

Abstract
Objectives
Tofacitinib is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis (RA). We report an integrated safety summary of tofacitinib from two phase I, nine phase II, six phase III and two long-term extension studies in adult patients with active RA.
Methods
Data were pooled for all tofacitinib-treated patients (data cut-off: 31 March 2015). Incidence rates (IRs; patients with event/100 patient-years) and 95% CIs are reported for adverse events (AEs) of interest.
Results
6194 patients received tofacitinib for a total 19 406 patient-years' exposure; median exposure was 3.4 patient-years. IR (95% CI) for serious AEs was 9.4 (9.0 to 9.9); IR for serious infections was 2.7 (2.5 to 3.0). IR for (all) herpes zoster was 3.9 (3.6 to 4.2); IR for disseminated or multidermatomal herpes zoster was 0.3 (0.2 to 0.4). IR for opportunistic infections (excluding tuberculosis) was 0.3 (0.2 to 0.4) and was 0.2 (0.1 to 0.3) for tuberculosis. IR for malignancies (excluding non-melanoma skin cancer (NMSC)) was 0.9 (0.8 to 1.0); NMSC IR was 0.6 (0.5 to 0.7). IR for gastrointestinal perforations was 0.1 (0.1 to 0.2). Analysis of IR for serious infections, herpes zoster and malignancies by 6-month intervals did not reveal any notable increase in IR with longer-duration tofacitinib exposure.
Conclusion
This analysis of tofacitinib exposure up to 8.5 years allowed estimation of safety events with improved precision versus previous tofacitinib reports. AEs were generally stable over time; no new safety signals were observed compared with previous tofacitinib reports.
Trial registration numbers
NCT01262118, NCT01484561, NCT00147498, NCT00413660, NCT00550446, NCT00603512, NCT00687193, NCT01164579, NCT00976599, NCT01059864, NCT01359150, NCT00960440, NCT00847613, NCT00814307, NCT00856544, NCT00853385, NCT01039688, NCT00413699, NCT00661661; Results.
Keywords: Rheumatoid Arthritis, Treatment, Cardiovascular Disease, Infections, Tuberculosis
Introduction
Tofacitinib is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis (RA). Tofacitinib has demonstrated efficacy and manageable safety in patients with active RA in phase I–III trials and long-term extension (LTE) studies.1–18 Results from these studies and adverse event (AE) profiles of other disease-modifying antirheumatic drugs (DMARDs) informed the selection of safety events of special interest: serious infection events (SIEs), opportunistic infections (OI; including tuberculosis (TB)), herpes zoster (HZ), malignancies, cardiovascular events and gastrointestinal (GI) perforations.
We report for the first time integrated data based on cumulative tofacitinib exposure throughout the RA development programme. This analysis extends previous safety reports for tofacitinib which focused on specific AEs and includes up to 8.5 years of tofacitinib exposure, allowing estimation of rates for safety events of interest with improved precision versus previous reports, and novel methods to examine dose-related AEs.
Patients and methods
Studies
Data were pooled from patients with RA treated with tofacitinib in phases I–III and LTE studies (see online supplementary table S1). All studies were completed by 31 March 2015 except LTE study NCT00413699. LTE data collection and analyses are ongoing (database not locked; some values may change in final locked database).
Patients (aged ≥18 years) had an active RA diagnosis based on the American College of Rheumatology 1987 revised criteria19 and active disease at screening and baseline.1–18 Key exclusion criteria included untreated infection with Mycobacterium tuberculosis or clinically significant infection, and history of malignancy except adequately treated squamous cell or basal cell skin cancer or cervical carcinoma in situ.1–18
Studies were conducted in compliance with the Declaration of Helsinki, International Council for Harmonisation Guidelines for Good Clinical Practice and local country regulations. The study protocol was approved by the Institutional Review Board or Independent Ethics Committee at each centre. Patients provided written informed consent.
Dosing
Patients received tofacitinib 1, 3, 5, 10, 15 or 30 mg twice daily or 20 mg once daily, as monotherapy or with background DMARDs (see online supplementary table S1). Upon entering LTE studies, patients from phase II and III studies received tofacitinib 5 and 10 mg twice daily, respectively, regardless of index study treatment. Within the LTE, tofacitinib could be increased to 10 mg twice daily for inadequate response or reduced to 5 mg twice daily for safety reasons.
Because patients could change doses between index and LTE studies and within LTE, dose was categorised using two methods. The primary analysis used the average dosing algorithm in which patients were assigned to average tofacitinib 5 or 10 mg twice daily if the average daily dose at end of enrolment up to the cut-off date was <15 or ≥15 mg, respectively. The constant dosing algorithm was used in a sensitivity analysis. Only patients exposed to a constant tofacitinib dose of 5 or 10 mg twice daily without prior exposure to a different tofacitinib dose or adalimumab were included in the algorithm. Person-time with exposure to constant tofacitinib 5 or 10 mg twice daily would not sum to the overall exposure, because patients had exposure censored upon tofacitinib dose change.
Data collection, coding and adjudication
The safety database included patients receiving ≥1 tofacitinib dose.
Data were collected for all treatment-emergent AEs and serious AEs (SAEs) and coded using Medical Dictionary for Regulatory Activities (MedDRA) V.18.0. Details of comorbidities were requested at baseline.
An external, independent committee of infectious disease experts blindly adjudicated and classified all SIEs and all events of possible OIs occurring in the tofacitinib RA development programme. SIEs were defined as requiring hospitalisation or parenteral antimicrobial therapy, or otherwise meeting SAE criteria; patients with SIEs were discontinued from the study. HZ involving >2 adjacent dermatomes or with disseminated disease were considered OIs. TB screening was performed as previously described.20
Malignancy data were adjudicated as previously described.21
GI perforations were blindly adjudicated by two sponsor-independent reviewers (US board-certified practising gastroenterologists). SAEs in the clinical and safety databases that might reflect a GI perforation (see online supplementary appendix) were reviewed. Status was determined by two reviewers, with any differences resolved by a third reviewer. Clinical events reflecting an opening in the GI tract, including those associated with appendicitis and diverticulitis, were classified as confirmed GI perforations. Incidence rate (IR) of deaths occurring within 30 days after tofacitinib discontinuation was calculated.
annrheumdis-2016-210457supp001.pdf (447.3KB, pdf)
Statistical analysis
Safety analyses were based on observed cases. Crude IRs per 100 patient-years were calculated for patients receiving tofacitinib and those in average or constant 5 or 10 mg twice daily groups. IRs for each AE were obtained by dividing the number of first-time occurrences of the AE over a time interval, by the total duration of study treatment exposure censored at time of first event, death or discontinuation from study time interval. An exact Poisson 95% CI adjusted for exposure time was calculated for IRs.
Standardised incidence ratios (SIRs) were calculated as the ratio of observed AEs to those in the US National Cancer Institute Surveillance and Epidemiology and End Results (SEER) database, 1992–201122; 95% CIs for SIRs were calculated following a Poisson distribution.
TB rates were stratified by geographical background rates using the WHO incidence categorisation of low, intermediate and high.23
To assess whether IRs increased over time, rates were examined within 6-month intervals of tofacitinib exposure. To investigate whether the hazard for developing a malignancy was constant over time, the probability distribution of time to first event was analysed using a Kaplan-Meier curve and cumulative hazard function. Patients were censored at their last day of tofacitinib exposure.
A Cox regression model evaluated risk factors for SIEs, HZ and OIs excluding TB; backward selection (stay fixed at 15%) was used to screen baseline factors: age, gender, region, body mass index (BMI), smoking history, RA disease duration, line of therapy, diabetes, chronic obstructive pulmonary disease (COPD), DAS-4(CRP), methotrexate use, glucocorticoid dose groups, absolute lymphocyte count (ALC), rheumatoid factor and Health Assessment Questionnaire-Disability Index (HAQ-DI) score to model the time to event from the first tofacitinib dose.
Results
Patients and tofacitinib exposure
Six thousand one hundred and ninety-four patients received tofacitinib. Overall, 4794 (77.4%), 4032 (65.1%), 3351 (54.1%) and 2489 (40.2%) patients received tofacitinib for >12, 24, 36 and 48 months; overall median exposure was 3.38 patient-years. Demographics and disease characteristics were similar between groups (table 1).
Table 1.
Patient demographics and baseline disease characteristics
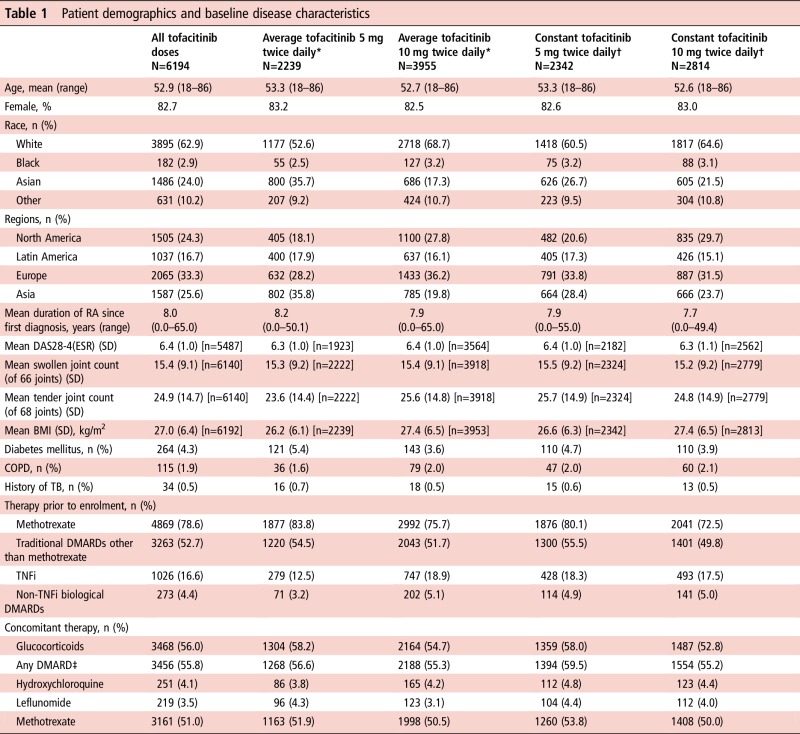
|
All groups are based on tofacitinib exposure data (not tofacitinib patient-level data).
*Average dosing was based on average daily dose: patients receiving <15 mg/day were assigned to the 5 mg twice daily group; patients receiving ≥15 mg/day were assigned to the 10 mg twice daily group.
†Constant dosage without prior exposure to another tofacitinib dose or adalimumab during the study; patients who switched doses were not included in this group.
‡Most common DMARDs are listed.
BMI, body mass index; COPD, chronic obstructive pulmonary disease; DAS28-4(ESR), disease activity score in 28 joints, erythrocyte sedimentation rate; DMARD, disease-modifying antirheumatic drug; RA, rheumatoid arthritis; TB, tuberculosis; TNFi, tumour necrosis factor inhibitor.
AEs and SAEs
The most common AEs (all causality) were nasopharyngitis, upper respiratory tract infection and urinary tract infection (UTI); the most common System Organ Class of SAEs was infections and infestations.
IRs for AEs, discontinuations due to AEs, SAEs and deaths were similar for average and constant tofacitinib 5 and 10 mg twice daily (table 2). The most common causes of death were infections, cardiovascular events and malignancies.
Table 2.
IRs (patients with events/100 patient-years; 95% CI) of AEs and SAEs (all-cause)
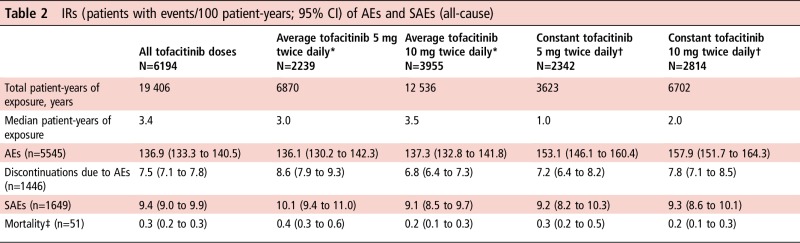
|
*Average dosing was based on average daily dose: patients receiving <15 mg/day were assigned to the 5 mg twice daily group; patients receiving ≥15 mg/day were assigned to the 10 mg twice daily group.
†Constant dosage without prior exposure to another tofacitinib dose or adalimumab during the study; patients who switched doses were not included in this group.
‡Within 30 days of last dose of study drug.
AE, adverse event; IR, incidence rate; n, unique number of patients with event; SAE, serious AE.
Serious infections
The most common types were pneumonia, HZ, UTI and cellulitis. IRs did not increase with longer treatment (figure 1A). IRs of SIEs and deaths due to infections by average and constant dose had overlapping 95% CIs (table 3).
Figure 1.

Continued
Table 3.
IRs of infections or malignancies, patients with events/100 patient-years (95% CI)
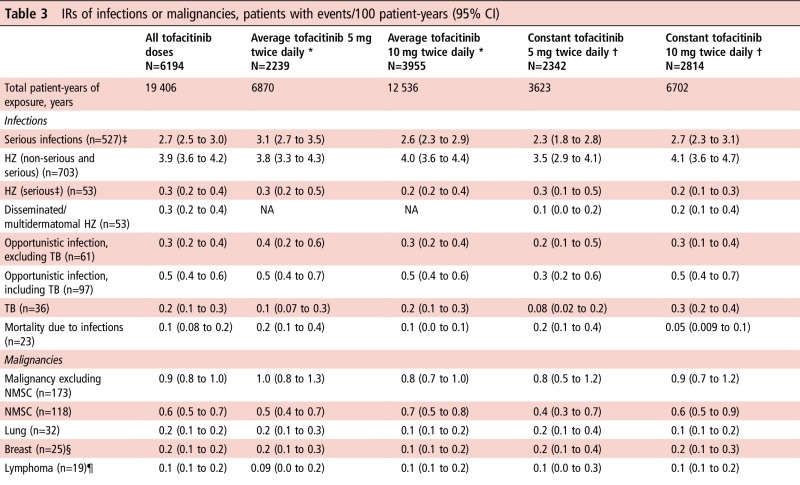
|
*Average dosing was based on average daily dose: patients receiving <15 mg/day were assigned to the 5 mg twice daily group; patients receiving ≥15 mg/day were assigned to the 10 mg twice daily group.
†Constant dosage without prior exposure to another tofacitinib dose or adalimumab during the study; patients who switched doses were not included in this group.
‡Defined as requiring hospitalisation or parenteral antimicrobial therapy, or otherwise meeting SAE criteria.
§IR calculated for female patients only; N [total patient-years' exposure]: N=5125 [16 077] (all tofacitinib); N=1863 [5701] (average tofacitinib 5 mg twice daily); N=3262 [10 377] (average tofacitinib 10 mg twice daily); N=1935 [2984] (constant tofacitinib 5 mg twice daily); N=2335 [5608] (constant tofacitinib 10 mg twice daily).
¶Lymphoproliferative disorders/lymphoma.
HZ, herpes zoster; IR, incidence rate; n, unique number of patients with event; NA, not available; NMSC, non-melanoma skin cancer; SAE, serious adverse event; TB, tuberculosis.
Figure 1.
IRs for (A) SIE, (B) HZ (non-serious and serious) and (C) OI (excluding tuberculosis) over time for all tofacitinib doses. HZ, herpes zoster; IR, incidence rate; OI, opportunistic infection; SIE, serious infection event.
Based on Cox regression analysis, baseline glucocorticoid dose >0–<7.5 and ≥7.5 mg/day (selected based on clinical relevancy and sample size) were associated with increased risk of SIEs (HRs (95% CI) 1.6 (1.3 to 2.0) and 1.7 (1.3 to 2.2), respectively, vs no glucocorticoid use; p=0.0001) (figure 2A). Other significant baseline risk factors were higher age, presence of COPD, higher HAQ-DI score, higher BMI, prior confirmed post-baseline lymphopenia (<500 cells/µL), diabetes, female gender, line of therapy (3rd vs 2nd line), geographical region (Asia, Europe and Latin America, each vs US/Canada) and time-varying tofacitinib dose (referent to 5 mg twice daily) (all p<0.05) (figure 2A).
Figure 2.

HRs of potential risk factors for events of serious infection (A), herpes zoster (B) and opportunistic infections excluding tuberculosis (C)—results from multivariable Cox regression models in the phases I–III and LTE studies *Medical history and/or complication of COPD. †In Unit=x, ‘x’ is the change in the continuous variable corresponding to which the change in hazards is observed. ‡Based on exposure period before lymphopenia <500 cells/µL versus exposure period after lymphopenia <500 cells/µL. COPD, chronic obstructive pulmonary disease; HAQ-DI, Health Assessment Questionnaire-Disability Index; LA, Latin America; LTE, long-term extension.
Six patients had lymphopenia <500 cells/µL, with a crude IR (95% CI) for SIEs of 8.3 (3.0 to 18.1), and 115 patients had lymphopenia ≥500–<1000 cells/µL, with a crude IR (95% CI) of 3.4 (2.8 to 4.1). Inclusion of confirmed ALC<500 cells/µL as a time-varying categorical covariate in a multivariable Cox regression model showed an increased risk of SIEs in the period following confirmed ALC <500 cells/µL (HR (95% CI) 2.5 (1.1 to 5.7)) versus the period before confirmed ALC <500 cells/µL. Evaluation of a threshold of ALC <1000 cells/µL (exposure period prior to lymphopenia <1000 cells/µL vs exposure period after lymphopenia <1000 cells/µL) showed a HR (95% CI) of 1.3 (1.0 to 1.6) (p=0.02), suggesting a trend towards increasing risk with lower lymphocyte counts. The 500 cells/µL threshold is recommended in the product label as the discontinuation criterion.
Herpes zoster
Overall, 703 patients developed HZ; IRs for the first occurrence of non-serious or serious HZ had overlapping 95% CIs for average and constant tofacitinib 5 and 10 mg twice daily (table 3). IR analysis by 6-month intervals did not reveal increasing IRs with longer exposure (figure 1B).
Most HZ cases (92%) involved one dermatome; IR (95% CI) of disseminated/multidermatomal HZ was 0.3 (0.2 to 0.4). Serious HZ was reported in 53 patients. HZ IRs (95% CI) were higher in Asia (5.9 (5.2 to 6.6)) than other regions (see online supplementary table S2).
Baseline glucocorticoid doses >0–<7.5 mg/day and ≥7.5 mg/day were associated with increased HZ (HR (95% CI) 1.5 (1.3 to 1.9) and 1.4 (1.1 to 1.8), respectively, vs no glucocorticoid use; p<0.0001) (figure 2B). Other significant risk factors were baseline age, geographical region, smoking history (ex-smoker and smoker, each vs never smoked) and time-varying tofacitinib dose (all p<0.05) (figure 2B).
Opportunistic infections
OIs excluding TB were reported in 61 patients (see online supplementary table S3), and OIs including TB in 97 patients; 95% CIs for IRs with average and constant tofacitinib 5 and 10 mg twice daily overlapped (table 3). IRs of OIs excluding TB did not increase with longer exposure (figure 1C). Thirty-one OI events were serious.
Baseline age, geographical region and time-varying tofacitinib dose were associated with increased OIs excluding TB (all p<0.05) (figure 2C).
Tuberculosis
Active TB was reported in 36 patients, four had latent TB at screening with a history of adequate treatment. TB IRs were similar for average and constant tofacitinib 5 and 10 mg twice daily (table 3). Pulmonary and non-pulmonary TB occurred in 17 and 19 patients, respectively. Most cases (28/36) occurred in geographical regions endemic for TB (see online supplementary table S4). At screening, 301 patients had latent TB in the phase I–III studies. Of these, 23 had untreated or inadequately treated latent TB, and were treated with isoniazid and permitted to enrol in the study after ≥1 month of treatment. None of these 301 patients developed active TB.
Malignancies
Malignancies (excluding non-melanoma skin cancer, NMSC) occurred in 173 patients and NMSC in 118 patients; analysis of IRs by dose revealed widely overlapping 95% CIs (table 3).
Geographical variation in the NMSC distribution was observed (see online supplementary table S5). Analyses of IRs by 6-month intervals did not reveal any trend (figure 3A, B). A constant hazard over time for developing a malignancy was seen before month 60 (see online supplementary figure S1). Estimation beyond month 60 was less precise due to small patient numbers and limited patient-years of exposure.
Figure 3.

IRs for malignancies excluding NMSC (A) and (B) NMSC over time for all tofacitinib doses. IR, incidence rate; NMSC, non-melanoma skin cancer.
Age-adjusted and sex-adjusted SIR (95% CI) for all malignancies (excluding NMSC) versus SEER among tofacitinib-treated patients was 1.0 (0.8 to 1.1). SIRs (95% CI) for lymphoma, lung cancer and breast cancer were 2.6 (1.6 to 4.1), 1.4 (1.0 to 2.0) and 0.5 (0.3 to 0.7), respectively.
GI perforations
Twenty-two patients experienced GI perforations. IRs (95% CI) were 0.11 (0.07 to 0.17) overall (0.07 (0.02 to 0.17) and 0.14 (0.08 to 0.22) for average 5 and 10 mg twice daily; 0.00 (0.00 to 0.10) and 0.15 (0.07 to 0.27) for constant 5 and 10 mg twice daily). Perforations occurred in the large bowel, excluding anus and rectum (n=13), gastroduodenal area (n=3), small bowel (n=1), anus and rectum (n=2) and undetermined locations (n=3). All received concomitant therapy with non-steroidal anti-inflammatory drugs (NSAIDs) or corticosteroids. Ten patients received NSAIDs and corticosteroids; nine NSAIDs alone and three chronic corticosteroid therapy alone. Thirteen patients had a history of diverticulitis or diverticulosis and two additional patients had a history of gastric ulcers.
Discussion
This analysis presents an integrated view of safety data across the tofacitinib RA development programme. Types and rates of AEs were similar to those observed in phase III trials, with no evidence of directional trends with longer-term tofacitinib exposure through 8.5 years.
SIEs are an identified risk with immunomodulatory medications in RA, including tofacitinib and biological DMARDs (bDMARDs). IRs were consistent with those reported in phase III tofacitinib trials.12–17 24 SIE IRs for bDMARDs in RA clinical trials range from 3.0 to 5.5 per 100 patient-years,25 and are similar to those reported with tumour necrosis factor inhibitors (TNFi) in RA registries (3.2–4.6 per 100 patient-years).26–28 IRs with tofacitinib were generally consistent with IRs with bDMARDs.25–28 Previous studies of registry data revealed a decrease in SIEs with TNFi over time, likely due to discontinuation in patients at increased risk of SIEs, and reduction in risk associated with improvement in function and decreased glucocorticoid use.29–31 In contrast, analyses of data from open-label LTE studies suggest that the SIE risk remains stable over time.32 33
Although an increase in SIEs was not detected here, the studies discontinued patients who developed SAEs, which may have depleted patients at risk of recurrent SAEs. Only time to first event, and not second and subsequent events were analysed.
Previous analyses of the tofacitinib RA development programme identified increased rates of HZ with tofacitinib versus placebo, with greater age and Asian locations identified as risk factors.34 Here, most HZ cases remained non-serious allowing study continuation; approximately 8% of patients with HZ experienced disseminated/multidermatomal HZ. HZ risk is elevated in patients with RA versus the general population;35 reports are conflicting regarding risk with bDMARDs.36 37 Rate comparisons between tofacitinib and bDMARDs are limited by lack of directly comparable data, but indirect comparisons indicate that IRs appear higher with tofacitinib versus other agents.37 However, rates of disseminated/multidermatomal HZ and serious HZ were low and comparable with rates of multidermatomal and ophthalmic HZ reported with bDMARDs or conventional DMARDs.37
Rates of fungal and viral OIs were similar to those previously reported,20 suggesting OIs are a risk with tofacitinib, although it is unclear whether risk differs from that associated with bDMARDs. Precise estimations of differences in OI risk among RA therapies are limited by heterogeneous definitions of OIs,38 geographical variability and lack of head-to-head studies with sufficient power to detect differences.
A higher incidence of TB has been observed in patients with RA versus the general population, and in those receiving TNFi.39 40 Here, the elevation in TB IR with tofacitinib was within the range for bDMARDs.33 41 TB rates reflected geographical background TB prevalence.
IR of malignancies (excluding NMSC) here was 0.89, similar to those observed in previous tofacitinib studies.18 21 Patients with RA are at higher risk of developing some malignancies than the general population.42 43 IRs and SIRs for malignancies (excluding NMSC) reported here are in the range reported with bDMARDs.33 44–46 Analysis of IR of malignancies by 6-month interval exposure revealed variability, whereas analysis of the probability distribution of time to first malignancy event revealed a constant hazard over time. Of note, no real dose difference was observed. Although SIRs for malignancies (excluding NMSC) by 6-month interval exposure were not evaluated, SIRs were stable over time in an analysis of malignancy data (to 10 April 2013) from 14 tofacitinib studies.21 Despite this, vigilance should be exercised when evaluating malignancy risk with long-term exposure.
Cardiovascular safety data for tofacitinib pooled from phase III and LTE studies (data cut-off: 10 April 2013) have been published.47 Similar findings were reported in this analysis of pooled data from phases I–III and LTE studies (data cut-off: 31 March 2015; see online supplementary appendix).
GI perforations are a known risk in patients with RA, especially in patients treated with NSAIDS or glucocorticoids.48 GI perforations with tocilizumab had an IR of 0.3 events/100 patient-years,49 and an observational study revealed an IR of 0.1 events/100 patient-years for TNFi.50 Another study showed higher GI perforation rates with bDMARDs and concomitant glucocorticoids (0.1 events/100 patient-years), versus bDMARDs without glucocorticoids (0.05 events/10 patient-years), indicating glucocorticoid use as a risk factor.51 GI perforations IRs here are within the range reported; most patients with GI perforations had underlying risk factors (eg, glucocorticoids and/or NSAIDs).
Comparison of IRs by dose is limited by several factors. One factor is the imbalance between the tofacitinib 5 and 10 mg twice daily doses in the LTE studies, due to the fact that patients were not randomised to treatment. Instead, patients from phase II and III studies were transitioned into the LTE on 5 and 10 mg twice daily, respectively, except for patients from China and Japan who initiated treatment with tofacitinib 5 mg twice daily per protocol. Therefore, differences in the chronology of LTE study initiation and patient numbers from phase II versus phase III led to a longer median duration of exposure with 5 mg twice daily, but higher overall patient-years' exposure with 10 mg twice daily. This, coupled with differences in geographical regions between trials, preclude definitive dose comparisons. This analysis is also limited by exclusion of patients upon development of a SAE and censoring at time of first event, meaning healthier patients remain at later time points. This limits our ability to evaluate potential changes in SAE rates over time with greater cumulative tofacitinib exposure. Furthermore, the average dosing approach used in the primary analysis did not consider the actual dose at the time of AE. As any method of dose categorisation in this population would have drawbacks, we used constant dosing in a sensitivity analysis to give a more complete picture. Comparisons with placebo were reported for the placebo-controlled phases II and III tofacitinib index studies;3–7 12–16 however, duration of treatment with placebo was short, and patient-years of exposure to placebo was limited, therefore we have not included placebo data in our analysis.
This report describes the tofacitinib safety profile across the RA clinical programme to 31 March 2015, with >6000 patients treated for ≤8.5 years. These data represent the most comprehensive view of long-term safety to date, and reveal a stable AE profile versus controlled studies and earlier analyses of LTE data. Ongoing comparative clinical studies and post-marketing surveillance will provide further information on the tofacitinib risk profile in the clinical trial and real-world settings.
Acknowledgments
Editorial support under the direction of the authors was provided by Kate Silverthorne on behalf of Complete Medical Communications and funded by Pfizer Inc. Pinaki Biswas, an employee of Pfizer Inc, provided statistical support for these analyses.
Footnotes
Contributors: SBC made substantial contributions to the study conception and design and analysis and interpretation of the data; was involved in drafting the article and revising it critically for important intellectual content; and approved the final version to be published. YT, JRC, KK and RR made substantial contributions to the study conception and design, acquisition of data, and analysis and interpretation of the data; were involved in drafting the article and revising it critically for important intellectual content; and approved the final version to be published. XM made substantial contributions to the analysis and interpretation of the data; was involved in drafting the article and revising it critically for important intellectual content; and approved the final version to be published. EBL, CC-S, LW and JW made substantial contributions to acquisition of data and analysis and interpretation of the data; were involved in drafting the article and revising it critically for important intellectual content; and approved the final version to be published. PN made substantial contributions to acquisition of data; was involved in drafting the article and revising it critically for important intellectual content; and approved the final version to be published. KLW, KT, RDM and JG made substantial contributions to the analysis and interpretation of the data; were involved in drafting the article and revising it critically for important intellectual content; and approved the final version to be published.
Funding: This study was sponsored by Pfizer Inc.
Competing interests: SBC, KLW, CC-S and JW have served as consultants for, and have received speaker fees and honoraria from, Pfizer Inc. YT has served as a consultant for, and has received speaker fees and honoraria from, AbbVie, Asahi-kasei, Astellas Pharma, BMS, Chugai Pharma, Daiichi Sankyo, Eli Lilly, GlaxoSmithKline, Janssen, Mitsubishi-Tanabe, Pfizer Inc, Sanofi, Takeda, Teijin and YL Biologics. XM has served as a consultant for, and has received speaker fees and honoraria from, BMS, GlaxoSmithKline, Pfizer Inc and UCB. EBL has served as a consultant for Pfizer Inc. KT, RDM, JG, KK and LW are employees and shareholders of Pfizer Inc. RR was an employee of Pfizer Inc at the time these analyses were conducted and holds stock/stock options in Pfizer Inc.
Patient consent: Obtained.
Ethics approval: Multiple Ethics Committees/Institutional Review Boards approved the studies. Additional details available upon request.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Charles-Schoeman C, Fleischmann R, Davignon J, et al. . Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol 2015;67:616–25. 10.1002/art.38974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kremer JM, Kivitz AJ, Simon-Campos JA, et al. . Evaluation of the effect of tofacitinib on measured glomerular filtration rate in patients with active rheumatoid arthritis: results from a randomised controlled trial. Arthritis Res Ther 2015;17:95 10.1186/s13075-015-0612-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kremer JM, Bloom BJ, Breedveld FC, et al. . The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum 2009;60:1895–905. 10.1002/art.24567 [DOI] [PubMed] [Google Scholar]
- 4. Kremer JM, Cohen S, Wilkinson BE, et al. . A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum 2012;64:970–81. 10.1002/art.33419 [DOI] [PubMed] [Google Scholar]
- 5. Fleischmann R, Cutolo M, Genovese MC, et al. . Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum 2012;64:617–29. 10.1002/art.33383 [DOI] [PubMed] [Google Scholar]
- 6. Tanaka Y, Suzuki M, Nakamura H, et al. . Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res (Hoboken) 2011;63:1150–8. 10.1002/acr.20494 [DOI] [PubMed] [Google Scholar]
- 7. Tanaka Y, Takeuchi T, Yamanaka H, et al. . Efficacy and safety of tofacitinib as monotherapy in Japanese patients with active rheumatoid arthritis: a 12-week, randomized, phase 2 study. Mod Rheumatol 2015;25:514–21. 10.3109/14397595.2014.995875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conaghan PG, Østergaard M, Bowes MA, et al. . Comparing the effects of tofacitinib, methotrexate and the combination, on bone marrow oedema, synovitis and bone erosion in methotrexate-naive, early active rheumatoid arthritis: results of an exploratory randomised MRI study incorporating semiquantitative and quantitative techniques. Ann Rheum Dis 2016;75:1024–33. 10.1136/annrheumdis-2015-208267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyle DL, Soma K, Hodge J, et al. . The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis 2015;74:1311–16. 10.1136/annrheumdis-2014-206028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McInnes IB, Kim HY, Lee SH, et al. . Open-label tofacitinib and double-blind atorvastatin in rheumatoid arthritis patients: a randomised study. Ann Rheum Dis 2014;73:124–31. 10.1136/annrheumdis-2012-202442 [DOI] [PubMed] [Google Scholar]
- 11. Winthrop KL, Silverfield J, Racewicz A, et al. . The effect of tofacitinib on pneumococcal and influenza vaccine responses in rheumatoid arthritis. Ann Rheum Dis 2016;75:687–95. 10.1136/annrheumdis-2014-207191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burmester GR, Blanco R, Charles-Schoeman C, et al. . Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 2013;381:451–60. 10.1016/S0140-6736(12)61424-X [DOI] [PubMed] [Google Scholar]
- 13. van der Heijde D, Tanaka Y, Fleischmann R, et al. . Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum 2013;65:559–70. 10.1002/art.37816 [DOI] [PubMed] [Google Scholar]
- 14. Fleischmann R, Kremer J, Cush J, et al. . Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012;367:495–507. 10.1056/NEJMoa1109071 [DOI] [PubMed] [Google Scholar]
- 15. Kremer J, Li ZG, Hall S, et al. . Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med 2013;159:253–61. 10.7326/0003-4819-159-4-201308200-00006 [DOI] [PubMed] [Google Scholar]
- 16. van Vollenhoven RF, Fleischmann R, Cohen S, et al. . Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012;367:508–19. 10.1056/NEJMoa1112072 [DOI] [PubMed] [Google Scholar]
- 17. Lee EB, Fleischmann R, Hall S, et al. . Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014;370:2377–86. 10.1056/NEJMoa1310476 [DOI] [PubMed] [Google Scholar]
- 18. Wollenhaupt J, Silverfield J, Lee EB, et al. . Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J Rheumatol 2014;41:837–52. 10.3899/jrheum.130683 [DOI] [PubMed] [Google Scholar]
- 19. Arnett FC, Edworthy SM, Bloch DA, et al. . The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. 10.1002/art.1780310302 [DOI] [PubMed] [Google Scholar]
- 20. Winthrop KL, Park SH, Gul A, et al. . Tuberculosis and other opportunistic infections in tofacitinib-treated patients with rheumatoid arthritis. Ann Rheum Dis 2016;75:1133–8. 10.1136/annrheumdis-2015-207319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Curtis JR, Lee EB, Kaplan IV, et al. . Tofacitinib, an oral Janus kinase inhibitor: analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann Rheum Dis 2016;75:831–41. 10.1136/annrheumdis-2014-205847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Howlader N, Noone AM, Krapcho M, et al. . SEER Cancer Statistics Review, 1975–2011. http://seer.cancer.gov/csr/1975_2011/ (accessed 11 Jan 2017).
- 23. World Health Organization. Global tuberculosis control: WHO report 2011. http://whqlibdoc.who.int/publications/2011/9789241564380_eng.pdf (accessed 11 Jan 2017).
- 24. Cohen S, Radominski SC, Gomez-Reino JJ, et al. . Analysis of infections and all-cause mortality in Phase II, Phase III, and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Ann Rheum Dis 2014;66:2924–37. 10.1002/art.38779 [DOI] [PubMed] [Google Scholar]
- 25. Strand V, Ahadieh S, French J, et al. . Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res Ther 2015;17:362 10.1186/s13075-015-0880-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Atzeni F, Sarzi-Puttini P, Botsios C, et al. . Long-term anti-TNF therapy and the risk of serious infections in a cohort of patients with rheumatoid arthritis: comparison of adalimumab, etanercept and infliximab in the GISEA registry. Autoimmun Rev 2012;12:225–9. 10.1016/j.autrev.2012.06.008 [DOI] [PubMed] [Google Scholar]
- 27. Galloway JB, Hyrich KL, Mercer LK, et al. . Anti-TNF therapy is associated with an increased risk of serious infections in patients with rheumatoid arthritis especially in the first 6 months of treatment: updated results from the British Society for Rheumatology Biologics Register with special emphasis on risks in the elderly. Rheumatology (Oxford) 2011;50:124–31. 10.1093/rheumatology/keq242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Dartel SA, Fransen J, Kievit W, et al. . Predictors for the 5-year risk of serious infections in patients with rheumatoid arthritis treated with anti-tumour necrosis factor therapy: a cohort study in the Dutch Rheumatoid Arthritis Monitoring (DREAM) registry. Rheumatology (Oxford) 2013;52:1052–7. 10.1093/rheumatology/kes413 [DOI] [PubMed] [Google Scholar]
- 29. Strangfeld A, Eveslage M, Schneider M, et al. . Treatment benefit or survival of the fittest: what drives the time-dependent decrease in serious infection rates under TNF inhibition and what does this imply for the individual patient? Ann Rheum Dis 2011;70:1914–20. 10.1136/ard.2011.151043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Curtis JR, Patkar N, Xie A, et al. . Risk of serious bacterial infections among rheumatoid arthritis patients exposed to tumor necrosis factor alpha antagonists. Arthritis Rheum 2007;56:1125–33. 10.1002/art.22504 [DOI] [PubMed] [Google Scholar]
- 31. Toh S, Li L, Harrold LR, et al. . Comparative safety of infliximab and etanercept on the risk of serious infections: does the association vary by patient characteristics? Pharmacoepidemiol Drug Saf 2012;21:524–34. 10.1002/pds.3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weinblatt ME, Bathon JM, Kremer JM, et al. . Safety and efficacy of etanercept beyond 10 years of therapy in North American patients with early and longstanding rheumatoid arthritis. Arthritis Care Res (Hoboken) 2011;63:373–82. 10.1002/acr.20372 [DOI] [PubMed] [Google Scholar]
- 33. Burmester GR, Panaccione R, Gordon KB, et al. . Adalimumab: long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn's disease. Ann Rheum Dis 2013;72:517–24. 10.1136/annrheumdis-2011-201244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winthrop KL, Yamanaka H, Valdez H, et al. . Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Ann Rheum Dis 2014;66:2675–84. 10.1002/art.38745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smitten AL, Choi HK, Hochberg MC, et al. . The risk of herpes zoster in patients with rheumatoid arthritis in the United States and the United Kingdom. Arthritis Rheum 2007;57:1431–8. 10.1002/art.23112 [DOI] [PubMed] [Google Scholar]
- 36. Winthrop KL, Baddley JW, Chen L, et al. . Association between the initiation of anti-tumor necrosis factor therapy and the risk of herpes zoster. JAMA 2013;309:887–95. 10.1001/jama.2013.1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strangfeld A, Listing J, Herzer P, et al. . Risk of herpes zoster in patients with rheumatoid arthritis treated with anti-TNF-alpha agents. JAMA 2009;301:737–44. 10.1001/jama.2009.146 [DOI] [PubMed] [Google Scholar]
- 38. Winthrop KL, Novosad SA, Baddley JW, et al. . Opportunistic infections and biologic therapies in immune-mediated inflammatory diseases: consensus recommendations for infection reporting during clinical trials and postmarketing surveillance. Ann Rheum Dis 2015;74:2107–16. 10.1136/annrheumdis-2015-207841 [DOI] [PubMed] [Google Scholar]
- 39. Arkema EV, Jonsson J, Baecklund E, et al. . Are patients with rheumatoid arthritis still at an increased risk of tuberculosis and what is the role of biological treatments? Ann Rheum Dis 2015;74:1212–17. 10.1136/annrheumdis-2013-204960 [DOI] [PubMed] [Google Scholar]
- 40. Baronnet L, Barnetche T, Kahn V, et al. . Incidence of tuberculosis in patients with rheumatoid arthritis. A systematic literature review. Joint Bone Spine 2011;78:279–84. 10.1016/j.jbspin.2010.12.004 [DOI] [PubMed] [Google Scholar]
- 41. Wolfe F, Michaud K, Anderson J, et al. . Tuberculosis infection in patients with rheumatoid arthritis and the effect of infliximab therapy. Arthritis Rheum 2004;50:372–9. 10.1002/art.20009 [DOI] [PubMed] [Google Scholar]
- 42. Franks AL, Slansky JE. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res 2012;32:1119–36. [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas E, Brewster DH, Black RJ, et al. . Risk of malignancy among patients with rheumatic conditions. Int J Cancer 2000;88:497–502. [DOI] [PubMed] [Google Scholar]
- 44. Weinblatt ME, Moreland LW, Westhovens R, et al. . Safety of abatacept administered intravenously in treatment of rheumatoid arthritis: integrated analyses of up to 8 years of treatment from the abatacept clinical trial program. J Rheumatol 2013;40:787–97. 10.3899/jrheum.120906 [DOI] [PubMed] [Google Scholar]
- 45. Bykerk VP, Cush J, Winthrop K, et al. . Update on the safety profile of certolizumab pegol in rheumatoid arthritis: an integrated analysis from clinical trials. Ann Rheum Dis 2015;74:96–103. 10.1136/annrheumdis-2013-203660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007;56:2886–95. 10.1002/art.22864 [DOI] [PubMed] [Google Scholar]
- 47. Charles-Schoeman C, Wicker P, Gonzalez-Gay MD, et al. . Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral Janus kinase inhibitor. Semin Arthritis Rheum 2016;46:261–71. 10.1016/j.semarthrit.2016.05.014 [DOI] [PubMed] [Google Scholar]
- 48. Curtis JR, Lanas A, John A, et al. . Factors associated with gastrointestinal perforation in a cohort of patients with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:1819–28. 10.1002/acr.21764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van Vollenhoven RF, Ronald F, Keystone E, et al. . Gastrointestinal safety in patients with rheumatoid arthritis treated with tocilizumab: data From Roche clinical trials [abstract]. Arthritis Rheum 2009;60(Suppl 10):1613 10.1002/art.24941 [DOI] [Google Scholar]
- 50. Závada J, Lunt M, Davies R, et al. . The risk of gastrointestinal perforations in patients with rheumatoid arthritis treated with anti-TNF therapy: results from the BSRBR-RA. Ann Rheum Dis 2014;73:252–5. 10.1136/annrheumdis-2012-203102 [DOI] [PubMed] [Google Scholar]
- 51. Curtis JR, Xie F, Chen L, et al. . The incidence of gastrointestinal perforations among rheumatoid arthritis patients. Arthritis Rheum 2011;63:346–51. 10.1002/art.30107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2016-210457supp001.pdf (447.3KB, pdf)