

Abstract
Dysregulation of skeletal remodeling is a component of renal osteodystrophy. Previously, we showed that activin receptor signaling is differentially affected in various tissues in chronic kidney disease (CKD). We tested whether a ligand trap for the activin receptor type 2A (RAP-011) is an effective treatment of the osteodystrophy of the CKD-mineral bone disorder. With a 70% reduction in the glomerular filtration rate, CKD was induced at 14 weeks of age in the ldlr−/− high fat-fed mouse model of atherosclerotic vascular calcification and diabetes. Twenty mice with CKD, hyperphosphatemia, hyperparathyroidism, and elevated activin A were treated with RAP-011, wherease 19 mice were given vehicle twice weekly from week 22 until the mice were killed at 28 weeks of age. The animals were then evaluated by skeletal histomorphometry, micro-computed tomography, mechanical strength testing, and ex vivo bone cell culture. Results in the CKD groups were compared with those of the 16 sham-operated ldlr−/− high fat-fed mice. Sham-operated mice had low-turnover osteodystrophy and skeletal frailty. CKD stimulated bone remodeling with significant increases in osteoclast and osteoblast numbers and bone resorption. Compared with mice with CKD and sham-operated mice, RAP-011 treatment eliminated the CKD-induced increase in these histomorphometric parameters and increased trabecular bone fraction. RAP-011 significantly increased cortical bone area and thickness. Activin A-enhanced osteoclastogenesis was mediated through p-Smad2 association with c-fos and activation of nuclear factor of activated T cells c1 (NFATc1). Thus, an ActRIIA ligand trap reversed CKD-stimulated bone remodeling, likely through inhibition of activin-A induced osteoclastogenesis.
Keywords: activin A, activin receptor type IIA, chronic kidney disease, renal osteodystrophy, signaling
Renal osteodystrophy is a component of the chronic kidney disease-mineral bone disorder (CKD-MBD) syndrome. It causes a high incidence of skeletal fractures and contributes to the high mortality rates associated with kidney diseases.1 Following the naming of the CKD-MBD syndrome,2 it became apparent that the syndrome had an earlier onset in the course of kidney disease than the hyperparathyroidism and calcitriol deficiency that had previously been linked to renal osteodystrophy.3,4 Characterization of CKD-MBD in early CKD demonstrated onset in stage 2 CKD both clinically and in translational models.4–8 In early CKD, the CKD-MBD was characterized by arterial vascular cell dedifferentiation/calcification, loss of klotho, increased fibroblast growth factor 23 (FGF-23) secretion, and an osteodystrophy.7 Progress in understanding the causes of the CKD-MBD has been made,6,9–11 but the nature of early osteodystrophy and its causes are mostly unknown.
We have shown that CKD directly produces an osteodys-trophy.12 In C57BL/6J mice subjected to renal ablation, with phosphate, calcium, parathyroid hormone (PTH), and calcitriol at normal levels, a low-turnover osteodystrophy, the adynamic bond disorder, was produced.12 We next showed that renal injury produced circulating Wnt inhibitors and inhibited the bone formation rate. Renal injury stimulated several kidney Wnts (portmanteau of Wingless and Integrated) and Wnt inhibitors among the nephrogenic factors reactivated in renal repair.13,14 The Wnt inhibitors are circulating factors, and the family includes the Dickkopfs (Dkk), whose levels are increased in the circulation in CKD.9,14 Neutralization of a key Wnt inhibitor elevated in the circulation in CKD, Dkk1, was efficacious in treating the CKD-MBD,9 by inhibiting CKD-induced vascular dedifferentiation, vascular calcification, and increasing bone formation in the renal osteodys-trophy.9 That kidney-disease produced Wnt inhibitors are a factor in the osteodystrophy of early CKD is in agreement with clinical and translational studies from de Oliveira et al.,10 Sabbagh et al.,11 and Moe et al.15
We have also shown that changes in transforming growth factor-β (TGF-β) superfamily function are involved in the pathogenesis of the CKD-MBD and that they may be upstream of Wnt activation in CKD.12,16 Bone morphogenetic protein 7 (BMP-7) deficiency during development causes renal hypoplasia/dysplasia and skeletal patterning abnormalities,17 and it is lost in kidney disease.18,19 In a diabetic CKD model, BMP-7 administration prevented vascular cell dedifferentiation and calcification20 and stimulated bone formation in low-turnover osteodystrophy.12 When BMP-7 was administered to a model of high-turnover renal osteodystrophy, it further increased bone formation rates and increased bone mass.21 BMP-7, like all TGF-β family ligands, binds to type 2 receptors, which associate and activate type 1 receptors, initiating signal transduction. Besides TGF-β receptor II and bone morphogenetic protein type 2 receptor, the activin receptor type 2A (ActRIIA) and activin receptor type 2B are the most important type 2 receptors of the superfamily, and ActRIIA is a BMP-7 receptor.22 Activin A inhibits BMP-7 signaling by binding ActRIIA.23 We recently reported the effects of a ligand trap for ActRIIA on CKD-stimulated renal fibrosis and aortic calcification.16 We found that CKD decreased aortic vascular smooth muscle cell ActRIIA levels, and ActRIIA signaling was decreased by CKD in our diabetic model.16 The ActRIIA ligand trap increased aortic ActRIIA signaling measured by Smad activation, blocked CKD-stimulated vascular smooth muscle osteoblastic transition, and decreased atherosclerotic vascular calcification. In the kidney, we found that the ligand trap increased renal αklotho expression. Furthermore, in the kidney, ActRIIA levels were not changed by CKD, and the ligand trap decreased renal ActRIIA signaling. The ligand trap also decreased renal Wnt activation and decreased circulating Dkk1. Renal fibrosis and proteinuria were decreased by the ActRIIA ligand trap. Thus, key components of the CKD-MBD, vascular calcification and decreased klotho, were affected by the ligand trap. Here we report the effects of the ligand trap on the skeleton, PTH, and FGF-23 components of the CKD-MBD in the mice from the Agapova et al.16 study.
The model of CKD that we studied involves the most common cause of CKD, type 2 diabetes, which is associated with an osteodystrophy independent of CKD, and is characterized by low bone turnover and skeletal frailty despite normal bone density by dual-energy x-ray absorptiome-try.24,25 This characterization is similar to that of low-turnover renal osteodystrophy and prompts us to question how early CKD affects the osteodystrophy of type 2 diabetes. Our murine model of type 2 diabetes, atherosclerosis, and atherosclerotic calcification in ldlr−/− high fat-fed mice harbors a low-turnover baseline osteodystrophy,7,9 and circulating Wnt inhibitors contribute to the effects of CKD.9 Because the ActRIIA ligand trap decreased circulating Dkk1, we focused on factors of the TGF-β family produced by kidney disease that circulate and may perturb normal physiologic systemic processes. Here we demonstrate in our model that CKD stimulated bone remodeling and osteoclast stimulation is inhibited by the ActRIIA ligand trap.
RESULTS
To analyze the role of ActRIIA in the CKD-MBD, we utilized a ligand trap consisting of the murine extracellular domain of ActRIIA fused to a murine IgG-Fc fragment. The experimental design of the ligand trap experiments in the high fat–fed ldlr−/− mouse with ablative CKD is shown in Supplementary Figure S1.
Baseline osteodystrophy harbored by sham-operated Idlr−/− mice
We first characterized the state of the skeleton in the baseline control sham-operated mice because high-fat feeding26 and the high fat-fed ldlr−/− mouse27,28 have been shown to produce a low-turnover osteodystrophy. The high fat–fed ldlr−/− mouse has insulin resistance that progresses to type 2 diabetes by 28 weeks of age.20,29 The mice (sham, the sham-operated group in these studies) harbor an osteodystrophy characterized histomorphometrically by relatively maintained osteoclast numbers/surfaces compared with wild-type (WT) C57B6J mice (Figure 1). However, osteoblast numbers/surfaces, bone formation rates, and osteoblast efficiency were significantly decreased in sham mice (Figure 2), and osteoid volume and surfaces were significantly decreased in sham mice as was the mineral apposition rate, but only a trend toward decreased mineralization lag time was found (Figure 3). The decrease in osteoblast number in the sham mice is in agreement with our recent studies,9 but it is more pronounced than in our earlier studies.30,31 The basis for this phenotypic change in the sham mice is not known, but was associated with the change to blinded histomorphometry performed in Dr. Malluche’s laboratory. Although trabecular bone architecture was not significantly altered in the sham mice (Figure 4), total area and cortical bone area were decreased in the femoral midshaft (Figure 5). The effects of diabetes and the lipodystrophy on the mechanical properties of long bones assessed by 4-point bending of femora demonstrated a profound decrease in fracture resistance. Structural differences (decreases in elastic, postyield and total displacement and work) were driven by tissue effects. In the tissue, there were significant decreases in strength (yield and ultimate stress) (Supplementary Figure S2) and strain that resulted in decreased resilience and toughness (Supplementary Figure S2) in the femora of sham mice compared with those of WT mice.
Figure 1. Effects of chronic kidney disease (CKD) and RAP-011 on osteoclasts and bone resorption.
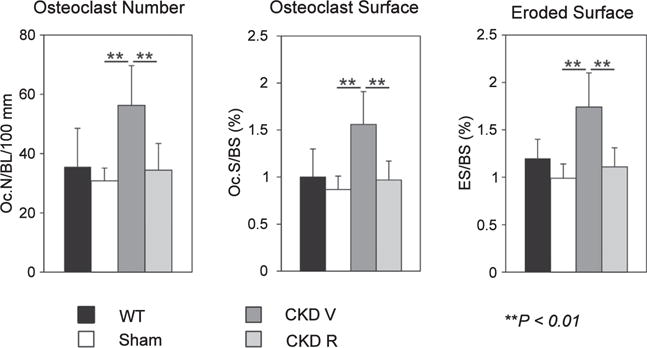
CKD stimulated osteoclast number (Oc.N.) per bone length (BL) and osteoclast surface per bone surface (Oc.S./BS) along with eroded surface per bone surface (ES/BS) in the distal femoral metaphysis. RAP-011 treatment reversed the effects of CKD. **P < 0.01 for comparisons between sham and chronic kidney disease mice treated with vehicle (CKD V) and between CKD V and CKD R. CKD R, CKD+RAP-011; WT, wild type.
Figure 2. Osteoblasts and bone formation in the various groups of mice.

The sham (ldlr−/− high-fat fed) mice had reduced osteoblast number, surfaces, and bone formation rate (BFR) in the distal femoral metaphysis. BFR per osteoblast (BFR/Ob) was decreased in the sham mice. Chronic kidney disease (CKD) stimulated osteoblast number (Ob.N.) per bone length (BL) and osteoblast surface per bone surface (Ob.S./BS). There was a trend of CKD to increase BFR/BS. BFR/Ob was decreased by CKD. RAP-011 treatment reversed the effects of CKD on osteoblast numbers and surface, decreased BFR/BS, and increased BFR/Ob. **P < 0.01 for comparisons between wild type (WT) and sham, between sham and chronic kidney disease mice treated with vehicle (CKD V), and between CKD V and CKD R. CKD R, CKD+RAP-011.
Figure 3. Osteoid and mineralization in the various groups of mice.
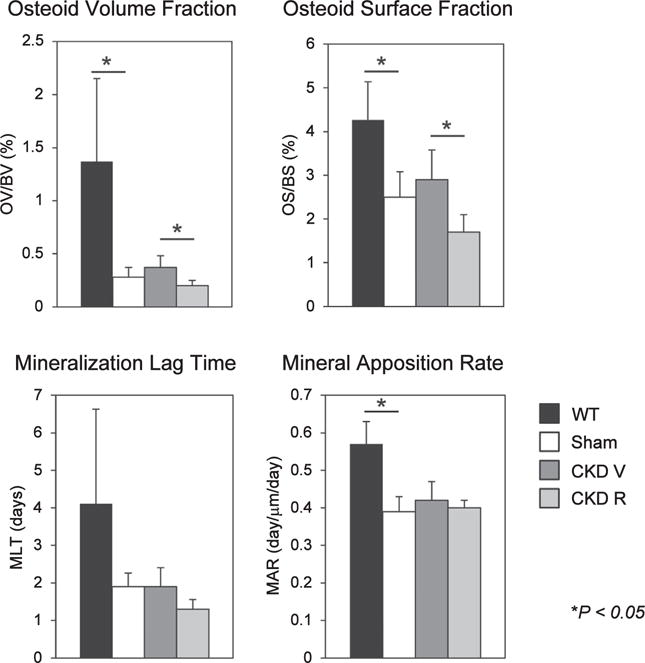
Osteoid volume (OV), osteoid surface (OS), and mineral apposition rate (MAR) were decreased in the sham (Ldlr−/− high-fat fed) mice. Chronic kidney disease (CKD) did not affect these parameters compared with sham mice. RAP-011 treatment decreased OV and OS compared with chronic kidney disease mice treated with vehicle (CKD V). **P < 0.01 for comparisons between wild type (WT) and sham and between CKD V and CKD R. CKD R, CKD+RAP-011.
Figure 4. Micro computed tomography analysis of trabecular bone in the distal femoral metaphysis.
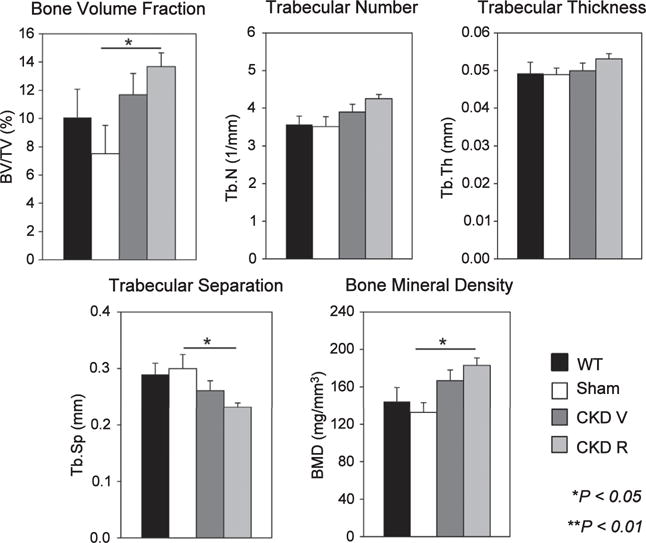
Chronic kidney disease (CKD) increased trabecular bone volume (BV/TV) and bone mineral density (BMD). RAP-011 treatment increased BV/TV and trabecular number (Tb.N) and decreased trabecular separation (Tb.Sp.). CKD R, CKD+RAP-011; CKD V, chronic kidney disease mice treated with vehicle; Tb.Th, trabecular bone thickness; WT, wild type. **P < 0.01 for comparisons between sham and CKD R.
Figure 5. Micro computed tomography analysis of cortical bone.
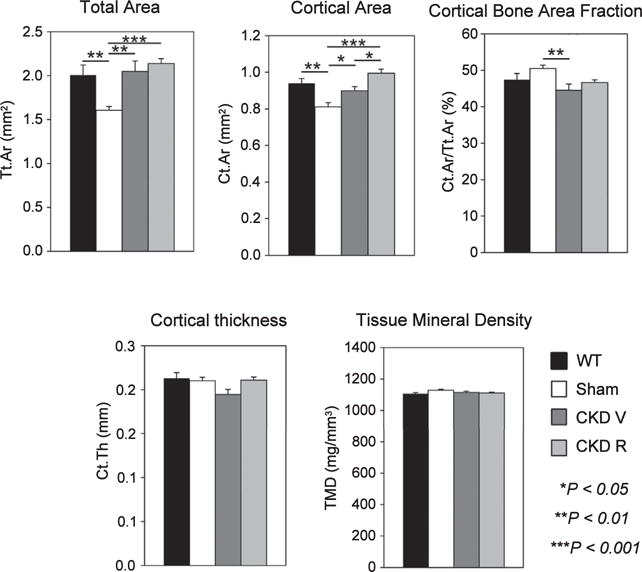
Total and cortical bone area were decreased in the sham (ldlr−/− high-fat fed) mice. Chronic kidney disease increased total cortical area (Tt.Ar), decreased cortical bone area fraction (Ct.Ar/Tt.Ar) and decreased cortical bone thickness (Ct.Th). RAP-011 treatment increased cortical bone area (Ct.Ar) and cortical thickness (Ct.Th). CKD R, CKD+RAP-011; TMD, tissue mineral density; WT, wild type *P < 0.05 for comparisons between sham and CKD V and between CKD V, chronic kidney disease mice treated with vehicle (CKD V) and CKD R; **P < 0.01 for comparisons between WT and sham and between sham and CKD V; ***P < 0.001 for comparisons between sham and CKD R.
Effects of CKD on the osteodystrophy harbored by sham-operated ldlr−/− mice
In addition to the background low-turnover osteodystrophy of the sham high fat-fed ldlr−/− mice, we induced kidney injury and nephrectomy producing repair/hypertrophy of the remnant kidney and CKD. In previous studies, when we induced milder CKD with reduction in glomerular filtration rate (GFR) of only 30% to 40%, the low-turnover osteodystrophy of the sham mice (cited previously) persisted despite CKD, and PTH levels were not elevated.9 In the studies reported here, with the addition of CKD and the reduction in GFR of 70%, the low-turnover state of the high fat-fed ldlr−/− skeleton was overcome by stimulation of bone remodeling (Figures 1–3).
As shown in Figure 1, the CKD mice treated with vehicle (CKD V) had significant increases in osteoclast numbers and surfaces and in eroded surfaces. The CKD mice had a significantly increased osteoblast number and surface compared with sham mice but not to the full correction levels of WT mice (Figure 2). The bone formation rate (BFR) per bone surface, although trending greater than that of the sham, remained depressed compared with WT mice. There was a decrease in osteoblast efficiency (BFR/osteoblast number) (Figure 2). Mineralizing surfaces were increased by CKD (Table 1). Mineralization lag time and mineral apposition rate were not affected by CKD (Figure 3). The histomorphometric parameters of trabecular morphology were confirmed by micro computed tomography (CT) analysis (Figure 4). CKD-induced a trend toward increased bone volume (bone volume/total volume) and increased bone mineral density in the trabeculae. In addition, CKD increased total area and decreased cortical bone area fraction in the femora (Figure 5). The change in bone turnover produced by CKD resulted in increases in deformability and energy dissipation at both the structural and tissue levels, indicative of an increase in resistance to fracture compared with the diabetic sham mice (Figure 6).
Table 1.
Effect of chronic kidney disease and RAP-011 on mineralizing surfaces
| WT | Sham = 16 | CKD V = 19 |
CKD R = 20 |
|
|---|---|---|---|---|
| Mineralizing surface [dLS + (sLS/2)]/BS-% | 6.88 | 11.09a | 7.34 | |
| Mean ± SE | 0.68 | 1.42 | 0.98 |
BS, bone surface; CKD R, CKD+RAP-011; CKD V, CKD mice treated with vehicle; dLS, double-labeled surface; sLS/2, single-labeled surface; WT, wild type.
P < 0.01 compared with sham.
Figure 6. Effects of chronic kidney disease (CKD) and RAP-011 on mechanical properties of femurs.

At the tissue level, CKD resulted in increased toughness (through increased total strain) without a change in strength. There were no significant effects of RAP-011 on mechanical testing parameters compared with CKD. P < 0.026 for comparisons between sham and chronic kidney disease mice treated with vehicle (CKD V).
Effects of the ActRIIA ligand trap on trabecular bone histomorphometry, micro CT, and mechanical strength testing of bones from CKD RAP-011 -treated mice
Treatment with the ActRIIA ligand trap, RAP-011 (CKD R), decreased osteoclast number and osteoclast surfaces and eroded surfaces (Figure 1), as well as osteoblast numbers and surfaces (Figure 2) compared with vehicle-treated CKD mice. Overall BFRs were decreased by RAP-011, but bone formation rate per osteoblast was increased by RAP-011 treatment (Figure 2). RAP-011 decreased osteoid volume and surfaces (Figure 3), but did not affect mineralization lag time. The histomorphometric effects of RAP-011 treatment were confirmed by micro CT, showing that RAP-011 increased bone volume and trabeculae number while decreasing trabecular separation (Figure 4). In the femoral midshaft, RAP-011 increased cortical bone area and cortical thickness compared with femora from CKD V (Figure 5). The trend toward increased deformability and energy dissipation in the fracture-resistance studies produced by RAP-011 treatment were similar to the CKD V mice but did not achieve significance (Figure 6).
Effects of the ActRIIA ligand trap on PTH levels, serum chemistries, and FGF-23 levels
The effects of RAP-011 on osteoclast and osteoblast surfaces and BFRs are surprising, especially because the ActRIIA ligand trap had no effects on plasma PTH levels (Figure 7), which are the supposed means of regulating osteoclast and osteoblast numbers and stimulating high-turnover osteodystrophy in CKD.32 This means that an ActRIIA ligand participated in the stimulation of bone remodeling, probably through the direct effects of osteoclast stimulation. Treatment with RAP-011 had no effect on the serum phosphate or calcium (Table 2), in agreement with the lack of effect on plasma PTH levels (Figure 7). The CKD mice were hyperphosphatemic (Table 2), which occurs somewhat earlier than the onset of hyperphosphatemia in human stage 4 CKD. Plasma FGF-23 levels were stimulated by CKD and were further increased in the RAP-011-treated group (Figure 7).
Figure 7. Effects of chronic kidney disease (CKD) and RAP-011 on parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF-23) levels of the various mouse groups.
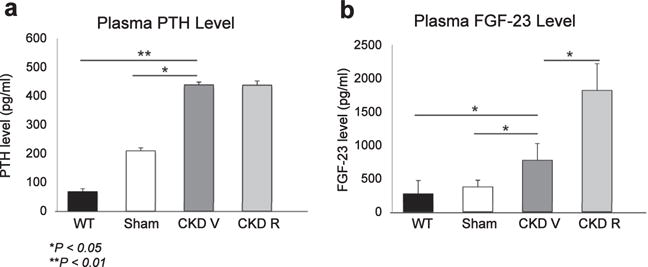
(a) CKD induced significant increases in PTH levels, which were not affected by treatment with RAP-011. (b) CKD induced significant increases in the FGF-23 levels, which were further increased in the RAP-011-treated mice. Data are mean ± SD. *P < 0.05 for comparisons between sham and chronic kidney disease mice treated with vehicle (CKD V), wild-type (WT) mice and CKD V, and CKD V and CKD R; **P < 0.01 for comparison between WT and CKD V. CKD R, CKD+RAP-011.
Table 2.
Serum biochemical parameters in the various groups of animals
| Parameter | Wild type | Sham | CKD V | CKD R |
|---|---|---|---|---|
| Mouse strain | C57/BJ6 | ldlr−/− | ldlr−/− | ldlr−/− |
| Diet | Chow | High fat | High fat | High fat |
| Surgery | None | Sham | CKD | CKD |
| Weeks postnatal | 28 | 28 | 28 | 28 |
| Treatment | None | None | Vehicle | RAP-011 |
| No. | 12 | 15 | 14 | 15 |
| BUN (mg/dl) | 24.0 ± 4.6 | 20.6 ± 3.7 | 37.7 ± 7.6a | 36.5 ± 5.8a |
| Ca (mg/di) | 8.3 ± 1.8 | 8.9 ± 0.9 | 9.4 ± 0.8 | 8.8 ± 0.3 |
| Phosphorus (mg/di) | 8.9 ± 0.2 | 7.9 ± 2.3 | 11.0 ± 1.6a | 11.8 ± 1.2a |
BUN, blood urea nitrogen; Ca, calcium; CKD R, chronic kidney disease receptor; CKD V, chronic kidney disease mice treated with vehicle.
P < 0.05, CKD V and CKD R compared with sham. Inulin clearances in the mice were reported in Agapova et al.16
Effects of activin A on ActRIIA levels and signaling during osteoclastogenesis
The dramatic effects of the ActRIIA ligand trap on bone turnover in the CKD mice naturally raised the question of the state of ActRIIA signaling in skeletal cells of the CKD mice and the nature of the ligand whose effects were affected by the trap. We recently reported that CKD increases circulating levels of activin A while not affecting follistatin or follistatin-like 3 levels.16 We showed that the CKD mice harbor decreased levels of ActRIIA in the aorta and consequent decreased vascular ActRIIA signaling.16 Interestingly, kidney ActRIIA levels were not affected and renal ActRIIA signaling was increased in CKD.16 The ligand that most likely produced these effects was activin A produced by renal interstitial cells. Because bone-remodeling cycles begin with osteoclast stimulation, we studied the effects of activin A on osteoclastogenesis in vitro. Using bone marrow mononuclear cells (BMMCs), we demonstrated that activin A stimulates RANK ligand (RANKL)-induced osteoclastogenesis.33 Treatment of BMMCs with the receptor activator of nuclear factor-κB ligand (RANKL) is a standard technique of osteoclast production in vitro. As shown in Figure 8, activin A stimulation of BMMCs from WT mice enhanced RANKL-stimulated osteoclastogenesis, as shown by the levels of markers of mature or activated osteoclasts, nuclear factor of activated T cells c1 (NFATc1, an osteoclast transcription factor), cathepsin K, and integrin subunit β3 at day 4 of BMMC stimulation with RANKL. d The mechanism of activin A effects on osteoclastogenesis are unknown. In Figure 9, using c-fos and p-Smad 2 antibodies in chromatin immunoprecipitation assays), we show that c-fos forms a complex with the NFATc1 promoter during RANKL stimulation, but during activin A plus RANKL, both stimulated p-Smad2 and c-fos associated with the NFATc1 promoter forming a synergistic complex on NFATc1 (Figure 9), in agreement with studies showing direct association with c-Fos and p-Smad2.34
Figure 8. Effects of activin A on osteoclastogenesis.

Treatment of bone marrow mononuclear cells with RANK ligand (RANKL) for 4 days as described in Methods was used to induce differentiation to osteoclasts. Osteoclast production, which we have previously reported on,35 was marked by induction of the specific transcription factor nuclear factor of activated T cells c1 (NFATc1), and the osteoclast phenotype differentiation markers cathepsin K and integrin subunit β3. Treatment with activin A enhanced RANKL-stimulated osteoclastogenesis and induction of NFATc1, cathepsin K, and integrin β3. Activin A treatment did not affect levels of activin receptor type 2A (ActRIIA), and increased p-Smad2 associated with NFATc1, as shown in Figure 9. D, day; M-CSF, macrophage colony-stimulating factor.
Figure 9. Mechanism of activin A stimulation of osteoclastogenesis.
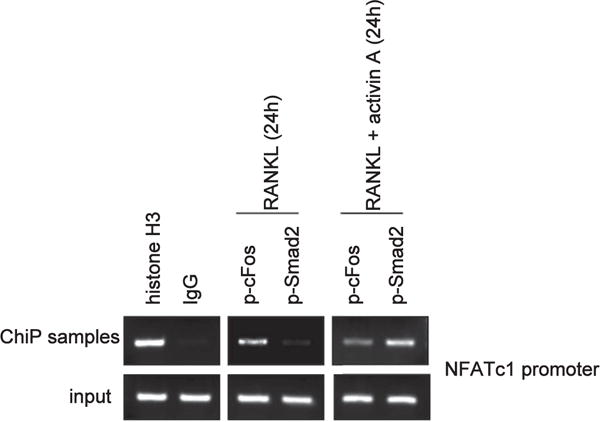
Chromatin immunoprecipitation (ChIP) with antibodies to histone H3 (positive control), p-cFos, and p-Smad2 in bone marrow mononuclear cells revealed the nuclear factor of activated T cells c1 (NFATc1) promoter associated with p-cFos in RANK ligand (RANKL)-treated cells.34 Treatment with RANKL plus activin A revealed both p-cFos and p-Smad2 associated with the NFATcl promoter.
DISCUSSION
Our data demonstrate that CKD, with a GFR reduction of 70% in a diet-induced diabetic mouse model, is associated with a transition in skeletal remodeling from a low-turnover to a higher turnover state with increases in osteoclast and osteoblast number/surface in the presence of elevated PTH. The mechanical effect of this change on remodeling was an increase in compliance without a loss of strength. Treatment with an ActRIIA ligand trap reversed the transition toward higher turnover remodeling and increased trabecular and cortical bone volume without affecting PTH levels. RAP-011 had no effect on bone mechanical properties, but the trend toward an increase in compliance (without loss of strength) suggested a mechanism of action distinct from the mechanism by which CKD induced turnover may have affected bone mechanical properties. Although our data do not prove the identity of the ligand responsible for the effects of the ActRIIA ligand trap, the primary ActRIIA ligand, activin A, is a lead candidate. For example, circulating activin A levels were increased in CKD.16 Here, in in vitro studies, activin A enhanced RANKL-stimulated osteoclastogenesis in bone marrow mononuclear cells, in agreement with previous studies,33,35 through p-Smad2 association with c-Fos on the NFATcl promoter. This finding is in agreement with recent studies of Omata et al.34
Our results are also supported by previous studies36 in which Smad2 activation was shown to stimulate bone remodeling in CKD independent of PTH. Liu et al.36 published results differ from ours in that the pathway to Smad2 activation was through the TGF-β type 2 receptor activated by TGF-β and not through ActRIIA, as in our results. However, despite the difference in the mechanism of Smad2 activation, our results are in line with those of Liu et al.36 regarding the downstream effects of Smad2 inhibition—that of increased cortical and trabecular bone volume due to decreased osteoclast-mediated bone resorption and remodeling. The decrease in osteoclastogenesis and remodeling stimulated by RAP-011 returned osteoblast number to the levels in sham mice and inhibited BFRs. However, bone formation per osteoblast was increased by RAP-011, reversing the CKD-induced inhibition. This may be due to decreased efficiency of the proliferating osteoblasts stimulated by CKD compatible with effects of the TGF-β family, including ActRIIA signaling, to stimulate osteoblast proliferation but restrict osteoblast maturation.37
Studies by Sabbagh et al.11 demonstrated repression of osteocytic Wnt/β-catenin signaling early in CKD and demonstrated an association with increased osteoclast activity independent of PTH. They attributed the decrease in bone formation produced by an antibody to TGF-β to inhibition of TGF-β receptor signaling in osteoblasts.36 By immunohistochemistry, they localized p-Smad2/3 to osteoblasts and bone marrow, but in our studies of BMMC differentiation to osteoclasts, p-Smad2 was clearly present in osteoclasts. It is unclear whether the increase in TGF-β and TGF-βR1 shown by Sabbagh et al. were directly associated with the p-Smad2/3 immunohistochemistry results. Our results have a clear focus on the osteoclast because the stimulation of bone formation produced by RAP-011 in studies of nondiabetic chow-fed mice38 were blocked in our ldlr−/− high fat-fed mice.
The fact that early CKD causes a renal osteodystrophy was also studied in a rodent model by Moe et al.39 The authors used a polycystic kidney disease model in rats and demonstrated that at a 50% reduction in GFR in a slowly evolving kidney disease, there is a stimulation of osteoclastogenesis and bone turnover that produced mechanical weakening of long bones. Our results extend these studies by showing that factors in addition to PTH contribute to the stimulation of osteoclastogenesis and bone turnover in early CKD. The studies by Moe et al., Sabbagh et al., and Liu et al. and our results are in agreement that CKD with GFR reductions >50% causes a stimulation in bone remodeling through activating osteoclastogenesis, an effect mediated through Smad 2 activation, but attributed in the past to PTH. This apparent contradiction may be related to the interaction of the PTH receptor I and TGF-β receptor II.40
Results of studies by Lotinun et al.41 and Pearsall et al.38 demonstrating that RAP-011, or its human equivalent ACE-011, stimulated bone formation in monkeys, murine models, and humans with osteoporosis, differ from our findings. However, it is likely that activin A signaling is affected by environmental differences induced by high fat feeding-induced diabetes producing resistance to ActRIIA inhibition by RAP-011 in stimulating bone formation. Recent studies also suggest that activin A stimulation of osteoclastogenesis in vitro is mediated through stromal cells in the isolated bone marrow mononuclear cell preparation.42 However, our data showing activin A stimulation of c-fos activation of NFATc1 through a cooperative c-fos-p-Smad2 complex is an osteoclast precursor cell-specific function. The actions of activin A on osteoclastogenesis and osteoclast function require additional study.
A limitation of the study is the low-turnover osteodystrophy found in the sham animals due to a decreased osteoblastogenesis response to bone resorption secondary to adipogenesis, similar to hyperlipidemic human type 2 diabetes in humans. The specific relationship of our model to type 2 diabetes produces questions regarding application of the findings to nondiabetic renal osteodystrophy. Another limitation of our studies is the severe skeletal frailty produced in the sham animals by the genotype and high-fat feeding. This may have served to diminish the effects of CKD on skeletal frailty.
In conclusion, with the common clinical background of high fat feeding-induced diabetes and hyperlipidemia, CKD (70% reduction in GFR) stimulated transition of a low-turnover osteodystrophy toward a higher turnover state due to osteoclastogenesis stimulated by p-Smad2, which was prevented by treatment with an ActRIIA ligand trap. The lead candidate ActRIIA ligand, activin A, may have been produced in the kidney as we have previously shown16 and affected bone remodeling from the circulation. These results support modulation of ActRIIA signaling as a new therapeutic target in CKD.
METHODS
The studies reported here used methodology previously reported,9,16 and detailed methods are found in the Supplementary Material. Briefly, the animals used were those previously described, using animal protocols approved by the Washington University Animal Studies Committee.16 Four groups of male mice were used in this study (Supplementary Figure 1). The skeletons of the 22-week-old CKD mice reported by Agapova et al.16 were not analyzed, but readers should consider this study as a treatment, not a prevention, trial. In other words, the effects of CKD were allowed to develop from 14 to 22 weeks before institution of the vehicle or RAP-011 treatment. The mice reported here also differ from the Dkk1 study9 in that their CKD was worse (GFR reductions equivalent to those seen in human CKD stage 3 compared with GFR reductions in Fang et al.9 equivalent to human CKD stage 2), and euthanasia was at 28 weeks instead of 22 weeks as in Fang et al. In Fang et al., CKD did not increase osteoclast and osteoblast numbers, and the low-turnover osteodystrophy of the sham animals persisted after the induction of mild CKD. The ldlr–/– high fat-fed mice with CKD that received subcutaneous injections of RAP-011 (Celgene, Summit, NJ), 10 mg/kg, were treated twice a week beginning at 22 weeks until they were killed at 28 weeks (CKD R). Inulin clearances were performed at 26 weeks if mice were killed at 28 weeks, according to manufacturer instructions (BioPal Inc., Worcester, MA). Serum was analyzed on the day of blood draw for blood urea nitrogen, calcium, and phosphate by standard autoanalyzer laboratory methods performed by our animal facility. Plasma PTH and FGF-23 levels were determined by enzyme-linked immunosorbent assays. EDTA based platelet-poor plasma samples were made by 2-step centrifugation at 6000 rpm for 5 minutes and 14,000 rpm for 2 minutes (at 4 °C). Samples were stored at −20 °C or below until used. Plasma FGF-23 levels were measured using a mouse C-terminal kit (Immunotopics, San Clemente, CA) and intact PTH levels (ALPCO Diagnostic, Salem, NH). Enzyme-linked immunosorbent assay was performed according to the manufacturers’ recommendations. The absorbance was read at 450 nm using a microplate reader (VERSAmax Molecular Devices, Molecular Devices, LLC, Sunnyvale, CA).
Chromatin immunoprecipitation assays
The cells were treated with RANKL (100 ng/ml) with or without activin A (100 ng/ml) for 24 hours. Chromatin immunoprecipitation assay was performed with the simple chromatin immunoprecipitation plus enzymatic chromatin IP kit (Cell Signaling, Danvers, MA) according to the manufacturer’s suggestions using antibodies against histone H3 (1:50 Cell Signaling), normal rabbit IgG (1:100 Cell Signaling), p-c-Fos (1:50 Cell Signaling), or p-Smad2 (1:50 Cell signaling). The purified DNA was analyzed by polymerase chain reaction using primers that detect sequences containing the mouse NFATc1 promoter (forward: 5-CCGGGACGCCCATGCAATCTGTTAGTAATT-3, and reverse: 5-GCGGGTGCCCTGAGAAAGCTACTCTCCCTT-3). All primers were synthesized by Integrated DNA Technologies (Coralville, IA).
Statistics
One-way analysis of variance statistical analysis of the data was performed using SigmaPlot version 12.5 (Systat Software Inc., San Jose, CA), and the differences in the mean values among the treatment groups were considered statistically significant at P < 0.05. The normality of the data was assessed by the Shapiro-Wilk normality test. The Holm-Sidak method was used for all pairwise multiple comparison procedures. All data are expressed as mean ± SE. The differences between groups in the mechanical bending data were assessed by post hoc Tukey tests. Data for all groups represent a number of 7 to 20.
Supplementary Material
Figure S1. Experimental design of RAP-011 effects on the chronic kidney disease-mineral bone disorder in ldlr−/− high fat-fed mice. Mice in 4 groups were fed the high-fat diet beginning at 12 weeks of age. At 12 weeks, either sham operation (SO) or electrocautery cortical injury (EC) was performed. At 14 weeks, either SO or contralateral nephrectomy (NX) was performed. At 22 weeks of life, vehicle treatment or RAP-011, 10 mg/kg s.c. twice weekly, was instituted. WT, wild-type mice on chow diet for normal reference levels. Sham, sham-operated ldlr−/− high fat-fed mice; CKD V, CKD ldlr–/– high fat fed-mice vehicle treated; CKD R, CKD ldlr−/− high fat fed-mice RAP-011 treated; WT, Sham, CKD V, and CKD R mice were killed at 28 weeks of age. CKD R, CKD V, chronic kidney disease mice treated with vehicle. CKD R, CKD+RAP-011.
Figure S2. Effects of low-density lipoprotein receptor (ldlr) deficiency and high-fat feeding on mechanical properties of the femur. At the tissue level, properties were decreased across the board in ldlr−/− high fat-fed mice (sham) compared with wild-type mice fed chow. Decreased properties include strength (yield and ultimate stress), deformability (strain), and resilience/toughness.
Acknowledgments
These studies were supported by NIH grants RO1 DK070790, and RO1 DK089137 (KAH) and RO1 DK080770 (HM), and P30 AR057235. Additional support was in the form of an IST from Celgene. Special thanks to Helen Odle for assistance with the manuscript.
Footnotes
DISCLOSURE
KAH and HM have received research support from Celgene, and KAH and HM have served on the Advisory Board of Celgene. WS and VS are Celgene employees. All the other authors declared no competing interests. Celgene is considering developing the human homolog of RAP-011, sotatercept, for progression of renal disease and the CKD-MBD.
Supplemental Methods
Supplementary material is linked to the online version of the paper at www.kidney-international.org.
References
- 1.Hruska KA, Choi ET, Memon I, et al. Cardiovascular risk in chronic kidney disease (CKD), the CKD-mineral bone disorder (CKD-MBD) Pediatr Nephrol. 2010;25:769–778. doi: 10.1007/s00467-009-1337-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from kidney disease: Improving Global Outcomes (KDIGO) Kidney Int. 2006;69:1945–1953. doi: 10.1038/sj.ki.5000414. [DOI] [PubMed] [Google Scholar]
- 3.Hruska KA, Mathew S. The chronic kidney disease mineral bone disorder (CKD-MBD) In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Seventh. Washington, DC: American Society of Bone and Mineral Research; 2008. [Google Scholar]
- 4.Pereira RC, Juppner H, Azucena-Serrano CE, et al. Patterns of FGF-23, DMP1 and MEPE expression in patients with chronic kidney disease. Bone. 2009;45:1161–1168. doi: 10.1016/j.bone.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang Y, Zhang Y, Mathew S, et al. Early chronic kidney disease (CKD) stimulates vascular calcification (VC) and decreased bone formation rates prior to positive phosphate balance. J Am Soc Nephrol. 2009;20:36A. [Google Scholar]
- 6.Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124–136. doi: 10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang Y, Ginsberg C, Sugatani T, et al. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int. 2014;85:142–150. doi: 10.1038/ki.2013.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wesseling-Perry K, Pereira RC, Tseng C-H, et al. Early skeletal and biochemical alterations in pediatric chronic kidney disease. Clin J Am Soc Nephrol. 2012;7:146–152. doi: 10.2215/CJN.05940611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang Y, Ginsberg C, Seifert M, et al. CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J Am Soc Nephrol. 2014;25:1760–1763. doi: 10.1681/ASN.2013080818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Oliveira RB, Graciolli FG, dos Reis LM, et al. Disturbances of Wnt/β-catenin pathway and energy metabolism in early CKD: effect of phosphate binders. Nephrol Dial Transplant. 2013;28:2510–2517. doi: 10.1093/ndt/gft234. [DOI] [PubMed] [Google Scholar]
- 11.Sabbagh Y, Graciolli FG, O’Brien S, et al. Repression of osteocyte Wnt/β-catenin signaling is an early event in the progression of renal osteodystrophy. J Bone Miner Res. 2012;27:1757–1772. doi: 10.1002/jbmr.1630. [DOI] [PubMed] [Google Scholar]
- 12.Lund RJ, Davies MR, Brown AJ, Hruska KA. Successful treatment of an adynamic bone disorder with bone morphogenetic protein-7 in a renal ablation model. J Am Soc Nephrol. 2004;15:359–369. doi: 10.1097/01.asn.0000109671.99498.08. [DOI] [PubMed] [Google Scholar]
- 13.Surendran K, McCaul SP, Simon TC. A role for Wnt-4 in renal fibrosis. Am J Physiol Renal Physiol. 2002;282:F431–F441. doi: 10.1152/ajprenal.0009.2001. [DOI] [PubMed] [Google Scholar]
- 14.Surendran K, Schiavi S, Hruska KA. Wnt-dependent-β-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. J Am Soc Nephrol. 2005;16:2373–2384. doi: 10.1681/ASN.2004110949. [DOI] [PubMed] [Google Scholar]
- 15.Moe SM, Chen NX, Newman CL, et al. Anti-sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J Bone Miner Res. 2015;30:499–509. doi: 10.1002/jbmr.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agapova OA, Fang Y, Sugatani T, et al. Ligand trap for the activin type IIA receptor protects against vascular disease and renal fibrosis in mice with chronic kidney disease. Kidney Int. 2016;89:1231–1243. doi: 10.1016/j.kint.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo G, Hofmann C, Bronckers AL, et al. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 1995;9:2808–2820. doi: 10.1101/gad.9.22.2808. [DOI] [PubMed] [Google Scholar]
- 18.Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol. 2001;12:2392–2399. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Chen Q, Simon TC, et al. Bone morphogenetic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003;63:2037–2049. doi: 10.1046/j.1523-1755.2003.00035.x. [DOI] [PubMed] [Google Scholar]
- 20.Davies MR, Lund RJ, Hruska KA. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J Am Soc Nephrol. 2003;14:1559–1567. doi: 10.1097/01.asn.0000068404.57780.dd. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez EA, Lund RJ, Martin KJ, et al. Treatment of a murine model of high-turnover renal osteodystrophy by exogenous BMP-7. Kidney Int. 2002;61:1322–1331. doi: 10.1046/j.1523-1755.2002.00258.x. [DOI] [PubMed] [Google Scholar]
- 22.Perron JC, Dodd J. ActRIIA and BMPRII type II BMP receptor subunits selectively required for Smad4-independent BMP7-evoked chemotaxis. PLoS One. 2009;4:e8198. doi: 10.1371/journal.pone.0008198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsen OE, Wader KF, Hella H, et al. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun Signal. 2015;13:27. doi: 10.1186/s12964-015-0104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farr JN, Khosla S. Determinants of bone strength and quality in diabetes mellitus in humans. Bone. 2016;82:28–34. doi: 10.1016/j.bone.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shanbhogue VV, Mitchell DM, Rosen CJ, Bouxsein ML. Type 2 diabetes and the skeleton: new insights into sweet bones. Lancet Diabetes Endocrinol. 2016;4:159–173. doi: 10.1016/S2213-8587(15)00283-1. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Almeida M, Weinstein RS, et al. High fat diet-induced skeletal inflammation inhibits Wnt signaling, Wnt ligand expression and bone formation in ApoE null mice. Am J Physiol Endocrinol Metab. 2016;310:E762–E773. doi: 10.1152/ajpendo.00501.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sage AP, Lu J, Atti E, et al. Hyperlipidemia induces resistance to PTH bone anabolism in mice via oxidized lipids. J Bone Miner Res. 2011;26:1197–1206. doi: 10.1002/jbmr.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okayasu M, Nakayachi M, Hayashida C, et al. Low-density lipoprotein receptor deficiency causes impaired osteoclastogenesis and increased bone mass in mice because of defect in osteoclastic cell-cell fusion. J Biol Chem. 2012;287:19229–19241. doi: 10.1074/jbc.M111.323600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Towler DA, Bidder M, Latifi T, et al. Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J Biol Chem. 1998;273:30427–30434. doi: 10.1074/jbc.273.46.30427. [DOI] [PubMed] [Google Scholar]
- 30.Davies MR, Lund RJ, Mathew S, Hruska KA. Low turnover osteodystrophy and vascular calcification are amenable to skeletal anabolism in an animal model of chronic kidney disease and the metabolic syndrome. J Am Soc Nephrol. 2005;16:917–928. doi: 10.1681/ASN.2004100835. [DOI] [PubMed] [Google Scholar]
- 31.Mathew S, Lund R, Strebeck F, et al. Reversal of the adynamic bone disorder and decreased vascular calcification in chronic kidney disease by sevelamer carbonate therapy. J Am Soc Nephrol. 2007;18:122–130. doi: 10.1681/ASN.2006050490. [DOI] [PubMed] [Google Scholar]
- 32.KDIGO Clinical practice guideline for the diagnosis, evaluation prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD) Kidney Int Suppl. 2009;76(suppl 113):S1–S130. doi: 10.1038/ki.2009.188. [DOI] [PubMed] [Google Scholar]
- 33.Sugatani T, Alvarez UM, Hruska KA. Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J Cell Biochem. 2003;90:59–67. doi: 10.1002/jcb.10613. [DOI] [PubMed] [Google Scholar]
- 34.Omata Y, Yasui T, Hirose J, et al. Genomewide comprehensive analysis reveals critical cooperation between Smad and c-Fos in RANKL-induced osteoclastogenesis. J Bone Miner Res. 2015;30:869–877. doi: 10.1002/jbmr.2418. [DOI] [PubMed] [Google Scholar]
- 35.Gaddy-Kurten D, Coker JK, Abe E, et al. Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology. 2002;143:74–83. doi: 10.1210/endo.143.1.8580. [DOI] [PubMed] [Google Scholar]
- 36.Liu S, Song W, Boulanger JH, et al. Role of TGF-β in a mouse model of high turnover renal osteodystrophy. J Bone Miner Res. 2014;29:1141–1157. doi: 10.1002/jbmr.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alliston T, Choy L, Ducy P, et al. TGF-β-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. 2001;20:2254–2272. doi: 10.1093/emboj/20.9.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearsall RS, Canalis E, Cornwall-Brady M, et al. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc Natl Acad Sci USA. 2008;105:7082–7087. doi: 10.1073/pnas.0711263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moe SM, Radcliffe JS, White KE, et al. The pathophysiology of early-stage chronic kidney disease-mineral bone disorder (CKD-MBD) and response to phosphate binders in the rat. J Bone Miner Res. 2011;26:2672–2681. doi: 10.1002/jbmr.485. [DOI] [PubMed] [Google Scholar]
- 40.Qiu T, Wu X, Zhang F, et al. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat Cell Biol. 2010;12:224–234. doi: 10.1038/ncb2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lotinun S, Pearsall RS, Davies MV, et al. A soluble activin receptor type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone. 2010;46:1082–1088. doi: 10.1016/j.bone.2010.01.370. [DOI] [PubMed] [Google Scholar]
- 42.Fowler TW, Kamalakar A, Akel NS, et al. Activin A inhibits RANKL-mediated osteoclast formation, movement and function in murine bone marrow macrophage cultures. J Cell Sci. 2015;128:683–694. doi: 10.1242/jcs.157834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Experimental design of RAP-011 effects on the chronic kidney disease-mineral bone disorder in ldlr−/− high fat-fed mice. Mice in 4 groups were fed the high-fat diet beginning at 12 weeks of age. At 12 weeks, either sham operation (SO) or electrocautery cortical injury (EC) was performed. At 14 weeks, either SO or contralateral nephrectomy (NX) was performed. At 22 weeks of life, vehicle treatment or RAP-011, 10 mg/kg s.c. twice weekly, was instituted. WT, wild-type mice on chow diet for normal reference levels. Sham, sham-operated ldlr−/− high fat-fed mice; CKD V, CKD ldlr–/– high fat fed-mice vehicle treated; CKD R, CKD ldlr−/− high fat fed-mice RAP-011 treated; WT, Sham, CKD V, and CKD R mice were killed at 28 weeks of age. CKD R, CKD V, chronic kidney disease mice treated with vehicle. CKD R, CKD+RAP-011.
Figure S2. Effects of low-density lipoprotein receptor (ldlr) deficiency and high-fat feeding on mechanical properties of the femur. At the tissue level, properties were decreased across the board in ldlr−/− high fat-fed mice (sham) compared with wild-type mice fed chow. Decreased properties include strength (yield and ultimate stress), deformability (strain), and resilience/toughness.