binding domain and the active site of EGFR were studied extensively in order to understand the structural changes. All the parameters investigated in the present study revealed considerable changes in the studied structures, and the knowledge gained from this may be used to develop novel kinase inhibitors that will be effective irrespective of the structural alterations in kinase domain.
Keywords: non-small cell lung cancer, epidermal growth factor receptor, mutation, T790M, molecular dynamics simulation
Introduction
Lung cancer is the most commonly diagnosed type of cancer, with 1.82 million new cases diagnosed in 2012 and 1.6 million associated mortalities annually, which makes it the most common cause of cancer-associated mortality worldwide (1). Lung cancer has two primary types, small-cell lung carcinoma and non-small cell lung carcinoma (NSCLC), with NSCLC being more prevalent (2). The 5-year survival rate following initial diagnoses of patients with NSCLC is only 5–15%, which means it is the most lethal type of cancer (3). The treatment of NSCLC was linked with targeting the epidermal growth factor receptor (EGFR) by Wrann et al (4) over four decades ago. Inhibition of the EGFR signaling pathway by kinase inhibitors and monoclonal antibodies is a viable option for therapy of NSCLC (5–7).
The kinase inhibitor response to patients with NSCLC is linked to mutations in the EGFR protein, and the mutations of EGFR are classified into activating mutations and second mutations; the latter are associated with lung cancer and the former mutations cause drug resistance (8–10). The efficacy of kinase inhibitors is associated with the mutational change from threonine to methionine at amino acid position 790 (T790M); this mutation is associated with disease resistance by sterically blocking kinase inhibitors, including gefitinib and erlotinib (11–13). The location of this threonine at position 790 acts as a gatekeeper, as it is located at the entrance of a hydrophobic groove at the back of the adenosine triphosphate binding pocket (14).
Atomic insight into altered architecture due to mutations using molecular dynamic simulation (MDS) is a practice currently in use (15,16). The present study used MDS to investigate the anomalies in the gatekeeper region due to mutation from threonine to methionine. The X-ray crystallographic structure of the wild-type EGFR kinase domain with PDB ID no. 2GS2 (17) was selected for the MDS analysis. This structure and the architecture of its mutated forms were analyzed using Gromacs inbuilt tools. In order to understand the effect of mutation on the flexibility of the two structures, principle component analysis and free energy landscape analysis were performed.
Materials and methods
Protein preparation
The crystallographic structure of the tyrosine kinase domain of EGFR was retrieved from the Research Collaboratory for Structural Bioinformatics protein data bank (http://www.rcsb.org/pdb/home/home.do) and the structure with PDB ID 2GS2 (17) was used in the present study. The structure was energy minimized prior to and following insertion of mutations using the Swiss Protein Data Bank viewer (18). A total of two structures were generated; the first was the wild-type EGFR tyrosine kinase domain and the second was the tyrosine kinase domain with T790M drug-resistant mutation.
Molecular dynamics simulation
Gromacs version 4.6.6 package (19) was developed for analysis of the bimolecular systems of proteins, DNA and lipids in order to investigate the architecture of the tyrosine kinase domain of EGFR. All three systems were analyzed under a GROMOS96 43a1 force field (20). The EGFR tyrosine kinase domain was placed in a rectangular box of 15 Å marginal radius and the protein domain under investigation was placed in the center. Subsequently, the box was filled with water using the TIP3P model (21) and the system was made neutral using the Genion tool of the Gromacs package. The two systems generated were subjected to a force of 100 kcal/mol for 5,000 steps, during which the solvent molecules were relaxed and the solutes were restrained to their original position. In order to regulate the temperature inside the system, the Berendsen temperature coupling method (22) was used and the system was maintained under 1 atm pressure with allowed compressibility ranging from 4.5×10−5 atm. The system was energy minimized twice prior to position restraint simulation for 5 nsec. Following this step, the system was subjected to a 50 nsec MDS run and the results were saved following every 2 psec. In order to evaluate the alterations in architecture of the tyrosine kinase domain of EGFR protein tools, g_rms, g_rmsf, g_sas, g_hbond, g_gyrate, g_rama, g_rmsdist, g-sham and do_dssp were used. The results were visualized using Pymol (Schrödinger, Inc., New York, NY, USA) (23) and VMD (University of Illinois at Urbana-Champaign, Champaign, IL, USA) (24), and the graphs were plotted using the Grace GUI toolkit version 5.1.19 (Oregon Graduate Institute of Science and Technology, Hillsboro, OR, USA) (25).
Results
General structural changes in Kinase domain of EGFR by T790M
In order to investigate the effect of T790M mutations on the tyrosine kinase domain, the present study used the X-ray crystallographic method and the structures were subjected to MDS. The results were analyzed to investigate the anomaly in the architecture of the kinase domain. The protein structure and its function are interlinked and any change in the protein structure affects its function. In the case of EGFR, the mutations that were analyzed in the present study were responsible for the drug failure.
The pictorial representation of MDS of the two structures under observation are presented in Fig. 1; the structures under observation were stable throughout the process. The moviemaker from Pymol viewer suite was used to make the movie of the three runs with each frame of the movie retrieved following 100 psec (the movies are available on request). The basic visualization demonstrated the change in the area of the domain.
Figure 1.

Pictorial representations of the molecular dynamic simulation run of the two structures from 0–50 nsec.
In order to investigate the intricate details of the tyrosine kinase domain architecture alterations due to the mutations analyzed, the present study used various Gromacs inbuilt tools. g_rmsf was used to evaluate the root mean square fluctuation of the residues of the tyrosine kinase domain. The results are presented in Fig. 2A. T790M mutation from rmsf analysis demonstrated a distinct pattern compared with the wild-type, and the residues of importance are marked in Fig. 2A. The g_rmsf tool was also used to determine the b-factor to a file with the average coordinates; the results are presented in Fig. 2B. The spectrum ranges from blue to white to red, indicating the fluctuation in the region, with blue being the least and red being the most flexible. The mutant domain revealed the most fluctuation and the wild-type domain demonstrated the least.
Figure 2.

(A) RMSF of the EGFR wild-type (black) and mutant (red). The important residues of ATP binding and active site are marked. (B) The b-factor implicated on the average structures of the wild-type and mutant EGFR, clearly indicating the mutant structure to be more fluctuating in nature. RMSF, root mean square fluctuation; ATP, adenosine triphosphate; EGFR, epidermal growth factor receptor.
Root mean square deviation (RMSD) was evaluated using the g_rms tool, and the tool was used to compare the structures by evaluating their RMSD values. The mutant structure revealed the maximum deviation, as presented in Fig. 3A. g_rmsdist determined the RMSD of atom distances; the wild-type domain of the structure was the most stable and the mutant was the most unstable, as presented in Fig. 3B.
Figure 3.

(A) RMSD of the EGFR wild-type (black) and mutant (white), which demonstrated that the wild-type attained stability after 10 nsec and the mutant structure revealed fluctuation throughout the run. (B) The RMS distribution peak clearly demonstrated that the wild-type (black) was more stable in terms of RMS and a.u., whereas the mutant was highly unstable. RMSD, root-mean-square deviation; a.u., atomic unit.
The g_gyrate tool was used to determine the radius of gyration of the atoms in the three structures under observation, and the mutant structure revealed the maximum deviation. The mutant domain was demonstrated to decrease in the radius of gyration following a 25-nsec run (Fig. 4), indicating the decrease in size. The same trend was observed when total solvent accessible surface area (SASA) was evaluated using the g_sas tool. SASA results (Fig. 5) revealed that the mutation had a contrasting effect on the tyrosine kinase domain of EGFR, with mutation decreasing the size of the domain, hence a smaller SASA.
Figure 4.
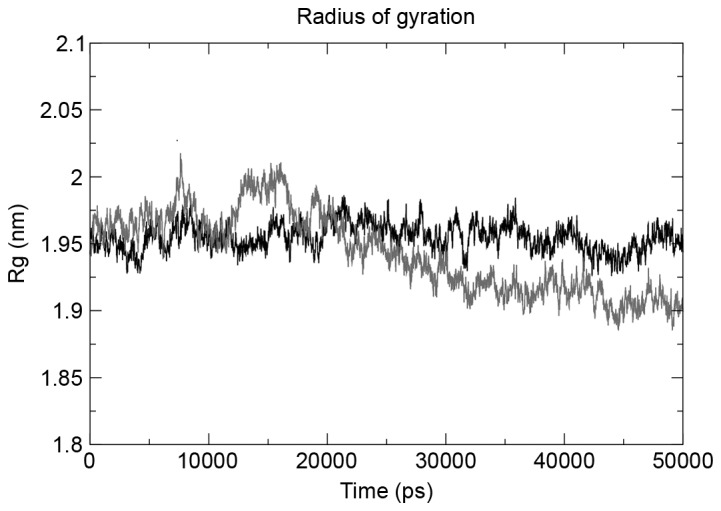
Radius of gyration plot of the two structures for a 50-nsec run demonstrated the mutant structure decreased its size following a 20-nsec run and continued to decrease until 50 nsec. Rg, radius of gyration.
Figure 5.
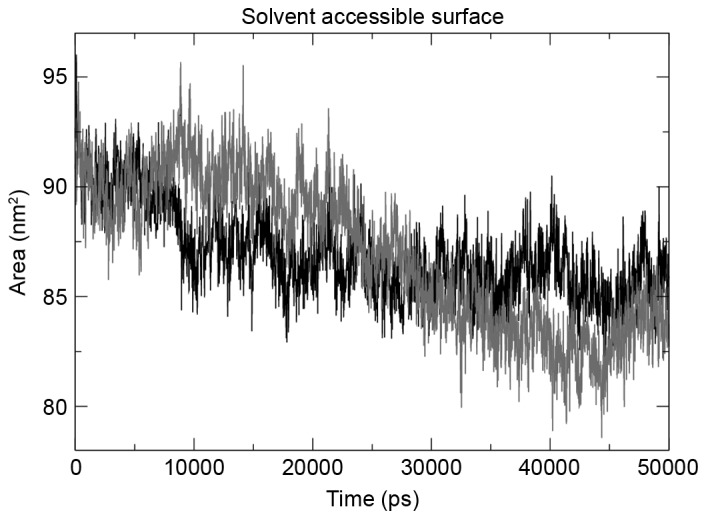
Solvent accessible surface area of the mutant structure revealed a decrease compared with the wild-type. The decrease was observed following a 30-nsec run.
Intra- and interhydrogen bond pattern analysis
The intrahydrogen bond pattern of the two structures under observation was investigated using the g_hbond tool of the Gromacs package. The wild-type structure formed a mean of 203 hydrogen bonds per time frame out of a possible 164,227, and the mutant formed a mean of 201 hydrogen bonds per time frame out of possible 166,173. Fig. 6A presents a pictorial representation of the mean number of hydrogen bonds per time frame within the domains. Fig. 6B presents the hydrogen bond pattern between the protein and water; the results showed 519 and 520 bonds for the wild-type and mutant structures, respectively.
Figure 6.

(A) Intra protein hydrogen bond pattern of wild-type (and mutant revealed that the mutant had a smaller number throughout the run. The mean number of hydrogen bonds for the mutant was 201 bonds per frame and for the wild-type was 203 bonds per frame. (B) This figure presents the protein-water hydrogen bond pattern of the two structures under observation.
Secondary structure analysis
The secondary structure architecture of the protein was investigated for alterations in the layout of the domain in general. The average structure of the EGFR tyrosine kinase domain was retrieved from the two simulations, and the structures were analyzed for secondary structure architecture layout. Fig. 7 presents the three secondary structure layouts, where clear alterations can be observed in the mutant layouts when compared with the wild-type. To investigate the secondary structure changes over time, the do_dssp tool was used and the structures were retrieved following every 100 psec; the results are presented in Fig. 8.
Figure 7.

Average secondary structure layout of the simulated structures following a 50-nsec run. The mutant structure demonstrated a considerable alteration in its secondary structure architecture.
Figure 8.
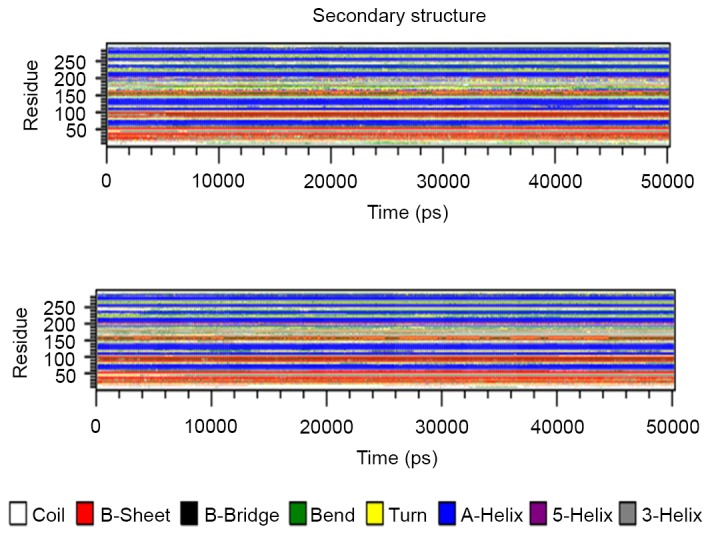
Secondary structure layout of the two structures generated using do_dssp. The structures for the analysis were captured every 100 psec.
Discussion
Following mutation there is an alteration in amino acids and their basic features of size, charge and hydrophobicity value. These changes induce protein architectural alterations, which in turn affect its function, as reported by Chikan et al (15). In the present study, the change from threonine to methionine also induced these alterations; methionine is a larger residue with a greater hydrophobic property compared with wild-type threonine. Due to these changes, the mutant structure demonstrated considerable alterations in the secondary structure architecture, which revealed its effect on EGFR kinase domain size and its total polar solubility. Each of these properties demonstrated a reduced value compared with that of the wild-type structure.
The threonine at position 790 was conserved, but a few other residue types were observed at this position, including methionine. This means that homologous proteins exist with the same residue type as the mutant at this position, and this mutation is possibly not damaging to the protein. The threonine also forms hydrogen bonds with arginine at position 776; with the alteration in properties of the residue this intrahydrogen bond formation is lost, which induces the variation of intrahydrogen bond pattern in the core of the kinase domain, a change that may have an effect on tyrosine kinase inhibitor (TKI) failure. Threonine 790, the ‘gatekeeper’ residue in EGFR, has previously been suggested to cause resistance by sterically blocking the binding of known TKIs and is sensitive to structurally similar irreversible inhibitors (11). The data from the present study suggested that the intrahydrogen bond pattern in the core of the kinase domain can be a factor triggering selective inhibition of EGFR by TKIs.
The structural insight obtained regarding the kinase domain of EGFR due to T790M mutation may pave the way for novel TKI developments. Previous studies have used computational approaches to look into novel TKI development; the approach has been used to propose lead compounds that exhibit more efficient binding with mutated EGFR (26,27). Zhao et al (26) reported on T790M and L858R mutations in EGFR and their results are in accordance with the findings from the present study. The present study proposes that changing the intrahydrogen bond pattern in the core of the kinase domain serves as a base for structure-based drug sensitivity in EGFR. This may provide a basis on which novel TKI development can be achieved, where the stable mutant structure from the simulation trajectory can act as a template and novel TKI can be screened over it. The possible TKI developed using the structure-based drug designing method may inhibit the kinase activity without any effect from drug resistant mutations (26,27).
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Longo D, Fauci A, Kasper D, Hauser S, Jameson J, Loscalzo J. Harrison's Principles of Internal Medicine. 18th. McGraw-Hill Professional; New York, NY: 2011. [Google Scholar]
- 3.Dacic S, Flanagan M, Cieply K, Ramalingam S, Luketich J, Belani C, Yousem SA. Significance of EGFR protein expression and gene amplification in non-small cell lung carcinoma. Am J Clin Pathol. 2006;125:860–865. doi: 10.1309/H5UW6CPCWWC92241. [DOI] [PubMed] [Google Scholar]
- 4.Wrann MM, Fox CF. Identification of epidermal growth factor receptors in a hyperproducing human epidermoid carcinoma cell line. J Biol Chem. 1979;254:8083–8086. [PubMed] [Google Scholar]
- 5.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 6.Prewett M, Rockwell P, Rockwell RF, Giorgio NA, Mendelsohn J, Scher HI, Goldstein NI. The biologic effects of C225, a chimeric monoclonal antibody to the EGFR, on human prostate carcinoma. J Immunother Emphasis Tumor Immunol. 1996;19:419–427. doi: 10.1097/00002371-199611000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Kris MG, Natale RB, Herbst RS, Lynch TJ, Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: A randomized trial. JAMA. 2003;290:2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 8.Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 9.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 10.Johnson BE, Jänne PA. Epidermal growth factor receptor mutations in patients with non-small cell lung cancer. Cancer Res. 2005;65:7525–7529. doi: 10.1158/0008-5472.CAN-05-1257. [DOI] [PubMed] [Google Scholar]
- 11.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP; Proc Natl Acad Sci USA; 2008; pp. 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 14.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib; Proc Natl Acad Sci USA; 2005; pp. 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chikan N, Bukhari S, Shabir N, Amin A, Shafi S, Qadri RA, Patel TN. Atomic insight into the altered O6-Methylguanine-DNA methyltransferase protein architecture in gastric cancer. PLoS One. 2015;10:e0127741. doi: 10.1371/journal.pone.0127741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bukhari S, Mokhdomi TA, Chikan NA, Amin A, Qazi H, Wani SH, Wafai AH, Tyub S, Mustafa F, Mir MS, et al. Affinity proteomics led identification of vimentin as a potential biomarker in colon cancers: Insights from serological screening and computational modelling. Mol Biosyst. 2015;11:159–169. doi: 10.1039/C4MB00506F. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 19.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 20.Schuler LD, Daura X, Van Gunsteren WF. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J Comput Chem. 2001;22:1205–1218. doi: 10.1002/jcc.1078. [DOI] [Google Scholar]
- 21.Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298K. J Phy Chem. 2001;105:9954–9960. doi: 10.1021/jp003020w. [DOI] [Google Scholar]
- 22.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem phys. 1984;81:3684. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 23.DeLano WL. http://www.pymol.org/ [May 30;2017 ];The PyMOL molecular graphics system. 2002
- 24.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27-28. [DOI] [PubMed] [Google Scholar]
- 25.Turner PJ. XMGRACE, Version 5.1.19. Center for Coastal and Land-Margin Research. Oregon Graduate Institute of Science and Technology; Beaverton, OR: 2005. [Google Scholar]
- 26.Zhao FL, Yang GH, Xiang S, Gao DD, Zeng C. In silico analysis of the effect of mutation on epidermal growth factor receptor in non-small-cell lung carcinoma: From mutational analysis to drug designing. J Biomol Struct Dyn. 2017;35:427–434. doi: 10.1080/07391102.2016.1146165. [DOI] [PubMed] [Google Scholar]
- 27.Doss GP, Rajith B, Chakraborty C, NagaSundaram N, Ali SK, Zhu H. Structural signature of the G719S-T790M double mutation in the EGFR kinase domain and its response to inhibitors. Sci Rep. 2014;4:5868. doi: 10.1038/srep05868. [DOI] [PMC free article] [PubMed] [Google Scholar]