

Abstract
Next generation sequencing technologies have recently been applied to characterize mutational spectra of the heterogeneous population of viral genotypes (known as a quasispecies) within HIV-infected patients. Such information is clinically relevant because minority genetic subpopulations of HIV within patients enable viral escape from selection pressures such as the immune response and antiretroviral therapy. However, methods for quasispecies sequence reconstruction from next generation sequencing reads are not yet widely used and remains an emerging area of research. Furthermore, the majority of research methodology in HIV has focused on 454 sequencing, while many next-generation sequencing platforms used in practice are limited to shorter read lengths relative to 454 sequencing. Little work has been done in determining how best to address the read length limitations of other platforms.
The approach described here incorporates graph representations of both read differences and read overlap to conservatively determine the regions of the sequence with sufficient variability to separate quasispecies sequences. Within these tractable regions of quasispecies inference, we use constraint programming to solve for an optimal quasispecies subsequence determination via vertex coloring of the conflict graph, a representation which also lends itself to data with non-contiguous reads such as paired-end sequencing. We demonstrate the utility of the method by applying it to simulations based on actual intra-patient clonal HIV-1 sequencing data.
1. Introduction
Viral sequencing has played an important role in both the study of HIV and the treatment of HIV patients. Viral genotyping using Sanger sequencing is now integrated into HIV patient care in the developed world and aids the determination of treatment regimens for new patients as well as patients failing treatment [1–3]. Global databases of HIV sequences have been established [4–6] and serve to monitor the global diversity of viral genotypes, provide a reference for sequence interpretation, and catalyze discoveries related to sequence evolution and genotype-phenotype associations. In our own work we have used these databases to study the impact of different genetic backgrounds on the evolution of HIV-1 drug resistance [7–9]. As such, the field of HIV drug resistance serves as a model for the application of sequencing technologies in clinical practice.
However, standard sequencing methodologies are limited in their ability to characterize the viral population within an infected individual. The high rate of turnover and error-prone genome replication process of HIV leads to an intra-patient viral population that consists of genetically distinct subpopulations [10–14]. Thus the viral population is often described as a quasispecies [15]. Viral populations that circulate at low levels, termed minor variants, are clinically relevant because drug-resistant subpopulations may be selected upon treatment [16–18]. These minor variants are often undetectable using standard sequencing protocols. Specialized techniques such as clonal and single genome sequencing (SGS) have been developed to obtain sequences of minor variant subpopulations within patients [19–22]. SGS, in particular, was designed to address and minimize sequencing artifacts such as recombination of quasispecies sequences during PCR [19]. However, these methods require significant expertise and investment.
Next Generation Sequencing (NGS) is a term applied to a variety of recent sequencing platforms which sequence samples at high depth of coverage at relatively low monetary and training cost. The depth of coverage allows for the detection of minority variant subpopulations with prevalences < 20% [19]. Its application to HIV has been an active area of research in recent years [18,23–30]. Nevertheless, in most research settings, NGS data have primarily been applied in a restricted manner - reads are mapped to an HIV-1 reference genome and the mutational composition of each sequence position is examined independently. Determination of mutational linkage and the reconstruction of individual quasispecies sequences are active areas of research [31–36]. Analysis tools to address these problems will aid in our ability to understand and interpret the vast amounts of data produced when applying these new sequencing technologies to HIV.
1.1. Quasispecies sequence reconstruction
Any two virus particles with differing genomes may be considered as originating from genotypically distinct subpopulations. However, this view of quasispecies subpopulations is not especially useful in practice since it is relatively uncommon for viral sequencing to examine full genomes.
More commonly, viral genotypes are defined in terms of clinically-relevant regions or positions of the sequence. Studies of HIV drug resistance typically focus on pol gene sequences. One may further narrow the sequence positions under consideration depending on the question of interest. For example, studies of resistance to first-line regimens may focus on the reverse transcriptase sequence. In clinical settings, genotypes may be defined with respect to known drug resistance mutation positions.
The quasispecies reconstruction problem is to construct a representation of the sequences underlying subpopulation sequences from the partial information provided by sequencing reads. In many cases, the relative population frequencies of each sequence are also inferred. Reads represent sampled sequence fragments of one of the underlying quasispecies sequences. Quasispecies reconstruction is closely related to haplotype assembly - the construction of haplotypes of a diploid organism from sequencing read fragments. Several approaches to quasispecies reconstruction have been proposed thus far [31–36], each of which has introduced mathematical representations which have provided new insights on the problem. Related problems have also arisen in genome assembly [37–43] and metagenomics [44]. The bipartition approach to haplotype assembly [45,46] served as an inspiration for the approach taken in this paper.
Due to the complex error profile of next generation sequencing, error correction is a chief concern. Errors and biases arise for a multitude of reasons, including recombination during PCR, selection of primers, and sequence-specific errors [47–49]. Successful quasispecies reconstruction requires robust error correction in addition to reconstruction algorithms. Attempts to address these errors include both analysis approaches [23,48] as well as tagging protocols [30,50,51].
Although robust error correction procedures are an important step in applying any method in practice, the focus of the current study is primarily to present a new approach to representation and quasispecies reconstruction. The approach is influenced by practical motivations – conservative reconstruction in the presence of shorter reads and the need for representations which encompass non-contiguous reads. Non-contiguous reads include paired-end sequencing and some third generation sequencing technologies [52]. While the majority of next-generation sequencing research in the HIV field has focused on Roche 454 Sequencing, Illumina sequencing platforms are more widely available. One challenge in interpreting Illumina reads is that reads are generally shorter – ranging from 35 bp contiguous reads to 2 × 150 bp paired-end reads in most cases (with some newer platforms capable of up to 2 × 250 bp). By contrast, 454 sequencing reads can obtain contiguous reads ranging from 400 bp to 800 bp reads. With shorter reads, there may be conserved regions of the genome which are not spanned by any read regardless of the depth of coverage. A objective of our approach, is to conservatively determine where subpopulation sequence distinctions are well defined by the data and to reconstruct subsequences if reconstruction on the larger sequence is indeterminate and likely to introduce false recombinants. Our approach is to use a representation which reflects the relative tractability of different regions of the sequence and focus on reconstructions where there are data to support it. We refer to the method as QColors since it uses a graph coloring representation to perform the reconstruction of quasispecies subsequences from NGS reads.
2. Method
As suggested in the introduction, we adopt a different problem formulation than previous approaches to quasispecies reconstruction:
QUASISPECIES SUBSEQUENCE RECONSTRUCTION PROBLEM. Given a set of reads r1, …, rn, find subsets of reads where quasispecies sequence distinctions can be inferred while minimizing the introduction of false recombinants. Within these read subsets, partition reads into a parsimonious generating set of subsequences.
Our approach was motivated by the problem that short read lengths can render full-length reconstructions indeterminate and a method which produces too many false recombinants will not be useful for scientists. Furthermore, to our knowledge, there have not been representations of quasispecies reconstruction which allow for non-contiguous reads. A summary of our approach to this problem is outlined in Algorithm 1. The algorithm and simulations were implemented in C++ in conjunction with the Gecode constraint programming library [53]. Details of the method are described in the following sections.
Algorithm 1.
QColors.
| Map reads to the reference sequence |
| Construct conflict Gc = (V, Ec) and overlap Go = (V, Eo) graphs |
| Determine connected subgraphs of Go and Gc using a |
| Depth-first traversal |
| for each connected subgraph in Go do |
| for each connected subgraph in Gc do |
| Define the neighborhood conflict graph, G(V′, E′) |
| with and |
| Find a homomorphic reduction G(V′, E′) → H |
| Find maximal cliques of H |
| Solve for the optimal coloring (QS sequence assignment) of H with clique and pairwise constraints |
| end for |
| end for |
2.1. Read mapping
Reconstruction of quasispecies sequences differs from more generic metagenomics problem formulations [44] since a well-defined reference genome for all sequence fragments is available. A scanning analysis of the HXB2 HIV-1 reference genome [54] shows that 95% of reads can be uniquely mapped to the full genome using k-mers as short as 10 bp. Since mapping is generally not a significant source of error for pol due to the size of the gene sequence, mapping is assumed to be unambiguous for the purpose of these simulations.
With sufficiently high coverage depth, the identity of sequence characters which exhibit no variation across the quasispecies sequences can be easily obtained directly from the mapped reads. Thus the remaining problem is to assign reads with sequence variation to an appropriate quasispecies sequence.
2.2. Definitions of the overlap graph and the conflict graph
We can represent relationships between reads as two complementary graphs – the overlap graph and the conflict graph.
A read conflict occurs when two reads have inconsistent sequences within an overlapping region. The conflict graph is defined as Gc = (V, Ec) consisting of vertices, V, representing reads and edges, Ec, with pairs of vertices connected by an edge eij iff reads represented by vertices vi and vj overlap and conflict.
The overlap graph is defined as Go = (V, Eo) consisting of the same set of vertices V (again representing reads) and edges Eo which represent consistent (non-conflicting) relationships between overlapping reads. Pairs of vertices, vi and vj are connected by an edge eij iff reads represented by the vertices sufficiently overlap and do not conflict. An input parameter is used to determine the minimum number of overlapped positions between two reads for an edge to exist. This input parameter affects the conservativeness of the reconstruction procedure, and should be as high as the sequencing parameters (coverage, insert sizes) allow.
Unlike the overlap graph, there is no minimum number of overlapped positions required for a conflict graph edge because a conflict invalidates the possibility that two reads originate from the same quasispecies sequence, while agreement between reads does not prove that they originate from the same quasispecies sequence. Although the objective here is to describe the reconstruction approach rather than error correction strategies, for experimentally-derived data, the error profile can potentially be used to suggest a less-stringent conflict definition (e.g. more than one mutation between reads).
In the limit that reads span the entire sequence, Gc will be the graph complement implied by Go, but this is not generally true if reads are shorter than the length of the sequence.
2.3. Reconstructing quasispecies from reads
Reconstructing quasispecies sequences from NGS reads is informed by both differences between reads (to distinguish sequences) and overlaps between reads (to extend sequences). In terms of the graph representations, quasispecies sequences can be inferred where connected subgraphs of the conflict and overlap graphs intersect.
We refer to these subgraphs of the conflict graph which include only vertex intersections to an overlap graph as a neighborhood conflict graph, G(V′, E′) (see Algorithm 1). Conceptually, these represent clusters of reads which are related by both mutational differences and overlaps. For a constant depth of coverage, longer reads will lead to sequence regions which encompass larger spans of the sequence, while for shorter read lengths, inferences may only be made on pockets of variation within the genome.
Within the neighborhood conflict graph the objective is to partition the vertices into a minimal number of non-conflicting independent sets. This is equivalent to a vertex graph coloring problem, which is known to be NP-Hard [55], but with data reduction and a constraint programming (CP) formulation, useful solutions can be obtained within acceptable computation times. This framework naturally permits paired-end and other non-contiguous reads, as the discontinuous character of the reads does not change the construction of the conflict and overlap graphs.
The set of vertices in this graph is further reduced by collapsing redundant reads. Redundancy is defined in terms of the graph data structures rather than the span of the reads. Due to the distances between variable positions in the sequence, it is common for reads to span different sequence positions, yet share the same set of edges. Groups of such vertices are collapsed into a single vertex to create a homomorphism of the neighborhood conflict graph. A proper coloring of a homomorphism is also a valid coloring of the original graph [56]. In the limit that the read length is the length of the sequence and coverage depth is high, this simple reduction will produce a complete graph in which each vertex corresponds to a distinct quasispecies sequence.
We use a Bron-Kerbosch algorithm to identify maximal cliques in the graph [57]. This provides a lower bound to the chromatic number of the CP [56] and also introduces a distinctness constraint on vertices within cliques. The colors of the maximal clique are also assigned a fixed set of colors 1…s where s is the size of the maximal clique, reducing the domain of the search space by eliminating equivalent solutions. Constraint programming was implemented using C++ and the Gecode constraint programming library [53]. A branch and bound best solution search [58] was used with the number of colors as a cost function along with the following constraints:
C1: The colorings of the maximal clique in H are fixed as 1, …, s, where s is the size of the maximal clique.
C2: All colors of cliques in H are distinct
C3: Colors of vertices connected by edges in H are distinct.
Once a coloring of H is obtained, these quasispecies sequence assignments are propogated back to the reads modeled by H to obtain quasispecies assignments for each read. Conserved sequence positions are also added to generate model sequences. Figure 1 illustrates a toy example of how reads encode conflict and overlap graphs.
Fig. 1.
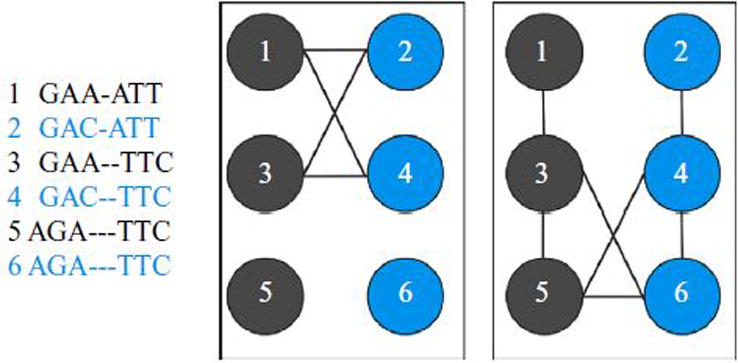
A toy example of 6 paired-end reads with inserts ranging from 1–3 bp (left) of 2 quasispecies sequences (represented by blue and gray). The conflict graph (middle) and an overlap graph with an overlap threshold of 5 (right) are shown. Reads 1, 2, 3, and 4, define a neighborhood conflict graph for which 1 and 3 are assigned a single color and 2 and 4 are assigned a second color. Characters in reads 5 and 6 exhibit no conflicts, reflecting conserved positions which are included in all quasispecies sequences.
2.4. Simulation and evaluation
A read sampling simulator was written in C++ to evaluate the reconstruction. The simulation allows for either contiguous or paired-end fragments to be sampled from an underlying set of known quasispecies sequences. Quasispecies sequences were obtained from a clonal sequencing study of the HIV-1 pol gene by Bacheler et al. [59] available online at the Los Alamos HIV Database [60]. Initial simulations were performed to examine the qualitative characteristics of conflict graphs as read characteristics were varied.
Simulations of 10,000 150 × 2 bp paired-end reads with inserts varying uniformly from 0 to 50 bp were sampled from clonal sequences of 5 patients (patient ID P00001, P00003, P00005, P00021, P00026) with relatively high numbers of clonal sequences available (49, 33, 42, 58, and 62, respectively) [59]. Although the size of the simulation is small compared to the number of raw reads which are feasible by NGS, multiplexing and error correction procedures, would be expected to reduce the size of the read set by several orders of magnitude. An overlap threshold of 295 bp was used to construct the overlap graph. Reconstructed quasispecies subsequences were then compared to the generating set of quasispecies sequences to evaluate the reconstruction. Additionally, a simulation of 5,000 250 × 2 bp (overlap threshold 485 bp) paired-end reads with inserts varying from 0 to 100 bp was sampled from clonal sequences of patient ID P00003 to approximate different sampling conditions (sparser, longer, paired-end reads such as obtained from a MiSeq).
3. Results
To test the utility of this approach, clonal sequences [59] were used to simulate sampling and test quasispecies sequence reconstruction. These sequences consist of 984 bp from the pol gene region of the HIV-1 genome.
Figure 2 shows examples of conflict graphs using a small number of reads for clarity. Connected components in the conflict graph reflect sequence regions of pol for which variation distinguishes reads in different quasispecies sequences. In situations where reads are shorter than a conserved portion of the sequence, distinct connected components in the conflict graph will result (Fig. 2 top left), independent of depth of coverage. Inferences can be made within local regions, but a larger reconstruction from read consistency alone will have degenerate solutions (many of which will be false recombinants) unless additional assumptions regarding long-range characteristics are incorporated.
Fig. 2.

Conflict graphs using read lengths of short (50 bp top left, 100 bp top right) and long (300 bp bottom left, 600 bp bottom right) contiguous reads sampled from Patient ID P00001 in [59]. Vertices in the graph represent reads while edges represent conflicting sequences between overlapping reads. Only a small number of samples was used to generate these graphs for the sake of clarity. Colors shown correspond to the underlying quasispecies sequences used to generate the graph.
Simulations of paired end reads were performed on clonal sequencing data of five patients from [59] under the same sampling conditions (see methods). A summary of the simulations are shown in table 1.
Table 1.
Summary of simulation results (no. is an abbreviation for “number”)
| Patient ID | No. reconstructed QS subsequences | Matched reconstructed QS subsequences | Uniquely matched reconstructed QS subsequences | Unmatched reconstructed QS subsequences | Smallest reconstruction (no. positions) | Largest reconstruction (no. positions) |
|---|---|---|---|---|---|---|
| P00001 | 36 | 32 | 16 | 4 | 869 | 884 |
| P00003 | 167 | 167 | 48 | 0 | 890 | 914 |
| P00003* | 102 | 102 | 51 | 0 | 914 | 933 |
| P00005 | 60 | 54 | 36 | 6 | 878 | 912 |
| P00021 | 114 | 113 | 36 | 1 | 829 | 877 |
| P00026 | 33 | 33 | 10 | 0 | 828 | 848 |
The first column indicates the patient ID according to the Bacheler dataset. The second column indicates the number of subsequence reconstructions generated by the method. The third column indicates how many of the reconstructed subsequences correspond to subsequences of actual QS sequences. The fourth column indicates how many of the reconstructed subsequences uniquely match an actual QS sequence. The fourth column indicates how many reconstructed subsequences do not match any actual QS sequence (i.e. were incorrectly inferred recombinants). The rightmost two columns indicate the number of positions inferred for the smallest and largest reconstructions. P0003* indicates a simulation performed under different sampling conditions – with longer reads and inserts, but at lower coverage (see methods).
Viral sequence diversity and the phylogenetic characteristics of the viral population varies substantially between patients and these differences lead to variability in the reconstruction outcome. However in all simulations only a minority of inferred subsequences corresponded to incorrect recombinations of reads. Of the 36 quasispecies subsequences reconstructed for patient P00001, 32/36 (89%) represent subsequences of actual quasispecies sequences, 16 of which map uniquely to a single actual quasispecies sequence, while 4 reconstructed sequences did not correspond to an actual quasispecies sequence. Likewise, of the 60 model quasispecies subsequences reconstructed for P00005 (Fig. 3), 54/60 (90%) represent subsequences of actual quasispecies sequences, 36 of which map uniquely to a single actual quasispecies sequence, while 6 of reconstructed sequences did not correspond to a actual quasispecies sequence.
Fig. 3.
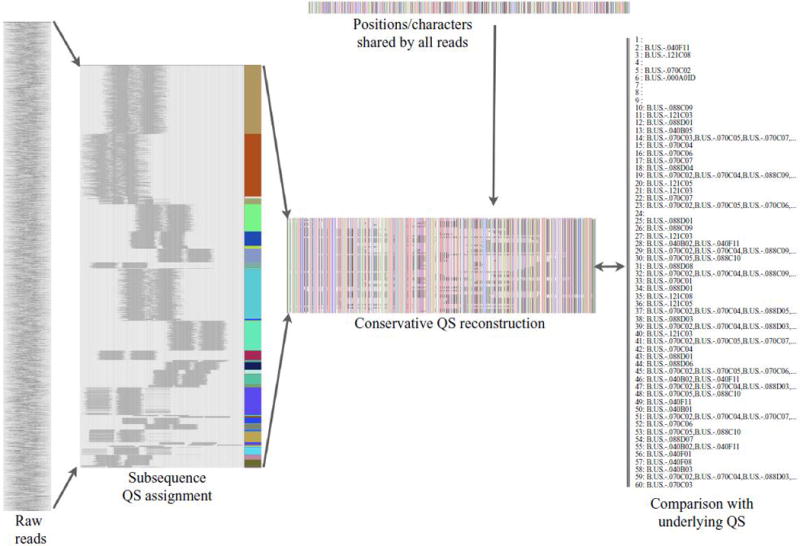
The QS reconstruction pipeline can be seen as a data reduction which aims to limit false explanatory sequences. The process starts with raw reads (left). These are aggregated into tractable quasispecies subsequences supported by read conflicts and overlap, as discussed in the methods. Using the mapped reads, sequence positions which are perfectly conserved across reads (top) are also incorporated to construct an explanatory set of quasispecies subsequences (center, labeled “conservative quasispecies reconstruction”, each row corresponds to the sequence obtained from a set of non-conflicting reads, columns correspond to sequence positions, and colors correspond to sequence characters – A = red, C = green, G = blue, T = white, undetermined = gray). Reconstruction is conservative in that the majority of these subsequences match at least one true underlying sequence (54/60 for P00005, shown in this figure). 36 of these quasispecies sequences contain sufficient information to map uniquely to an underlying quasispecies sequence.
An additional simulation was performed on patient P00003 to explore the impact of sequencing conditions with sparser sampling and longer paired-end inserts and reads (indicated as P00003* in the table). Longer reads lead to more informative reconstructions, even with sparser sampling. Specifically, the subsequence reconstructions were merged into a smaller number of reconstructions (from 167 to 102) which were generally longer (ranging from 914–933 instead of 890–914), none of which were false recombinants. Slightly more of these reconstructions could be uniquely associated with a single underlying sequence (From 48 to 51).
4. Discussion
Here we have developed and tested a method which contributes several new perspectives to the quasispecies reconstruction problem. First, we described a representation for read relationships which is amenable to the analysis of non-contiguous reads (e.g. paired end sequencing). QColors allows for the analysis of non-contiguous reads because the method does not rely on the ordering of the reads with respect to the genome, only the consistency/conflict between reads. Second, we infer quasispecies sequences within tractable domains even if short read lengths render full-length sequence reconstructions indeterminate. Third, we devise a method of data reduction and graph coloring using constraint programming to group reads into non-conflicting sets of quasispecies subsequences. We demonstrate the utility of the method using simulations based on data from clonal sequencing experiments.
Previous methods for quasispecies sequence reconstruction have different objectives from the current method. For example, ShoRAH [34] efficiently estimates quasispecies sequence frequencies and incorporates error-correction, but is intended for longer, contiguous reads. Without paired-end information, sequences reconstructed from ShoRAH with 150 bp reads on three patients (P00001, P00003, and P00005) resulted in reconstructed sequences which differred from the generating sequences with a hamming distance of 27 or more, reflecting differences in its aims and target platform. Future approaches may combine the error-correction and frequency estimation functionality of other methods, while incorporating representations and algorithms which model information from discontiguous reads as shown here. Since QColors generates subsequence reconstructions from short, non-contiguous reads, it may also be used as a preprocessing step which complements other approaches requiring longer, contiguous reads for global sequence reconstruction.
Additional challenges remain before the current method can be widely adopted. Most importantly is to connect these methods to robust experimental and analytical error correction procedures [48,50,61] and testing with simulated sequencing error models [62]. Second, improving the performance of the optimization procedure will allow for denser reconstructions with larger numbers of reads (current analyses require several hours of runtime on a standard laptop computer). Consideration of alternative constraints, probabilistic formulations, or metaheuristics in place of constraint programming should be considered. Third, while the motivation for this work was rooted in a practical application, an examination of graph coloring theory in the context of quasispecies conflict graphs would be beneficial. Further development of QColors, as well as alternative quasispecies reconstruction approaches will ultimately aid in broadening the applicability of NGS to viral sequencing in research and clinical practice.
Acknowledgments
This work was supported by the National Institute of Allergy And Infectious Diseases at the National Institutes of Health grants number R01AI66922 and P30AI042853 for Dr. Huang, Dr. Kantor, Ms. DeLong, and Ms. Schreier. Austin Huang was also funded by a NIH T32 postdoctoral fellowship, T32DA13911-10. Thanks to Derek Aguiar for useful discussions of constraint programming and Bjarni Halldorsson for early discussions on graph coloring extensions to haplotype phasing.
Footnotes
Copyright of In Silico Biology is the property of IOS Press and its content may not be copied or emailed to multiple sites or posted to a listserv without the copyright holder’s express written permission. However, users may print, download, or email articles for individual use.
References
- 1.Johnson VA, Brun-Vzinet F, Clotet B, Gnthard HF, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD. Update of the drug resistance mutations in HIV-1: december 2009. Clinical Infectious Diseases. 2008;47:266–285. [Google Scholar]
- 2.Bennett DE, Camacho RJ, Otelea D, Kuritzkes DR, Fleury H, Kiuchi M, Heneine W, Kantor R, Jordan MR, Schapiro JM, et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS One. 2009;4(3) doi: 10.1371/journal.pone.0004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan PA, Kantor R. Transmitted drug resistance in nonsubtype b HIV-1 infection. HIV Therapy. 2009;3(5):447–465. doi: 10.2217/hiv.09.30. [Online]. Available: http://www.futuremedicine.com/doi/abs/10.2217/hiv.09.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic acids research. 2003;31(1):298. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Kumarasamy N, Ditangco R, Kamarulzaman A, Lee CK, Li PC, Paton NI, Phanuphak P, Pujari S, Vibhagool A. The TREAT asia HIV observational database: baseline and retrospective data. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2005;38(2):174. doi: 10.1097/01.qai.0000145351.96815.d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Oliveira T, Shafer RW, Seebregts C. Public database for HIV drug resistance in southern africa. Nature. 2010;464(7289):673–673. doi: 10.1038/464673c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantor R, Katzenstein DA, Efron B, Carvalho AP, Wynhoven B, Cane P, Clarke J, Sirivichayakul S, Soares MA, Snoeck J, Pillay C, Rudich H, Rodrigues R, Holguin A, Ariyoshi K, Bouzas MB, Cahn P, Sugiura W, Soriano V, Brigido LF, Grossman Z, Morris L, Vandamme A, Tanuri A, Phanuphak P, Weber JN, Pillay D, Harrigan PR, Camacho R, Schapiro JM, Shafer RW. Impact of HIV-1 subtype and antiretroviral therapy on protease and reverse transcriptase genotype: Results of a global collaboration. PLoS Med. 2005;2(4):e112. doi: 10.1371/journal.pmed.0020112. [Online]. Available: http://dx.doi.org/10.1371%2Fjournal.pmed.0020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang A, Hogan J, Istrail S, Kantor R. Stratification by HIV-1 subtype does not eliminate systematic geographical variation. Antiviral Therapy. 2010;15(Suppl 2):A161. [Google Scholar]
- 9.Huang A, Hogan JW, Istrail S, DeLong A, Katzenstein DA, Kantor R. First-line antiretroviral treatment effects on resistance mutation prevalence in HIV-1 subtypes. 6th IAS Conference on HIV Pathogenesis, Treatment and Prevention; Rome, Italy. 2011. [Google Scholar]
- 10.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373(6510):123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 11.Mansky LM. Forward mutation rate of human immunodeficiency virus type 1 in a t lymphoid cell line*. AIDS research and human retroviruses. 1996;12(4):307–314. doi: 10.1089/aid.1996.12.307. [DOI] [PubMed] [Google Scholar]
- 12.Goodenow M, Huet T, Saurin W, Kwok S, Sninsky J, Wain-Hobson S. HIV-1 isolates are rapidly evolving quasispecies: evidence for viral mixtures and preferred nucleotide substitutions. JAIDS Journal of Acquired Immune Deficiency Syndromes. 1989;2(4):344. [PubMed] [Google Scholar]
- 13.Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267(5197):483. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 14.Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nature Reviews Genetics. 2009;10(8):540–550. doi: 10.1038/nrg2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boerlijst MC, Bonhoeffer S, Nowak MA. Viral quasispecies and recombination. Proceedings: Biological Sciences. 1996;263(1376):1577–1584. [Google Scholar]
- 16.Briones C, de Vicente A, Molina-Pars C, Domingo E. Minority memory genomes can influence the evolution of HIV-1 quasispecies in vivo. Gene. 2006;384:129–138. doi: 10.1016/j.gene.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 17.Briones C, Domingo E. Minority report: hidden memory genomes in HIV-1 quasispecies and possible clinical implications. AIDS Rev. 2008;10:93109. [PubMed] [Google Scholar]
- 18.Tsibris AMN, Korber B, Arnaout R, Russ C, Lo CC, Leitner T, Gaschen B, Theiler J, Paredes R, Su Z. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PloS one. 2009;4(5):e5683. doi: 10.1371/journal.pone.0005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT, Dewar RL, Metcalf JA, Hammer S, Mellors JW, Coffin JM. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in Treatment-Experienced patients are missed by standard genotype analysis. J Clin Microbiol. 2005;43(1):406–413. doi: 10.1128/JCM.43.1.406-413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wirden M, Malet I, Derache A, Marcelin AG, Roquebert B, Simon A, Kirstetter M, Joubert LM, Katlama C, Calvez V. Clonal analyses of HIV quasispecies in patients harbouring plasma genotype with K65R mutation associated with thymidine analogue mutations or L74V substitution. AIDS. 2005;19(6):630. doi: 10.1097/01.aids.0000163942.93563.fd. [DOI] [PubMed] [Google Scholar]
- 21.Lu J, Deeks SG, Hoh R, Beatty G, Kuritzkes BA, Martin JN, Kuritzkes DR. Rapid emergence of enfuvirtide resistance in HIV-1-infected patients: results of a clonal analysis. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2006;43(1):60. doi: 10.1097/01.qai.0000234083.34161.55. [DOI] [PubMed] [Google Scholar]
- 22.Kassaye S, Lee E, Kantor R, Johnston E, Winters M, Zijenah L, Mateta P, Katzenstein D. Drug resistance in plasma and breast milk after single-dose nevirapine in subtype c HIV type 1: population and clonal sequence analysis. AIDS research and human retroviruses. 2007;23(8):1055–1061. doi: 10.1089/aid.2007.0045. [DOI] [PubMed] [Google Scholar]
- 23.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome research. 2007;17(8):1195. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Archer J, Braverman MS, Taillon BE, Desany B, James I, Harrigan PR, Lewis M, Robertson DL. Detection of low-frequency pretherapy chemokine (CXC motif) receptor 4-using HIV-1 with ultra-deep pyrosequencing. AIDS (London, England) 2009;23(10):1209. doi: 10.1097/QAD.0b013e32832b4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beerenwinkel N, Zagordi O. Ultra-deep sequencing for the analysis of viral populations. Current Opinion in Virology. doi: 10.1016/j.coviro.2011.07.008. In Press, Corrected Proof. [Online]. Available: http://www.sciencedirect.com/science/article/pii/S1879625711000629. [DOI] [PubMed]
- 26.Lataillade M, Chiarella J, Yang R, Schnittman S, Wirtz V, Uy J, Seekins D, Krystal M, Mancini M, McGrath D. Prevalence and clinical significance of HIV drug resistance mutations by Ultra-Deep sequencing in Antiretroviral-Nave subjects in the CASTLE study. PloS one. 2010;5(6):e10952. doi: 10.1371/journal.pone.0010952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simen BB, Braverman M, Abbate I, Aerssens J, Bidet Y, Bouchez O, Gabriel C, Izopet J, Kessler H, Radonic A, Metzner K, Paredes R, Recordon-Pinson P, Sakwa J, Schmitz-Agheguian G, Daumer M. A multicentre collaborative study on HIV drug resistance testing using 454 massively parallel pyrosequencing. Antiviral Therapy. 2010;15(Supp 2):A37. [Google Scholar]
- 28.Margeridon-Thermet S, Shulman NS, Ahmed A, Shahriar R, Liu T, Wang C, Holmes SP, Babrzadeh F, Gharizadeh B, Hanczaruk B. Ultra-deep pyrosequencing of hepatitis b virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)treated patients and NRTI-naive patients. Journal of Infectious Diseases. 2009;199(9):1275. doi: 10.1086/597808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mild M, Hedskog C, Jernberg J, Albert J. Performance of ultra-deep pyrosequencing in analysis of HIV-1 pol gene variation. PLoS One. 2011;6(7):e22741. doi: 10.1371/journal.pone.0022741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a primer ID. Proceedings of the National Academy of Sciences. 2011;108(50):20 166–20 171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eriksson N, Pachter L, Mitsuya Y, Rhee SY, Wang C, Gharizadeh B, Ronaghi M, Shafer RW, Beerenwinkel N. Viral population estimation using pyrosequencing. PLoS Computational Biology. 2008;4(5):e1000074. doi: 10.1371/journal.pcbi.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westbrooks K, Astrovskaya I, Campo D, Khudyakov Y, Berman P, Zelikovsky A. HCV quasispecies assembly using network flows. in Proceedings of the 4th international conference on Bioinformatics research and applications Springer-Verlag. 2008:159–170. [Google Scholar]
- 33.Astrovskaya I, Tork B, Mangul S, Westbrooks K, Mandoiu I, Balfe P, Zelikovsky A. Inferring viral quasispecies spectra from 454 pyrosequencing reads. BMC Bioinformatics. 2011;12(Suppl 6):S1. doi: 10.1186/1471-2105-12-S6-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zagordi O, Bhattacharya A, Eriksson N, Beerenwinkel N. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics. 2011;12(1):119. doi: 10.1186/1471-2105-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prosperi MC, Salemi M. QuRe: software for viral quasispecies reconstruction from next-generation sequencing data. Bioinformatics. 2012;28(1):132–133. doi: 10.1093/bioinformatics/btr627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mancuso N, Tork B, Mandoiu II, Zelikovsky A, Skums P. Viral quasispecies reconstruction from amplicon 454 pyrosequencing reads. Proc 1st Workshop on Computational Advances in Molecular Epidemiology. 2011:94–101. [Google Scholar]
- 37.Pop M, Salzberg SL, Shumway M. Genome sequence assembly: Algorithms and issues. Computer. 2002;4(5):47–54. e5683. [Google Scholar]
- 38.Istrail S, Sutton GG, Florea L, Halpern AL, Mobarry CM, Lippert R, Walenz B, Shatkay H, Dew I, Miller JR, et al. Whole-genome shotgun assembly and comparison of human genome assemblies. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):1916. doi: 10.1073/pnas.0307971100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Idury RM, Waterman MS. A new algorithm for DNA sequence assembly. Journal of Computational Biology. 1995;2(2):291306. doi: 10.1089/cmb.1995.2.291. [DOI] [PubMed] [Google Scholar]
- 40.Sundquist A, Ronaghi M, Tang H, Pevzner P, Batzoglou S. Whole-genome sequencing and assembly with high-throughput, short-read technologies. PLoS One. 2007;2(5):e484. doi: 10.1371/journal.pone.0000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de bruijn graphs. Genome research. 2008;18(5):821. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pevzner PA, Tang H, Waterman MS. An eulerian path approach to DNA fragment assembly. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(17):9748. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaisson MJ, Pevzner PA. Short read fragment assembly of bacterial genomes. Genome Research. 2008;18(2):324. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laserson J, Jojic V, Koller D. Genovo: De novo assembly for metagenomes. Research In Computational Molecular Biology Springer. 2010:341–356. doi: 10.1089/cmb.2010.0244. [DOI] [PubMed] [Google Scholar]
- 45.Lippert R, Schwartz R, Lancia G, Istrail S. Algorithmic strategies for the single nucleotide polymorphism haplotype assembly problem. Briefings in bioinformatics. 2002;3(1):23. doi: 10.1093/bib/3.1.23. [DOI] [PubMed] [Google Scholar]
- 46.Halldrsson BV, Aguiar D, Istrail S. Haplotype phasing by Multi-Assembly of shared haplotypes: Phase-Dependent interactions between rare variants. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2011:88. doi: 10.1142/9789814335058_0010. [DOI] [PubMed] [Google Scholar]
- 47.Willerth SM, Pedro HAM, Pachter L, Humeau LM, Arkin AP, Schaffer DV, Vartanian JP. Development of a low bias method for characterizing viral populations using next generation sequencing technology. PloS one. 2010;5(10):255–264. doi: 10.1371/journal.pone.0013564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zagordi O, Klein R, Daumer M, Beerenwinkel N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Research. 2010;38(21):7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, Ishikawa S, Linak MC, Hirai A, Takahashi H. Sequence-specific error profile of illumina sequencers. Nucleic Acids Research. 2011;39(13):e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jabara C, Jones C, Anderson J, Swanstrom R. Accurate sampling and deep sequencing HIV-1 protease using primer ID. 18th Conference on Retroviruses and Opportunistic Infections; Boston, MA. 2011. [Google Scholar]
- 51.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proceedings of the National Academy of Sciences. 2011;108(23):9530. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ritz A, Bashir A, Raphael BJ. Structural variation analysis with strobe reads. Bioinformatics. 2010;26(10):1291. doi: 10.1093/bioinformatics/btq153. [DOI] [PubMed] [Google Scholar]
- 53.Schulte C, Lagerkvist M, Tack G. Gecode. Software download and online material. at the website: http://www.gecode.org.
- 54.Korber B, Foley BT, Kuiken CL, Pillai SK, Sodroski JG. Numbering positions in HIV relative to HXB2CG. Human retroviruses and AIDS. 1998;3:102–111. [Google Scholar]
- 55.Garey MR, Johnson DS. Computers and Intractability: A Guide to the Theory of NP-completeness. WH Freeman & Co; Gordonsville, VA: 1979. [Google Scholar]
- 56.Jensen TR. Graph coloring problems. John Wiley & Sons; Hoboken, NJ: 1994. [Google Scholar]
- 57.Bron C, Kerbosch J. Algorithm 457: finding all cliques of an undirected graph. Communications of the ACM. 1973;16(9):575–577. [Google Scholar]
- 58.Schulte C. Programming constraint inference engines. Principles and Practice of Constraint Programming-CP97. 1997;1330:519–533. Available: http://www.springerlink.com/content/d2811540pk74p9u2/ [Google Scholar]
- 59.Bacheler LT, Anton ED, Kudish P, Baker D, Bunville J, Krakowski K, Bolling L, Aujay M, Wang XV, Ellis D. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrobial agents and chemotherapy. 2000;44(9):2475. doi: 10.1128/aac.44.9.2475-2484.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korber BT, Brander C, Haynes BF, Koup R, Moore JP, Walker BD, Watkins DI. HIV Molecular Immunology Compendium 2006/2007. Los Alamos National Laboratory; Los Alamos, NM: 2007. [Google Scholar]
- 61.Zagordi O, Geyrhofer L, Roth V, Beerenwinkel N. Deep sequencing of a genetically heterogeneous sample: local haplotype reconstruction and read error correction. Journal of Computational Biology. 2010;17(3):417–428. doi: 10.1089/cmb.2009.0164. [DOI] [PubMed] [Google Scholar]
- 62.Richter D, Ott F, Auch A, Schmid R, Huson D. Metasima sequencing simulator for genomics and metagenomics. PloS one. 2008;3(10):e3373. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]