

Abstract
Despite over 60 years of research on antiviral drugs, very few are FDA approved to treat acute viral infections. Rift Valley fever virus (RVFV), an arthropod borne virus that causes hemorrhagic fever in severe cases, currently lacks effective treatments. Existing as obligate intracellular parasites, viruses have evolved to manipulate host cell signaling pathways to meet their replication needs. Specifically, translation modulation is often necessary for viruses to establish infection in their host. Here we demonstrated phosphorylation of p70 S6 kinase, S6 ribosomal protein, and eIF4G following RVFV infection in vitro through western blot analysis and in a mouse model of infection through reverse phase protein microarrays (RPPA). Inhibition of p70 S6 kinase through rapamycin treatment reduced viral titers in vitro and increased survival and mitigated clinical disease in RVFV challenged mice. Additionally, the phosphorylation of p70 S6 kinase was decreased following rapamycin treatment in vivo. Collectively these data demonstrate modulating p70 S6 kinase can be an effective antiviral strategy.
1. Introduction
Bunyarviruses, a family of enveloped, single stranded, tripartite, negative sense RNA viruses, cause a variety of globally important diseases including hemorrhagic fever. Members of this family include Crimean-Congo hemorrhagic fever virus, Hantaan virus, and Rift Valley fever virus (RVFV) (Bouloy and Weber, 2010; Elliott, 1990 ; Ikegami et al., 2009). Of particular note is RVFV, an arthropod-borne virus that has left its original Sub-Saharan niche and has traversed the African continent and Arabian Peninsula (Boshra et al., 2015; Centers for Disease and Prevention, 2000; Chevalier, 2013; Gaudreault et al., 2015; Golnar et al., 2014). Outbreaks have been as large as tens to hundreds of thousands of cases (Abdel-Wahab et al., 1978; Himeidan et al., 2014) with a case fatality rate of 0.5 to 2% (Madani et al., 2003). In the worst outbreaks, case fatality rates have been close to 30% (Himeidan et al., 2014). Survivors of RVFV infection suffer several long-term sequelae including neurologic or ocular disease (Al-Hazmi et al., 2003; Kahlon et al., 2010; Madani et al., 2003; Mohamed et al., 2010). Additionally, the socioeconomic impact of these outbreaks is devastating with the loss of livestock, revenue, and the unraveling of local communities from such hardship (Chengula et al., 2013; Peyre et al., 2015; Sindato et al., 2011). The United States (US) has reservoir mosquito species for RVFV, Aedes/Culex genera, and amplifying hosts, cattle/sheep/goats, in plentiful supply, thus setting the stage for potential introduction into the US (Gaudreault et al., 2015; Golnar et al., 2014).
West Nile and Zika viruses are harbingers of the widespread impact of arboviral diseases; therefore it is imperative that effective antivirals are developed in the event of an inadvertent or intentional RVFV outbreak. The magnitude of this threat has been recognized by the National Institutes of Allergy and Infectious Disease in categorizing RVFV as a Category A agent. Currently, there are no FDA-approved vaccines or antivirals for the prevention or treatment of RVFV; it is crucial this shortfall is addressed to prepare the US for potential importation or introduction and to provide therapies in currently affected regions.
Existing as obligate intracellular parasites, viruses have evolved to manipulate host cell signaling pathways to meet their replication needs (Diamond and Farzan, 2013; Flint SJR, 2015a; Walsh et al., 2013). Lacking proteins necessary for viral protein production, viruses parasitize the host cell translational machinery (Flint SJR, 2015a; Walsh et al., 2013). Because of this requirement, a main cellular defense mechanism is translational shutoff (Flint SJR, 2015a; Walsh et al., 2013). Translation is tightly controlled within each cell and is modulated based on internal and external signals. One of the central protein pathways controlling translation is the mTORC1-p70 S6 kinase axis. This axis regulates initiation of translation following cell stimulation by growth factors, amino acids, or mitogens. p70 S6 kinase (p70 S6K), is the smaller of two S6K1 isoforms, p70 S6K and p85 S6K (Dowling et al., 2010; Ferrari et al., 1991; Pullen et al., 1998; Pullen and Thomas, 1997). p70 S6K is a modular kinase with four distinct areas, with activation controlled through the phosphorylation of amino acids in the catalytic, linker, and autoinhibitory pseudosubstrate domains (Pullen et al., 1998; Pullen and Thomas, 1997). Once activated, p70 S6K phosphorylates several substrates, including S6 ribosomal protein, which is a part of the 40S ribosomal subunit and important in modulation of translation (Dufner and Thomas, 1999; Fenton and Gout, 2011; Ferrari et al., 1991; Flotow and Thomas, 1992; Pullen and Thomas, 1997; Sonenberg and Hinnebusch, 2009). Another key regulator of translation just downstream of p70 S6K is eIF4G, a scaffold protein for the recruitment of translation initiation proteins. This protein, competes with the negative regulator of translation, 4E-BP (eukaryotic translation initiation factor 4E-binding protein 1), for the same binding sites on eIF4E. eIF4G binding to eIF4E contributes to translation activation, whereas when 4E-BP is in complex with eIF4E a decrease in overall translation occurs (Flint SJR, 2015a; Sonenberg and Hinnebusch, 2009).
In order to determine activation or inhibition of the p70 S6K pathway following RVFV infection, we examined phosphorylation sites on p70 S6K as well as on two downstream proteins S6 ribosomal protein and eIF4G, in vitro and in vivo. We found phosphorylation of p70 S6K, S6 ribosomal protein, and eIF4G following RVFV infection in vitro and in vivo. We used the mTORC-p70 S6 kinase axis translation inhibitor, rapamycin, to determine in vitro and in vivo efficacy of this drug against RVFV. In our in vitro model, rapamycin decreased viral protein levels as well as levels of infectious virus. Rapamycin treatment resulted in a 50% increase in survival in RVFV infected BALB/c mice in both low and high infectious dose viral challenge experiments. Elucidating cell signaling modulation following RVFV infection and demonstrating efficacy with an FDA approved drug is a promising development for future therapeutic use against this devastating viral hemorrhagic fever.
2. Materials and Methods
2.1 Cell Culture
H2.35 (ATCC, CRL-1995) cells were maintained in Dulbecco’s low glucose modified minimum essential medium + GlutaMAX with 200 nM dexamethasone and 4.0% heat inactivated fetal bovine serum (FBS). Vero cells (ATCC, CCL-81) were maintained in Dulbecco’s modified minimum essential medium (DMEM) supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. H2.35 cells were maintained at 33°C with 10% CO2 while Vero cells were maintained at 37°C with 5% CO2.
2.2 Viruses
Recombinant (r)MP12 virus was rescued and titered as previously described (Benedict et al., 2015; Ikegami et al., 2006; Kalveram et al., 2013). RVFV ZH501 was obtained from Drs. Stuart Nichol and Pierre Rollin, Centers for Disease Control and Prevention. Upon receipt, the virus was passaged once in Vero cells and sucrose purified prior to use (Benedict et al., 2015).
For viral infections of H2.35 cells (mouse hepatocytes), cells were cultured in 12 well plates and grown to 80–90% confluency. Cells were serum starved for 72 hours to synchronize cells in G0/G1 phase (Baer et al., 2012). The next day, cells were infected with either MP12 or ZH501 virus at a multiplicity of infection (MOI) of 5. Cells were incubated for 1 hour at 33°C and at 10% CO2, with rocking of cells every 15 minutes. Infectious media was removed, cells washed once with phosphate buffered saline (PBS) without Ca2+ and Mg2+, and complete media added. The cells were left to incubate and collected using the appropriate method for downstream applications at the specified time points.
2.3 Western Blot
Protein lysates were collected and analyzed by western blot as previously described (Austin et al., 2012). In brief, primary antibodies against p70 S6 Kinase (Thr389) (Cell Signaling Technology 9205), S6 ribosomal protein (Ser235/236) (Cell Signaling Technology, 4856), eIF4G (Ser1108) (Cell Signaling Technology, 2441), RVFV MP12 Antibody (IBT Bioservices 04-0001), or HRP-conjugated actin (Abcam, ab49900) were diluted 1:1000 in 5% bovine serum albumin in 1× TBS with 0.1% Tween-20 solution followed by the addition of the appropriate secondary antibody. The western blots were visualized by chemiluminescence using SuperSignal West Femto Maximum Sensitivity Substrate kit (ThermoScientific) and a Bio Rad Molecular Imager ChemiDoc XRS system (Bio-Rad) (Austin et al., 2012; Baer et al., 2012).
2.4 Reverse Phase Protein Microarray
Liver and/or spleen from mice were harvested on the appropriate day post-infection. Sections were placed in blue lysis buffer [T-PER (Pierce), 10% TCEP (Pierce), 2X SDS tris-glycine loading buffer (Invitrogen)] homogenized, spun at 12,000 rpm, heated at 100°C for 15 minutes, and removed from containment for RPPA analysis. Viral inactivation was validated through plaque and cytopathic effect assays prior to removal of samples from containment. RPPA were constructed initially with undiluted lysates for total protein concentration with Sypro Ruby Protein Blot stain (Invitrogen). The lysates were diluted to 0.5mg/mL total protein prior to printing the samples onto nitrocellulose coated slides (Grace Bio-Labs, Bend, OR) for analysis. RPPA construction and staining was done as previously described (Popova et al., 2010). For each antibody (Supplemental Figure 2A, B), the average pixel intensity value for negative control (staining with secondary antibody only) was subtracted from the average pixel intensity value for that specific antibody and then normalized to pyruvate dehydrogenase. Pyruvate dehydrogenase was used to normalize each spot on the RPPA due to its normal data distribution, as determined via histogram and Shapiro-Wilk test, and its low standard deviation across samples. All relative intensity values were scaled from 0–1 for comparing data between endpoints (Chiechi et al., 2012).
2.5 Plaque Assay
Extracellular supernatants were collected at the indicated time points and stored at −80°C. Viral titers were determined by plaque assay using Vero cells as previously described (Baer and Kehn-Hall, 2014).
2.6 qRT-PCR
Cells were plated in 12-well plates and mock-infected or infected with RVFV MP12 or ZH501. Supernatants and cells were collected at their respective timepoints and intracellular RNA was extracted using the RNeasy Mini Kit (Qiagen) according to manufacturer’s protocol. Extracellular RNA was extracted from supernatants using the MagMAX™-96 Viral RNA Isolation Kit (Thermo Fisher Scientific AM1836) according to manufacturer’s protocol. Absolute quantification of RVFV genomic copies was determined by RT-quantitative (q)PCR as previously published (Shafagati et al., 2013).
2.7 Immunohistochemistry
Tissue sections (5μm) were deparaffinized in xylene, rehydrated in graded ethanol (100%, 95%, and 70%) and rinsed in water. Tissue sections were incubated in 1× Rodent Decloaker (Biocare Medical; SKU: RD913) for 15 minutes at 110ºC. Slides were then washed (Dako K8007) and blocked in 3% hydrogen peroxide for 5 minutes, followed by another wash and a serum free protein block (Dako X0909) of 5-minutes duration. Slides were incubated with the primary rabbit polyclonal antibody against RVFV MP12 vaccine strain (IBT Bioservices, MD, 04-0001) at a dilution of 1:1000 for 30 minutes. The sections were then rinsed and incubated for thirty minutes with the peroxidase-labeled polymer (secondary antibody). The sections were rinsed, covered and incubated with substrate chromogen solution for five minutes. The sections were then rinsed, stained with hematoxylin, and rinsed again. Sections were then dehydrated and cover-slipped.
For detection of phospho-p70 S6 Kinase, a rabbit polyclonal against p70 S6 Kinase (Thr421) (Thermo Fisher Scientific (PA5-36835) was used at a dilution of 1:100, for 60 minutes at room temp. After deparaffinization and rehydration as above, antigen retrieval was performed in pH 9 solution for 60 minutes at 95°C. Tissue sections were stained on a Dako Autostainer using the Envision+HRP detection system following manufacturer’s directions and cover-slipped.
2.8 Animal studies
For the validation experiments and the efficacy studies, six to eight week old female BALB/c mice were obtained from either The Jackson Laboratory (low infectious dose/study 1) or Envigo Laboratories (high infectious dose /study 2). Mice were infected with RVFV ZH501 by sub-cutaneous injection. Infectious dose was verified by performing a plaque assay on the viral inoculum and is noted in the figure legends. In the efficacy studies, mice were pretreated 1h prior to infection, and each day thereafter with 10 mg/kg of rapamycin or solvent control intraperitoneally until day 10 (low dose infection/study 1) or day 14 (high infectious dose /study 2). Weight and clinical score were monitored daily. Mice scoring above a 5 on the clinical scoring scale were monitored at least 2 times a day, with both AM and PM checks done. All personnel checking mice were unblinded.
All animal experiments were carried out in animal bio-safety level 3 (BSL-3) facilities at George Mason University’s National Center for Biodefense and Infectious Diseases Biomedical Research Laboratory in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals following GMU approved IACUC protocols.
2.9 Cytotoxic concentration 50 (CC50) assays
Cells were plated in a white-walled 96-well plate and allowed to incubate at 33°C and 10% CO2 overnight. H2.35 cells were treated with drug concentrations starting at 100 micromolar (μM) with 1:2 serial dilutions down to 1.56 μM, and drugs were incubated with cells for 24 hours. At 24 hours, all cells were analyzed using Cell Titer-Glo Cell Luminescent Viability Assay (Promega, G7570) according to vendor’s instructions. This assay measures relative ATP levels. Briefly, an equal volume of room temperature media and Cell Titer-Glo reagent were added to the cells. The plate was shaken for 2 min on an orbital shaker followed by a 10 minute room temperature incubation. Viability was detected via luminescence detection using the DTX 880 multimode detector (Beckman Coulter) and percent viability was calculated relative to the DMSO control.
2.10 Effective Concentration 50 (EC50) Assays
Cells were plated in a white-walled 96-well plate and allowed to incubate at 33°C and 10% CO2 overnight. H2.35 cells were pre-treated for 1 hour with drug concentrations starting at 100 μM with 1:2 serial dilutions down to 1.56 μM. Cells were then infected with RVFV MP12 ΔNSs-Luc (MP12 lacking the NSs gene and replaced by a gene encoding Renilla luciferase) at a multiplicity of infection (MOI) of 1. Cells were incubated for 1 hour at 33°C and at 10% CO2. Infectious media was removed, cells washed once with PBS without Ca2+ and Mg2+, and drug was re-applied. Cells were analyzed at 18 hpi using Renilla-Glo® Luciferase Assay System (Promega) according to vendor’s instructions. Briefly, an equal volume of room temperature media and Renilla-Glo reagent was added to the cells. The plate was shaken for 2 min on an orbital shaker followed by a 10 minute room temperature incubation. Luminescence was detected via luminescence detection using the DTX 880 multimode detector (Beckman Coulter) and percent luminescence was calculated relative to the DMSO control.
2.11 Treatments
Rapamycin was obtained from LC Laboratories, catalog #R-5000. For in vitro studies, H2.35 cells were treated for 1 hour with DMSO or rapamycin (10 or 30 μM). After pretreatment, drug was removed, and cells were infected for 1 hour. After washing out virus inoculum, cells were cultured in complete media with rapamycin and cells collected at the respective assay time points.
For in vivo efficacy studies, rapamycin was dissolved in bio-grade 100% ethanol at 20 mg/ml concentration and aliquoted for daily use. Rapamycin treated mice received 10 μl of 10mg/kg mixture daily while the control group received 10 μl of vehicle (0.25% PEG, .25% Tween 80 in ultrapure water) only. Each mouse was given an intraperitoneal dose of either rapamycin or vehicle daily, beginning on day 0 at approximately 1 hour prior to infection and continuing daily until day 10 in the low infectious dose/study 1 (Figure 6) and day 14 in the high infectious dose/study 2 (Figure 7).
Figure 6. In vivo efficacy of rapamycin, low dose study.

Female BALB/c mice, 6–8 weeks of age, were randomly distributed into treatment groups consisting of 10 mice per group. Mice were infected via the subcutaneous route with a low dose of RVFV ZH501 (150 PFU/mouse). Health scores and weights were assessed daily. The endpoint was day 16, death, or euthanasia. The survival curves are shown in panels A, % of weight maintained over the course of the study is shown in panel B, and the clinical scoring chart for the vehicle control group and rapamycin treated group are shown in panel C. Log-rank (Mantel-Cox) test was used to determine statistical significance of the survival curve and a two-way ANOVA analysis was used to evaluate the change in weight loss, *p-value ≤ 0.05.
Figure 7. In vivo efficacy of rapamycin, high dose study.

Female BALB/c mice, 6–8 weeks of age, were randomly distributed into treatment groups consisting of 10 mice per group. Mice were infected via the subcutaneous route with a high dose of RVFV ZH501 (1,500 PFU/mouse). Health scores and weights were assessed daily. The endpoints were day 21, death, or euthanasia. The survival curve is shown in panels A, % of weight maintained over the course of the study is shown in panel B, and the clinical scoring chart for the vehicle control group and rapamycin treated group are shown in panel C. Log-rank (Mantel-Cox) test was used to determine statistical significance of the survival curve and a two-way ANOVA analysis was used to evaluate the change in weight loss, *p-value ≤ 0.05.
2.12 Statistics
Log-rank statistical analysis was used to evaluate the survival curve of each study. A two-way ANOVA was used to determine statistical significance of weight change, viral titers in vivo, and p70 S6K signaling in vivo. All other statistical analysis was calculated using unpaired, two-tailed student t-tests using Graphpad Prism software (version 7). RPPA analysis (Figure 5) was performed using RAS template version (Chiechi et al., 2012).
Figure 5. p70 S6K pathway is activated following RVFV infection in vivo.

(A) BALB/c mice (groups of 3) were subcutaneously infected with RVFV ZH501 (1000 PFU/mouse). Control mice were sacrificed on day 0 and infected mice on days 3, 4, and 5. Liver and spleen were collected and placed in blue lysis buffer. Tissues were homogenized, spun, transferred to new tubes, heated at 100°C for 15 minutes, and then frozen at −80°C prior to RPPA analysis. Error bars represent standard error of the mean; n=3. (B) and (C) Immunohistochemistry for p70S 6K (Thr421) was performed as an additional determinant of activation of p70 S6K within the liver of infected mice. Panel B is an uninfected control and panel C is an infected day 4 sample. Arrows indicate an increase in p70 S6K staining.
3. Results
3.1 p70 S6K signaling is activated following RVFV infection in vitro
Previous studies have shown that there are alterations in phosphorylation status of p70 S6K in human small airway epithelial cells following RVFV infection (Popova et al., 2010). Work by Dr. Cherry’s group has also demonstrated that translational pathways are altered following RVFV infection (Hopkins et al., 2015). Given the importance of this pathway, follow-up studies were undertaken to determine if alterations in key signaling proteins were occurring following RVFV in a time dependent manner. A BALB/c mouse hepatocyte cell line (H2.35 cells) was chosen for analysis, as the liver is the primary target for RVFV infection (Al-Hazmi et al., 2003; Coetzer, 1982; Coetzer et al., 1982; Iwasa, 1959; Madani et al., 2003; Mims, 1957; Smith et al., 2010). To closely mimic hepatocytes in vivo, cells were serum starved for 72 hours to synchronize them to G0/G1 phase (Baer et al., 2012). Both the live attenuated strain of RVFV, MP12, (Figure 1A) and the virulent strain, ZH501 (Figure 2A), displayed robust replication in H2.35 cells. However, MP12 replication did lag behind ZH501 replication at 9 hours post infection (hpi). p70 S6K and its substrate target, S6 ribosomal protein, were phosphorylated following RVFV infection as was eIF4G, a major scaffolding protein required for translation (Flint SJR, 2015a; Gingras et al., 1999; Sonenberg et al., 1978) (Figure 1B–C, 2B–C). Phosphorylation of all three proteins was robust at later time points, e.g. 18 and 24 hpi. These results demonstrate the activation of p70 S6K signaling in mouse hepatocytes following RVFV infection.
Figure 1. Growth kinetics of MP12 and its effect on p70 S6K signaling in H2.35 cells.
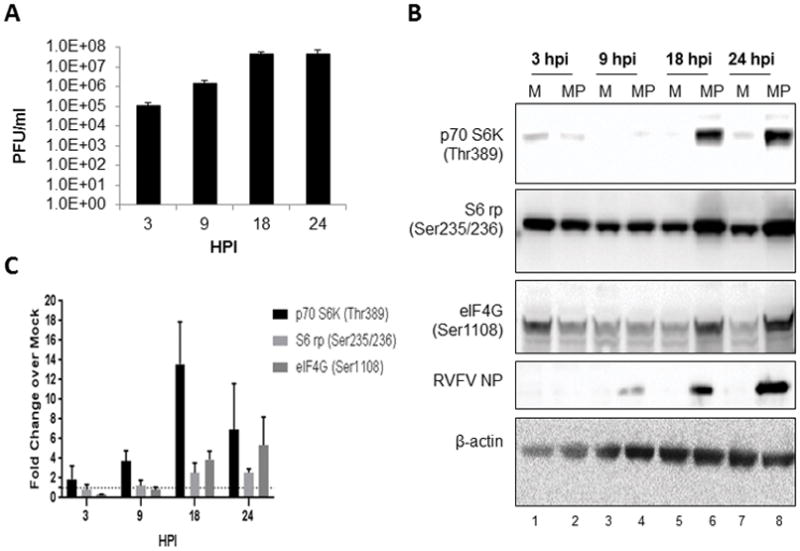
(A) H2.35 cells were infected with MP12 at an MOI 5.0. Cell lysates were collected 3, 9, 18 and 24 hpi and viral titers were measured by standard plaque assay (B) H2.35 cells were serum starved for 72 hours and then either mock-infected or infected with MP12 at an MOI 5.0. Cell lysates were collected at 3, 9, 18 and 24 hours post-infection and analyzed by western blot. Membranes were probed p70 S6K (Thr389), S6 ribosomal protein Ser235/236 [noted as S6 rp (Ser235/236)], eIF4G (Ser1108), RVFV nucleoprotein (NP), and β-actin as a loading control. (C) Data plotted represents fold change over mock from blots in (B). Mean values and standard deviations of the chemiluminescent signal intensity of three biological replicates are represented. All values were normalized to the loading control, β-actin, with concomitant background signal subtraction. Error bars represent the means ± SD; n=3.
Figure 2. Growth kinetics of ZH501 and its effect on p70 S6K signaling in H2.35 cells.
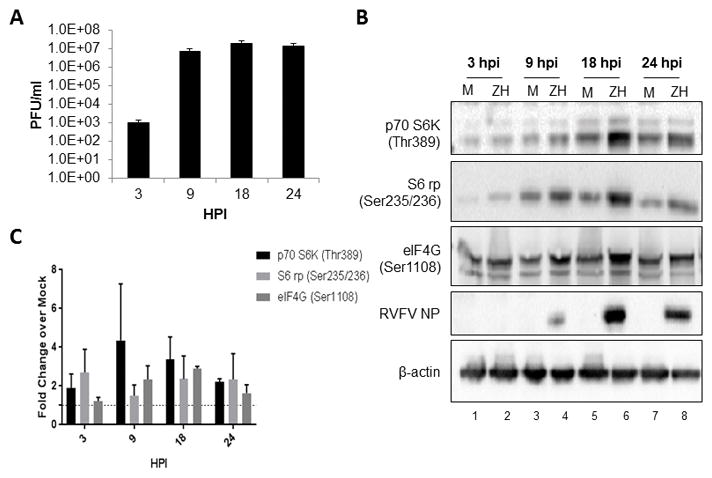
(A) H2.35 cells were infected with ZH501 at an MOI 5.0. Cell lysates were collected 3, 9, 18 and 24 hpi and viral titers were measured by standard plaque assay. (B) H2.35 cells were serum starved for 48–72 hours and then either mock-infected or infected with ZH501 at an MOI 5.0 for one hour. Cell lysates were collected at 3, 9, 18 and 24 hours post-infection and analyzed by western blot. Membranes were probed p70 S6K (Thr389), S6 rp (Ser235/236), eIF4G (Ser1108), RVFV NP, and β-actin as a loading control. (C) Data plotted represents fold change over mock from blots in (B). Mean values and standard deviations of the chemiluminescent signal intensity of three biological replicates are represented. All values were normalized to the loading control, β-actin, with concomitant background signal subtraction. Error bars represent the means ± SD; n=3.
3.2 Rapamycin inhibits p70 S6K signaling and RVFV infection in vitro
Our next step was to identify potential drug candidates to inhibit protein translation as a targeted therapeutic intervention. Rapamycin, an FDA-approved drug often used to prevent organ transplant rejection, was chosen because of its known safety profile (Alamo et al., 2009; Juwana et al., 2010; Koenig et al., 2012; Yang et al., 2015) and its documented inhibition of p70 S6K enzymatic activity on S6 ribosomal protein, and eIF4G (Chaturvedi et al., 2009; Polakiewicz et al., 1998; Raught et al., 2000). Rapamycin binds FKBP12, followed by binding to the mTORC1 complex, with subsequent inhibition of mTORC1 and its downstream substrates (Weichhart et al., 2015). CC50 and EC50 assays were performed to rule out mouse hepatocyte cell toxicity and determine an appropriate dose to use in our in vitro efficacy studies. H2.35 were very tolerant of rapamycin treatment, with only a ~20% decrease in viability observed at the highest concentration tested (100 μM), resulting in a CC50 of >100 μM (Supplemental Figure 1). Rapamycin displayed an EC50 of 11 μM against RVFV MP12-luc (Figure 3A). In order to further test drug efficacy in vitro, H2.35 cells were pre-treated with rapamycin and following MP12 infection, cells were cultured with complete media supplemented with the inhibitor until harvested. Protein phosphorylation (Figure 3B), infectious titers (Figure 3C–D), and intra-and extracellular viral RNA levels (Figure 3E–F) were evaluated at 18 hpi. Phosphorylation of p70 S6K (Thr 389), and downstream protein S6 ribosomal protein (Ser 235/235), as well as eIF4G (Ser 1108) dramatically decreased following treatment with rapamycin (Figure 3B). Additionally, the levels of RVFV N protein were decreased following rapamycin treatment (Figure 3B). The level of infectious viral titer was reduced by approximately 2 to 3 logs as the concentration of rapamycin was increased (Figure 3C). A decrease in viral titer was also observed with RVFV ZH501 (Figure 3D). RNA levels closely followed this trend as genomic copies per reaction decreased 2 to 3 logs as drug concentration increased (Figure 3E–F). A decrease in viral protein, RNA and infectious titers are consistent with inhibition of viral translation. These results demonstrate that rapamycin inhibition of p70 S6K decreases RVFV replication.
Figure 3. Rapamycin inhibits p70 S6K signaling and RVFV infection in vitro.
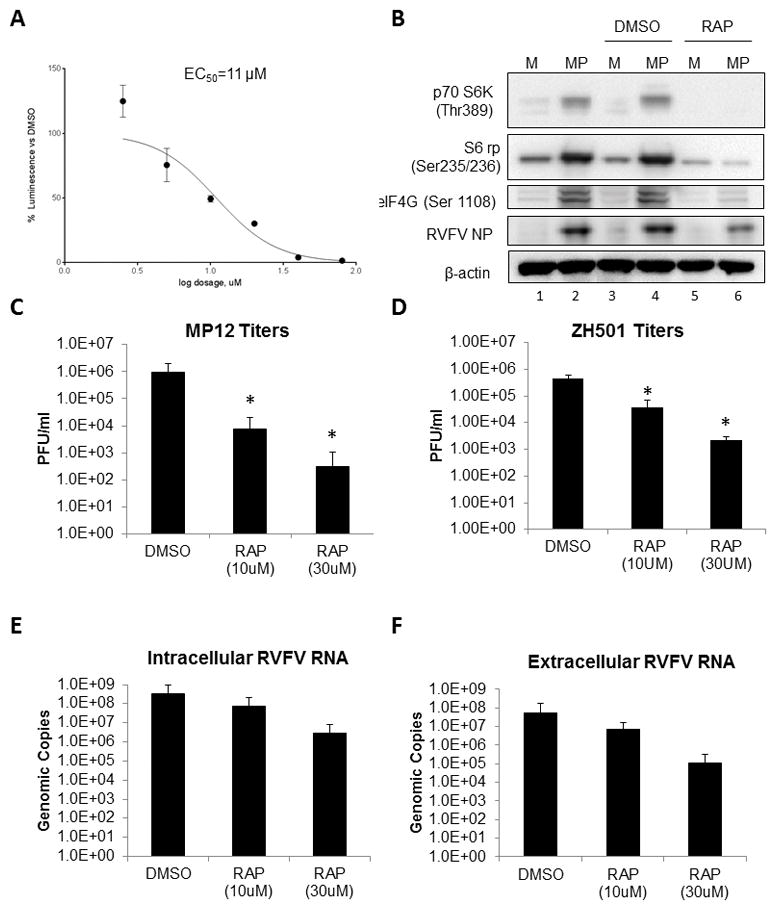
(A) To determine the EC50 of rapamycin (RAP), H2.35 cells were allowed to incubate at 33°C and 10% CO2 overnight. H2.35 cells were pre-treated for 1 hour with drug concentrations starting at 100 μM with 1:2 serial dilutions down to 1.56 μM. Cells were then infected with MP12 virus at a MOI of 1. Infectious media was removed, cells washed once and drug was re-applied. Cells were analyzed at 18 hpi using the Renilla Glo Assay. Luminescence is presented calculated relative to the DMSO control. (B) H2.35 cells were serum starved for 72 hours and then were pre-treated with either DMSO or 10 μM RAP. Cells were infected with MP12 (MOI 5) for one hour, followed by removal of viral inoculum, and addition of growth medium containing DMSO and 10 uM RAP. At 18 hpi, cell lysates were collected for western blot analysis. (C–D) H2.35 cells were serum starved for 72 hours and then were pre-treated with either DMSO, 10 μM RAP, or 30 μM RAP. Cells were infected at an MOI of 5 with (C) MP12 or (D) ZH501 for one hour, followed by removal of viral inoculum, and addition of growth medium containing DMSO, 10 μM RAP, or 30 μM RAP. At 18 hpi, supernatants were collected for standard plaque assay. The mean and SD (N=4) are plotted. For ZH501, 30 μM treatments, N=2. *p-value ≤ 0.05 (E) At 18 hpi, cell lysates were collected in RLT buffer for RNA extraction and qRT-PCR was performed to determine viral genomic copies (F); At 18 hpi, supernatants were collected andviral RNA extracted and quantified by qRT-PCR.(F). The mean and SD (N=5) are plotted for panels E and F.
3.3 RVFV animal model validation
In order to determine if these same cellular pathways were activated in vivo following RVFV infection, we first chose to validate viral kinetics within the BALB/c mouse model of infection (Smith et al., 2010) using the virulent ZH501 strain. Duly informed, we could assay protein phosphorylation on the days of peak viral levels. BALB/c mice were randomly assigned to groups of 3 and infected with 1,000 plaque forming units (PFU) of RVFV ZH501 strain subcutaneously in the right flank to mimic both the mosquito-borne route of infection and pathogenesis following mosquito transmission (Reed et al., 2013; Smith et al., 2010). Three uninfected control mice were sacrificed on day 0 and three infected mice were sacrificed on each day from days 1–8. Liver, spleen, brain, serum, and the right inguinal lymph node were harvested from each mouse and sections were either placed in 10% neutral buffered formalin for histologic evaluation or were used to determine viral titers in each organ.
Viral titers peaked 4 days post-infection (dpi) within the liver and within other tissues on 5 and 6 dpi (Figure 4A). Histologic examination revealed groups of hepatocytes with shrunken, hypereosinophilic cytoplasm and condensed, eccentrically placed nuclei (arrow) which is consistent with cells undergoing apoptosis (Figure 4B). Additionally, adjacent to these areas were nuclei containing 3 to 4 μm hypereosinophilic, intranuclear inclusions (circle) consistent with previously described RVFV protein inclusions (Daubney, 1931; Smith et al., 2010; Swanepoel and Blackburn, 1977). Histologic findings in the spleen consisted of minimal lymphocytolysis and lymphoid depletion on day 4 (data not shown). By 3 dpi, rare hepatocytes displayed intracytoplasmic RVFV immunohistochemical staining (Figure 4D), while 4 dpi this staining had spread throughout the liver to include large groups of cells displaying strong, specific, intracytoplasmic RVFV immunohistochemical staining (Figure 4E). Additionally, viral levels decreased in all organs on days 7 and 8 with the exception of the brain, which showed an increase in viral titers on day 8 (Figure 4A) indicative of the delayed-onset neurologic phase of the disease. These results support prior study results utilizing the BALB/c mouse model for RVFV infection (Smith et al., 2010).
Figure 4. RVFV model validation.
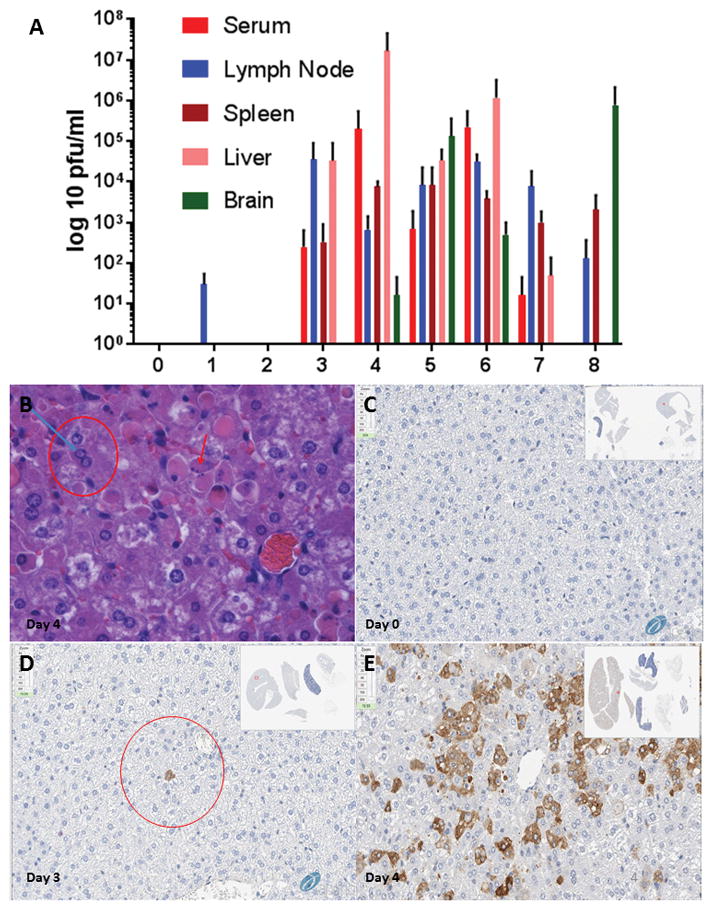
(A) BALB/c mice were randomly assigned to groups of 3 and infected with 1,000 PFU of RVFV ZH501 subcutaneously. Three uninfected control mice were sacrificed on day 0 with infected mice being sacrificed from days 1–8. Liver, spleen, brain, serum, and lymph node were harvested from each mouse and sections were either placed in 10% neutral buffered formalin for histologic evaluation or were used to determine viral titers in each organ via plaque assay. (B) Liver, Infected Mouse, Day 4: Histologic examination revealed groups of hepatocytes with shrunken, hypereosinophilic, cytoplasm and condensed, eccentrically placed nuclei (red arrow) consistent with groups of cells undergoing apoptosis. Adjacent to these areas were nuclei containing 3 to 4 um, hypereosinophilic, intranuclear inclusions (blue arrow/red circle) consistent with RVFV protein inclusions. (C) Uninfected Liver – Day 4, RVFV Immunohistochemistry. (D) Day 3 PI – rare hepatocytes (circle) displayed intracytoplasmic immunohistochemical staining. (E) Day 4 PI – positive staining throughout the liver to include large groups of cells displaying strong, specific, intracytoplasmic immunohistochemical staining.
3.4 p70 S6K signaling is activated in RVFV infected BALB/c mice
In order to evaluate in vivo translation modulation following RVFV infection, we used reverse phase protein microarrays (RPPA). RPPA has been used extensively in cancer research to quantify signal transduction kinases and their post-translationally modified forms (Espina et al., 2009; Espina et al., 2008; Petricoin et al., 2005; Wulfkuhle et al., 2008). Our goal was to use this same methodology to determine if p70 S6K signaling was altered following viral infection and identify and quantify potential therapeutic targets. The protein endpoints evaluated by RPPA are listed in Supplemental Figure 2.
BALB/c mice were randomly distributed into groups of 3 with subsequent serial sacrifice of control mice on day 0 and infected mice sacrificed on days 3, 4, and 5 to coincide with the peak in viral titers as noted from our validation experiment (Figure 4A). Liver and spleen were the focus of this study due to noted high viral levels in our previous experiment (Figure 4A). RPPA analysis was performed on the tissue lysates. All three phospho-protein targets which were phosphorylated in our in vitro model, p70 S6K (Thr389), S6 ribosomal protein (Ser235/236), and eIF4G (Ser1108), were phosphorylated in the liver following RVFV infection (Figure 5A). While the increase in phosphorylation events was not statistically significant, a time dependent increase in phosphorylation was observed. p70 S6K signaling in the spleen was not as clear-cut with significant variability occurring between samples, which suggest that either the pathway is not activated in the spleen following RVFV infection or there are high levels of constitutive activation that are complicating the analysis.
Immunohistochemistry (IHC) was also performed on p70 S6K (Thr421) as an additional determinant of upregulation within the liver of infected mice. An IHC compatible validated antibody for p70SK (Thr389) was not available, therefore we opted to evaluate p70 S6 (Thr421) because this particular phosphorylation site is in the pseudosubstrate domain, and once phosphorylated, is thought to activate p70 S6K (Dufner and Thomas, 1999; Polakiewicz et al., 1998; Pullen and Thomas, 1997). An increase in p70 S6K (Thr421) staining was noted on day 4 in comparison to mock, uninfected controls from day 0 (Figure 5B–C). A semi-quantitative analysis of the IHC p70 S6K staining confirmed that there was an increase in p70 S6K phosphorylation in liver tissue following RVFV infection (Supplemental Figure 3).
3.5 In vivo efficacy of rapamycin against RVFV challenge
The ability of rapamycin (10 mg/kg) to protect mice against a low infectious dose (~150 PFU/mouse) and a high infectious dose (~1500 PFU/mouse) RVFV challenge was tested. Female BALB/c mice, 6–8 weeks of age, were used in treatment groups consisting of 10 mice per treatment group. Health scores and weights were assessed daily.
In the low infectious dose study, all mice treated with of rapamycin survived until the end of the study. Five of ten mice succumbed to infection in the vehicle control group (Figure 6A), with one mouse displaying hind-limb paralysis on day 15 post-infection (Figure 6C). All rapamycin treated mice gained weight over the course of the study, while the vehicle control group had lower weight averages overall (Figure 6B). The change in weight was statistically different between the two groups. From a clinical perspective, both the vehicle control group and the rapamycin treated groups showed signs of illness during the acute and delayed stages of disease, mirroring the two stages of disease described by Smith et al. (Smith et al., 2010). However, the illness was more pronounced in both stages of disease in the vehicle control group versus the rapamycin treated mice (Figure 6C).
In the high infectious dose study, 6 of 10 mice treated with rapamycin survived until the end of the study (Day 21), with one mouse succumbing at 15 dpi (Figure 7A). This mouse, (Figure 7D, mouse 2) lost weight from day 8 onwards, but other than weight loss and a ruffled, wet hair coat, displayed no other overt clinical signs of illness per our in-house scoring system (Figure 7C). In the control group, 7 of 10 mice succumbed to infection by day 12, with two mice on days 8 and 9 showing neurologic illness manifesting as a head tilt and spinning (Figure 7C). All mice in the rapamycin treatment group gained weight until day 10, at which point the overall weight gain of this group decreased (Figure 7B). The change in weight was statistically different between the two groups. One notable difference was observed in the rapamycin treated group; this group displayed wet, ruffled hair coats beginning 10 dpi. In contrast, this ruffled, wet hair appearance was not observed in treated mice in the low infectious dose experiment. The leveling off of weight gain at day 10 also did not occur in the low infectious dose study (Figure 6B), in which treatments ended on day 10. In fact, at day 10 when treatments ended in the low infectious dose challenge study, weight steadily increased until study termination (Figure 6B). This suggests drug toxicity may have been an issue in the rapamycin treatment group in the high infectious dose study. In the high infectious dose study, rapamycin treatments were continued past day 10 in an attempt to prolong the potential therapeutic effect of rapamycin through the late/delayed stage neurologic phase of the disease, but it seems this extension of treatment had a somewhat deleterious effect when continued past 10 days.
From a clinical perspective, overall, rapamycin treated mice showed fewer signs of illness during the course of the study (Figure 7C). While neurologic disease was again noted in mice in the vehicle control group, at no time did any mice in the rapamycin treated group display signs of neurologic disease as seen in the vehicle control mice (Figure 7C). Collectively these results demonstrate rapamycin increased survival and mitigated overall clinical illness.
3.6 p70 S6K signaling is altered following rapamycin treatment in vivo
We wanted to determine what effect rapamycin would have on the activation status of the aforementioned p70 S6K and downstream proteins S6 ribosomal protein and eIF4G in BALB/c mice following RVFV challenge. During the high dose efficacy study, 20 mice (10 treated and 10 control mice) were serial sacrificed on days 4 and 6 with subsequent collection of liver, spleen, and serum to determine viral titers and the phosphorylation status of these proteins following rapamycin treatment. There was no difference in viral titers in the liver, spleen, or serum following rapamycin treatment at either time point analyzed (Supplemental Figure 4). However, there was an overall, albeit not statistically significant, increase in phosphorylated p70 S6K (Thr389) in the liver and spleen at 4 dpi (Figure 8A and B). The exact reason for this increase is unclear, but one possibility is a compensatory response. Additionally, there was a trend in decreasing levels of phosphorylated p70 S6K (Thr389) and S6 ribosomal protein (Ser235/236) detected in the liver and spleen of treated mice (Figure 8A–D) at 6 dpi, although this decrease only reached statistical significance in the spleen. eIF4G (Ser1108) phosphorylation was not altered by rapamycin treatment at 4 or 6 dpi. Collectively, these data suggest the p70 S6K signaling pathway is being modulated through the use of rapamycin and this modulation corresponds with decreased RVFV pathogenesis.
Figure 8. Pathway modulation screening in the liver and spleen following rapamycin treatment.

Female BALB/c mice, 6–8 weeks of age, were infected via the subcutaneous route with a high dose of RVFV ZH501 (1,500 PFU/mouse). Mice were randomly distributed into groups of 5 with subsequent serial sacrifice of vehicle treated control mice and rapamycin (10mg/kg) treated mice on days 4 and 6. Liver and spleen were collected and placed in blue lysis buffer for downstream RPPA analysis. Data plotted represents means and standard deviations from 5 animals per condition. Black circles and red squares represent vehicle treated or rapamycin treated mice, respectively. Phospho-protein levels in the liver (A, C, E) or spleen (B, D, F) of both vehicle control and rapamycin treated groups on days 4 and 6 post-infection are displayed. A two-way ANOVA analysis was used to evaluate changes in protein phosphorylation, *p-value ≤ 0.05.
4. Discussion
Despite intensive research efforts, there are only a handful of viruses for which effective therapeutics exist (Bonhoeffer et al., 1997; De Clercq, 2013; Drews, 2000; Flint SJR, 2015b). Most of these treatments are for chronic viral infections such as HIV and hepatitis C (Das and Arnold, 2013a, b; Flint SJR, 2015b; Halegoua-De Marzio and Hann, 2014; Manns and von Hahn, 2013). The necessity for and dearth of effective and approved treatments for acute viral infections was on full display after the Ebola outbreak in West Africa in 2014. The United States has also seen other mosquito-borne viruses (West Nile, dengue, and Zika viruses) emerge or re-emerge over the last twenty years, and effective antivirals against these viruses are direly needed as a matter of public health (Butterworth et al., 2016; Jones et al., 2016; Millman et al., 2016; Murray et al., 2010; Weaver et al., 2016). RNA viruses are inherently error prone, therefore they often quickly mutate in the presence of drugs that target viral proteins and viral enzymes (De Clercq, 2013; Flint SJR, 2015b). Viruses require the host cell molecular apparatus to replicate and survive, and our lab is focusing on host based therapeutics to leverage this viral need against them. We hypothesize it will be harder for viruses to mutate around drugs targeted at host cell pathways, and as such, drugs altering these pathways will be an effective therapeutic strategy. On-going studies in our laboratory will aid in confirming this hypothesis.
In virally infected cells, cell signaling subversion and switching on/off of pathways occurs to benefit the virus and its propagation. Our work shows that p70 S6K, S6 ribosomal protein and eIF4G are phosphorylated following RVFV in vitro and in vivo in a time-dependent fashion. Previous work has demonstrated that RVFV infection results in translational arrest, with host protein synthesis dramatically inhibited at later points following infection, but viral protein production was still readily detectable (Brennan et al., 2014). Another study found that 5′-TOP mRNAs, which tend to code for translational apparatus proteins, were targeted for degradation following RVFV infection (Hopkins et al., 2015). This study found that mTOR signaling was attenuated indirectly via decreased AKT phosphorylation, an upstream activator of mTOR, and through the reduction in phosphorylation of S6 ribosomal protein (Ser240/244) and 4EBP1 (Thr37/46) (Hopkins et al., 2015). These studies have somewhat conflicting results from the studies described within. One potential reason for this difference could be due to the cellular model of infection used. The work of Hopkins et al. was performed in vitro using both mouse embryonic fibroblasts (MEF) and a human osteosarcoma cell line (U2OS). In contrast, our studies were performed using a mouse hepatocyte cell line, which was chosen to analyze events occurring within the major target organ of the acute phase of RVFV infection. Moreover, our findings were validated in vivo. While MEFS and U2OS cells are both permissive and susceptible to infection, they may represent a different virus-host relationship than do hepatocytes in the face of RVFV infection (Yan and Chen, 2012). There is also the possibly that our observed phosphorylation of p70 S6K may be contributing to modulation of other cellular processes apart from direct translational regulation, such as cell size, glucose homeostasis, and transcription (Ruvinsky and Meyuhas, 2006).
Once we found the upregulation of p70 S6K in our cell culture and in vivo models, our goal was to find an FDA-approved drug effective against p70 S6K. Because FDA-approved drugs have undergone extensive safety and pharmacologic testing, repurposing these drugs for other uses is a potentially quicker and more cost effective drug development strategy than developing new antivirals (Benedict et al., 2015; Madrid et al., 2013; NIH, 2016). Rapamycin, a drug commonly used to prevent organ transplant rejection, was identified as a promising candidate because it causes inhibition of p70 S6K, S6 ribosomal protein, and eIF4G (Blommaart et al., 1995; Chaturvedi et al., 2009; FDA, 2017; Polakiewicz et al., 1998; Powers et al., 1999; Raught et al., 2000). Thus we hypothesized that rapamycin treatment would hinder RVFV pathogenesis. The overall trend of decreased p70 S6K signaling with concomitant decrease in RVFV pathogenesis following rapamycin treatment supported this hypothesis. In both the low and high infectious dose challenge studies, mice displayed a 50% increase in survival as well as statistically significant mitigation of weight loss.
Rapamycin is a commonly used drug to prevent organ transplant rejection, and it is effective at this task because it inhibits the adaptive immune response by inhibiting T and B cell proliferation and dendritic cell maturation (Thomson et al., 2009). What effect this immunomodulatory role had in this study is unclear. In many viral hemorrhagic fevers, excessive cytokine release and an overall over-exuberant inflammatory response are a large part of the pathogenesis of disease (Bray and Geisbert, 2005; Geisbert et al., 2003a; Geisbert et al., 2003b; Howerth, 2015; Sordillo and Helson, 2015), and some of the positive effects of rapamycin in this study could be due, in part, to modulation of this excessive release of inflammatory mediators from lymphocytes. Future studies will focus on the inflammatory environment in the context of RVFV infection following rapamycin treatment.
While we evaluated certain aspects of p70 S6K signaling following RVFV infection as well as the response to rapamycin in the in vivo model, conventional wisdom states that there may be additional molecular perturbations which we have not uncovered. There are several up- and downstream proteins which influence p70 S6K which we were not fully able to evaluate based on time and resource constraints. Examples include upstream kinases mTOR, AKT and PDK, and in future studies we hope to further evaluate these proteins during RVFV infection.
Additionally, rapamycin, while being a translation inhibitor through mTOR and subsequently p70 S6K, is also an autophagy activator. Inhibition of the mTOR pathway subsequent to rapamycin treatment has also been shown to increase longevity in mouse models of infection (Harrison et al., 2009), and rapamycin’s pro-autophagic stimulus may play a role (Madeo et al., 2015). The positive effect autophagy may have contributed in overall mouse survival in the course of this study is also unknown and warrants further investigation. Autophagy, apoptosis, and p70 S6K signaling have been shown to be interconnected in virally infected cells (Datan et al., 2014). For RVFV specifically, activation of autophagy was found to be restrictive (Moy et al., 2014). Future work will focus on determining how/if autophagy is modulated in vivo following RVFV infection as well as determining if manipulation of autophagy can be used in an antiviral fashion.
The activation status of one downstream protein we evaluated, eIF4G, an important scaffolding protein for cap-dependent translation, was unaffected following rapamycin treatment in vivo. One hypothesis involves the kinase p90RSK (90-kDa ribosomal protein S6 kinase). p90RSK may be important in activating the eIF4G scaffold protein as it is in cancer cells treated with rapamycin (Chaturvedi et al., 2009). p90RSK is a multi-domain kinase with two non-identical, functional kinase domains and a carboxy-terminal docking site (Fisher and Blenis, 1996) with several important activation sites including Ser380, Thr359, Ser363, and Thr573 (Dalby et al., 1998). Phosphorylation of these sites follows various growth signals, serum stimulation, oncogenic perturbation, or hormonal stimuli through the action of extracellular signaling-regulated kinases (Dalby et al., 1998; Fisher and Blenis, 1996; Romeo et al., 2012). Interestingly, p90RSK selectively and only phosphorylates S6 ribosomal protein at Ser235 and Ser236, and it does so at a much lower level than p70 S6K (Roux et al., 2007). p90RSK was shown to be activated in response to RVFV infection in vitro (Popova et al., 2010), and the role it plays in RVFV infection needs to be explored. Future studies will focus on determining if this kinase is upregulated in vivo and if modulation of its activity can alter the outcome on RVFV production.
5. Conclusion
Antiviral drug therapy greatly lags behind antibacterial therapy in many ways. In order to address this problem, we sought a new strategy to tackle an old problem. We used a novel methodology, RPPA, to identify the molecular pathway perturbations during viral infection in vivo and monitor these pathways in response to treatment. To the best of our knowledge, this is the first time RPPA has been employed to determine the response to antiviral treatment via cell signaling pathway modulation in an in vivo model. Such a novel use of these protein arrays points towards an exciting new way of testing and evaluating antiviral drug efficacy.
Additionally, our strategy of targeting host cell pathways to combat viral infections shows promise based on the significant increase in survival and mitigation of clinical illness shown in RVFV infected mice pre-treated with rapamycin. It is especially noteworthy that efficacy against RVFV was shown using an FDA approved drug, thus increasing the chances of future use of this therapy. This exciting new workflow of antiviral drug development will hopefully lead to new treatment breakthroughs and hope for many patients suffering from acute viral infections. In this instance, we noted modulation of p70 S6K signaling following infection. By using rapamycin as a roadblock in this kinase’s activation, viral replication was impeded with an increase in overall survival. With refinement of this treatment strategy, more effective interventions may be on the horizon.
Supplementary Material
Cells were plated in a white-walled 96-well plate and allowed to incubate at 33°C and 10% CO2 overnight. H2.35 cells were treated with drug concentrations starting at 100 μM with 1:2 serial dilutions down to 1.56 μM, and drugs were incubated with cells for 24 hours. At 24 hours, all cells were analyzed using Cell Titer-Glo Cell Luminescent Viability Assay (Promega, G7570) according to vendor’s instructions. Viability was detected via luminescence detection using the DTX 880 multimode detector (Beckman Coulter) and percent viability was calculated relative to the DMSO control. The mean and SD (N=3) are plotted. The CC50 was found to be > 100 μM.
(A) For each translation signaling endpoint, the company name, catalog number, lot number, and dilution used are listed. (B) Antibodies used for RPPA were validated by western blot analysis. Uninfected, serum starved, H2.35 cells were probed at 0 HR for (I) p70 S6K (Thr389), (II) S6 rp (Ser235/236), (III) eIF4G (Ser1108) to validate each antibody. MagicMark™ XP Western Protein Standard was used to denote molecular weight. Lane III was cut at approximately 40kd, so no bands are visible beyond this point.
Quantification of the p70S 6K (Thr421) immunohistochemistry was performed as an additional determinant of activation of p70 S6 kinase within the liver of infected mice. Positive stained cells within ten high powered fields were counted for both day 4, RVFV infected mouse liver and for day 0, uninfected mouse liver. An increase in p70 S6 kinase staining was noted on day 4 in comparison to mock, uninfected controls from day 0. Error bars represent the means ± SD. *** P ≤ 0.001
BALB/c mice were randomly distributed into groups of 5 with subsequent serial sacrifice of vehicle treated control mice and rapamycin (10 mg/kg) treated mice on days 4 and 6. Liver, spleen and serum were collected. A portion of the liver and spleen were homogenized in DMEM. Serum and homogenized tissues were assayed for infectious titers by plaque assay (panels A, C, and E). A portion of the liver, spleen and serum was placed in Trizol LS for subsequent RNA extraction and quantification via qRT-PCR (panels B, D, and F). Data plotted represents means and standard deviations from 5 animals per condition. Black circles and red squares represent vehicle treated or rapamycin treated mice, respectively.
Highlights.
p70 S6 kinase, S6 ribosomal protein, and eIF4G are phosphorylated following RVFV infection in vitro and in vivo.
Rapamycin treatment reduced viral replication and decreased phosphorylation of these proteins in vitro.
Rapamycin treatment increased survival and reduced clinical disease in RVFV challenged mice.
Phosphorylation of p70 S6 kinase was decreased following rapamycin treatment in vivo.
Acknowledgments
Funding
This work was funded through an NIH research grant 1R15AI100001–01A1 and a Defense Threat Reduction Agency (DTRA) grant HDTRA1-13-1-0006 to KK. DTRA does not have any role in the design of the study and collection, analysis, and interpretation of data and nor in writing the manuscript.
The authors would like to thank Drs. Stuart Nichol and Pierre Rollin, CDC, for providing RVFV ZH501. The authors would also like to thank Dr. Shinji Makino, University of Texas Medical Branch, Galveston, TX, for the RVFV MP12 reverse genetics system and for the RVFV MP12 ΔNSs-Luc virus. Sarah Norris, Chief of Biostatistics at USAMRIID, was instrumental in helping with the statistical analysis of our in vivo studies. The authors would like to give a special thanks to the entire National Center for Biodefense and Infectious Diseases, George Mason University, Manassas, VA for the support to conduct and complete the described experiments, especially those done in BSL-3.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Wahab KS, El Baz LM, El-Tayeb EM, Omar H, Ossman MA, Yasin W. Rift Valley Fever virus infections in Egypt: Pathological and virological findings in man. Trans R Soc Trop Med Hyg. 1978;72:392–396. doi: 10.1016/0035-9203(78)90134-7. [DOI] [PubMed] [Google Scholar]
- Al-Hazmi M, Ayoola EA, Abdurahman M, Banzal S, Ashraf J, El-Bushra A, Hazmi A, Abdullah M, Abbo H, Elamin A, Al-Sammani el T, Gadour M, Menon C, Hamza M, Rahim I, Hafez M, Jambavalikar M, Arishi H, Aqeel A. Epidemic Rift Valley fever in Saudi Arabia: a clinical study of severe illness in humans. Clin Infect Dis. 2003;36:245–252. doi: 10.1086/345671. [DOI] [PubMed] [Google Scholar]
- Alamo JM, Barrera L, Casado MD, Bernal C, Marin LM, Suarez G, Sanchez-Moreno L, Jimenez R, Suarez-Grau JM, Sousa JM, Cordero E, Gomez-Bravo MA. Efficacy, tolerance, and safety of mammalian target of rapamycin inhibitors as rescue immunosuppressants in liver transplantation. Transplant Proc. 2009;41:2181–2183. doi: 10.1016/j.transproceed.2009.06.083. [DOI] [PubMed] [Google Scholar]
- Austin D, Baer A, Lundberg L, Shafagati N, Schoonmaker A, Narayanan A, Popova T, Panthier JJ, Kashanchi F, Bailey C, Kehn-Hall K. p53 Activation following Rift Valley fever virus infection contributes to cell death and viral production. PLoS One. 2012;7:e36327. doi: 10.1371/journal.pone.0036327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer A, Austin D, Narayanan A, Popova T, Kainulainen M, Bailey C, Kashanchi F, Weber F, Kehn-Hall K. Induction of DNA damage signaling upon Rift Valley fever virus infection results in cell cycle arrest and increased viral replication. J Biol Chem. 2012;287:7399–7410. doi: 10.1074/jbc.M111.296608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict A, Bansal N, Senina S, Hooper I, Lundberg L, de la Fuente C, Narayanan A, Gutting B, Kehn-Hall K. Repurposing FDA-approved drugs as therapeutics to treat Rift Valley fever virus infection. Front Microbiol. 2015;6:676. doi: 10.3389/fmicb.2015.00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–2326. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer S, May RM, Shaw GM, Nowak MA. Virus dynamics and drug therapy. Proc Natl Acad Sci U S A. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshra H, Truong T, Babiuk S, Hemida MG. Seroprevalence of Sheep and Goat Pox, Peste Des Petits Ruminants and Rift Valley Fever in Saudi Arabia. PLoS One. 2015;10:e0140328. doi: 10.1371/journal.pone.0140328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloy M, Weber F. Molecular biology of rift valley Fever virus. Open Virol J. 2010;4:8–14. doi: 10.2174/1874357901004010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M, Geisbert TW. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int J Biochem Cell Biol. 2005;37:1560–1566. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Brennan B, Welch SR, Elliott RM. The consequences of reconfiguring the ambisense S genome segment of Rift Valley fever virus on viral replication in mammalian and mosquito cells and for genome packaging. PLoS Pathog. 2014;10:e1003922. doi: 10.1371/journal.ppat.1003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth MK, Morin CW, Comrie AC. An Analysis of the Potential Impact of Climate Change on Dengue Transmission in the Southeastern United States. Environ Health Perspect. 2016 doi: 10.1289/EHP218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease C, Prevention. Update: outbreak of Rift Valley Fever--Saudi Arabia, August-November 2000. MMWR Morb Mortal Wkly Rep. 2000;49:982–985. [PubMed] [Google Scholar]
- Chaturvedi D, Gao X, Cohen MS, Taunton J, Patel TB. Rapamycin induces transactivation of the EGFR and increases cell survival. Oncogene. 2009;28:1187–1196. doi: 10.1038/onc.2008.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chengula AA, Mdegela RH, Kasanga CJ. Socio-economic impact of Rift Valley fever to pastoralists and agro pastoralists in Arusha, Manyara and Morogoro regions in Tanzania. Springerplus. 2013;2:549. doi: 10.1186/2193-1801-2-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier V. Relevance of Rift Valley fever to public health in the European Union. Clin Microbiol Infect. 2013;19:705–708. doi: 10.1111/1469-0691.12163. [DOI] [PubMed] [Google Scholar]
- Chiechi A, Mueller C, Boehm KM, Romano A, Benassi MS, Picci P, Liotta LA, Espina V. Improved data normalization methods for reverse phase protein microarray analysis of complex biological samples. Biotechniques. 2012;0:1–7. doi: 10.2144/000113926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzer JA. The pathology of Rift Valley fever. II. Lesions occurring in field cases in adult cattle, calves and aborted foetuses. Onderstepoort J Vet Res. 1982;49:11–17. [PubMed] [Google Scholar]
- Coetzer JA, Ishak KG, Calvert RC. Sequential development of the liver lesions in new-born lambs infected with Rift Valley fever virus. II. Ultrastructural findings. Onderstepoort J Vet Res. 1982;49:109–122. [PubMed] [Google Scholar]
- Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J Biol Chem. 1998;273:1496–1505. doi: 10.1074/jbc.273.3.1496. [DOI] [PubMed] [Google Scholar]
- Das K, Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr Opin Virol. 2013a;3:111–118. doi: 10.1016/j.coviro.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K, Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr Opin Virol. 2013b;3:119–128. doi: 10.1016/j.coviro.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datan E, Shirazian A, Benjamin S, Matassov D, Tinari A, Malorni W, Lockshin RA, Garcia-Sastre A, Zakeri Z. mTOR/p70S6K signaling distinguishes routine, maintenance-level autophagy from autophagic cell death during influenza A infection. Virology. 2014;452–453:175–190. doi: 10.1016/j.virol.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubney RD, Hudson JR, Garnham PC. Enzootic hepatitis or Rift Valley Fever. Journal of Pathology and Bacteriology. 1931;86:530–532. [Google Scholar]
- De Clercq E. Antivirals: past, present and future. Biochem Pharmacol. 2013;85:727–744. doi: 10.1016/j.bcp.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol. 2013;13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804:433–439. doi: 10.1016/j.bbapap.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res. 1999;253:100–109. doi: 10.1006/excr.1999.4683. [DOI] [PubMed] [Google Scholar]
- Elliott RM. Molecular biology of the Bunyaviridae. J Gen Virol. 1990;71:501–522. doi: 10.1099/0022-1317-71-3-501. [DOI] [PubMed] [Google Scholar]
- Espina V, Liotta LA, Petricoin EF., 3rd Reverse-phase protein microarrays for theranostics and patient tailored therapy. Methods Mol Biol. 2009;520:89–105. doi: 10.1007/978-1-60327-811-9_7. [DOI] [PubMed] [Google Scholar]
- Espina V, Wulfkuhle J, Calvert VS, Liotta LA, Petricoin EF., 3rd Reverse phase protein microarrays for theranostics and patient-tailored therapy. Methods Mol Biol. 2008;441:113–128. doi: 10.1007/978-1-60327-047-2_8. [DOI] [PubMed] [Google Scholar]
- FDA. US Food and Drug Administration. US Food and Drug Administration; 2017. [Google Scholar]
- Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43:47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- Ferrari S, Bandi HR, Hofsteenge J, Bussian BM, Thomas G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J Biol Chem. 1991;266:22770–22775. [PubMed] [Google Scholar]
- Fisher TL, Blenis J. Evidence for two catalytically active kinase domains in pp90rsk. Mol Cell Biol. 1996;16:1212–1219. doi: 10.1128/mcb.16.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint SJRR, VR, Rall GF, Skalka AM. Protein Synthesis, Principles of Virology, Fourth Edition. ASM Press; Washington, DC: 2015a. pp. 348–379. [Google Scholar]
- Flint SJR, RV, Rall GF, Skalka AM. Antiviral Drugs, Principles of Virology. 4. ASM Press; Washington DC: 2015b. pp. 282–314. [Google Scholar]
- Flotow H, Thomas G. Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J Biol Chem. 1992;267:3074–3078. [PubMed] [Google Scholar]
- Gaudreault NN, Indran SV, Bryant PK, Richt JA, Wilson WC. Comparison of Rift Valley fever virus replication in North American livestock and wildlife cell lines. Front Microbiol. 2015;6:664. doi: 10.3389/fmicb.2015.00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Hensley LE, Larsen T, Young HA, Reed DS, Geisbert JB, Scott DP, Kagan E, Jahrling PB, Davis KJ. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol. 2003a;163:2347–2370. doi: 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E, Hensley LE. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. 2003b;163:2371–2382. doi: 10.1016/S0002-9440(10)63592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Golnar AJ, Turell MJ, LaBeaud AD, Kading RC, Hamer GL. Predicting the mosquito species and vertebrate species involved in the theoretical transmission of Rift Valley fever virus in the United States. PLoS Negl Trop Dis. 2014;8:e3163. doi: 10.1371/journal.pntd.0003163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halegoua-De Marzio D, Hann HW. Then and now: the progress in hepatitis B treatment over the past 20 years. World J Gastroenterol. 2014;20:401–413. doi: 10.3748/wjg.v20.i2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeidan YE, Kweka EJ, Mahgoub MM, El Rayah el A, Ouma JO. Recent outbreaks of rift valley Fever in East Africa and the middle East. Front Public Health. 2014;2:169. doi: 10.3389/fpubh.2014.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins KC, Tartell MA, Herrmann C, Hackett BA, Taschuk F, Panda D, Menghani SV, Sabin LR, Cherry S. Virus-induced translational arrest through 4EBP1/2-dependent decay of 5′-TOP mRNAs restricts viral infection. Proc Natl Acad Sci U S A. 2015;112:E2920–2929. doi: 10.1073/pnas.1418805112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerth EW. Cytokine release and endothelial dysfunction: a perfect storm in orbivirus pathogenesis. Vet Ital. 2015;51:275–281. doi: 10.12834/VetIt.593.2854.1. [DOI] [PubMed] [Google Scholar]
- Ikegami T, Narayanan K, Won S, Kamitani W, Peters CJ, Makino S. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog. 2009;5:e1000287. doi: 10.1371/journal.ppat.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S. Rescue of infectious rift valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J Virol. 2006;80:2933–2940. doi: 10.1128/JVI.80.6.2933-2940.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa S. Multiplication of Rift Valley fever virus in human liver cell culture with special reference to production complement fixing antigen. Jpn J Exp Med. 1959;29:323–334. [PubMed] [Google Scholar]
- Jones JM, Lopez B, Adams L, Galvez FJ, Nunez AS, Santillan NA, Plante L, Hemme RR, Casal M, Hunsperger EA, Munoz-Jordan J, Acevedo V, Ernst K, Hayden M, Waterman S, Gomez D, Sharp TM, Komatsu KK Dengue Investigative T, Arizona Department of Health S, Yuma County Public Health Services District A, Division of Vector-Borne Diseases N.C.f.E, Zoonotic Infections Diseases CDCSJPR, University of A, Division of Global M, Quarantine N.C.f.E, Zoonotic Infectious Diseases CDC, Epidemiology Elective Program CDC. Binational Dengue Outbreak Along the United States-Mexico Border - Yuma County, Arizona, and Sonora, Mexico, 2014. MMWR Morb Mortal Wkly Rep. 2016;65:495–499. doi: 10.15585/mmwr.mm6519a3. [DOI] [PubMed] [Google Scholar]
- Juwana YB, Rasoul S, Ottervanger JP, Suryapranata H. Efficacy and safety of rapamycin as compared to paclitaxel-eluting stents: a meta-analysis. J Invasive Cardiol. 2010;22:312–316. [PubMed] [Google Scholar]
- Kahlon SS, Peters CJ, Leduc J, Muchiri EM, Muiruri S, Njenga MK, Breiman RF, White AC, Jr, King CH. Severe Rift Valley fever may present with a characteristic clinical syndrome. Am J Trop Med Hyg. 2010;82:371–375. doi: 10.4269/ajtmh.2010.09-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalveram B, Lihoradova O, Indran SV, Lokugamage N, Head JA, Ikegami T. Rift Valley fever virus NSs inhibits host transcription independently of the degradation of dsRNA-dependent protein kinase PKR. Virology. 2013;435:415–424. doi: 10.1016/j.virol.2012.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig MK, Hebert AA, Roberson J, Samuels J, Slopis J, Woerner A, Northrup H. Topical rapamycin therapy to alleviate the cutaneous manifestations of tuberous sclerosis complex: a double-blind, randomized, controlled trial to evaluate the safety and efficacy of topically applied rapamycin. Drugs R D. 2012;12:121–126. doi: 10.2165/11634580-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani TA, Al-Mazrou YY, Al-Jeffri MH, Mishkhas AA, Al-Rabeah AM, Turkistani AM, Al-Sayed MO, Abodahish AA, Khan AS, Ksiazek TG, Shobokshi O. Rift Valley fever epidemic in Saudi Arabia: epidemiological, clinical, and laboratory characteristics. Clin Infect Dis. 2003;37:1084–1092. doi: 10.1086/378747. [DOI] [PubMed] [Google Scholar]
- Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125:85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid PB, Chopra S, Manger ID, Gilfillan L, Keepers TR, Shurtleff AC, Green CE, Iyer LV, Dilks HH, Davey RA, Kolokoltsov AA, Carrion R, Jr, Patterson JL, Bavari S, Panchal RG, Warren TK, Wells JB, Moos WH, Burke RL, Tanga MJ. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One. 2013;8:e60579. doi: 10.1371/journal.pone.0060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns MP, von Hahn T. Novel therapies for hepatitis C - one pill fits all? Nat Rev Drug Discov. 2013;12:595–610. doi: 10.1038/nrd4050. [DOI] [PubMed] [Google Scholar]
- Millman AJ, Esposito DH, Biggs HM, Decenteceo M, Klevos A, Hunsperger E, Munoz-Jordan J, Kosoy OI, McPherson H, Sullivan C, Voorhees D, Baron D, Watkins J, Gaul L, Sotir MJ, Brunette G, Fischer M, Sharp TM, Jentes ES. Chikungunya and Dengue Virus Infections Among United States Community Service Volunteers Returning from the Dominican Republic, 2014. Am J Trop Med Hyg. 2016;94:1336–1341. doi: 10.4269/ajtmh.15-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mims CA. Rift Valley fever virus in mice. VI. Histological changes in the liver in relation to virus multiplication. Aust J Exp Biol Med Sci. 1957;35:595–604. doi: 10.1038/icb.1957.61. [DOI] [PubMed] [Google Scholar]
- Mohamed M, Mosha F, Mghamba J, Zaki SR, Shieh WJ, Paweska J, Omulo S, Gikundi S, Mmbuji P, Bloland P, Zeidner N, Kalinga R, Breiman RF, Njenga MK. Epidemiologic and clinical aspects of a Rift Valley fever outbreak in humans in Tanzania, 2007. Am J Trop Med Hyg. 2010;83:22–27. doi: 10.4269/ajtmh.2010.09-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano MV, Yagi Y, Fitzgerald KA, Stanger BZ, Soldan SS, Cherry S. Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity. 2014;40:51–65. doi: 10.1016/j.immuni.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KO, Mertens E, Despres P. West Nile virus and its emergence in the United States of America. Vet Res. 2010;41:67. doi: 10.1051/vetres/2010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH; Services, U.S.D.o.H.a.H, editor. National Center for Advancing Translational Sciences. National Institutes of Health; Bethesda, MD: 2016. p. Repurposing Drugs. [Google Scholar]
- Petricoin EF, 3rd, Bichsel VE, Calvert VS, Espina V, Winters M, Young L, Belluco C, Trock BJ, Lippman M, Fishman DA, Sgroi DC, Munson PJ, Esserman LJ, Liotta LA. Mapping molecular networks using proteomics: a vision for patient-tailored combination therapy. J Clin Oncol. 2005;23:3614–3621. doi: 10.1200/JCO.2005.02.509. [DOI] [PubMed] [Google Scholar]
- Peyre M, Chevalier V, Abdo-Salem S, Velthuis A, Antoine-Moussiaux N, Thiry E, Roger F. A Systematic Scoping Study of the Socio-Economic Impact of Rift Valley Fever: Research Gaps and Needs. Zoonoses Public Health. 2015;62:309–325. doi: 10.1111/zph.12153. [DOI] [PubMed] [Google Scholar]
- Polakiewicz RD, Schieferl SM, Gingras AC, Sonenberg N, Comb MJ. mu-Opioid receptor activates signaling pathways implicated in cell survival and translational control. J Biol Chem. 1998;273:23534–23541. doi: 10.1074/jbc.273.36.23534. [DOI] [PubMed] [Google Scholar]
- Popova TG, Turell MJ, Espina V, Kehn-Hall K, Kidd J, Narayanan A, Liotta L, Petricoin EF, 3rd, Kashanchi F, Bailey C, Popov SG. Reverse-phase phosphoproteome analysis of signaling pathways induced by Rift valley fever virus in human small airway epithelial cells. PLoS One. 2010;5:e13805. doi: 10.1371/journal.pone.0013805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JF, Tischler AS, Cherington V. Discordant effects of rapamycin on proliferation and p70S6 kinase phosphorylation in normal and neoplastic rat chromaffin cells. Neurosci Lett. 1999;259:137–140. doi: 10.1016/s0304-3940(98)00936-7. [DOI] [PubMed] [Google Scholar]
- Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- Pullen N, Thomas G. The modular phosphorylation and activation of p70s6k. FEBS Lett. 1997;410:78–82. doi: 10.1016/s0014-5793(97)00323-2. [DOI] [PubMed] [Google Scholar]
- Raught B, Gingras AC, Gygi SP, Imataka H, Morino S, Gradi A, Aebersold R, Sonenberg N. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 2000;19:434–444. doi: 10.1093/emboj/19.3.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed C, Lin K, Wilhelmsen C, Friedrich B, Nalca A, Keeney A, Donnelly G, Shamblin J, Hensley LE, Olinger G, Smith DR. Aerosol exposure to Rift Valley fever virus causes earlier and more severe neuropathology in the murine model, which has important implications for therapeutic development. PLoS Negl Trop Dis. 2013;7:e2156. doi: 10.1371/journal.pntd.0002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo Y, Zhang X, Roux PP. Regulation and function of the RSK family of protein kinases. Biochem J. 2012;441:553–569. doi: 10.1042/BJ20110289. [DOI] [PubMed] [Google Scholar]
- Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Sindato C, Karimuribo E, Mboera LE. The epidemiology and socio-economic impact of rift valley fever epidemics in Tanzania: a review. Tanzan J Health Res. 2011;13:305–318. doi: 10.4314/thrb.v13i5.1. [DOI] [PubMed] [Google Scholar]
- Smith DR, Steele KE, Shamblin J, Honko A, Johnson J, Reed C, Kennedy M, Chapman JL, Hensley LE. The pathogenesis of Rift Valley fever virus in the mouse model. Virology. 2010;407:256–267. doi: 10.1016/j.virol.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Morgan MA, Merrick WC, Shatkin AJ. A polypeptide in eukaryotic initiation factors that crosslinks specifically to the 5′-terminal cap in mRNA. Proc Natl Acad Sci U S A. 1978;75:4843–4847. doi: 10.1073/pnas.75.10.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordillo PP, Helson L. Curcumin suppression of cytokine release and cytokine storm. A potential therapy for patients with Ebola and other severe viral infections. In Vivo. 2015;29:1–4. [PubMed] [Google Scholar]
- Swanepoel R, Blackburn NK. Demonstration of nuclear immunofluorescence in Rift Valley fever infected cells. J Gen Virol. 1977;34:557–561. doi: 10.1099/0022-1317-34-3-557. [DOI] [PubMed] [Google Scholar]
- Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Mathews MB, Mohr I. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol. 2013;5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, Shi PY, Vasilakis N. Zika virus: History, emergence, biology, and prospects for control. Antiviral Res. 2016;130:69–80. doi: 10.1016/j.antiviral.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichhart T, Hengstschlager M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. 2015;15:599–614. doi: 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulfkuhle JD, Speer R, Pierobon M, Laird J, Espina V, Deng J, Mammano E, Yang SX, Swain SM, Nitti D, Esserman LJ, Belluco C, Liotta LA, Petricoin EF., 3rd Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J Proteome Res. 2008;7:1508–1517. doi: 10.1021/pr7008127. [DOI] [PubMed] [Google Scholar]
- Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. 2012;13:214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Yang L, Yang X, Shi X, Wang J, Liu Y, Ju J, Zou L. Efficacy and safety of a mammalian target of rapamycin inhibitor in pediatric patients with tuberous sclerosis complex: A systematic review and meta-analysis. Exp Ther Med. 2015;9:626–630. doi: 10.3892/etm.2014.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells were plated in a white-walled 96-well plate and allowed to incubate at 33°C and 10% CO2 overnight. H2.35 cells were treated with drug concentrations starting at 100 μM with 1:2 serial dilutions down to 1.56 μM, and drugs were incubated with cells for 24 hours. At 24 hours, all cells were analyzed using Cell Titer-Glo Cell Luminescent Viability Assay (Promega, G7570) according to vendor’s instructions. Viability was detected via luminescence detection using the DTX 880 multimode detector (Beckman Coulter) and percent viability was calculated relative to the DMSO control. The mean and SD (N=3) are plotted. The CC50 was found to be > 100 μM.
(A) For each translation signaling endpoint, the company name, catalog number, lot number, and dilution used are listed. (B) Antibodies used for RPPA were validated by western blot analysis. Uninfected, serum starved, H2.35 cells were probed at 0 HR for (I) p70 S6K (Thr389), (II) S6 rp (Ser235/236), (III) eIF4G (Ser1108) to validate each antibody. MagicMark™ XP Western Protein Standard was used to denote molecular weight. Lane III was cut at approximately 40kd, so no bands are visible beyond this point.
Quantification of the p70S 6K (Thr421) immunohistochemistry was performed as an additional determinant of activation of p70 S6 kinase within the liver of infected mice. Positive stained cells within ten high powered fields were counted for both day 4, RVFV infected mouse liver and for day 0, uninfected mouse liver. An increase in p70 S6 kinase staining was noted on day 4 in comparison to mock, uninfected controls from day 0. Error bars represent the means ± SD. *** P ≤ 0.001
BALB/c mice were randomly distributed into groups of 5 with subsequent serial sacrifice of vehicle treated control mice and rapamycin (10 mg/kg) treated mice on days 4 and 6. Liver, spleen and serum were collected. A portion of the liver and spleen were homogenized in DMEM. Serum and homogenized tissues were assayed for infectious titers by plaque assay (panels A, C, and E). A portion of the liver, spleen and serum was placed in Trizol LS for subsequent RNA extraction and quantification via qRT-PCR (panels B, D, and F). Data plotted represents means and standard deviations from 5 animals per condition. Black circles and red squares represent vehicle treated or rapamycin treated mice, respectively.