

Abstract
Metabolic inflammation is closely associated with hyperlipidemia and cardiovascular disease. However, the underlying mechanisms are not fully understood. The current study established that cAMP-responsive-element-binding protein H (CREBH), an acute-phase transcription factor, enhances very-low-density lipoprotein (VLDL) assembly and secretion by upregulating apolipoprotein B (apoB) expression, and contributes to metabolic inflammation-associated hyperlipoproteinemia induced by TNFα, lipopolysaccharides (LPS), and a high-fat diet (HFD) in mice. Specifically, overexpression of CREBH significantly induced mRNA and protein expression of apoB in McA-7777 cells. Luciferase assay further revealed that the presence of CREBH could significantly increase the activities of the apoB gene promoter. In contrast, genetic depletion of CREBH in mice resulted in significant reduction in expression of hepatic apoB mRNA. Challenging mice with an acute fat load led to upregulation of triglyceride (TG)-rich lipoprotein secretion in wildtype mice, but not in CREBH-null mice. TNFα treatment activated hepatic CREBH expression, which in turn enhanced hepatic apoB biosynthesis and VLDL secretion. Metabolic inflammation induced by LPS or HFD also resulted in overproduction of apoB and hyperlipoproteinemia in wildtype mice, but not in CREBH-null mice. This study demonstrates that CREBH could be a mediator between metabolic inflammation and hepatic VLDL overproduction in chronic metabolic disorders. This novel finding establishes CREBH as the first transcription factor that regulates apoB expression on the transcriptional level and the subsequent VLDL biosynthesis in response to metabolic inflammation. The study also provides novel insight into the pathogenesis of hyperlipidemia in metabolic syndrome.
Keywords: CREBH, metabolic inflammation, TNFα, VLDL, apoB, metabolic syndrome
Introduction
Hyperlipidemia is a major complication of insulin-resistant conditions, such as obesity and type 2 diabetes, and contributes to an increased risk of cardiovascular disease (CVD) [1–3]. One key mechanism underlying hyperlipidemia in metabolic syndrome is the overproduction of apolipoprotein B (apoB), the essential structural protein of the triglyceride (TG)-rich lipoproteins, i.e. very-low-density lipoproteins (VLDL) and chylomicrons [1]. Clinically, apoB is more closely associated with central adiposity, insulin resistance and inflammation than low-density lipoprotein (LDL) cholesterol and non-high-density lipoprotein (HDL) cholesterol (the cholesterol from lipoproteins other than HDL). Thus, the abundance of apoB in circulation has been proposed to be a better biomarker than LDL cholesterol and non-HDL cholesterol for identifying a subgroup of individuals with elevated cardiovascular risk [4, 5]. In this regard, it is clearly important to understand how apoB is upregulated in metabolic syndrome. ApoB is an amphipathic glycoprotein that is synthesized and secreted from the small intestinal and hepatic endoplasmic reticulum (ER) where TG-rich lipoproteins are assembled [6]. Biosynthesis of apoB is modulated by both insulin and metabolic stress signaling [7–9]. Insulin inhibits apoB expression whereas lipid-induced ER stress increases apoB secretion [7, 10].
The development of metabolic syndrome is closely associated with the activation of a low-grade, chronic inflammation orchestrated by the metabolic system in response to excessive supply of nutrients, energy, or other metabolites [11]. Ample evidence further shows that lipogenic diets, such as high-fat and high-fructose diets, induce secretion of pro-inflammatory cytokines, TNFα and IL-6, in humans and animal models [12]. While it is now well-appreciated that inflammation significantly contributes to the perturbations of lipid and lipoprotein metabolism observed in obesity and insulin resistance [10, 13–17], the underlying mechanism of how inflammatory cytokines regulate the biosynthesis of apoB and overproduction of VLDL is not fully understood.
The acute-phase response (APR) is a systemic inflammatory component of innate immunity [18, 19]. During APR, a series of changes in lipid and lipoprotein metabolism occur, including the increase of VLDL and TG and the decrease of HDL, LDL, and total cholesterol in circulation [20]. ApoB has been proposed to be involved in the acute-phase inflammation. A three-year follow-up study found that increased plasma ceruplasmin, a positive acute-phase reactant, was associated with an almost three-fold increase in the risk of future major adverse cardiovascular events. The contribution of ceruplasmin to the risk of CVD was associated with abnormal lipoprotein profile and other factors [21].
The cAMP-responsive binding protein (CREBH) is an ER-bound transcription factor activated by pro-inflammatory cytokines, including TNFα and IL-1, and is critical for the active expression of APR genes, such as serum amyloid P-component (SAP) and C-reactive protein (CRP) [22]. Similar to the tissue distribution of apoB, CREBH is dominantly expressed in the liver and small intestine [22]. There is reduced expression of apoB mRNA in the fetal livers of CREBH-depleted mice (CREBH-null) as compared to their wildtype (WT) littermates [22]. However, whether CREBH directly regulates apoB biosynthesis and how CREBH and apoB contribute to the hyperlipidemia induced by metabolic inflammation remains unclear.
In this study, we hypothesized that CREBH is a transcription factor that positively regulates mRNA and protein expression of apoB and contributes to the metabolic inflammation-induced VLDL overproduction in chronic metabolic disorders. We tested our hypothesis by generating a series of mouse models with metabolic inflammation induced by TNFα, lipopolysaccharide (LPS), and high-fat feeding in the WT and CREBH-null mice. We further used an in vitro cell culture system treated with pro-inflammatory cytokines to corroborate our findings. Our results demonstrate that CREBH is the missing link between metabolic inflammation, hepatic VLDL-apoB overproduction, and hyperlipidemia. This novel finding reveals an unprecedented mechanistic link by which metabolic inflammation activates CREBH to upregulate expression of apoB, which subsequently leads to the abnormal lipoprotein biogenesis that contributes to the hyperlipoproteinemia associated with metabolic syndrome.
Materials and Methods
Cell culture, treatments and transfection
Rat hepatoma cell lines, McA-RH7777 (McA) (ATCC) cells, were maintained in DMEM containing 10% FBS at 37°C, 5% CO2, respectively. Cells were seeded one day before the treatment. Cells were then fasted for 12 h followed by 20ng/mL TNFα for 6 and 12 h treatment. Total RNAs, whole cell lysates and cell culture media were collected at the end points of each treatment as described below. Plasmid pFlag-CREBH WT and pFlag-CREBH-DN were kindly provided by Dr. Randal J. Kaufman (Howard Hughes Medical Institute, University of Michigan Medical Center, Ann Arbor, Michigan, USA) and were previously described [22]. For cell transfection, 2μg of plasmid DNAs were transfected into McA cells as previously described [10].
Animal protocols
All animal experiments were approved by the University of Nebraska-Lincoln Institutional Animal Care and Use Committee and were performed conform the NIH guidelines (Guide for the care and use of laboratory animals). Mice used in this study were 10–16 weeks old. CREBH-null mice with exons 4–7 of the CREBH gene deleted were previously described [23]. Animals were housed on alternating 12h light and dark cycles with free access to food and water. For high fat feeding studies, WT and CREBH-null mice were randomly divided into four groups and given either a standard chow (Envigo, Madison, Wisconsin. #2016), or a HFD (60% caloric from fat, Dyets #103938) for 7 weeks. Upon completion of the 7 weeks feeding protocol, blood samples and liver tissues were collected for further analysis after 12h fasting. For TNFα administration experiment, both WT and CREBH-null mice were divided into two groups and mice were then fasted overnight for 6 h with free access to water. Mice were treated with either recombinant mouse TNFα (12μg/200g body weight, ip. injection) or saline control. Plasmas and livers were collected at 5h post-treatment. For LPS treatment, LPS was suspended in sterile pyrogen-free 0.9% NaCl (Abbott Laboratories, North Chicago, IL). WT and CREBH-null were given a single injection of LPS (2 μg/g body weight, ip.). Food was removed after LPS injection. Plasmas and livers were collected 10h after LPS treatment for further analysis. Tissue collections were performed at the end points of each experiment, as follows: animals were anesthetized using isoflurane (3% mixed with oxygen), tissues were excised and flash frozen in liquid nitrogen, and livers were then homogenized in solubilization buffer for further analysis as described previously [24, 25].
In vivo TG-rich lipoprotein collection assay
Mice were fasted for 12h and blood was collected via tail vein bleed (100μL) to determine baseline (0h) apoB levels. Mice were then administrated a dose of olive oil (200 μL) through oral gavage followed by treated with 20% poloxamer (500mg/kg, ip.) at 20min after fat gavage. Blood was collected at 3h via cardiac puncture (500μL). Plasma apolipoprotein levels were determined by immunoblotting analysis.
In vivo VLDL collection assay
Mice were fasted for 5h and blood was collected via tail vein bleed (100μL) to determine baseline (0h) apoB levels. Mice were then treated with 20% poloxamer (500mg/kg, ip.). Blood was collected at 3h via cardiac puncture (500μL). Plasma VLDL-apoB was determined by immunoblotting analysis.
Immunoblot analysis
Immunoblotting was performed as previously described [17]. The following antibodies were used: anti-CREBH (Santa Cruz, USA), anti-apoB, anti-apoE and albumin (Nittobo America), anti-phospho-Stat-3, anti-Stat-3 and anti-CRP (Cell signaling, USA); all antibodies were used at a final concentration of 0.1–1μg/mL. After incubation with the appropriate horseradish peroxidase-conjugated secondary antibody (1:5,000 dilution; GE Healthcare, UK), proteins were visualized by enhanced chemiluminescence (ECL) according to the manufacturer’s instructions (Amersham Biosciences, Pittsburgh, PA, USA).
Luciferase activity assay
McA cells were co-transfected with a CREBH-WT or a dominant negative function of CREBH (CREBH-DN) cDNA plus the firefly luciferase reporter gene under the control of human apoB gene promoter for 36h. Cell lysates were examined for firefly luciferase activities using the Dual-Luciferase® Reporter Assay System (Promega) and normalized to renilla luciferase activities as previously described [10].
Plasma lipid measurement
Plasma TG and cholesterol analyses were performed using an Enzymatic/GPO endpoint method (Pointe Scientific, Canton, MI) as per the manufacturer’s instructions and as previously described [17].
Liver lipid extraction and TG and cholesterol mass measurement
Liver lipid extraction and analysis were performed as previously described [17]. Briefly, approximately 300mg of liver tissue were added to 20 volumes of 2:1 chloroform: methanol mixture and incubated for 24h at room temperature. Following the incubation period 0.2 volumes of 0.9% NaCl were added to the solvent mixture. The samples were thoroughly vortexed, then centrifuged at 2,000 rpm for 3 min. The upper aqueous phase was removed and the solvent layer was allowed to evaporate. The dried lipid was resuspended in 1 mL of 100% ethanol. TG and cholesterol concentrations were determined using an Enzymatic/GPO endpoint method (Pointe Scientific, Canton, MI) as per the manufacturer’s instructions. Lipid data are expressed in milligrams of lipid per gram of liver tissue.
RNA isolation and qRT-PCR
Total RNA was isolated from mouse liver tissues and cells using TRIzol (Life Technologies, Grand Island, NY). First strand cDNA was synthesized with oligo (dT) and random primers using a High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Life Technologies). PCR reactions were carried out using SYBR Green PCR Master Mix (Applied Biosystems, Life Technologies). Relative quantities of mRNA were calculated from threshold cycle (CT) values with the comparative CT method, using 18S rRNA as an internal reference. Primer sequences are provided upon request.
Statistical analysis
Data obtained by densitometry or fluorography were evaluated using one-way ANOVA (GraphPad Prism 5, La Jolla, CA, USA). Post-test analysis was performed to determine the significance between groups, using unpaired two-way Student t-tests. All results are presented as means ± SEM. Asterisks (* or **) indicate statistically significant differences of P<0.05 or P <0.01, respectively, compared to control.
RESULTS
CREBH upregulates apoB mRNA expression which facilitates apoB secretion
Expression of apoB mRNA in mouse fetal liver was previously reported to be significantly reduced upon depletion of CREBH [22]. This finding prompted us to investigate whether CREBH exerts a regulatory effect on cellular apoB expression. We transfected McA cells, a rat hepatoma cell line, with a plasmid cDNA-encompassed CREBH-WT or a control empty vector (mock) to overexpress CREBH for 48h. qRT-PCR analysis revealed that cellular apoB mRNA expression was significantly enhanced in the presence of CREBH (Figure 1A). In contrast, overexpression of CREBH did not increase mRNA expression of the microsomal triglyceride transfer protein (MTP), an important chaperone involved in apoB lipidation during VLDL assembly (Figure 1A). To further investigate whether inhibition of CREBH activity will suppress apoB expression, plasmid cDNAs of CREBH-WT and a dominant negative CREBH (CREBH-ND) were transiently expressed in McA cells for 48h, followed by immunoblotting analysis for the secreted VLDL-apoB in the culture media. As shown in Figure 1B, secreted apoB was significantly increased in the CREBH-WT-expressing cells compared to the mock transfected cells (Figure 1B). In contrast, expression of CREBH-DN inhibited VLDL-apoB secretion (Figure 1B). These data suggest a positive relationship between CREBH activity and the abundance of cellular apoB mRNA and the secretion of VLDL-apoB protein. Sequence analysis identified one or two CRE-binding elements located in the promoter region of mouse and human apoB genes, respectively (Supplemental Figure 1). To further confirm that CREBH regulates apoB on the transcriptional level, we co-transfected McA cells with the CREBH-WT or CREBH-DN along with a luciferase construct under the control of human apoB gene promoter for 36h. Luciferase activity was significantly higher in the CREBH-WT transfected cells compared to the CREBH-DN transfected cells (Figure 1C). This result indicates that overexpression of CREBH-WT stimulated apoB-luciferase expression. Together, these data indicate that CREBH positively regulates apoB mRNA and protein expression.
Figure 1. CREBH upregulates apoB mRNA expression which facilitates apoB secretion.

(A and B) McAcells were seeded onto 6-well plates, and then transfected with 2μg an empty vector p3×FLAG-CMV7.1 (Mock), the vector carrying an open reading frame of WT CREBH (CREBH WT) or a dominant negative CREBH (CREBH DN), respectively. 48h post-transfection, cells and media were collected for protein and mRNA analysis. (A) mRNA of apoB and MTP in the transfected cells determined by qRT-PCR. (B) Immunoblotting analysis of apoB in the culture media. Right panel shows quantification of apoB protein signal intensity to loading control protein albumin. (C) McA cells were co-transfected with a CREBH WT or a dominant negative CREBH (CREBH DN) cDNA plus the firefly luciferase reporter gene under the control of human apoB gene promoter for 36h. Cell lysates were examined for firefly luciferase activities and normalized to renilla luciferase activities. The data represent one out of three independent experiments (n=6–9/group). Results are shown as mean ± SEM. *P < 0.05 and **P < 0.01 versus controls.
Impairment of CREBH signaling compromises VLDL and chylomicron secretion
To further investigate the association between CREBH and apoB in lipoprotein metabolism in vivo, we determined levels of hepatic apoB mRNA in the WT and CREBH-null mice. Because CREBH is activated by nutritional starvation, we fasted the mice for 12h prior to harvesting liver tissues to measure the hepatic apoB mRNA. As shown in Figure 2A, expression of apoB mRNA was significantly reduced in CREBH-null mice compared to their WT littermates (Figure 2A). In contrast, depletion of CREBH did not significantly alter MTP mRNA expression (Supplemental Figure 2). To determine whether reduced apoB mRNA in the CREBH-null mice compromises hepatic VLDL biogenesis during the fasting state, two groups of WT and CREBH-null mice were subjected to an in vivo VLDL collection assay as described in Materials and Methods. This assay uses poloxamer (500 mg/kg, ip.) to inhibit the activity of lipoprotein lipase, which prevents hydrolysis of lipoproteins in circulation. Blood samples were collected at 0h and 3h after poloxamer treatment. Analysis of plasma apoB protein levels revealed that expression of apoB protein was comparable between the WT and CREBH-null mice at the baseline (0h) (Figure 2B, upper panels). Poloxamer treatment induced comparable accumulation of plasma TG and cholesterol in both WT and CREBH-null mice (Suppl. figures 3A and B). However, plasma VLDL-apoB in CREBH-null mice did not parallel the increase of secreted lipids at 3h, but instead was significantly reduced compared to WT controls (Figure 2B, lower panels), indicating defective VLDL-apoB biosynthesis in the CREBH-null mice. To further investigate the secretion of chylomicrons upon challenging the mice with an acute fat load (a nutritional state that mimics the postprandial period), two groups of WT and CREBH-null mice were given an oral fat load (200 μL olive oil) followed a 12h fast. Mice were then treated with poloxamer (500mg/kg, ip.) 20min after the fat load. Blood samples were collected at 0h and 3h after poloxamer injection. Analysis of plasma apoB protein levels showed no significant difference in apoB protein levels between the WT and CREBH-null mice at baseline (0h) (Figure 2C). However, challenging mice with a fat load markedly increased plasma apoB100 and B48 in the WT mice but not the CREBH-null mice, indicated by the significantly lessened secretion of both apoB100 and B48 (Figure 2C). No significant changes were observed in apolipoprotein E (apoE), and another hepatic-secreted protein, albumin (Figure 2C). Consistently, the fold change of plasma TG contents in the WT mice were significantly higher than that in the CREBH-null mice (Figure 2D). Together, these results show that impairment of CREBH activity inhibits apoB mRNA expression and compromises the assembly and secretion of VLDL and chylomicrons in the fasting state and in response to acute fat challenge.
Figure 2. Impairment of CREBH signaling compromises VLDL and chylomicron secretion.
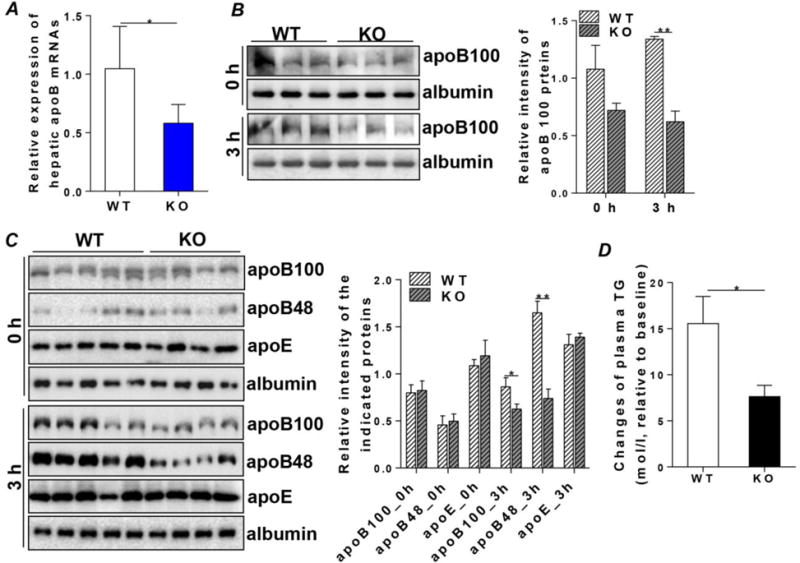
(A) mRNA in the livers of WT and CREBH-null (KO) mice after a 12h fast. (B) Blood was collected from WT and KO mice following a 5h fast for baseline control (0h). Mice were then treated with poloxamer (500mg/kg, ip. injection). Mice were sacrificed at 3h after poloxamer treatment. Blood samples were collected for immunoblotting analysis of plasma apoB100 and albumin [upper panels: baseline (0h), lower panels: 3h]. (C) Blood was collected from WT and KO mice following a 12h fast. Mice were then given an oral fat load (200μL olive oil) and treated with poloxamer (500mg/kg, IP injection.) at 20min post-fat load. Mice were sacrificed at 3h after poloxamer treatment. Blood samples were collected for immunoblotting analysis of plasma apoB100, B48, apoE and albumin at baseline (0h) and 3h. Right panels in (B) and (C) show quantification of the indicated proteins to loading control protein albumin. (D) Changes of plasma TG contents between baseline (0h) and 3h in (C). Results are shown as mean ± SEM, n=5–6/group. *P < 0.05 and **P < 0.01 versus controls.
CREBH mediates the inflammatory signaling pathways involved in overproduction of hepatic VLDL-apoB
CREBH has been shown to be involved in regulation of acute-phase gene expression [22]. To investigate whether CREBH is involved in metabolic inflammation-associated VLDL overproduction and the subsequent hyperlipidemia, McA cells were treated for 6h with TNFα (20ng/mL), a pro-inflammatory cytokine that is usually elevated in subjects with metabolic syndrome. Expressions of both full-length CREBH (CREBH-F) and activated CREBH N-terminal CREBH (CREBH-N) were significantly induced by TNFα treatment (Figure 3A). More importantly, activation of CREBH by TNFα at 6h post-treatment was followed by a marked increase in cellular apoB mRNA at 12 h post-treatment (Figures 3A and B). The increase of apoB mRNA enhanced secretion of VLDL, indicated by a higher level of secreted VLDL-apoB detected in the cell culture media of TNFα-treated cells (Figure 3C). To further corroborate the regulatory effect of TNFα on the CREBH-apoB axis, we pretreated two sets of McA cells with TNFα (20μg/mL) or mock (saline) for 6h to activate the cellular CREBH. The pretreated cells were then transfected with a luciferase construct under the control of apoB gene promoter. Cells were further incubated with TNFα or mock for additional 36 h. Determination of the luciferase activity showed that TNFα stimulated significantly higher expression of luciferase activity of apoB reporter compared to the mock treated cells (Figure 3D), indicating that TNFα-CREBH signaling stimulates apoB expression. Together, these data suggest that CREBH may mediate the inflammatory cytokine signaling to overexpression of VLDL-apoB in metabolic inflammation.
Figure 3. CREBH mediates the inflammatory signaling pathways involved in overproduction of hepatic VLDL-apoB.

McA cells were fasted for 12h followed by treated with TNFα (20ng/mL) for 6 and 12h respectively. Cell lysates and total RNA were prepared for the following experiments. (A) Immunoblotting analysis of cellular full length CREBH (CREBH-F) and N-terminal of CREBH (CREBH-N) after 6h TNFα treatment. (B) Cellular apoB mRNA determined by qRT-PCR at 12h post-treatment. (C) Immunoblotting analysis of apoB in the culture media of McA cells after 12h TNFα treatment. (D) McA cells were pre-treated with mock (saline) or TNFα (20ng/mL) for 6h to activate CREBH. Cells were then transfected with a firefly luciferase reporter gene under the control of human apoB gene promoter while continuing with TNFα treatment. 36h post-transfection, cell lysates were examined for firefly luciferase activities and normalized to renilla luciferase activities. The data represent one out of three independent experiments (n=6–9/group). Right panels in (A) and (C) show quantification of CREBH or apoB protein signal intensity to loading control proteins. Results are shown as mean ± SEM. *P < 0.05 and **P < 0.01 versus controls.
Depletion of CREBH diminishes TNFα-induced VLDL-apoB overproduction in vivo
We next employed the CREBH-null mouse model to assess the regulatory role of CREBH on inflammation-induced VLDL secretion in vivo. WT and CREBH-null mice were treated with either vehicle (saline) or recombinant mouse TNFα (12μg/200g body weight) following a 6h fast. Livers were collected 5h after treatment. TNFα treatment significantly induced mRNA and protein expression of CREBH in the livers of WT mice compared to the vehicle-treated mice (Figures 4A and B). Activation of CREBH by TNFα was accompanied by a significant upregulation of apoB mRNA and protein in the WT mice (Figures C and D). In contrast, TNFα failed to enhance apoB expression in CREBH-null mice (Figures 4C and D), suggesting that CREBH is integral in mediating the regulatory effect of TNFα on apoB mRNA transcription. Upon examining the liver lipid contents, we found that TNFα induced significant accumulation of TG, but not cholesterol, in both WT and CREBH-null mice (Figures 4E and F). The increases of both apoB and lipid substrates resulted in increased secretion of plasma VLDL-apoB in the WT but not the CREBH-null mice (Suppl. figure 4). These observations further support the notion that depletion of CREBH compromises hepatic apoB mRNA and protein expression, leading to defective VLDL assembly and secretion in response to pro-inflammatory cytokine TNFα treatment.
Figure 4. Depletion of CREBH diminishes TNFα induced VLDL-apoB overproduction in vivo.
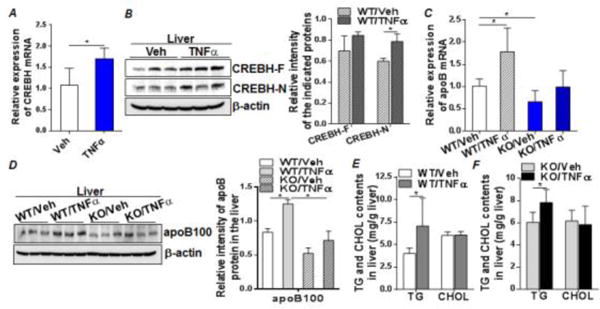
WT and KO mice were fasted for 6h. Mice were then given either recombinant mouse TNFα (12μg/200g body weight) or vehicle (Veh), saline, as control via ip. injection. Liver tissues were collected at 5 h post-treatment and liver lysates were prepared for analysis. (A and B) mRNA and protein expression of CREBH in the vehicle-treated and TNFα-treated WT mice. (C) ApoB mRNA in the liver tissues of WT and KO mice treated with vehicle or TNFα. (D) Protein expression of apoB in the livers of WT and KO mice treated with vehicle or TNFα. (E and F) Liver TG and cholesterol (CHOL) contents in the WT (E) or KO (F) mice treated with vehicle or TNFα. Right panels in (B) and (D) show quantification of CREBH or apoBprotein signal intensity to loading control protein β-actin. Results are shown as mean ± SEM, n=5–6/group. *P < 0.05 versus controls.
The CREBH-apoB signaling pathway mediates metabolic inflammation induced by LPS and HFD to VLDL overproduction
Bacterial LPS are capable of activating the APR, and CREBH has been reported to mediate LPS-induced acute-phase gene expression, including SAP and CRP [26]. Plasma LPS are moderately increased with consumption of HFD and are partially responsible for the development of HFD-induced metabolic syndrome [27]. To determine the potential role of the CREBH-apoB axis in LPS- and HFD-induced metabolic inflammation and the subsequent systemic hyperlipidemia, we treated WT and CREBH-null mice with vehicle (saline) or LPS (2μg/g body weight) and collected livers 10h after treatment. Analysis of hepatic CREBH showed that LPS treatment induced CREBH mRNA and protein expression, which was associated with a significant upregulation of apoB mRNA in WT mice (Figures 5B and C). In contrast, in CREBH-null mice, apoB mRNA expression was not induced by LPS (Figure 5C), indicating that an intact CREBH is essential for hepatic apoB mRNA expression in response to LPS treatment.
Figure 5. The CREBH-apoB signaling pathway mediates LPS induced inflammation to VLDL overproduction.
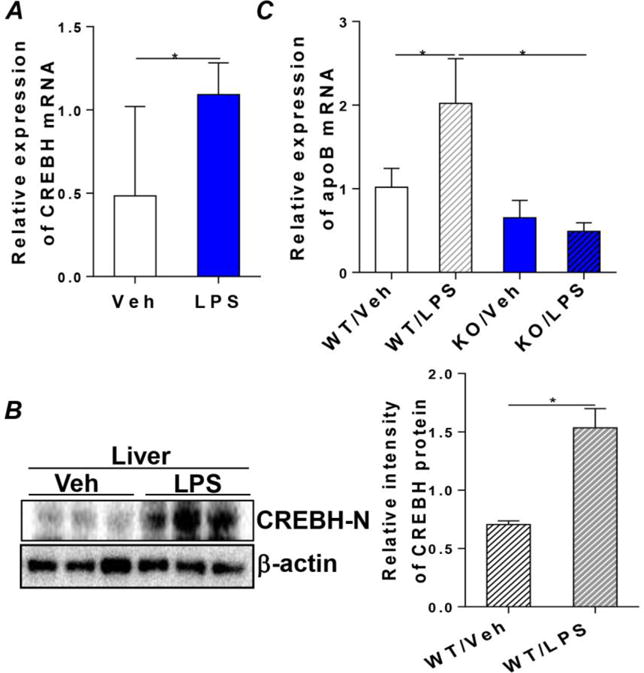
LPS was suspended in sterile pyrogen-free 0.9% NaCl. WT and KO mice were given a single dose of LPS (2μg/g body weight) or control vehicle (saline) through ip. injection. Liver tissues were collected 10h after LPS treatment and total RNA and liver lysates were prepared for analysis as follows. (A and B) mRNA and protein levels of CREBH in the vehicle or LPS treated WT mice. Right panel shows quantification of CREBH-N protein signal intensity to loading control protein β-actin. (C) mRNA expression of apoB in the WT and KO mice treated with vehicle or LPS. Results are shown as mean ± SEM, n=5–6/group. *P < 0.05 versus controls.
Next, we wanted to assess the impact of CREBH on HFD-induced hyperlipidemia. We fed the WT and CREBH-null mice with either chow or a HFD (60 percent calories from fat) for 7 weeks. High-fat feeding induced significant elevation of plasma TNFα in the WT mice (WT/HFD) compared to the chow-fed WT mice (WT/Chow) (Figure 6A). CREBH knockout diminished the responsive induction of TNFα in response to HFD (Figures 6A). This phenotype was associated with significant upregulation of hepatic CREBH mRNA and apoB protein expression in the WT but not the CREBH-null mice (Figures 6B and C). HFD further activated the hepatic de novo lipid synthesis in both WT and CREBH-null mice, evidenced by the accumulation of TG and cholesterol in the hepatocytes (Figure 6D). Increased hepatic lipid substrates usually would enhance assembly and secretion of VLDL into the circulation. However, this phenotype was observed only in the WT mice (Figure 6E). In the CREBH-null mice, despite the higher lipid levels in the hepatocytes, plasma VLDL-apoB was not increased in parallel (Figure 6E), suggesting a lack of proper VLDL assembly and secretion. Because CREBH has been reported to enhance expression of APR genes, such as CRP and SAP [22], we then tested the association between CREBH, apoB, and CRP in HFD-induced chronic metabolic inflammation. Measuring plasma CRP showed that an HFD induced a significant increase of CRP in the WT mice (Suppl. Figure 5A). Knockout of CREBH reduced plasma CRP levels, which remained at these lower levels when challenged with an HFD (Suppl. Figure 5A). These changes were similar to the pattern of plasma VLDL-apoB. Also investigated was whether the important acute-phase transcription factor Stat3 [28] plays a role in the CREBH-apoB signaling. CREBH depletion diminished HFD-induced Stat3 phosphorylation (Suppl. Figure 5B), suggesting that full activation of Stat3 depends on an intact signaling of CREBH. Together, these data suggest that CREBH depletion diminished metabolic inflammation induced by LPS and HFD. Reduced hepatic apoB biosynthesis and the subsequent defective VLDL-apoB secretion in CREBH-null mice were associated with downregulation of plasma CRP in response to nutritional lipid overload.
Figure 6. The CREBH-apoB signaling pathway mediates metabolic inflammation induced by HFD to VLDL overproduction.
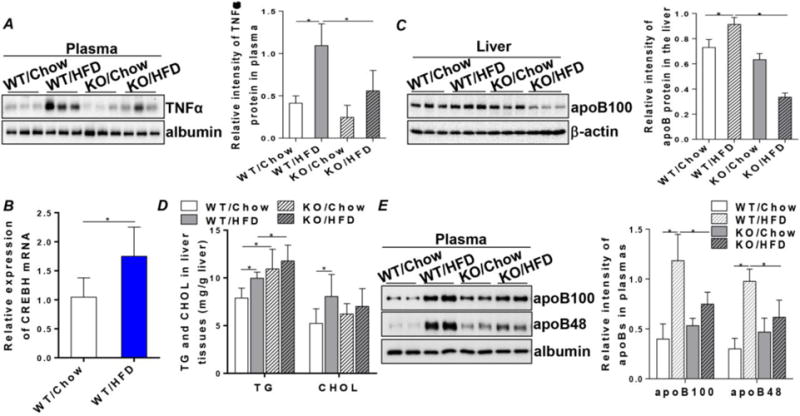
WT and KO mice were fed with either a chow or a HFD (60% caloric from fat, Dyets #103938) for 7 weeks. Blood samples and liver tissues were collected at the endpoint for analysis. (A) Immunoblotting of plasma TNFα. (B) mRNA expression of hepatic CREBH in the chow or high-fat fed WT mice. (C) Immunoblotting of apoB in the liver tissues of WT and KO mice fed chow or HFD. (D) TG and CHOL contents in liver tissues. (E) Immunoblotting of apoB100 and B48 in the plasmas of WT and KO mice fed chow or HFD. Right panels in (A), (C) and (E) show quantifications of TNFα or apoB protein signal intensity to the control proteins. Results are shown as mean ± SEM, n=5–6/group. *P < 0.05 versus controls.
Discussion
Chronic metabolic diseases, including obesity, insulin resistance, and diabetes, are commonly associated with chronic inflammation and hepatic overproduction of apoB, leading to pathological lipoprotein profiles such as VLDL overproduction and hyperlipidemia. Although perturbations of hepatic apoB metabolism have been well- accepted as a key contributing factor for the aberrant secretion of VLDL, the mechanistic link between metabolic inflammation and apoB overproduction is largely unknown. The assembly and secretion of VLDL take place in the hepatic ER and are regulated by multiple metabolic factors, including the abundance of apoB, the activity of MTP, and the availability of lipid substrates [29]. It is generally thought that under most metabolic stimuli, VLDL-apoB secretion is regulated post-transcriptionally [30], either through stabilization by binding to lipid substrates or degradation by proteasomal pathways [31]. In this study, we identified CREBH as the first cellular transcription factor that enhances apoB mRNA expression and translation via association with the CRE binding element in the promoter region of the apoB gene. We also showed for the first time that maintaining proper activity of CREBH is essential for VLDL-apoB biosynthesis and the subsequent transportation of hepatic lipids from hepatocytes to the circulation under physiological conditions. This is essential for maintaining hepatic lipid metabolic homeostasis in both fasting and postprandial states. When CREBH is constantly hyperactivated, such as during insulin resistance and metabolic inflammation, it enhances transcription and translation of hepatic apoB, leading to pathologic overproduction of VLDL and hyperlipidemia. This notion was further supported by the evidence obtained from CREBH-null mice, which showed that genetic depletion of CREBH significantly diminished the secretion of VLDL-apoB from hepatocytes in response to the inflammatory stimuli induced by TNFα and high-fat feeding, leading to lipid accumulation in the liver.
In addition to serving as a central organ for lipid metabolism, the liver also functions as an important constituent of the immune system in systemic inflammatory response. Hepatocytes are the major source of acute-phase proteins, the production of which is controlled by a variety of different cytokines released during the inflammatory process [28]. The Stat3- and NFκB-mediated signal transductions are critical components in the complex processing of intracellular signaling events. Expression of a subset of APR genes, including fibrinogens, α2-macroglobulin, or α1-antichymotrypsin, strictly depends on Stat3. This study identified the association of Stat3 with the CREBH-apoB axis and found that Stat3 is downstream of CREBH in the context of this work. How CREBH regulates expression of Stat3 and whether Stat3 plays a role in VLDL-apoB metabolism during metabolic inflammation requires further investigation.
APR induced by infection and inflammation causes a multitude of changes in the structure, composition, and function of lipoproteins. Many of these changes are similar to those observed during metabolic inflammation, which may be due to the likeness of the induced inflammatory cytokine profiles in these two inflammatory responses. CREBH has been shown to directly activate the transcription of acute-phase genes encoding SAP and CRP upon exposure to the pro-inflammatory cytokines IL-1β and IL-6 [22]. Although apoB has not been classified as an APR gene, an early and consistent increase of plasma VLDL has been observed during APR [32, 33]. Our study demonstrated that the CREBH-apoB axis is the signaling pathway that, at least partially, mediated upregulation of VLDL in APR induced by TNFα and LPS. In addition, this signaling pathway is also involved in mediating chronic metabolic inflammation that induces VLDL overproduction. Our group previously demonstrated that constant overproduction of apoB in hepatocytes stimulated by an atherogenic diet directly provokes hepatic ER stress and may contribute to the onset of hepatic insulin resistance [10]. CREBH is activated by ER stress [22, 34]. Therefore, a vicious cycle among metabolic inflammation, CREBH activation, VLDL overproduction, and ER stress could be developed, resulting in worsening of metabolic disorders.
CREBH is expressed dominantly both in the liver and small intestine. A recent study reported that overexpression of CREBH in the intestine prevents high-cholesterol diet-induced hypercholesterolemia by reducing Npc111 expression [35]. Thus, studying the impact of CREBH on VLDL biosynthesis in the intestine-specific or liver specific CREBH knockout mouse model may provide further insight into the association between CREBH and lipoprotein metabolism. In summary, this study demonstrated that the cAMP-signaling molecule CREBH transcriptionally regulates hepatic VLDL overproduction, which contributes to the hyperlipoproteinemia in metabolic inflammation associated disorders. This novel finding broadens the well-accepted concept that cellular abundance of apoB is regulated primarily at the co- and post-translational levels, as it demonstrated that expression of apoB is also regulated at the transcriptional level, at least in a state of metabolic stress. This novel mechanistic insight provides evidence that manipulating the CREBH-apoB axis may be effective in preventing and treating hyperlipidemia in metabolic syndrome.
Supplementary Material
Key Messages.
CREBH mediates inflammatory signaling to VLDL overproduction in metabolic stress.
Activation of CREBH in inflammation enhances mRNA and protein expression of apoB.
CREBH presents a potential novel therapeutic target for hyperlipoproteinemia.
Acknowledgments
We would like to thank Dr. Randal J. Kaufman (Howard Hughes Medical Institute, University of Michigan Medical Center, Ann Arbor, Michigan, USA) for kindly providing the cDNA constructs for pFlag-CREBH-WT and pFlag-CREBH-DN. This work was supported by an NIH grant P20 GM104320-01A, Hatch funds from USDA/NIFA and Layman funds from UNL to Q. Su.
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest related to the study.
References
- 1.Adiels M, Olofsson S-O, Taskinen M-R, Borén J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–1236. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- 2.Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 2011;22:353–363. doi: 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ginsberg HN, Zhang Y-L, Hernandez-Ono A. Regulation of plasma triglycerides in insulin resistance and diabetes. Arch Med Res. 2005;36:232–240. doi: 10.1016/j.arcmed.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Sattar N, Williams K, Sniderman AD, D’agostino R, Haffner SM. Comparison of the associations of apolipoprotein B and non-high-density lipoprotein cholesterol with other cardiovascular risk factors in patients with the metabolic syndrome in the Insulin Resistance Atherosclerosis Study. Circulation. 2004;110:2687–2693. doi: 10.1161/01.CIR.0000145660.60487.94. [DOI] [PubMed] [Google Scholar]
- 5.Sniderman A, Furberg C, Keech A, van Lennep JR. Apolipoproteins versus lipids as indices of coronary risk and as targets for statin treatment. The Lancet. 2003;361:777. doi: 10.1016/s0140-6736(03)12663-3. [DOI] [PubMed] [Google Scholar]
- 6.Banaszak LJ, Ranatunga WK. The assembly of apoB-containing lipoproteins: a structural biology point of view. Ann Med. 2008;40:253–267. doi: 10.1080/07853890701813070. [DOI] [PubMed] [Google Scholar]
- 7.Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest. 2008;118:316–332. doi: 10.1172/JCI32752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparks JD, Dong HH. FoxO1 and hepatic lipid metabolism. Curr Opin Lipidol. 2009;20:217. doi: 10.1097/MOL.0b013e32832b3f4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu W, Su Q, Rutledge AC, Zhang J, Adeli K. Glucosamine-induced endoplasmic reticulum stress attenuates apolipoprotein B100 synthesis via PERK signaling. J Lipid Res. 2009;50:1814–1823. doi: 10.1194/jlr.M800343-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su Q, Tsai J, Xu E, Qiu W, Bereczki E, Santha M, Adeli K. Apolipoprotein B100 acts as a molecular link between lipid — induced endoplasmic reticulum stress and hepatic insulin resistance. Hepatology. 2009;50:77–84. doi: 10.1002/hep.22960. [DOI] [PubMed] [Google Scholar]
- 11.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8:923–934. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basaranoglu M, Basaranoglu G, Sabuncu T, Sentürk H. Fructose as a key player in the development of fatty liver disease. World J Gastroenterol. 2013;19:1166–1172. doi: 10.3748/wjg.v19.i8.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dekker MJ, Su Q, Baker C, Rutledge AC, Adeli K. Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2010;299:E685–694. doi: 10.1152/ajpendo.00283.2010. [DOI] [PubMed] [Google Scholar]
- 14.McLaughlin T, Reaven G, Abbasi F, Lamendola C, Saad M, Waters D, Simon J, Krauss RM. Is there a simple way to identify insulin-resistant individuals at increased risk of cardiovascular disease? Am J Cardiol. 2005;96:399–404. doi: 10.1016/j.amjcard.2005.03.085. [DOI] [PubMed] [Google Scholar]
- 15.Qu S, Su D, Altomonte J, Kamagate A, He J, Perdomo G, Tse T, Jiang Y, Dong HH. PPARα mediates the hypolipidemic action of fibrates by antagonizing FoxO1. Am J Physiol Endocrinol Metab. 2007;292:E421–E434. doi: 10.1152/ajpendo.00157.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su Q, Rutledge AC, Dekker M, Adeli K. Apolipoprotein B: not just a biomarker but a causal factor in hepatic endoplasmic reticulum stress and insulin resistance. Clin Lipidol. 2010;5:267–276. [Google Scholar]
- 17.Su Q, Baker C, Christian P, Naples M, Tong X, Zhang K, Santha M, Adeli K. Hepatic mitochondrial and ER stress induced by defective PPARα signaling in the pathogenesis of hepatic steatosis. Am J Physiol Endocrinol Metab. 2014;306:E1264–E1273. doi: 10.1152/ajpendo.00438.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medzhitov R, Janeway CA. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 19.Yoo J-Y, Desiderio S. Innate and acquired immunity intersect in a global view of the acute-phase response. Proc Natl Acad Sci. 2003;100:1157–1162. doi: 10.1073/pnas.0336385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balci B. The modification of serum lipids after acute coronary syndrome and importance in clinical practice. Curr Cardiol Rev. 2011;7:272–276. doi: 10.2174/157340311799960690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziakas A, Gavrilidis S, Giannoglou G, Souliou E, Koskinas K, Gemitzis K, Hatzimiltiadis S, Efthimiadis G, Paraskevaidis S, Hatzitolios A, et al. Kinetics and prognostic value of inflammatory-sensitive protein, IL-6, and white blood cell levels in patients undergoing coronary stent implantation. Med Sci Monit. 2009;15:CR177–184. [PubMed] [Google Scholar]
- 22.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 23.Luebke - Wheeler J, Zhang K, Battle M, Si - Tayeb K, Garrison W, Chhinder S, Li J, Kaufman RJ, Duncan SA. Hepatocyte nuclear factor 4α is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology. 2008;48:1242–1250. doi: 10.1002/hep.22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen J, Tong X, Sud N, Khound R, Song Y, Maldonado-Gomez MX, Walter J, Su Q. Low-Density Lipoprotein Receptor Signaling Mediates the Triglyceride-Lowering Action of Akkermansia muciniphila in Genetic-Induced HyperlipidemiaHighlights. Arterioscler Thromb Vasc Biol. 2016;36:1448–1456. doi: 10.1161/ATVBAHA.116.307597. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Zhao M, Sud N, Christian P, Shen J, Song Y, Pashaj A, Zhang K, Carr T, Su Q. Glucagon regulates hepatic lipid metabolism via cAMP and Insig-2 signaling: implication for the pathogenesis of hypertriglyceridemia and hepatic steatosis. Sci Rep. 2016;6:32246. doi: 10.1038/srep32246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochrietor JD, Harrison KA, Zahedi K, Mortensen RF. Role of STAT3 and C/EBP in cytokine-dependent expression of the mouse serum amyloid P-component (SAP) and C-reactive protein (CRP) genes. Cytokine. 2000;12:888–899. doi: 10.1006/cyto.2000.0668. [DOI] [PubMed] [Google Scholar]
- 27.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 28.Bode JG, Albrecht U, Häussinger D, Heinrich PC, Schaper F. Hepatic acute phase proteins-regulation by IL-6-and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur J Cell Biol. 2012;91:496–505. doi: 10.1016/j.ejcb.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22–32. doi: 10.1194/jlr.r200014-jlr200. [DOI] [PubMed] [Google Scholar]
- 30.Blasiole DA, Davis RA, Attie AD. The physiological and molecular regulation of lipoprotein assembly and secretion. Mol Biosyst. 2007;3:608–619. doi: 10.1039/b700706j. [DOI] [PubMed] [Google Scholar]
- 31.Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50:S162–S166. doi: 10.1194/jlr.R800090-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahmed M, Jadhav A, Hassan A, Meng QH. Acute phase reactants as novel predictors of cardiovascular disease. ISRN Inflamm. 2012;2012 doi: 10.5402/2012/953461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 35.Kikuchi T, Orihara K, Oikawa F, Han SI, Kuba M, Okuda K, Satoh A, Osaki Y, Takeuchi Y, Aita Y, et al. Intestinal CREBH overexpression prevents high-cholesterol diet-induced hypercholesterolemia by reducing Npc1l1 expression. Mol Metab. 2016;5:1092–1102. doi: 10.1016/j.molmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.